
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew Leishmania donovani nucleoside hydrolase inhibitors from Brazilian flora†
Charlotte Nirma a,
Gregorio Torres Rangela,
Marina Amaral Alvesab,
Livia Marques Casanovaa,
Mayara Monteiro Moreiraa,
Luanna Monteiro Rodriguesa,
Lidilhone Hamerskia and
Luzineide Wanderley Tinoco*a
a,
Gregorio Torres Rangela,
Marina Amaral Alvesab,
Livia Marques Casanovaa,
Mayara Monteiro Moreiraa,
Luanna Monteiro Rodriguesa,
Lidilhone Hamerskia and
Luzineide Wanderley Tinoco*a
aUniversidade Federal do Rio de Janeiro, Instituto de Pesquisas de Produtos Naturais, Laboratório de Análise e Desenvolvimento de Inibidores Enzimáticos, Centro de Ciências da Saúde, 21941-902 Rio de Janeiro, RJ, Brazil. E-mail: lwtinoco@nppn.ufrj.br
bUniversidade Federal do Rio de Janeiro, Instituto de Ciências Biomédicas, Laboratório de Avaliação e Síntese de Substâncias Bioativas, Centro de Ciências da Saúde, 21941-902 Rio de Janeiro, RJ, Brazil
First published on 13th June 2019
Abstract
This study presents new inhibitors of the nucleoside hydrolase from Leishmania donovani (LdNH) with in vitro leishmanicidal activity. Biological screening of 214 Brazilian plant extracts was performed to select plants with enzyme inhibitory activity. Two plants were selected for their results, and for their lack of prior phytochemical description: Leandra amplexicaulis DC. (Melastomataceae) and Urvillea rufescens Cambess (Sapindaceae). Three flavonoids were isolated by bioguided fractionation of the hydroethanolic extracts: kaempferol 3-O-α-L-rhamnopyranoside (1) and kaempferol 3-O-β-D-xylopyranosyl-(1→2)-α-L-rhamnopyranoside (2) from L. amplexicaulis, as well as tricetin-4′-O-methyl flavone (3) from U. rufescens. These flavonoids showed inhibitory activities (IC50) of 197.4 μM (1), 74.7 μM (2) and 1.1 μM (3) on the LdNH. Their binding mode was proposed based on molecular docking with LdNH and by NMR Saturation Transfer Difference studies. Kinetic studies demonstrate that the most potent inhibitor (3) acts by uncompetitive inhibition. This study reports for the first time the inhibition of LdNH by naturally sourced flavonoids.
1. Introduction
Leishmaniasis is one of the most neglected diseases in the world. According to the World Health Organization (WHO), it is present in 98 countries, most of which are categorized as developing. This neglected disease represents a public health problem in tropical and subtropical regions of the world. WHO estimates 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of visceral leishmaniasis worldwide, with 20000 deaths per year1 caused by three species of protozoa: Leishmania donovani, Leishmania chagasi and Leishmania infantum.2 Development of alternative therapies to cure visceral leishmaniasis is urgently needed. Most of the drugs used, especially the pentavalent antimonials, are toxic and often ineffective due to parasite resistance.3 Additionally, amphotericin B, used when treatment with antimonials is inappropriate, is highly toxic and costly.4 The Leishmania donovani nucleoside hydrolase (LdNH) enzyme is a safe and effective target for the development of potential new drugs specific for the treatment of leishmaniasis.5,6
000 cases of visceral leishmaniasis worldwide, with 20000 deaths per year1 caused by three species of protozoa: Leishmania donovani, Leishmania chagasi and Leishmania infantum.2 Development of alternative therapies to cure visceral leishmaniasis is urgently needed. Most of the drugs used, especially the pentavalent antimonials, are toxic and often ineffective due to parasite resistance.3 Additionally, amphotericin B, used when treatment with antimonials is inappropriate, is highly toxic and costly.4 The Leishmania donovani nucleoside hydrolase (LdNH) enzyme is a safe and effective target for the development of potential new drugs specific for the treatment of leishmaniasis.5,6
LdNH hydrolyses N-riboglycoside bonds to provide the parasite with purine and pyrimidine bases for its DNA biosynthesis. Nucleoside hydrolase enzymes have not been identified in mammals, thus ensuring the selectivity of potential inhibitors. Moreover, LdNH shows high sequence similarity among different species of Leishmania and even other trypanosomatids.5,7,8 Due to the close genetic nature of different parasite species, this biochemical target could be conserved in other trypanosomatid parasites, allowing the discovery of a global antiprotozoal drug, with pan-trypanocidal activity.5
The purpose of our work is to isolate and characterize molecules from plants collected in the Atlantic Forest of the state of Rio de Janeiro (Brazil) capable of inhibiting the LdNH of Leishmania donovani. The use of plants for the discovery of new bioactive compounds, and the search for antileishmanial molecules from natural sources has been widely reported in the literature.9–13 A large percentage of these compounds were identified using in vitro assays against promastigotes or intracellular amastigotes of several species of Leishmania. However, to our knowledge there are no prior reports in which naturally occurring compounds were tested against the target LdNH.
This study presents the results of biological screening, guided by the inhibitory activity of LdNH, of 214 extracts of samples belonging to the I-FLORA project. I-FLORA is an interinstitutional and interdisciplinary project that aims to inventory the flora from Atlantic Forest of the state of Rio de Janeiro and add value to its diversity through chemical and biological studies.
Among the most active extracts, two were selected for phytochemical analysis, Leandra amplexicaulis and Urvillea rufescens. L. amplexicaulis, popular name “pixirica”,14,15 is a shrub distributed in the Atlantic Forest in the southeast (Minas Gerais, Espírito Santo, São Paulo, Rio de Janeiro) and south (Paraná, Santa Catarina) of Brazil. U. rufescens Cambess is a clambering plant native to the southeast and northeast regions of Brazil.16 To our knowledge, no phytochemical study has been carried out on either of these plants. Biodirected fractionation of the extracts allowed the isolation of three bioactive flavonoids. Flavonoids are already widely described for their bioactive properties, including those described as inhibitors of nucleoside hydrolases of Trichomonas vaginalis,17,18 as well as their leishmanicidal activities.11,19–21
To further elucidate the mechanism of action, we used molecular docking and NMR Saturation Transfer Difference (STD) experiments22 to analyze the interactions of the isolated active compounds with LdNH. These techniques provide insights into the nature of intermolecular interactions and can be used to map ligand–protein contacts.
2. Experimental
2.1 General experimental procedures
Optical rotation was measured with a Jasco P-2000 Polarimeter. NMR spectra were recorded using Agilent VNMRS-500 or 400 MR spectrometers. Chemical shifts (δ) are reported in ppm relative to the tetramethylsilane (TMS) resonance (0.00 ppm). High-resolution ESI-TOF-MS measurements were performed using a Shimadzu UPLC system equipped with a Poroshell ECC18 2.7 μm 2.1 × 100 mm column. The 7 min separation was carried out by isocratic elution using MeOH at a flow rate of 300 μL min−1. The UPLC was coupled to a Shimadzu time-of-flight (TOF) mass spectrometer equipped with an electrospray (ESI) interface. TLC analyses were conducted on 60A F254 Merck plates, visualized under UV light (254 and 365 nm) and revealed with iodine. Analytical and semi-preparative HPLC analyzes were performed with Varian or Shimadzu systems using a linear gradient. All solvents were HPLC grade (TEDIA®) and were prepared by addition of 0.05% acetic acid. Columns used for these experiments included Phenomenex Luna PhenylHexyl 5 μm 4.6 × 250 mm analytical columns and Phenomenex Luna PhenylHexyl 5 μm 10 × 250 mm and C18 5 μm 21.2 × 250 mm semi-preparative columns.2.2 Plant material and extraction
Plant species screened in the present study as part of the I-Flora project were collected in the Atlantic Forest from the state of Rio de Janeiro, Brazil from 2009 to 2012. Plant material (from branches, leaves or stems) was dried at room temperature for 48 h. Subsequently, the material was ground and macerated three times with ethanol/water 10% (v/v) at room temperature for 96 h: 48 h for the first extraction and 24 h the other two extractions.The screening step was performed with a total of 214 hydroethanolic extracts to evaluate their inhibitory activity for LdNH. Based on these initial results, two extracts were selected for bioguided fractionation: the extract of the flowers of Leandra amplexicaulis DC (Melastomataceae) and the leaf extract of Urvillea rufescens Cambess (Sapindaceae).
L. amplexicaulis was collected on April 15, 2012 in the Parque Nacional do Itatiaia, Itatiaia (−22.397309, −44.612180) in the state of Rio de Janeiro by Prof. Dr Marcelo Trovó Lopes de Oliveira from the Federal University of Rio de Janeiro. The plant was authenticated by Dr Trovó and a voucher specimen (RB639928) deposited at the herbarium of the Botanical Garden of Rio de Janeiro.
U. rufescens was collected in the Grumari Municipal Nature Park, Rio de Janeiro, Rio de Janeiro state (−23.054992, −43.549446), by Prof. Dr Genise Vieira Somner from the Federal Rural University of Rio de Janeiro (UFRRJ). The plant was authenticated by Dr Somner and a voucher specimen, (RBR 42621) was deposited at the herbarium of the Federal Rural University of Rio de Janeiro (UFRRJ).
2.3 Extraction and isolation of constituents
:2 MeOH/H2O followed by CH2Cl2 and 8:2 MeOH/H2O, and then AcOEt and 6:4 MeOH/H2O. The final AcOEt fraction (383.8 mg) proved to be the best source of LdNH inhibitory constituents. The AcOEt fraction was subjected to semi-preparative HPLC [column: PhenylHexyl; mobile phase: 20:80 CH3CN/H2O + 0.05% HOAc to 40:60 using a linear gradient over 20 min followed by 100% CH3CN for 5 min at a flow rate of 4 mL min−1 and UV detection at 210 nm] to yield four subfractions (A–D). Subfraction C showed the best inhibitory activity with 79% of the total inhibition. Fraction C (46.2 mg) was subjected to semi-preparative HPLC [column PhenylHexyl; mobile phase: 30:70 CH3CN/H2O + 0.05% HOAc to 60:40 using a linear gradient over 20 min at a flow rate of 2 mL min−1 and UV detection at 210 nm] to yield compounds 1 (6.1 mg, RT = 12 min) and 2 (14 mg, RT = 10 min).
Kaempferol 3-O-α-L-rhamnoside (1). Yellowish oil; [α]25D −116.9 (c 0.2, MeOH); 1H NMR (500 MHz, CD3OD): 7.77 (2H, d, J = 9 Hz, H-2′/H-6′), 6.94 (2H, d, J = 9 Hz, H-3′/H-5′), 6.38 (1H, d, J = 2 Hz, H-8), 6.21 (1H, d, J = 2 Hz, H-6), 5.38 (1H, d, J = 1.0 Hz, H-1′′), 4.22 (1H, dd, J = 2.7; 1.0 Hz, H-2′′), 3.70 (1H, m, H-3′′), 3.33 (1H, m, H-4′′), 3.32 (1H, m, H-5′′), 0.92 (3H, d, J = 6 Hz, H-6′′); 13C NMR (125 MHz, CD3OD): n.d. (C-4), 164.7 (C-7), 162.2 (C-5), 160.8 (C-4′), 157.8 (C-2), 157.6 (C-9), 134.8 (C-3), 131.5 (C-2′/C-6′), 120.7 (C-1′), 116.1 (C-3′/C-5′), 104.8 (C-10), 103.8 (C-1′′), 99.5 (C-6), 94.4 (C-8), 72.8 (C-5′′), 71.8 (C-3′′), 71.6 (C-4′′), 71.5 (C-2′′), 17.4 (C-6′′); HRMS-ESI [M − H]− m/z 431.0995 (theoretical mass for C21H19O10, 431.0984).
Kaempferol 3-O-β-D-xylopyranosyl-(1→2)-α-L-rhamnopyranoside (2). Yellowish oil; [α]25D −10 (c 0.16, MeOH); 1H NMR (500 MHz, CD3OD): 7.77 (2H, d, J = 8.6 Hz, H-2′/H-6′), 6.95 (2H, d, J = 8.6 Hz, H-3′/H-5′), 6.38 (1H, d, J = 1.9 Hz, H-8), 6.20 (1H, d, J = 1.9 Hz, H-6), 5.43 (1H, d, J = 1.3 Hz, H-1′′), 4.29 (1H, d, J = 7.6 Hz, H-1′′′), 4.20 (1H, dd, J = 3.6; 1.3 Hz, H-2′′), 3.83 (1H, dd, J = 9.9; 3.6 Hz H-3′′), 3.68 (1H, m, H-5′′′a), 3.67 (1H, m, H-5′′), 3.41 (1H, ddd, J = 11.1; 8.8; 5.4 Hz, H-4′′′), 3.34 (1H, d, J = 9.9 Hz, H-4′′), 3.28 (1H, d, J = 8.8 Hz, H-3′′′), 3.19 (1H, dd, J = 8.8; 7.6 Hz, H-2′′′), 3.09 (1H, dd, J = 11.1 Hz, H-5′′′b), 1.00 (3H, d, J = 6.2 Hz, H-6′′); 13C NMR (125 MHz, CD3OD): 178.10 (C-4), 164.49 (C-7), 161.72 (C-5), 160.24 (C-4′), 157.99 (C-2), 157.07 (C-9), 135.48 (C-3), 130.44 (C-2′/C-6′), 121.11 (C-1′), 115.15 (C-3′/C-5′), 104.61 (C-10), 106.32 (C-1′′′), 101.71 (C-1′′), 98.43 (C-6), 93.36 (C-8), 81.22 (C-2′′), 76.33 (C-3′′′), 73.85 (C-2′′′), 72.17 (C-4′′), 70.50 (C-3′′), 70.46 (C-5′′), 69.54 (C-4′′′), 65.66 (C-5′′′), 16.28 (C-6′′); HRMS-ESI [M − H]− m/z 563.1402 (theoretical mass for C26H27O14, 563.1406).
:2 MeOH/H2O followed by AcOEt and 6:4 MeOH/H2O. The final AcOEt fraction (2.04 g) proved to be the best source of LdNH inhibitory constituents. 243 mg of the AcOEt fraction was dissolved in 5 mL AcOEt/MeOH and centrifuged. The supernatant was introduced onto a Sephadex® LH20 column (77 × 3.5 cm) for size exclusion chromatography using 7:3 AcOEt/MeOH as the mobile phase. This separation produced 15 subfractions (A–N). Subfraction F showed the best inhibitory activity with 93.4% of the total inhibition. F (15.2 mg) was purified by analytical HPLC using a 250 × 4.6 mm Phenomenex® Phenyl Hexyl Luna column with 100 Å pore size and 5 μm particle size. The separation was carried out using isocratic elution with 50:50 (MeOH:H2O) to yield compound 3 (0.7 mg, RT: 14 min).As 3 presented high LdNH inhibitory activity, the purification process was modified to obtain greater quantities of the substance, enabling further assays. Thus, 500 mg of the AcOEt fraction was dissolved in 5 mL AcOEt/MeOH, and centrifuged. The supernatant was introduced onto a Sephadex® LH20 column (70 × 3.5 cm) eluted with 7:3 AcOEt/MeOH and then switched to 100% MeOH, which yielded five subfractions (I–V). Fraction V (65.5 mg) was enriched in the flavonoid of interest and was selected for further purification. Fraction V was introduced onto a reverse phase C-18 solid phase extraction cartridge (1 g; 6 mL; Bakerbond®) and eluted using a series of water/methanol solutions: 1:9 MeOH/H2O, 3:7 MeOH/H2O, 1:1 MeOH/H2O, 7:3 MeOH/H2O and MeOH. Thus, it was possible to obtain 7.7 mg of compound 3, which was eluted from the cartridge with 1:1 MeOH/H2O.
Tricetin 4-O-methyl-flavone (3). Yellowish oil. 1H NMR (400 MHz, CD3OD): 6.97 (2H, s, H-2′/H-6′), 6.52 (1H, s, H-3), 6.41 (1H, d, J = 1.9 Hz, H-8), 6.19 (1H, d, J = 1.9 Hz, H-6), 3.88 (3H, s, O–CH3); 13C NMR (100 MHz, CD3OD): 182.19 (C-4), 166.05 (C-7), 164.12 (C-2), 161.87 (C-5), 158.13 (C-9), 150.92 (C-3′/C-5′), 138.84 (C-4′), 126.39 (C-1′), 105.43 (C-2′/C-6′), 103.53 (C-3/C-10), 99.18 (C-6), 94.14 (C-8), 59.37 (O–CH3). ESI [M − H]− m/z 315.03 (theoretical mass for C16H12O7, 316.0583).
3. Results and discussion
As part of the I-Flora project, we investigated 214 hydroethanolic plant extracts for their LdNH inhibitory activity. Twenty-eight extracts inhibited LdNH by 50% or more at a concentration of 100 μg mL−1 (Fig. S1†). Due to the absence of prior phytochemical studies, extracts of the flowers of Leandra amplexicaulis DC (Melastomataceae) and the leaves of Urvillea rufescens Cambess (Sapindaceae) were selected from among the most active extract for further interrogation.Following the bioguided fractionation of L. amplexicaulis extract, two substances (1 and 2) were isolated and identified by 1D and 2D NMR and mass spectrometry (see Experimental section). Both molecules have an aglycone flavonoid structure substituted with one or two sugars (Fig. 1). Compound 1 has a kaempferol skeleton with a rhamnopyranosyl unit, suggested by a characteristic methyl doublet at δ 0.92 (J = 6 Hz). It was identified by comparison to literature data as kaempferol 3-O-α-L-rhamnopyranoside (afzelin).28
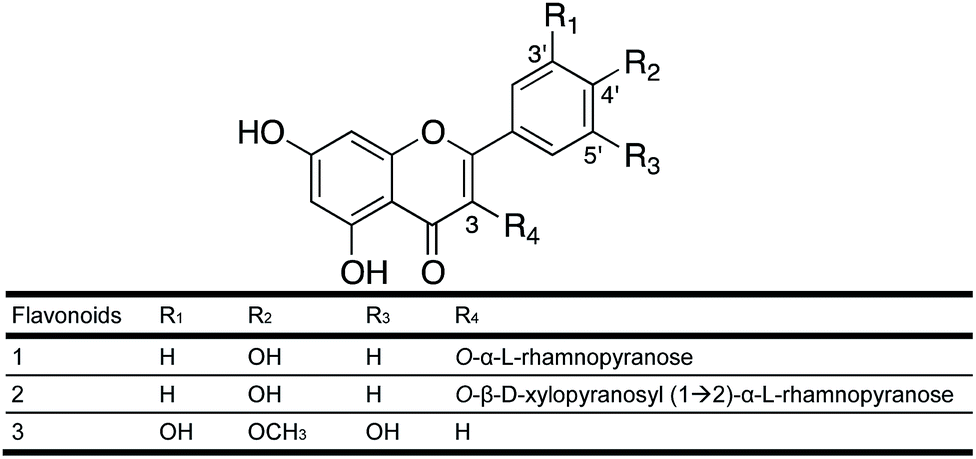 | ||
| Fig. 1 Molecular structure of the isolated flavonoids 1–3. | ||
The structure of compound 2 was elucidated based on 1H- and 13C-NMR data and mass spectrometry. The 1H and HSQC NMR spectra are available in the ESI (Fig. S2A and B†). Compound 2 also has a kaempferol skeleton with a rhamnose and a xylopyranose linked at C-3. It was identified as kaempferol 3-O-β-D-xylopyranosyl-(1→2)-α-L-rhamnopyranoside in accordance with literature data.29 This diglycosylated flavonol was first described in the leaves of Moghania faginea (Fabaceae),29 then from the medicinal plant Kalanchoe daigremontiana30 and subsequently from Pourthiaea villosa31 along with 1. The 3-O-glycosides of kaempferol are the most common group of flavonoids.
Following the bioguided fractionation of U. rufescens, compound 3 (Fig. 1) was isolated and identified by 1D and 2D NMR and mass spectrometry. The HSQC NMR spectrum is available in the ESI (Fig. S3†). According to these spectra and in comparison, with the literature, we concluded that the structure of compound 3 is a free aglycone named tricetin-4′-O-methyl flavone. This flavone was first isolated from Passiflora palmeri Rose (Passifloraceae)32 and then from Heliotropium pycnophyllum (Boraginaceae).33
The inhibitory activities (IC50) of compounds 1, 2 and 3 for LdNH were determined by spectroscopic analysis at 293 nm in an enzymatic assay coupled with xanthine oxidase. Both flavonoids with kaempferol structures showed moderate activities, 197.38 μM for 1 and 74.65 μM for 2. It is likely that the xylopyranosyl moiety present in compound 2 is responsible for its greater inhibitory activity compared to compound 1, which lacks this sugar. The leishmanicidal activity of 1 has already been reported, with IC50 = 70 μg mL−1 (162 μM) on the amastigote parasite Leishmania amazonensis.34 Thus, we can hypothesize that a possible mechanism for the leishmanicidal activity of the flavonoid is LdNH inhibition. Other mechanisms possibly contribute as well. Other studies report that compound 2 has anti-viral activity with an EC50 of 7.4 μg mL−1 against HSV-1 and 9.0 μg mL−1 against HSV-2.30 However, tricetin (3) which has only the aglicone showed higher inhibitory activity with an IC50 of 1.06 μM. The IC50 curves are shown in the ESI (Fig. S4†).
To understand the molecular-level interactions of flavonoids 1 and 2 with LdNH, the isolated compounds were analyzed by the NMR Saturation Transfer Difference experiment (NMR-STD).22 This pulse sequence allows the identification of the hydrogen atoms of the ligand that participate in interactions with LdNH. The intensities of the STD signals are directly proportional to the distances between the hydrogens of the ligands and the protein. Thus, the more intense STD signals indicate a greater proximity to the protein providing a map of the topology of the interaction.
For flavonoid 1 (Fig. 2A), the STD spectrum shows only weak resonances for the aromatic protons H-6 and H-8 of ring A, H-2′/H-6′ and H-3′/H-5′ of ring B, and the methyl protons of rhamnose moiety. For flavonoid 2 (Fig. 2B), the STD signals of the aromatic protons and rhamnose methyl group are much more intense than those observed for flavonoid 1. Furthermore, the signal at 1.00 ppm from the rhamnose methyl group was more intense than the signals of the other sugar protons, confirming the interaction of the rhamnose moiety with LdNH and suggesting that methyl group is closer to the enzyme. The flavonoid isolated from U. rufescens (compound 3) was not analyzed by NMR-STD due to aggregation in aqueous medium at the concentration required for the NMR studies (>200 μM). The sugar moiety is an important factor for the bioavailability of the flavonoid derivatives,35 but is absent in the flavonoid 3, which makes it less soluble in water than flavonoids 1 and 2. The NMR-STD data indicates that the sugar could be directly involved in the protein interaction, however, because the non-glycosylated flavonoid 3 has a much higher inhibitory potency than the glycosylated flavonoids, it is clear that the sugar is not essential for LdNH inhibition.
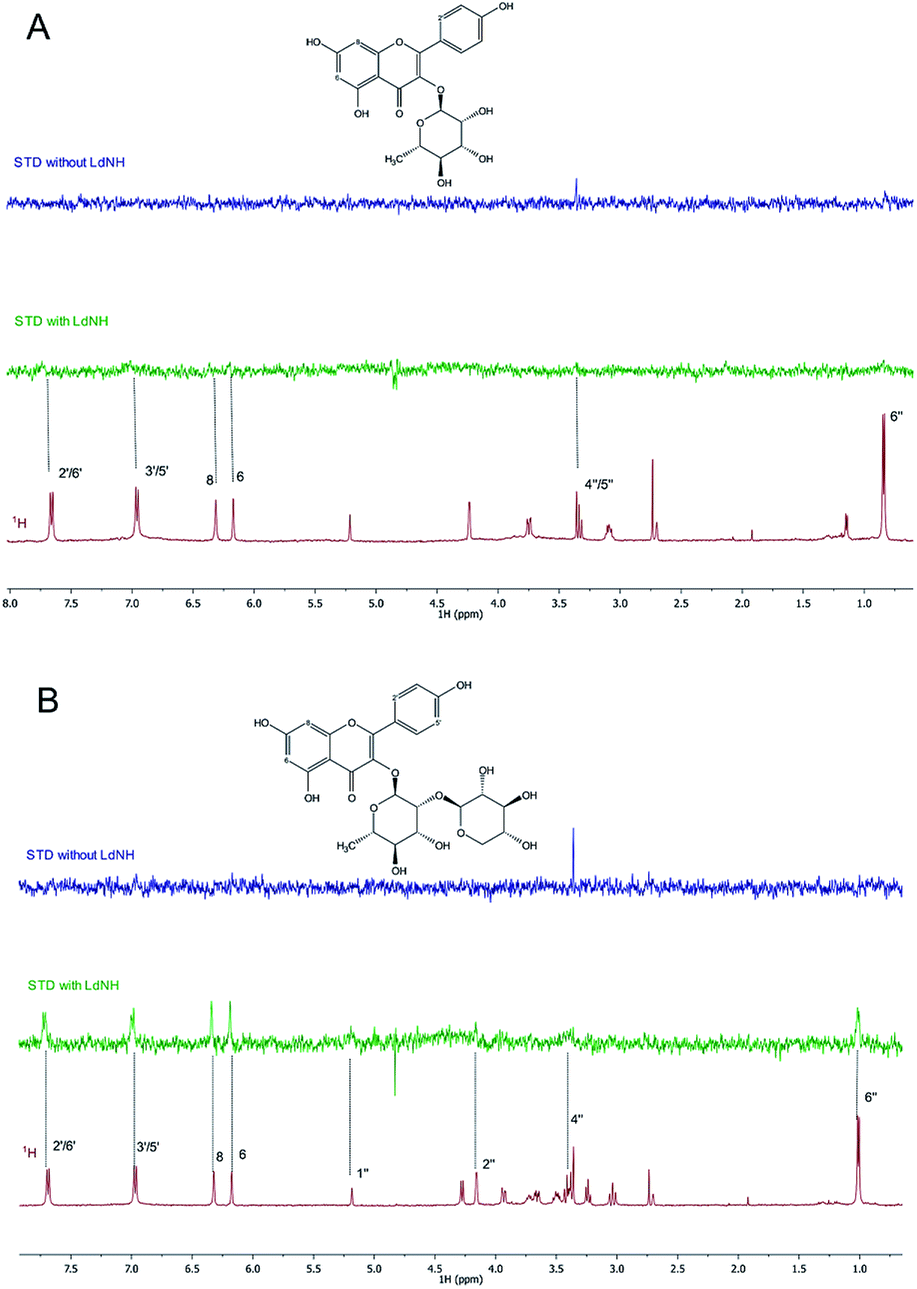 | ||
| Fig. 2 Representative results of the STD-NMR binding assay. (A) 1H NMR spectra (red) of compound 1 and its STD effects (green). (B) 1H NMR spectra (red) of compound 2 and its STD effects (green). As control, STD experiment was run for each sample in the absence of LdNH (blue). | ||
Considering that the substrates of LdNH are nucleosides, compounds containing a purine or pyrimidine ring bound to a ribose, suggests that an inhibitor containing a sugar moiety could function as a competitive inhibitor. As the non-glycosylated flavonoid 3 had the highest inhibitory potency, it leads us to believe that flavonoids 1 and 2 should inhibit LdNH by a mechanism different than flavonoid 3. In fact, in our previous work with molecular fragments, we identified for the first time a non-glycosylated inhibitor of the LdNH.24 This inhibitor was also the first to be described as an non-competitive inhibitor of this enzyme.
As flavonoid 3 was the most promising LdNH inhibitor, we carried out kinetics studies to determine the mechanism of inhibition of LdNH. In the first step, we examined the equilibrium enzyme–inhibitor complex (E–I) to evaluate the reversibility of inhibition. In this experiment, after the formation of the complex, a jump dilution was conducted and the rapid recovery of enzymatic activity was observed showing that flavonoid 3 acts as a reversible inhibitor of LdNH.
In the second step, to establish the type of inhibition presented by flavonoid 3, we investigated which of the classical models of inhibition is most consistent with our results. For this experiment, we analyzed kinetic data for three different concentrations of 3, with six different concentrations of inosine (Fig. 3). The treatment results were compared with those from control measurements performed in the absence of inhibitor. Non-linear regression was used to consider all data simultaneously and discriminate the inhibition mechanism using different parameters. An uncompetitive (or anti-competitive) model of inhibition statistically best described the results when compared to the competitive, non-competitive and mixed inhibition models. The parameters Vmax (mM min−1) = 191 (CI 95% = 167.8 to 214.2) and KM (mM) = 0.37 (CI 95% = 0.2428 to 0.5170) were also determined using global fitting of the data.
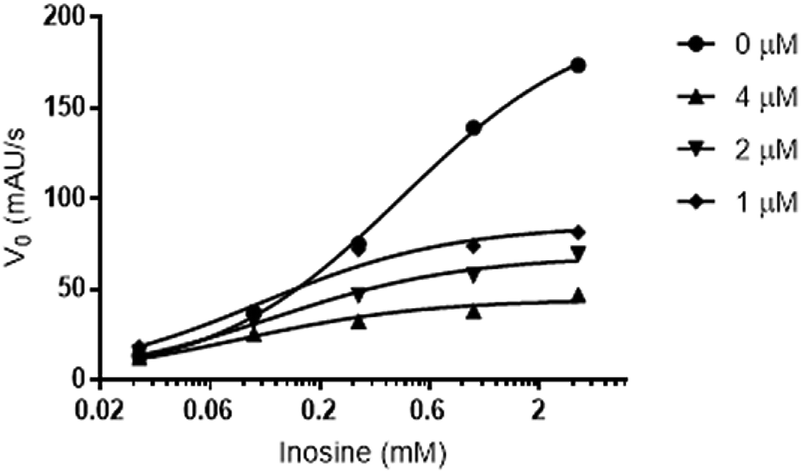 | ||
| Fig. 3 Concentration-effect curves for the enzymatic activity of LdNH in the presence of compound 3 with different inosine concentrations (0.03, 0.10, 0.30, 1.00 and 3.00 mM). The experiment was performed in the absence (control ●) or presence of increasing concentrations of compound 3 (1♦, 2▼ and 4▲ μM). | ||
Molecular docking was used to explore the interaction mode of flavonoid 3 with LdNH. These data can be used in the later stages for the structural optimization of this inhibitor to aid in the development of a prototype with inhibitory action of LdNH with greater potency and leishmanicidal action.
The homology model of LdNH developed and validated was previously published by Alves et al.24 and used in this stage of investigation. Given the uncompetitive inhibition mechanism of flavonoid 3, the model containing inosine was used to perform the docking assays in an exploratory manner without setting the docking space. For this, the LdNH model containing the complex with inosine and flavonoid 3, were submitted to the SwissDock server to identify the docking regions.
As observed in Fig. 4A, the three lowest energy docking poses of ligand–protein interaction are located in the same allosteric pocket. This allosteric pocket is located near the catalytic site, which is represented by the presence of inosine (orange) and the calcium ion (green sphere). The docking pose of the complex with the lowest intermolecular energy (−6.83 kcal mol−1) (Fig. 4B) shows that flavonoid 3 interacts with the LdNH residues of Ser-79, His-82, Glu-232 and Asn-77. Based on these results, our group is now working to optimize the structure of flavonoid 3 aiming to develop a compound with a greater number of interaction points with LdNH and to evaluate whether the structural changes promote an increase in the potency of the leishmanicidal effect.
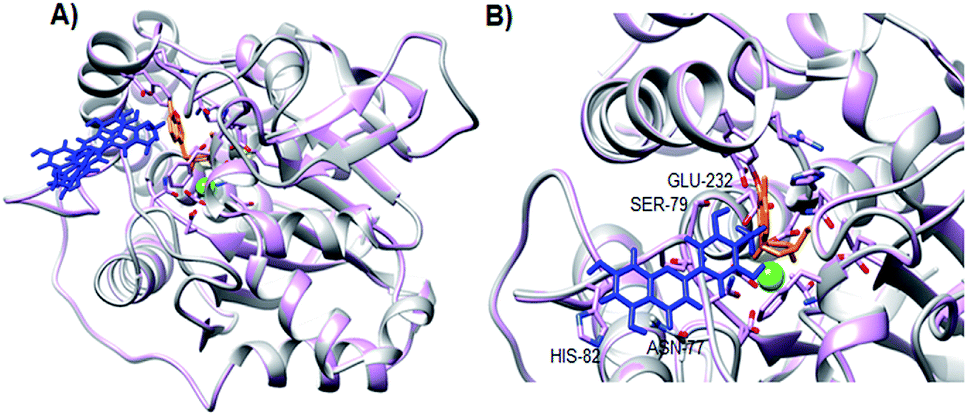 | ||
| Fig. 4 Docking of flavonoid 3 and LdNH-inosine complex. (A) Cartoon representation of LdNH including inosine (orange stick representation), calcium ion (green sphere) and the three lowest energy poses of flavonoid 3 (blue stick representation). (B) The best docking conformation of flavonoid 3 in the complex inosine-bound LdNH. Residues of LdNH within 4 Å of flavonoid 3 are indicated in the figure. | ||
4. Conclusions
This study demonstrates the great potential of the Brazilian flora in the supply of bioactive compounds, where 10% of the evaluated extracts presented an inhibitory potential of LdNH greater than 50%. As phytochemical studies of the selected plants were not found in the literature, we described for the first time the flavonoids kaempferol 3-O-α-L-rhamnopyranoside (1) and kaempferol 3-O-β-D-xylopyranosyl-(1→2)-α-L-rhamnopyranoside (2) for Leandra amplexicaulis DC as well as tricetin-4′-O-methyl flavone (3) for Urvillea rufescens Cambess. Although flavonoids have already been identified as leishmanicidal agents, this is the first time that this class of compounds has been described as inhibitors of LdNH. Flavonoid 3, with an IC50 of 1.06 μM and an uncompetitive inhibition mechanism opens a new perspective for the design of new and more potent inhibitors for this enzyme and for the treatment of leishmaniasis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (BR) Grant 303647/2015-9, and Fundação Carlos Chagas Filho de Amparo a Pesquisa no Estado do Rio de Janeiro – FAPERJ (BR) Grant E-26/111.483/2013. Authors would like to thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES for the scholarship to Gregorio Torres and Livia Casanova, and CNPq for the Fellowship 400095/2014-9 to Charlotte Nirma and scholarship to Mayara M. Moreira and Luanna M. Rodrigues. Our sincere thanks to Prof. Cynthia K. Larive (University of California-Riverside) for the careful review of the English language.References
- WHO, Second WHO report on neglected tropical diseases, 2013 Search PubMed.
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CrossRef CAS PubMed.
- A. Henderson and D. Jolliffe, Br. J. Clin. Pharmacol., 1985, 19, 73–77 CrossRef CAS PubMed.
- R. Sabra and R. A. Branch, Drug Saf., 1990, 5, 94–108 CrossRef CAS PubMed.
- L. Cui, G. Rajasekariah and S. K. Martin, Gene, 2001, 280, 153–162 CrossRef CAS PubMed.
- M. N. Rennó, T. C. C. França, D. Nico, C. B. Palatnik-De-Sousa, L. W. Tinoco and J. D. Figueroa-Villar, Eur. J. Med. Chem., 2012, 56, 301–307 CrossRef PubMed.
- T. C. C. França, M. D. R. M. Rocha, B. M. Reboredo, M. N. Rennó, L. W. Tinoco and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2008, 19, 64–73 CrossRef.
- W. Versées, K. Decanniere, E. Van Holsbeke, N. Devroede and J. Steyaert, J. Biol. Chem., 2002, 277, 15938–15946 CrossRef PubMed.
- R. S. Patil, M. S. Patil, S. S. Kshirsagar, P. S. Chaudari, J. P. Bayas and R. J. Oswal, World Journal of Public Health Sciences, 2012, 1, 7–22 Search PubMed.
- T. J. Schmidt, S. A. Khalid, A. J. Romanha, T. M. A. Alves, M. W. Biavatti, R. Brun and F. B. Da Costa, Curr. Med. Chem., 2012, 19, 2176–2228 CrossRef CAS PubMed.
- T. J. Schmidt, S. A. Khalid, A. J. Romanha, M. W. Biavatti, R. Brun, S. L. De Castro, V. F. Ferreira, L. L. Leon, N. P. Lopes, M. Niehues, I. V Ogungbe, A. M. Pohlit, M. T. Scotti, W. N. Setzer, M. Steindel and A. G. Tempone, Curr. Med. Chem., 2012, 19, 2128–2175 CrossRef CAS PubMed.
- P. E. Cockram and T. K. Smith, J. Nat. Prod., 2018, 81, 2138–2154 CrossRef CAS PubMed.
- A. S. Nagle, S. Khare, A. B. Kumar, F. Supek, A. Buchynskyy, C. J. N. Mathison, N. K. Chennamaneni, N. Pendem, F. S. Buckner, M. H. Gelb and V. Molteni, Chem. Rev., 2014, 114, 11305–11347 CrossRef CAS PubMed.
- Flora do Brasil 2020 Web page. http://www.floradobrasil.jbrj.gov.br, accessed Feb 27, 2019.
- E. A. De Camargo and R. Goldenberg, Iheringia, Ser. Bot., 2007, 62, 103–111 Search PubMed.
- A. Frazão, E. Genise and V. Somner, Hoehnea, 2016, 43, 437–459 CrossRef.
- R. Alam, A. T. Barbarovich, W. Caravan, M. Ismail, A. Barskaya, D. W. Parkin and B. J. Stockman, Chem. Biol. Drug Des., 2018, 92, 1736–1742 CrossRef CAS PubMed.
- S. Beck, S. N. Muellers, A. L. Benzie, D. W. Parkin and B. J. Stockman, Bioorg. Med. Chem. Lett., 2015, 25, 5036–5039 CrossRef CAS PubMed.
- M. Salem and K. Werbovetz, Curr. Med. Chem., 2006, 13, 2571–2598 CrossRef CAS PubMed.
- D. Tasdemir, M. Kaiser, R. Brun, V. Yardley, T. J. Schmidt, F. Tosun and P. Rüedi, Antimicrob. Agents Chemother., 2006, 50, 1352–1364 CrossRef CAS PubMed.
- T. Polonio and T. Efferth, Int. J. Mol. Med., 2008, 22, 277–286 CAS.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- M. A. Alves, C. Nirma, M. M. Moreira, R. O. Soares, P. G. Pascutti, F. Noël, P. R. R. Costa, C. M. R. Sant'Anna, E. J. Barreiro, L. M. Lima and L. W. Tinoco, RSC Adv., 2016, 6, 87738–87744 RSC.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, John Wiley & Sons, Inc., New Jersey, 1st edn, 2005 Search PubMed.
- C. Dalvit, J. Biomol. NMR, 1998, 11, 437–444 CrossRef CAS.
- A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS PubMed.
- L. Kerhoas, D. Aouak, A. Cingöz, J.-M. Routaboul, L. Lepiniec, J. Einhorn and N. Birlirakis, J. Agric. Food Chem., 2006, 54, 6603–6612 CrossRef CAS PubMed.
- H. Soicke, K. Görler and H. Waring, Planta Med., 1990, 56, 410–412 CrossRef CAS PubMed.
- F. G. G. Ürményi, G. do N. Saraiva, L. M. Casanova, A. dos S. Matos, L. M. de Magalhães Camargo, M. T. V. Romanos and S. S. Costa, Chem. Biodiversity, 2016, 13, 1707–1714 CrossRef PubMed.
- H. J. Lee, D. Ahn, E. B. Lee, T. G. Lee and D. K. Kim, Saengyak Hakhoechi, 2013, 44, 16–17 CAS.
- A. Ulubelen, T. J. Mabry, G. Dellamonica and J. Chopin, J. Nat. Prod., 1984, 47, 384–385 CrossRef CAS.
- E. Wollenweber, R. Wehde, M. Dörr and J. F. Stevens, Z. Naturforsch., C: J. Biosci., 2002, 57, 445–448 CAS.
- M. F. Muzitano, L. W. Tinoco, C. Guette, C. R. Kaiser, B. Rossi-Bergmann and S. S. Costa, Phytochemistry, 2006, 67, 2071–2077 CrossRef CAS.
- P. C. H. Hollman and I. C. W. Arts, J. Sci. Food Agric., 2000, 80, 1081–1093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and IC50 curves. See DOI: 10.1039/c9ra02382h |
| This journal is © The Royal Society of Chemistry 2019 |