
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of allyl alcohol as a method to valorise glycerol from the biodiesel production†
Michael Wormann and
Martin E. Maier *
*
Eberhard Karls Universität Tübingen, Institut für Organische Chemie, Auf der Morgenstelle 18, 72976 Tübingen, Germany. E-mail: martin.e.maier@uni-tuebingen.de
First published on 16th May 2019
Abstract
Reaction of triglycerides with trimethyl orthoformate in presence of camphorsulfonic acid (CSA) gave the fatty acid methyl esters (FAMEs, 4) in good yield. However, under these conditions, the protected glycerol could not be obtained. Formation of orthoesters 9 was possible in a separate reaction using very weak acidic conditions, namely catalytic amounts of pyridinium para-toluenesulfonate (PPTS). Subjecting the orthoesters 9 to thermolysis at 270 °C gave allyl alcohol (11) with good efficiency.
Introduction
Triglycerides, like vegetable oils or waste oils constitute an important source of natural and renewable organic matter. For example, the conversion of vegetable oils to biodiesel by transesterification with small alcohols like methanol or ethanol generates a clean-burning diesel additive. In 2009 around 104 thousand tons of oil equivalent were consumed.1 The problem is that the classical transesterification of vegetable oils leads to glycerol as by-product. While this triol can be used in polymers, in larger amounts it almost can be considered as waste. Therefore, conversion of glycerol to valuable small organic compounds by dehydration or deoxygenation reactions would enhance the effectiveness of the biodiesel process.2 Acrolein, epichlorohydrin, 1,2-propanediol or glycerol carbonate are chemicals obtained from glycerol.3 Quite recently it has been shown that deoxydehydration of glycerol and sugar alcohols can be achieved with rhenium based catalyst in presence of an reducing agent.4 Thus, glycerol yields allyl alcohol in these reactions. It is also possible to convert glycerol into allyl alcohol using formic acid at 230–240 °C.5 It was suggested that this reaction proceeds via an orthoester-type intermediate.An ideal scenario would be where the biodiesel production yields biodiesel as well as a valuable glycerol derivative in a single step. In fact, two processes of this type are known, where triglycerides are reacted with a methanol-containing compound to give biodiesel (fatty acid methyl ester, FAME) plus a valuable glycerol derivative (Scheme 1).6 In the so-called gliperol process, triglycerides are reacted with methyl acetate under acid catalysis to give triacetin and biodiesel in one step.7 A research group from Italy achieved the one-pot reaction of triglycerides with dimethyl carbonate to a mixture of FAME and glycerol carbonate under basic conditions.8
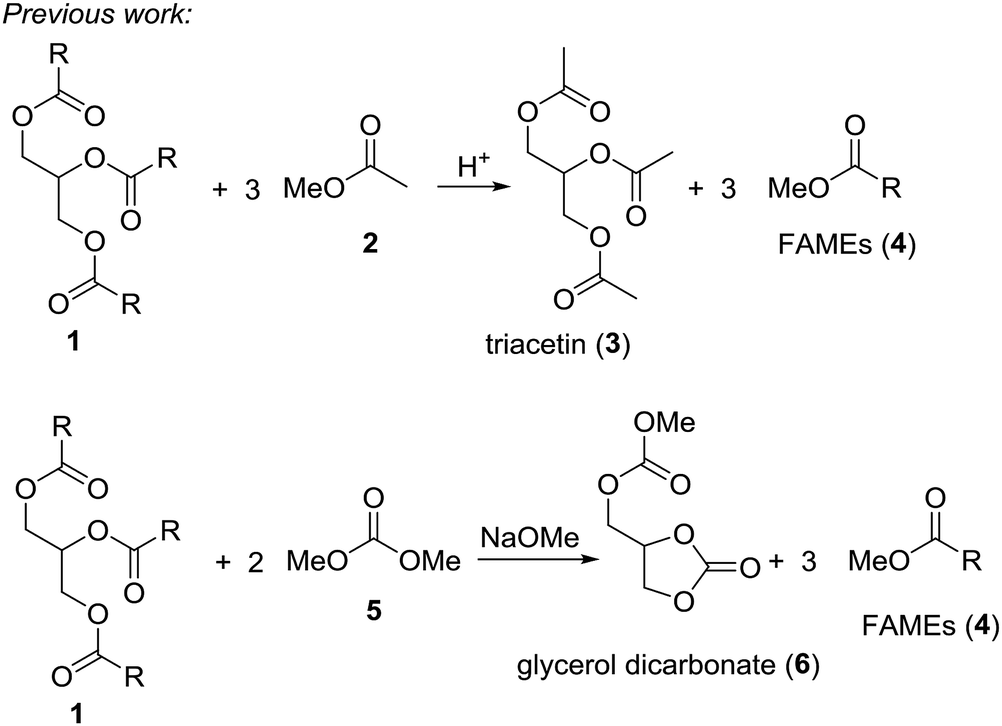 | ||
| Scheme 1 Known integrated processes for conversion of triglycerides (1) to FAMEs (4) and valuable glycerol derivatives 3 and 6 using methanol containing compounds methyl acetate (2) and dimethyl carbonate (5), respectively. | ||
Based on these examples we reasoned whether it would be possible to use other easily available methanol containing compounds that would convert triglycerides to biodiesel and an interesting glycerol derivative that would allow for easy separation of the mixture and further transformations of the glycerol derivative. In this context, we considered trimethyl orthoformate (Scheme 2). This compound is widely used in organic synthesis, mainly as C1 building block for heterocycle synthesis. With diols, the orthoformates react to cyclic orthoesters that can be fragmentated to an alkene, methanol or ethanol and carbon dioxide.9,10 Applied to glycerol this should open a route to allyl alcohol.
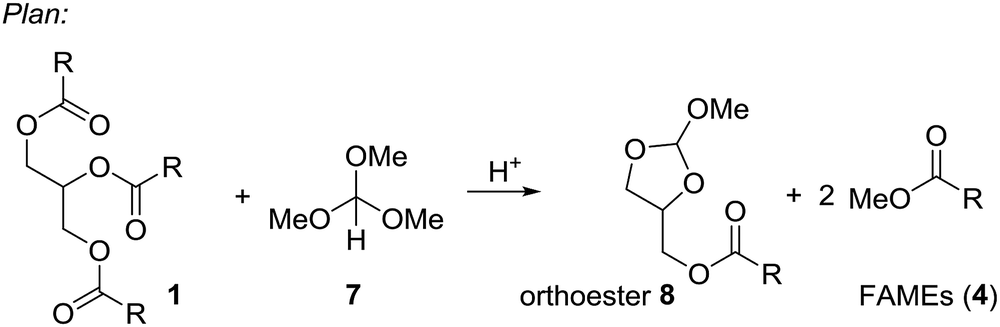 | ||
| Scheme 2 Plan for an integrated process to convert triglycerides (1) to FAMEs (4) and cyclic orthoester 8. | ||
Result and discussion
Initially we tried to react vegetable oil with an excess of trimethyl orthoformate (7) in presence of an organic acid to biodiesel (FAME) and the mixed orthoester 8 or 9 (Scheme 3).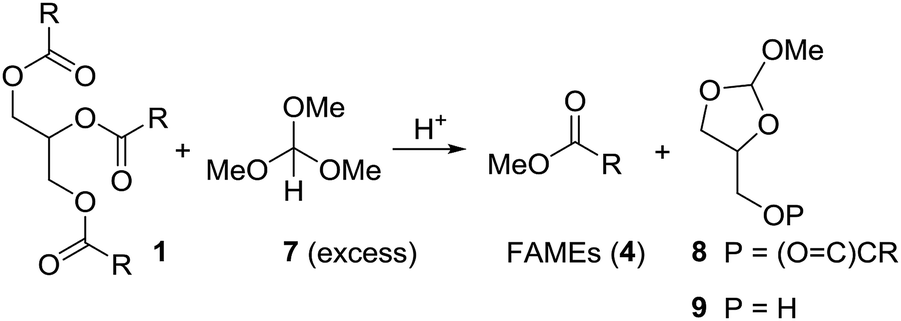 | ||
| Scheme 3 Attempts to convert triglycerides (1) to FAMEs (4) and cyclic orthoester 8 or 9 under acidic conditions using trimethyl orthoformate (7). | ||
Using the relatively weak acid pyridinium para-toluenesulfonate (PPTS) in catalytic amounts and keeping the reaction mixture at 100 °C for 10 h, neither gave 4 nor 8 or 9 (Table 1, entry 1). With para-toluenesulfonic acid (pTsOH·H2O, 1 equiv.) at 100 °C the mixture of 1 and 7 turned brown, but essentially full conversion to FAMEs (4) was observed (entry 2). Replacing pTsOH with methanesulfonic acid (MSA, 2 equiv.) resulted in a black mixture with many side products after 6 h at 100 °C (entry 3). Thus, this acid seems to be too strong. With trifluoroacetic acid (2 equiv.) the reaction does not go to completion (entry 4). Full conversion to FAMEs was observed with camphorsulfonic acid (CSA) (1.2 equiv.) at 100 °C (entry 5). However, in this case the mixture turned brown. Optimal conditions were found with camphorsulfonic acid (1.2 equiv.) and stirring of the mixture vegetable oil (1 equiv.), trimethyl orthoformate (5 equiv.) at 70 °C for 1.5 h (entry 6). Distillation of the mixture gave a first fraction (30–40 °C, 600 mbar) that consists of excess methyl formate and methanol. Upon cooling of the mixture, CSA crystallized and could be partly recovered by filtration. The FAMEs were obtained by vacuum distillation (100–120 °C, 1 × 10−2 mbar) as a colorless liquid. Glycerol remained in the distillation swamp. While this method of forming FAME might not be efficient from a commercial point of view, it does allow for partial recovery of methyl formate and methanol.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T/°C | t/h | Result |
| 1 | PPTS (cat.) | 100 | 10 | Green mixture, no products |
| 2 | pTsOH (1 equiv.) | 100 | 30 | Brown mixture, full conversion to 4 |
| 3 | MeSO3H (2 equiv.) | 100 | 6 | Black mixture, many side products |
| 4 | CF3CO2H (2 equiv.) | 100 | 6 | Equilibrium |
| 5 | CSA (1.2 equiv.) | 100 | 1.5 | Brown mixture, full conversion to 4 |
| 6 | CSA (1.2 equiv.) | 70 | 1.5 | Full conversion to 4 (99%) |
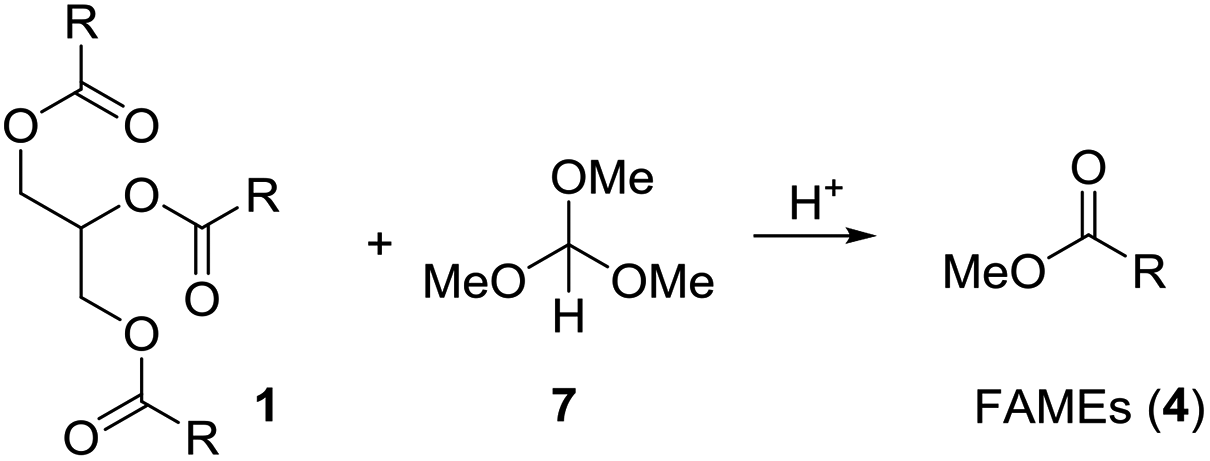
Table 2 shows the composition of the methyl esters, determined by gas chromatography. Thus, the olive oil that was used, mainly contains oleic acid (70%) and palmitic acid (21%). Linoleic acid and stearic acid are present in minor amounts.
| Entry | Fatty acid | Retention time/min | Percentage/% |
|---|---|---|---|
| 1 | Palmitic acid | 14.84 | 21 |
| 2 | Linoleic acid | 16.33 | 5 |
| 3 | Oleic acid | 16.45 | 70 |
| 4 | Stearic acid | 16.64 | 4 |
As we were not able to isolate orthoester derivatives from the reaction of the vegetable oil with trimethyl orthoformate, glycerol (10) was reacted separately with 7. Different conditions were screened (Table 3). A thermal reaction between glycerol and trimethyl orthoformate 7 (1.4 equiv.), both freshly distilled, left both molecules unchanged. Reaction of glycerol (10) with the orthoformate 7 (1.4 equiv.) in presence of catalytic amounts of acetic acid at 100 °C gave an unidentifiable mixture of products (entry 2). In presence of catalytic amounts of PPTS, glycerol (10) reacted with 7 to a mixture of the orthoesters 9a (trans) and 9b (cis) at 100 °C within 1 h. However, full conversion was not reached if only 1 equiv. of 7 were employed. Using a slight excess (1.4 equiv.) of 7 full conversion to the orthoesters 9 was observed within 1 h at 100 °C. The same result was obtained when the reaction mixture was kept for 2 h at room temperature (entry 5). The NMR spectra are included in the ESI.† The corresponding 6-membered orthoester was not observed.11 This is evident from the 13C NMR spectrum of 9a and 9b, which for each isomer of 9 shows 5 signals. Due to symmetry, the six-membered orthoester would have only 4 signals. The ratio of 9a (trans)/9b (cis) is about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The structures of the 2-methoxy-1,3-dioxolan-4-yl)methanol isomers 9a and 9b were assigned based on the chemical shift of the orthoformate proton (2-H). According to Hall et al.,11 2-H of the trans isomer resonates at lower field than the corresponding cis isomer. In DMSO-d6 solvent 2-H of 9a (trans) resonates at δ = 5.74 ppm, whereas 2-H of 9b (cis) resonates at δ = 5.73 ppm.
:1. The structures of the 2-methoxy-1,3-dioxolan-4-yl)methanol isomers 9a and 9b were assigned based on the chemical shift of the orthoformate proton (2-H). According to Hall et al.,11 2-H of the trans isomer resonates at lower field than the corresponding cis isomer. In DMSO-d6 solvent 2-H of 9a (trans) resonates at δ = 5.74 ppm, whereas 2-H of 9b (cis) resonates at δ = 5.73 ppm.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T/°C | t/h | Result |
| a A slight excess of orthoester 7 (1.4 equiv.) was used except for entry 3.b 1 equiv. of 7 was used. | ||||
| 1 | — | 100 | 24 | No reaction |
| 2 | AcOH (cat.) | 100 | 3 | Many side products |
| 3b | PPTS (cat.) | 100 | 1 | Product 9, but no full conversion |
| 4 | PPTS (cat.) | 100 | 1 | 9 (90%) |
| 5 | PPTS (cat.) | r.t. | 2 | 9 (yield > 90%) |
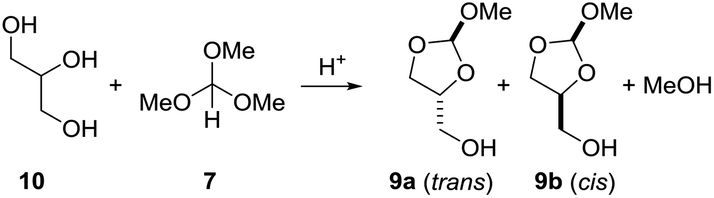
After having developed this simple procedure for the synthesis of orthoesters 9, we turned to their thermal fragmentation reaction. Heating of 9 to 170 °C in presence of catalytic amounts of CSA for 5 h gave only traces of allyl alcohol 11 (Scheme 4). It seemed that higher temperatures were necessary. Thus, keeping the mixture of 9 and CSA (cat.) for 2 h at 210 °C and then for further 2 h at 270 °C gave allyl alcohol 11 in 41% yield. The best results were obtained with catalytic amounts of PPTS and keeping the reaction mixture at 270 °C for 1.5 h. This way we were able to isolate allyl alcohol (11) in 76% yield. In praxis, it is best to use the crude mixture of orthoesters 9 directly for the pyrolysis. Most likely, the orthoester 9 disintegrates to a carbene and methanol, followed by decarboxylation. The methanol that is split off can be partly recovered as well.
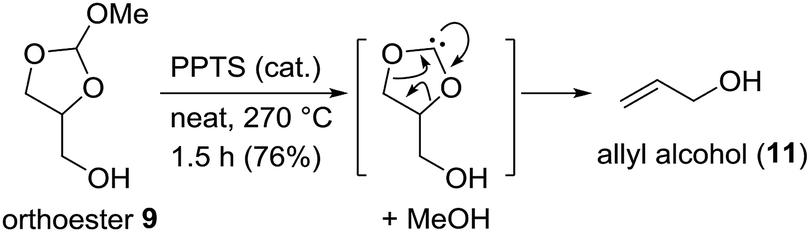 | ||
| Scheme 4 Pyrolysis of orthoesters 9 at elevated temperature to give allyl alcohol (11). | ||
Conclusions
We tried to implement an integrated process to convert triglycerides to FAMEs (4) and the orthoester 9. As we could show, the latter can be converted to allyl alcohol (11) by thermolysis at elevated temperature in presence of catalytic amounts of the weak acid catalyst PPTS. In contrast, the transesterification of triglycerides with trimethyl orthoformate (7) requires stronger acidic conditions. Thus, this reaction proceeds quite efficiently in the presence of stoichiometric amounts of CSA at 100 °C. However, under these conditions, it was not possible to isolate orthoester 9. In any case, we found a new way of generating FAMEs from triglycerides, and to convert glycerol to the valuable organic building block allyl alcohol (11). One should note that most of the methanol from the transesterification reaction and the orthoester formation using trimethyl orthoformate (7) can be recovered.Experimental
Materials, general methods and instrumentation
Glycerol and trimethyl orthoformate were purchased from Sigma-Aldrich (Merck). Olive oil was purchased in a local super market. All commercially available compounds (Acros and Merck) were used without purification. Solvents used in the reactions were purified before use. The progress of the reactions was followed by using TLC (POLYGRAM SIL G/UV254; petroleum ether/EtOAc). Flash chromatography was performed on silica gel Silica M, 0.04–0.063 mm, from Machery-Nagel GmbH & Co. KG, Germany. 1H (400.160 MHZ) and 13C (100.620 MHz) spectra were recorded on a Bruker Avance 400 III HD spectrometer in CDCl3 as solvent at room temperature. HRMS (ESI-TOF) analysis was performed on Bruker maXis 4G system. Gas chromatography was performed with a Hewlett Packard GC-MS system (HP 6890 Series, MSD 5973 Series); column: DB-5 (95% dimethylpolysiloxane, 5% phenylpolysiloxane, J&W Scientific (now Agilent), 13 m × 0.25 mm, film thickness dF = 0.1 μm, temperature gradient: 60 °C (3 min isotherm), 10 °C min−1 to 320 °C, 320 °C (10 min isotherm); carrier gas: He (1.2 mL min−1), split injection at 28 °C.Synthesis of fatty acid methyl esters (FAMEs)
A mixture of olive oil (5.00 mL, 6.93 mmol, 1 equiv.), trimethyl orthoformate (7) (2.82 mL, 25.9 mmol, 3.7 equiv.) and camphorsulfonic acid (1.93 g, 8.32 mmol, 1.2 equiv.) was stirred at 100 °C for 1.5 h under a nitrogen atmosphere. At this point all the olive oil had been transesterified to FAMEs. This crude product was purified by distillation in vacuo. A first fraction, consisting of methyl formate and methanol was collected between 30 and 40 °C at a pressure of 600 mbar. The distillation was then stopped and the mixture allowed to cool to room temperature. This led to precipitation of camphorsulfonic acid, which was collected by filtration (0.50 g) of the mixture through a glass frit. FAMEs (2.00 g, 99%) were obtained as a colorless liquid by distillation of the filtrate (100–120 °C, 1 × 10−2 mbar). The composition of the FAMEs was determined by gas chromatography (see Table 2).Synthesis of orthoesters 9
A mixture of glycerol (25.0 g, 0.27 mol, 1 equiv., commercial product), trimethyl orthoformate (41.5 mL, 0.38 mol, 1.4 equiv.) and a catalytic amount of pyridinium p-toluenesulfonate (PPTS, 0.20 g) was stirred at room temperature for 2 h. The produced MeOH was removed subsequently in vacuo. The resulting slurry of the crude orthoesters (36.5 g) could be used without any purification in the next step.For analytical purposes, K2CO3 (0.30 g) was added to the mixture before distilling it through a Vigreux-column (15 cm) in vacuo. After removal of MeOH (40 °C, 250 mbar), 28.2 g (78%) of a mixture of the two orthoesters 9a (trans) and 9b (cis) (74 °C, 1 × 10−3 mbar) was obtained as a colorless liquid. Distillation at temperatures higher than 150 °C should be avoided as this can cause polymerization of the mixture. The two orthoesters 9a,b are mixtures of trans/cis diastereomers.
1H NMR (400 MHz, DMSO-d6): Isomer 1, 9a (trans) δ = 5.74 (s, 1H, 2-H), 4.86 (t, J = 5.6 Hz, 1H, OH), 4.21 (dddd, J = 5.3, 5.3 Hz, 5.4 Hz, 6.8 Hz, 1H, 4-H), 4.00 (app. quint, J = 7.4 Hz, 1H, 5-H), 3.69 (dd, J = 5.4 Hz, J = 7.4 Hz, 1H, 5-H), 3.39 (app. t, J = 5.3, 5.6 Hz, 1H, CH2OH), 3.39 (app. t, J = 5.3, 5.6 Hz, 1H, CH2OH), 3.17 (s, 3H, OCH3). Isomer 2, 9b (cis) δ = 5.73 (s, 1H, 2-H), 4.85 (t, J = 5.6 Hz, 1H, OH), 4.05–4.12 (m, 1H, 4-H), 4.00 (app. quint, J = 7.4 Hz, 1H, 5-H), 3.65 (app. t, J = 7.4 Hz, 1H, 5-H), 3.53 (app. quint, J = 5.6 Hz, 1H, CH2OH), 3.45 (app. quint, J = 5.6 Hz, 1H, CH2OH), 3.18 (s, 3H, OCH3). 13C{1H} NMR (100 MHz, DMSO-d6): Isomer 1, 9a (trans) δ = 115.4 (C-2), 75.8 (C-4), 65.7 (C-5), 62.4 (CH2OH), 50.4 (OCH3). Isomer 2, 9b (cis) δ = 115.2 (C-2), 76.7 (C-4), 65.3 (C-5), 61.5 (CH2OH), 50.4 (OCH3). IR: 3469, 2942, 2839, 1447, 1370, 1205, 1136, 1077, 1031, 984, 921, 835 cm−1.
The peak assignment was done with the help of HSQC and H,H-COSY spectra.
HRMS (ESI-TOF): m/z [M + Na]+ calcd C5H10O4: 157.04713; found: 157.04717.
Synthesis of the allyl alcohol 11 from the orthoesters 9
A slurry of the crude orthoesters 9 (36.5 g), containing the pyridinium p-toluenesulfonate from the first step, was heated to 270 °C at atmospheric pressure in a distillation apparatus. The first fraction was MeOH (62 °C/atmospheric pressure), and after 45 min the allyl alcohol (64–140 °C/atmospheric pressure) was immediately produced under the release of CO2. The slightly yellow crude product, which nearly contains just the allyl alcohol, was purified by distillation. Because allyl alcohol will rapidly polymerize at temperatures above 45 °C, it was removed from the reaction mixture at room temperature in vacuo (0.5 mbar), whereby the product fractions where cooled with liquid nitrogen. Finally, to remove traces of MeOH, the allyl alcohol was concentrated at room temperature and 120 mbar for 30 min, yielding 11.9 g (76%) of allyl alcohol (11) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ = 5.94 (ddt, J = 5.1, 10.4 Hz, 17.2 Hz, 1H, 2-H), 5.23 (app. dq, J = 1.7, 17.2 Hz, 1H, 3-Ha), 5.09 (app. dq, J = 1.5, 10.4 Hz, 1H, 3-Hb), 4.08 (app. dt, J = 1.6, 5.1 Hz, 2H, 1-H). 13C{1H} NMR (100 MHz, CDCl3): δ = 137.2 (C-2), 115.0 (C-3), 63.6 (C-1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the State of Baden-Württembert, Germany and the DAAD (PPP Brasilien 2018, project-ID: 57391106) is gratefully acknowledged. We also thank Expedito Parente Jr, Fortaleza, Brazil for helpful discussions.Notes and references
- J. Malça and F. Freire, Renewable Sustainable Energy Rev., 2011, 15, 338–351 CrossRef.
- For some reviews, see: (a) M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed; M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef PubMed; I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef; S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef.
- E. Arceo, P. Marsden, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3357–3359 RSC.
- J. Calero, D. Luna, E. D. Sancho, C. Luna, F. M. Bautista, A. A. Romero, A. Posadillo, J. Berbel and C. Verdugo-Escamilla, Renewable Sustainable Energy Rev., 2015, 42, 1437–1452 CrossRef CAS.
- J. A. Kijenski, A. W. Lipkowski, W. Walisiewicz-Niedbalska, H. Gwardiak, K. Rozycki and I. Pawlak, Eur. Pat. Appl., EP 1580255 A1, 2005.
- (a) D. Fabbri, V. Bevoni, M. Notari and F. Rivetti, Fuel, 2007, 86, 690–697 CrossRef CAS; (b) L. S. Al-Saadi, V. C. Eze and A. P. Harvey, Front. Chem., 2018, 6, 625 CrossRef PubMed.
- (a) G. Crank and F. W. Eastwood, Aust. J. Chem., 1964, 17, 1392–1398 CrossRef CAS; (b) J. S. Josan and F. W. Eastwood, Carbohydr. Res., 1968, 7, 161–166 CrossRef CAS.
- See also: (a) P. Camps, J. Cardellach, J. Font, R. M. Ortuño and O. Ponsati, Tetrahedron, 1982, 38, 2395–2402 CrossRef CAS; (b) S. Hanessian, A. Bargiotti and M. LaRue, Tetrahedron Lett., 1978, 19, 737–740 CrossRef; (c) E. Block, in Olefin Synthesis by Deoxygenation of Vicinal Diols, ed. W. G. Dauben, John Wiley & Sons, New York, 1984, vol. 30, pp. 457–566 Search PubMed.
- Reaction of glycerol and triethyl orthoformate at 100 °C without a catalyst gives a mixture of the 5- and 6-membered orthoesters in a ratio of 76:24: Y. Yokoyama, A. B. Padias, F. De Blauwe and H. K. Hall, Macromolecules, 1980, 13, 252–261 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02338k |
| This journal is © The Royal Society of Chemistry 2019 |