
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysine-based dendrimer with double arginine residues†
Nadezhda N. Sheveleva *a,
Denis A. Markelova,
Mikhail A. Vovka,
Mariya E. Mikhailovaa,
Irina I. Tarasenkob,
Peter M. Tolstoya,
Igor M. Neelovc and
Erkki Lähderantad
*a,
Denis A. Markelova,
Mikhail A. Vovka,
Mariya E. Mikhailovaa,
Irina I. Tarasenkob,
Peter M. Tolstoya,
Igor M. Neelovc and
Erkki Lähderantad
aSt. Petersburg State University, 7/9 Universitetskaya Nab., St. Petersburg, 199034 Russia. E-mail: n.n.sheveleva@spbu.ru
bInstitute of Macromolecular Compounds, Russian Academy of Sciences, Bolshoi Prospect 31, V.O., St. Petersburg, 199004 Russia. E-mail: itarasenko@list.ru
cSt. Petersburg National Research University of Information Technologies, Mechanics and Optics (ITMO University), Kronverkskiy Pr. 49, St. Petersburg, 197101 Russia. E-mail: i.neelov@mail.ru
dDepartment of Physics, LUT University, Box 20, 53851 Lappeenranta, Finland. E-mail: erkki.lahderanta@lut.fi
First published on 7th June 2019
Abstract
Due to their well-defined structure, multivalency, biocompatibility, and low toxicity, lysine dendrimers can be used as safe and efficient nanocarriers for drug and gene delivery. One useful strategy for improving the gene delivery properties of dendrimers is modification with arginine amino acid (Arg) residues. Incorporation of Arg residues could be favorable for the enhancement in transfection efficiency of lysine based dendrimers. In this work, we have synthesized a new second-generation poly-L-lysine dendrimer with repeating units containing two linear Arg residues between neighboring lysine branching points (Lys-2Arg dendrimer) and studied its physicochemical properties. We confirmed the structure of Lys-2Arg dendrimer using various one- and two-dimensional 1H and 13C NMR spectroscopy methods. Comparison of T1H relaxation data for Lys-2Arg and Lys-2Lys dendrimers showed that the replacement of double Lys residues with double Arg residues resulted in a sharp decrease in the mobility of methylene groups in side segments and in the main chain of ε-Lys inner segments. We suggest that this unexpected effect is caused by a guanidine–guanidine pairing effect in water, which leads to entanglements between dendrimer branches.
Introduction
Dendrimers are nanosized artificial macromolecules with well-defined globular architecture that possess a variety of unique properties.1,2 They have a regular hyperbranched structure with a hydrophobic and/or hydrophilic interior, multivalency and extremely low polydispersity, which make them very suitable as nanocontainers for substances of different natures: from metal particles to genes and drugs.3–6 Every year, the number of papers dedicated to the study of the properties of dendrimers and their application in biomedicine and pharmaceutics increases.7,8 This trend demonstrates the importance of searching for dendrimers of new compositions and architectures to achieve safer and more efficient drug and gene delivery.5,9,10Among a wide variety of dendrimers, poly-L-lysine (PLL) dendrimers have attracted especial attention from researchers due to their high biocompatibility and relatively low toxicity. PLL dendrimers are modified with anionic, cationic and neutral amino acid residues to enhance their drug and gene delivery potential. The most common means of functionalization is through conjugation of other amino acid residues to the dendrimer terminal groups.11 The dendrimer interior can also be modified by incorporation of amino acid residues between inner lysine branching points.12
Functionalization of a PLL dendrimer with arginine (Arg) residues improves its transfection properties, thus increasing the charge of the dendrimer.11,12 Arginine residues are positively charged and protonated under biological conditions due to the presence of guanidine groups.13 Decoration of the dendrimer periphery with Arg residues improves its interaction with a negatively charged cell membrane, facilitating membrane penetration and enhancing transfection efficiency.5,9,14–18 It has been shown that guanidine groups have high binding ability to phosphate groups of DNA, which increases the condensation of DNA on the dendrimer surface.5,9,11 The presence of arginine moieties in peptides improves cellular uptake of siRNA.13,19 Additionally, when we introduce charged double Arg residues between inner Lys branching points of PLL dendrimer, we make its interior more charged and thus more hydrophilic. Due to this reason, such modified arginine-rich dendrimer is less suitable for encapsulation of hydrophobic drugs but is of great interest for gene delivery. Transfection is enhanced by increasing the cationic charge of dendrimer.19 In the work,20 dendrimers with inner segments containing lysine–leucine or arginine–leucine amino acid residues were synthesized and investigated. It was found that dendrimers with Arg residues in the inner segments exhibited three-fold higher DNA transfection than dendrimers with Lys residues.
The aim of the present work is to take the first steps in studying a new poly-L-lysine dendrimer containing double Arg residues with guanidine groups in side segments (Fig. 1 and 2). We have attached arginine residues to the inner segments, rather than to the terminal groups, of the dendrimer, i.e. inserting arginine residues in each inner dendrimer shells except the terminal shell. This structure makes a dendrimer more cationic and can contribute to a more uniform distribution of charged groups in the volume of the dendrimer that should result in better transfection efficiency.20 At the same time, Arg residues in dendrimer interior should be available to water and other small molecules because it is well known that about 70% of the volume inside the lysine dendrimers is occupied by water.21 We believe that the introduction of arginine into the dendrimer interior will significantly improve the delivery of RNA and DNA and promote the formation of a more stable complex between dendrimers and RNA/DNA molecules and an increase in their transfection. In this paper, we perform the synthesis of arginine-modified poly-L-lysine dendrimer as well as confirm and characterize its structure and physicochemical properties by 1H and 13C NMR spectroscopy.
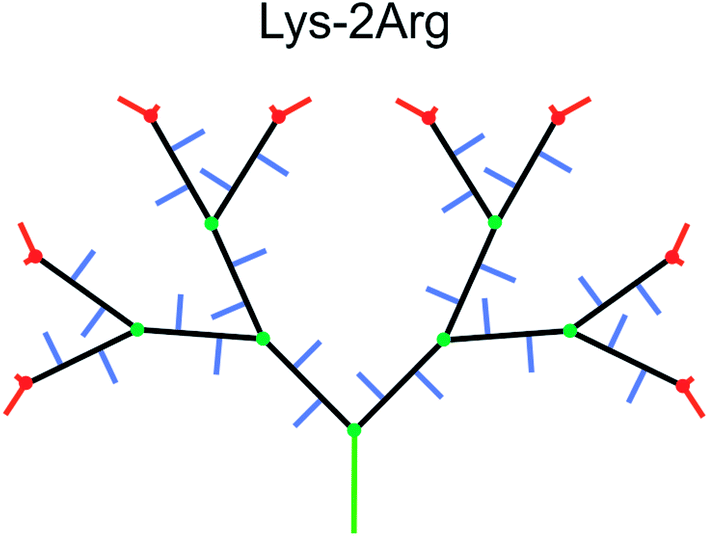 | ||
| Fig. 1 Schematic structure of the second generation Lys-2Arg dendrimer. Green color marks the core, black corresponds to the main chain, violet marks the side segments, and red corresponds to the terminal segments. Green and red circles show the Lys branching points in inner and terminal segments, respectively. | ||
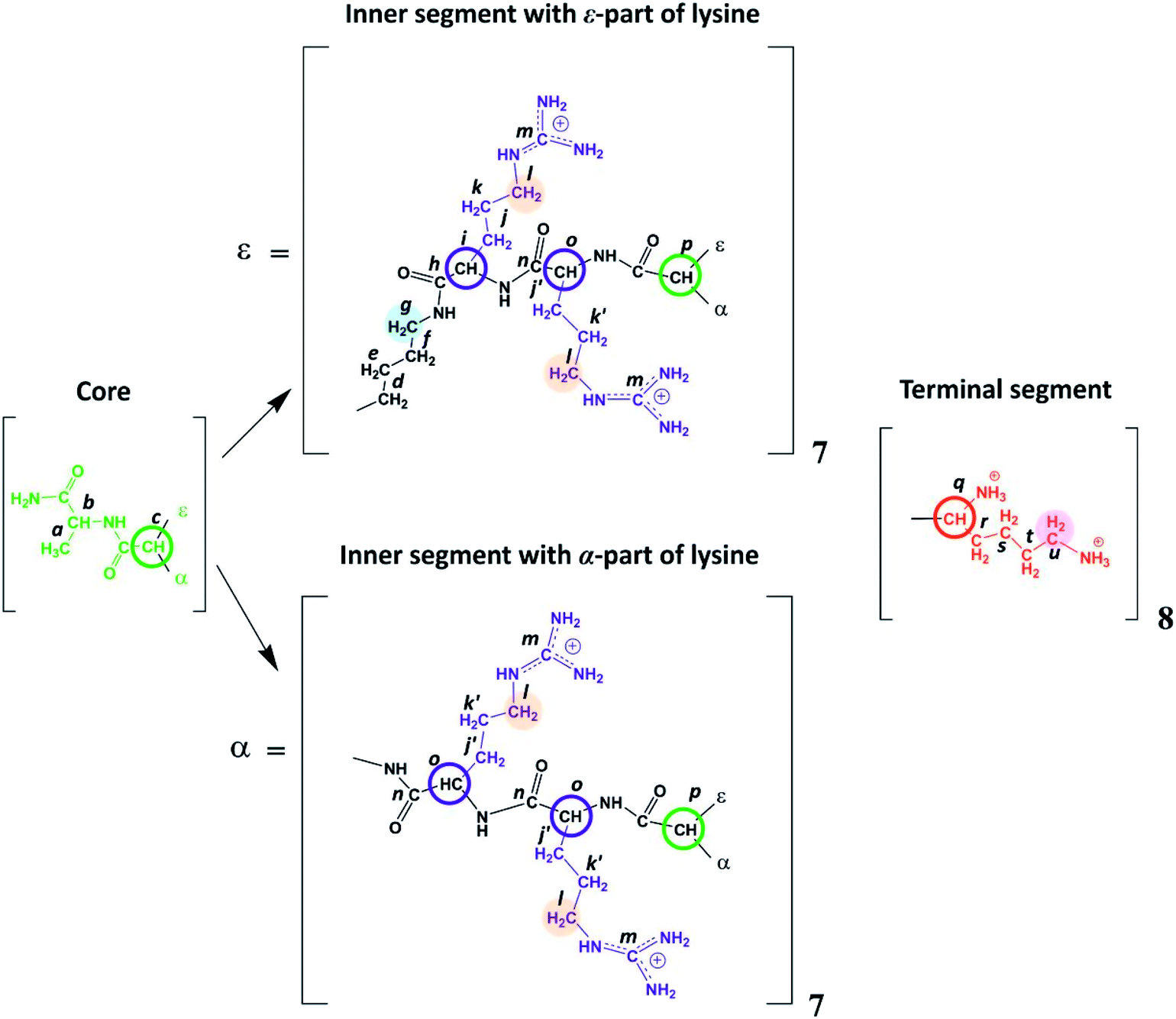 | ||
| Fig. 2 Chemical structure of Lys-2Arg dendrimer. The core of Lys-2Arg dendrimer consists of alanine amino acid residue. The inner segments contain ε- or α-part of Lys and two Arg residues. The side segments consist of ε-parts of Arg residues containing guanidine groups. The terminal segments contain Lys residues. Green color marks the core, black corresponds to the main chain, violet indicates the side segments, and red corresponds to the terminal segments. Green and red open circles show the Lys branching points in inner and terminal segments, respectively. Blue open circles show the CH groups of Arg. The CH2 groups of inner segment (i.e. of ε-part of branched Lys) connected to NH groups are highlighted by light blue, CH2 groups connected to NH groups in side segments are highlighted by orange, CH2 groups connected to protonated NH3+ groups in terminal segments (i.e. in ε-parts of terminal Lys) are highlighted by magenta. | ||
Experimental
Lysine-based second-generation dendrimer with inserted Arg residues (Lys-2Arg dendrimer) was synthesized by standard solid phase peptide synthesis (SPPS) (see ESI†). The structure of the investigated dendrimer is shown in Fig. 2. The core of Lys-2Arg dendrimer consists of alanine amino acid residue. The inner segments contain ε- or α-part of Lys and two Arg residues. The side segments consist of ε-parts of Arg residues containing guanidine groups. The terminal segments contain Lys residues. Lys-2Arg dendrimer has asymmetric branching.All NMR measurements were performed on a Bruker Avance III 500 MHz NMR (500 MHz and 126 MHz are frequencies for 1H and 13C, respectively) spectrometer equipped with a standard 5 mm BBFO direct observation probe and a Great 1/60A gradient system with a MIC S2 Diff/30 diffusive probe with a 1H convolution compensation coil (EVT). One-dimensional 1H and 13C NMR spectra were recorded. We also performed two-dimensional NMR experiments to confirm the Lys-2Arg dendrimer structure. 1H–1H COSY, 1H–13C HSQC, and HMBC spectra were obtained using standard pulse sequences. The 1H spin-lattice relaxation times, T1H, were acquired with an “inversion-recovery” pulse sequence modified by the destructive gradient pulses at the beginning of the sequence (“spoiler-recovery” sequence).22 The diffusion coefficient was measured by a stimulated echo pulse sequence with bipolar gradients to compensate for the effect of convection.23 We explored the signals from the CH2 groups chemically connected with N-atoms to study the orientational mobility in the dendrimer by NMR relaxation.
According to Fig. 2, Lys-2Arg dendrimer has the CH2–(N) groups inside the main chain, but only in those segments that contain the ε-part of lysine, and we consider them as “inner” groups in this study. Also, Lys-2Arg-dendrimer has the “side” CH2–(N) groups of Arg residues in side segments. Methylene groups connected with protonated NH3+ groups in the terminal segments are considered “terminal” ones.
Lys-2Arg dendrimer was dissolved in 0.154 M NaCl D2O (saline solution) at a concentration of about 1.50 g dl−1.
Results
1H and 13C spectral studies
There are three regions in the 1H NMR spectrum (Fig. 3), where peaks are observed: 4.39–3.89 ppm; 3.26–2.92 ppm and 2.03–1.22 ppm. In the first region (4.39–3.89 ppm), peaks 1–3 refer to CH groups in the dendrimer. The sum of the integrals for peaks 1–3 corresponds to the total content of protons in CH groups and has been taken as a reference. The other two regions belong to CH2 groups in different chemical environments. These are groups bonded to nitrogen-containing groups (peaks 4–6), and groups located in aliphatic parts (peaks 7–9). The integral intensity of peaks in these areas corresponds well to the chemical structure of the dendrimer (see Table 1).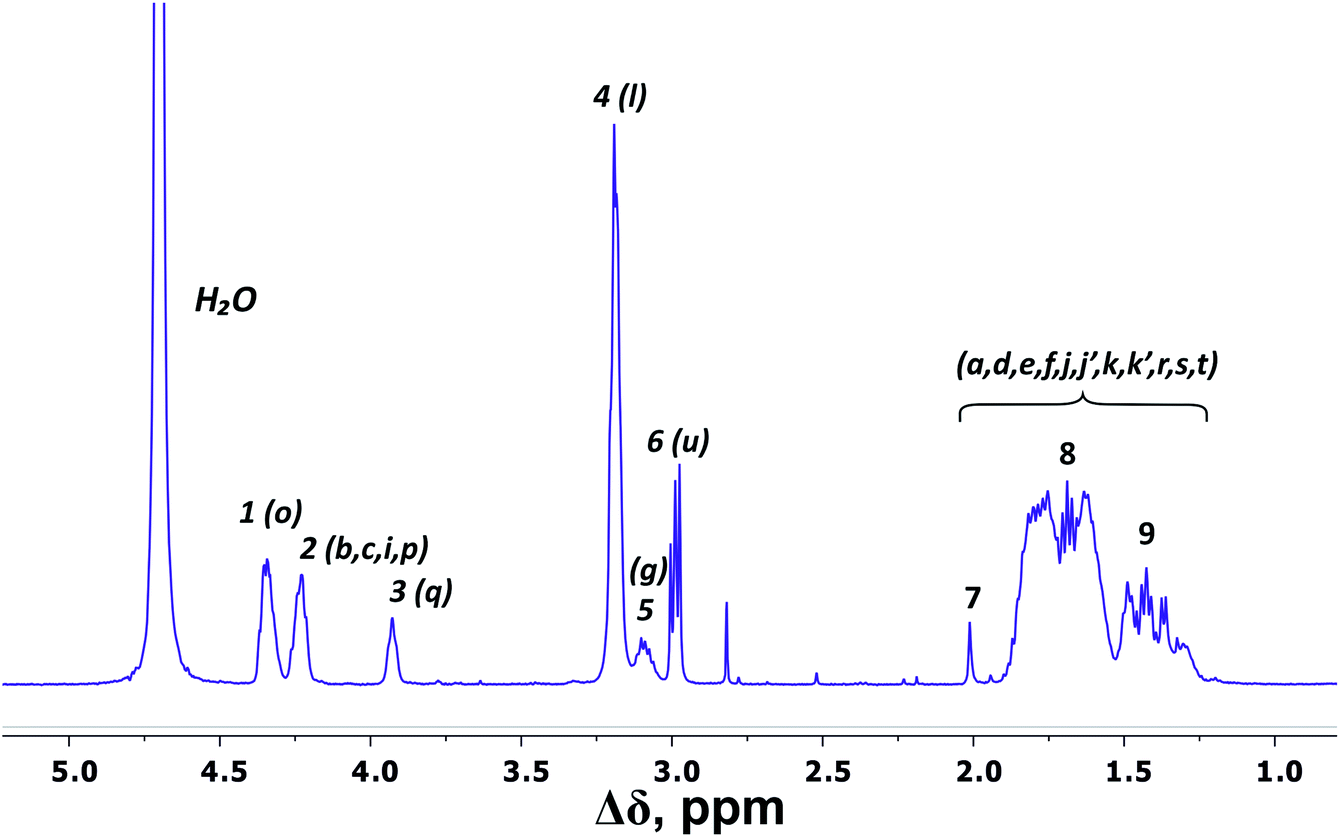 | ||
| Fig. 3 1H NMR spectrum of Lys-2Arg dendrimer at 298 K. The letter symbols correspond to the designations of the groups in Fig. 2. The attribution of peaks 1–9 is shown in Table 1. | ||
| Peak number | Type of group | Chemical shift, ppm | Integral value | Number of groups | Number of protons in groups | |
|---|---|---|---|---|---|---|
| a 92% side groups.b Located in core. | ||||||
| 1 | CH–(N) | 4.34 | 20.5 | 44.00 | 44 | 44 |
| 2 | CH–(N) | 4.23 | 16.3 | |||
| 3 | CH–(N) | 3.93 | 7.2 | |||
| 4 | CH2–(N) (side)a | 3.19 | 60.9 | 68.9 | 28 | 56 |
| 5 | CH2–(N) (inner) | 3.10, 3.19 | 8.0 | 7 | 14 | |
| 6 | CH2–NH3+ (terminal) | 2.99 | 16.2 | 16.2 | 8 | 16 |
| 7, 8, 9 | CH2, CH3b | 2.03–1.24 | 205.4 | 205.4 | 101 + 1b | 202 + 3b |
According to the chemical structure (Fig. 2), the dendrimer under consideration has several types of CH groups located: (1) at the Lys branching points between the inner segments; (2) at the side segments; (3) at the Lys branching points between the inner and the terminal segments. However, as will be shown below, there is no exact correspondence between peaks 1–3 and types of CH groups.
The electronegativity of a nitrogen atom influences protons in CH2 groups and leads to their increased chemical shift with respect to other CH2 groups. This means that peaks 4–6 in the range of 3.26–2.92 ppm belong to the CH2 groups attached to nitrogen (CH2–(N)). Three distinct peaks indicate the presence of CH2–(N) groups in three different chemical environments. According to the chemical structure, there are CH2–(N) groups in the main chain and in the side segments, and CH2–(N) groups bonded to the protonated NH3+ groups in the terminal segments (Fig. 2). The integral value of peak 6 is proportional to the number of protons in CH2–(N) groups in the terminal segments. Thus, peaks 4 and 5 refer to the side and the inner groups, respectively. Notice that the integral value of peak 5 is lower than the content of protons. Furthermore, the sum of the protons of peaks 4 and 5 is equal to 70 and the integral intensity is 68.9 (Table 1). We assume that peak 4 corresponds mainly to the signal from the side groups, and peak 5 corresponds to the signal from part of the protons of the inner groups. Then, using the 1H–13C HQSC, HMBC, and 1H–1H COSY experiments, we have shown that our assumption is true. For accuracy and consistency in using of terminology, we will associate peak 4 with the side groups (symbol l in Fig. 2 and 3), and peak 5 with the inner groups (symbol g in Fig. 2 and 3).
Peaks 7–9 are assigned to all other CH2 groups of Lys-2Arg dendrimer. Unfortunately, we were not able to carry out a detailed analysis in this region of the spectrum due to the overlapping of various peaks.
Fig. 4 shows the 13C NMR spectrum of Lys-2Arg dendrimer. According to the chemical structure, there are five types of 13C nuclei in the dendrimer: methyl (CH3), methylene (CH2), methine (CH), carbonyl (CO) and quaternary (C). The signals in the range of 173.5–172.9 ppm belong to carbon atoms located in carboxyl groups. The quaternary carbon of the guanidine group of arginine has a chemical shift equal to 156.71 ppm. The group of peaks in the range of 54–52.95 ppm refers to the carbon atoms in the CH groups. The next group of peaks in the range of 40.67–39 ppm refers to carbons of CH2 groups bonded to nitrogen atom. In the region of 31.25–21.20 ppm, there are peaks from carbons in the CH2 groups of the aliphatic part of the dendrimer.
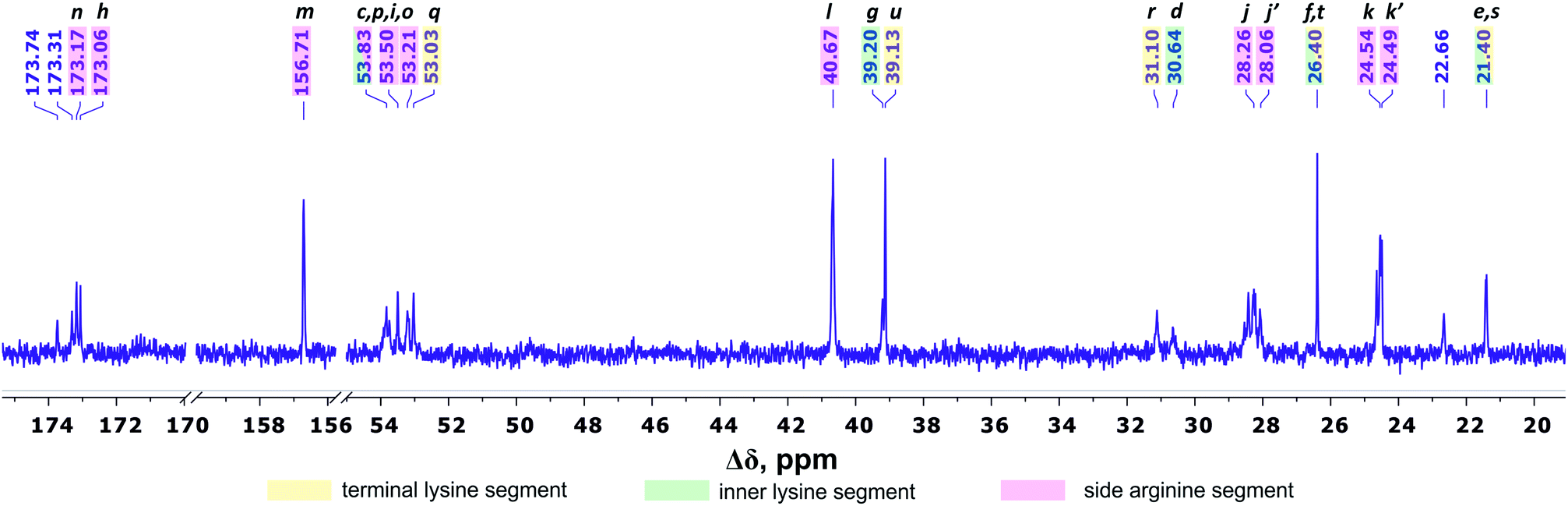 | ||
| Fig. 4 13C NMR spectrum of Lys-2Arg dendrimer at 298 K. The letter symbols correspond to the designations of the groups in Fig. 2. | ||
The primary analysis of the spectrum does not allow establishing the correspondence between peaks and carbon atoms of a particular segment. For accurate correlation of peaks in the 1H and 13C NMR spectra, we measured two-dimensional spectra. This allowed the possibility to determine a connection between the peaks in the spectrum of (i) hydrogen atoms in neighboring CHn groups (1H–1H COSY), (ii) carbons and hydrogens in one group (1H–13C HSQC), and (iii) carbons and protons in adjacent groups and groups separated by two to four chemical bonds (1H–13C HMBC).
The detailed analysis of two-dimensional spectra is provided in ESI.† Here, we present only the main results and conclusions. Cross peaks (3.19; 39.19) and (3.09; 39.19) on the 1H–13C HQSC spectrum of the dendrimer (Fig. 5) confirm that protons of the inner groups contribute to peaks 4 and 5 in Fig. 3 and Table 1. According to the 1H–1H COSY spectrum (Fig. 6), protons with chemical shifts of 3.09 and 3.19 ppm are in the same group. These two facts indicate that hydrogen nuclei in the same CH2 group have different chemical shifts (i.e., nonequivalent protons). In this case, the contribution of inner groups should be divided into two equal parts between peaks 4 and 5. According to Table 1, the calculated integral value of peak 5 is actually lower than the content of protons in the inner groups. Consequently, the part of the protons in the inner groups gives a signal at peak 4. The nonequivalence of these protons can be explained by their vicinity to the chiral carbon atom in the CH group of Arg (symbol i in Fig. 2). Despite chirality being present in most amino acids, we did not observe such an effect in the PLL dendrimers (with Lys-2Lys and Lys repeating units) that we studied earlier.24,25 It seems that features of the chemical structure of arginine contribute to the appearance of another peak, namely peak 5 (Fig. 3), in the range of the signal from CH2–(N) groups.
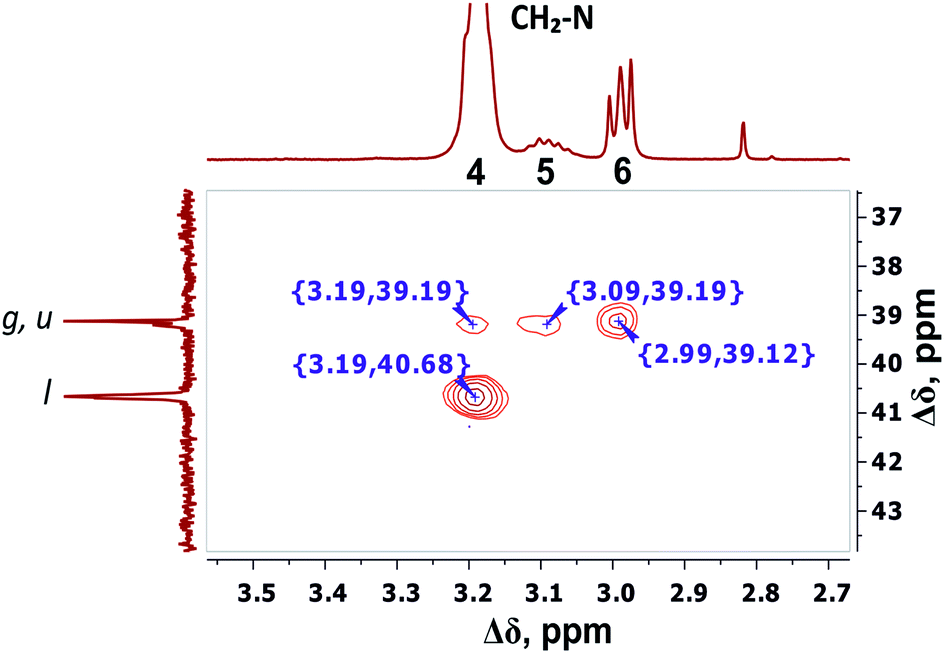 | ||
| Fig. 5 Two-dimensional 1H–13C HSQC spectrum of Lys-2Arg dendrimer in the range of 3.5–2.7 ppm at 298 K. | ||
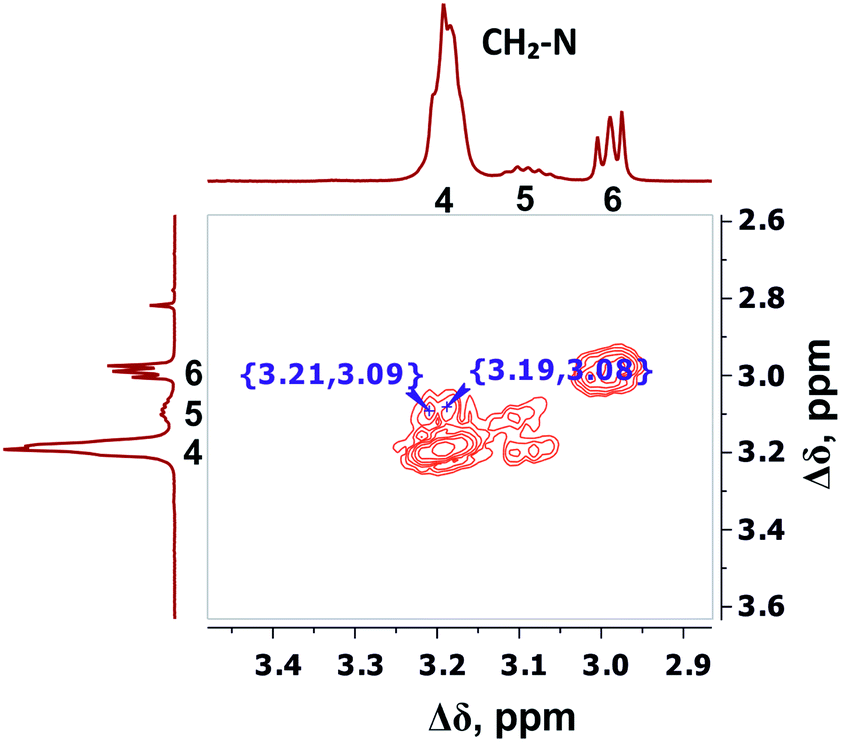 | ||
| Fig. 6 Two-dimensional 1H–1H COSY spectrum of Lys-2Arg dendrimer in the range of 3.4–2.9 ppm at 298 K. | ||
We conclude that 28 protons of CH groups of the side segments are distributed between peaks 1 and 2 as follows: seven protons (symbol i in Fig. 2) give a signal at peak 2, and all other protons (symbol o in Fig. 2) contribute to peak 1. According to the cross peak (4.25; 30.67) (Fig. 7), the protons of CH groups at Lys branching points between the inner segments (Fig. 2, green open circles) also contribute to the integral of peak 2.
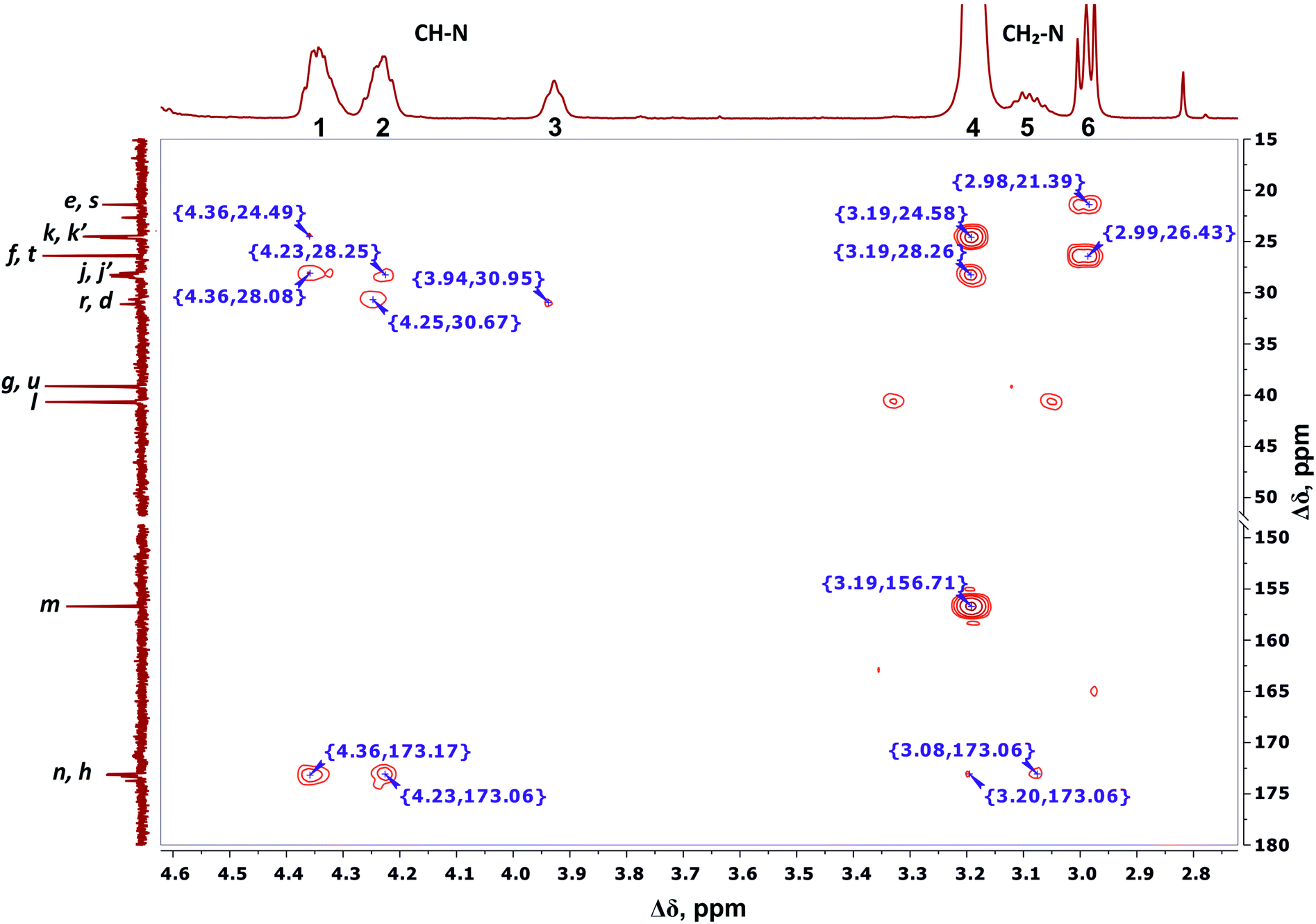 | ||
| Fig. 7 Two-dimensional 1H–13C HMBC spectrum of Lys-2Arg dendrimer in the range of 4.6–2.7 ppm at 298 K. The letter symbols correspond to the designations of the groups in Fig. 2. | ||
By analogy with CH2–(N) groups, we consider that the protons at peak 3 with the smallest chemical shift of 3.94 ppm belong to CH groups located at Lys branching points between the inner and the terminal segments (Fig. 2, red open circles).
According to 1H–13C HMBC data, the assignment of chemical shift at 156.71 ppm (symbol m in Fig. 4) to the carbon signal of the guanidine group is confirmed by the presence of cross-peak (3.19; 156.71) (Fig. 7) with protons in the side CH2–(N) groups in Arg residues. Also, the chemical shift (156.71 ppm) of carbon in the guanidine group is close to the chemical shift of carbon in the guanidine group in L-arginine (159.50 ppm).
Thus, the detailed analysis of the one-dimensional and two-dimensional NMR spectra on 1H and 13C nuclei make it possible to confirm the structure of Lys-2Arg dendrimer. Moreover, the ratio of CH, CH2–(N) and CH2 groups in the 1H NMR spectrum (Fig. 3) differs by less than 2% from the theoretically calculated one according to the structural formula (Fig. 2). Also, it is important to repeat that the inner groups possess chemically nonequivalent protons.
Diffusion
We estimated the hydrodynamic parameters of Lys-2Arg dendrimer. The diffusion coefficient is D = 1.02 × 1010 m2 s−1. The calculation details are presented in ESI.† This value allows us to determine the hydrodynamic radius by using the Stokes–Einstein eqn (1):
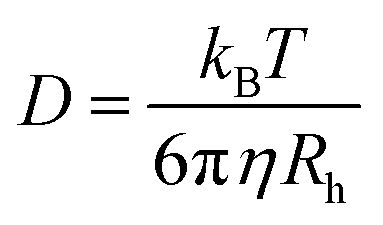 | (1) |
The calculated hydrodynamic radius from eqn (1) is 1.93 nm. It was found that the diffusion coefficient and hydrodynamic radius are the same as in Lys-2Lys dendrimer, which we obtained in our previous paper.24 In comparison with Lys-2Lys dendrimer, Lys-2Arg dendrimer has a slightly higher density equal to 0.35 g cm−3 (in contrast to 0.31 g cm−3). Therefore, we can conclude that the guanidine groups in side segments of Lys-2Arg dendrimer do not lead to significant changes in size and density of Lys-2Arg dendrimer in comparison with Lys-2Lys dendrimer.
1H NMR relaxation
Local orientational mobility in macromolecules can be studied by NMR relaxation measurements. For these studies, we considered the temperature dependence of spin-lattice relaxation for the peptide dendrimers. The spin-lattice relaxation rate for the dipole–dipole relaxation mechanism of 1H nuclei is written as24,26–30
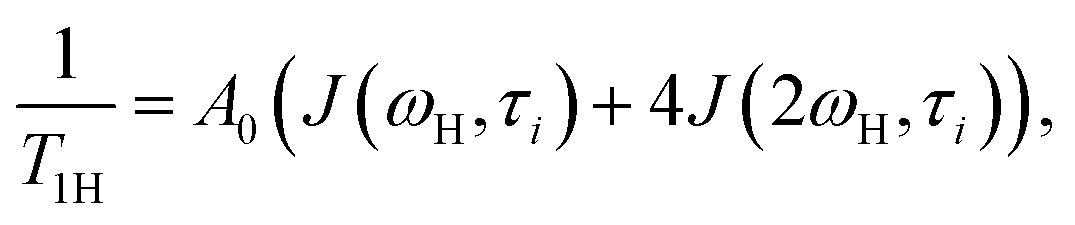 | (2) |
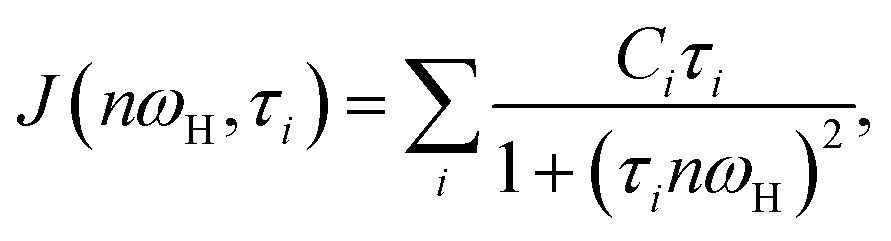 | (3) |
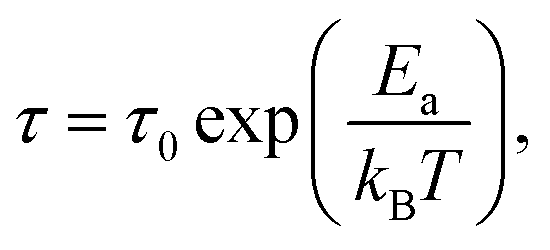 | (4) |
It is important to note that spectral density for different groups of a dendrimer macromolecule cannot be described in the framework of usual simplifications that take into account one or two correlation times. The developed theory of orientational mobility in dendrimers and hyperbranched macromolecules25,31–42 shows that local reorientation of a group in the dendrimer is determined by three main processes:
(i) Local reorientation of a given group which can be characterized by one relaxation time that is independent of the macromolecule size and the position of the group in the dendrimer;
(ii) Pulsation of branches/subbranches characterized by a set of relaxation times corresponding to the rotation as a whole of the pulsating branches/subbranches with various sizes;
(iii) The rotation of the dendrimer as a whole depends only on the size of the dendrimer.
We presented a detailed description of this theory in our recent review.43 It is important to note that the developed theory is in agreement with the experimental data, in particular for poly(acryl ether) dendrimers44,45 where it is possible to measure 1/T1H for different dendrimer shells.
Fig. 8 shows the temperature dependences of 1/T1H for the side (peak 4), inner (peak 5) and terminal groups (peak 6).
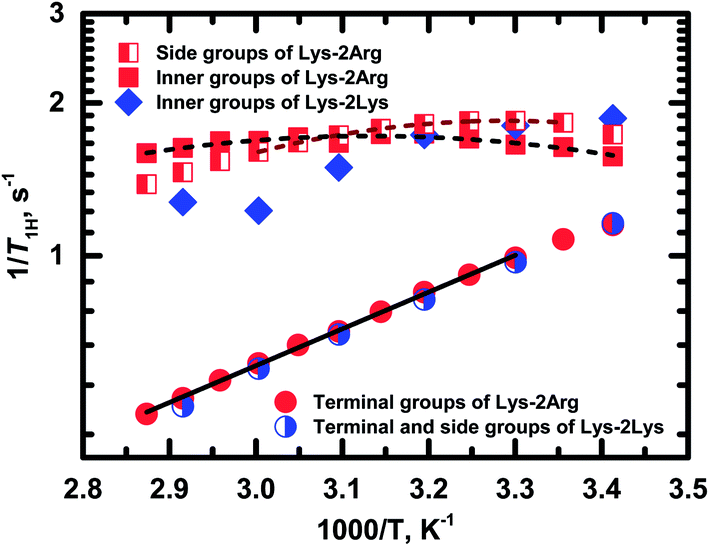 | ||
| Fig. 8 Temperature dependences of the spin-lattice relaxation rate, 1/T1H, for different groups in Lys-2Arg and Lys-2Lys dendrimers. Experimental data are shown as symbols. Dash and solid lines indicate the fitting of experimental data for Lys-2Arg dendrimer by eqn (2)–(4) and Arrhenius equation, respectively. | ||
Exponential growth of the 1/T1H dependence with decreasing temperature is observed for the terminal groups. The results of fitting by Arrhenius equation are shown in Table 2. Activation energy, Ea, of the terminal groups equals to 14 kJ mol−1 and is similar to the Ea value for the terminal groups in Lys dendrimer.38
| Type of CH2–(N) groups | A0 × 10−10 (±20%) | τ0 [ps] (±20%) | Ea [kJ mol−1] (±5%) | τc [ps] at 298 K |
|---|---|---|---|---|
a 92% side groups.b The absence of a maximum of dependence for the terminal groups does not allow extracting τc from this dependence. Thus, we used parameter A0 for the inner groups and calculated τ0 by 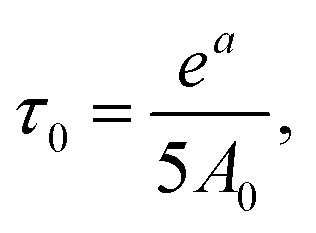 where a is a parameter of Arrhenius fitting. This equation can be obtained from eqn (2)–(4) when ωτc ≪ 1. In this table the values of τc are shown. where a is a parameter of Arrhenius fitting. This equation can be obtained from eqn (2)–(4) when ωτc ≪ 1. In this table the values of τc are shown. |
||||
| Sidea | 0.41 | 0.21 | 17 | 200 |
| Inner | 0.38 | 1.03 | 14 | 293 |
| Terminal | 0.38b | 0.21b | 14 | 60b |
In the case of the side and inner groups, the maxima of 1/T1H dependencies have appeared. The maximum for the inner groups is at a higher temperature (323 K) than for the side groups (303 K). To estimate the difference between correlation times of the side and inner groups, we approximated 1/T1H dependences by eqn (2)–(4) using the simplest approach with one correlation time, τc, which is an average relaxation time in the region of the 1/T1H maximum. Results of these fittings are shown in Table 2. The activation energy of the side groups is higher than Ea of inner groups but τc is shorter for the side groups. For illustration, we present the calculated values of τc at room temperature (Table 2). Apparently, all three processes contribute to the mobility of the inner groups; as a result, we see a wider maximum than for the side groups (Fig. 8). In the case of the side groups, the maximum should be determined only by the second process – branch pulsations. Also, an important result is that the correlation time for the terminal groups is significantly faster than τc for the side and inner groups. This result is caused by the fact that the correlation time depends only on the mobility of the terminal segment, i.e., the smallest possible dendrimer branch.43 The explanation of this behavior is presented below in Discussion.
Discussion
As mentioned above, the size of Lys-2Arg dendrimer does not change in comparison with Lys-2Lys dendrimer. However, NMR relaxation of the inner and the side groups in Lys-2Arg dendrimer changed dramatically. To illustrate these changes, we compare 1/T1H dependences for Lys-2Arg and Lys-2Lys dendrimers in Fig. 8. The temperature dependences of 1/T1H for the terminal groups of both dendrimers are practically identical and possess exponential growth with a decrease of temperature. This behavior is due to the high orientational mobility of the terminal groups in both dendrimers. The opposite is true for the side groups. In Lys-2Lys dendrimers, the side groups have the same high mobility as the terminal groups. In Lys-2Arg, the temperature dependence of 1/T1H for the side groups has the maximum at 303 K. The correlation time, τc, of the side groups is longer by several times than τc of the terminal groups. This demonstrates the low mobility of the side groups. It is important to note that the shift in the position of 1/T1H maximum to high temperatures means the slowing down of the orientational mobility. The inner groups in Lys-2Arg dendrimer have lower mobility than the side groups because the maximum is achieved at 323 K and τc is longer than τc of the side groups.According to the theory, the mobility of the side segments should be similar to the mobility of the terminal segments.24,40 This statement was confirmed by our recent experimental work using Lys-2Lys dendrimers.24 The opposing situation is in Lys-2Arg dendrimer, where the mobility of the side groups is similar to the mobility of the inner ones. This effect is probably connected with some specific feature of guanidine groups in the side segments of Lys-2Arg dendrimer.
The obvious explanation for this effect is that there are extra interactions between the guanidine groups, with each other or with other dendrimer parts. Unfortunately, we cannot establish the nature of these guanidine group interactions by using NMR methods. However, the interaction between guanidine groups in neighboring side segments occurs due to a like-charge arginine–arginine pairing effect.46–49 This effect is absent in aqueous poly-lysine containing ammonium-terminated side chains.46 Most likely, these interactions of guanidine groups of Arg residues hinder the orientational mobility of the side segments. This effect can cause a 1/T1H maximum for the mobility of the side groups. Moreover, any additional interactions between dendrimer branches slow down the reorientation of these branches. As shown earlier (see review43), the mobility of a branch – the pulsation of a branch – as a whole makes a significant contribution to the NMR relaxation of inner groups. Thus, the same guanidine interactions can lead to a shift of 1/T1H maximum to higher temperatures. It is important to note that NMR relaxation has been utilized for studies of structural properties of dendrimers; for example, see our works in ref. 50–52.
Additionally, we would like to draw attention to the nonequivalent protons in the inner groups, which were found in the analysis of the NMR spectra above. On the one hand, this nonequivalence can be explained by the chirality of lysine and arginine amino acids. On the other hand, it is more likely caused by a slowdown in the mobility of the side segments. Thus, the conformation in which hydrogen nuclei within the same CH2 group have different chemical shifts is the predominant conformation for Lys-2Arg dendrimer. This assumption is supported by the fact that there is no similar effect in Lys-2Lys dendrimer, in which the guanidine groups are “replaced” with NH2. However, we should highlight the difference between NMR spectroscopy and relaxation data. In NMR relaxation, the correlation time values for dendrimer groups do not exceed 1 ns. However, to detect the nonequivalence of the protons of the CH2 group in an NMR spectral experiment, the lifetime of this conformation on the order of milliseconds is required.
We plan further study of the influence of the guanidine–guanidine pairing effect on NMR relaxation in dendrimers by molecular dynamic simulations methods. We believe that the possibility of detecting this pairing effect in dendrimers using a relatively simple NMR relaxation method is of interest to biological applications.
Conclusions
In this work, we have synthesized and studied physicochemical properties of a new structure of second-generation poly-L-lysine dendrimer, in which segments (repeating units) between lysine branching points of the dendrimer consist of two linear Arg residues. This modification could be favorable for an increase of transfection by lysine based dendrimers that is important for biomedical applications.20Lys-2Arg dendrimer was synthesized by standard solid phase peptide synthesis. The structure of the dendrimer (see Fig. 1 and 2) was established by one and two dimensional 1H and 13C NMR spectroscopy, including 1H–1H COSY, 1H–13C HSQC, and HMBC techniques as well as relaxation (T1H) and diffusion measurements.
NMR spectroscopy studies allowed us not only to confirm the claimed structure but also to find the presence of nonequivalence of protons in the inner CH2–(N) groups, which is absent in similar Lys-2Lys dendrimer. Moreover, this circumstance should be taken into account in further study of peptide dendrimers. It will allow more accurate analysis of the 1H NMR spectra and avoid misinterpretation of the integral values. T1H relaxation measurements showed a non-typical sharp decrease in mobility of methylene groups in side segments. For typical dendrimer topology, the mobility of the side CH2–(N) groups should be close to fast mobility of the terminal CH2–(N) groups; for instance, see the results for Lys-2Lys dendrimer in ref. 24 In the case of Lys-2Arg dendrimer, the mobility of side segments is similar to the mobility of inner segments. Our estimation of the correlation times, τc, at room temperature for CH2–(N) groups in the inner, side, and terminal segments gave the following values: 290 ps, 200 ps, and 60 ps, respectively. This unexpected T1H behavior is most likely caused by well-known guanidine–guanidine pairing effect in water, which leads to entanglements between dendrimer branches. Thus, the unique structure of the dendrimer creates the possibility to identify the presence of entanglements inside the macromolecule using fairly simple measurements of NMR relaxation. We believe that this finding will be important not only to establish the structure of dendrimers but also to study a wider class of hyperbranched macromolecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NMR measurements were carried out in the Center for Magnetic Resonance of Research Park of St. Petersburg State University. This work is supported by the Russian Science Foundation (grants No. 19-13-00087). E. L. and N. N. S. are thankful for the Researcher Mobility Grant of the Academy of Finland (No. 318608) and the travel grant provided by Saint Petersburg State University (Event 6, ID 36154550). I. M. N. was supported by grant 08-08 of the Government of Russian Federation.References
- A. Samad, I. Alam and K. Saxena, Curr. Pharm. Des., 2009, 15, 2958–2969 Search PubMed.
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef.
- S. Mutalik, H. S. Parekh, Y. G. Anissimov, J. E. Grice and M. S. Roberts, Skin Pharmacol. Physiol., 2013, 26, 127–138 CrossRef CAS PubMed.
- B. Noriega-Luna, L. A. Godínez, F. J. Rodríguez, A. Rodríguez, G. Zaldívar-Lelo de Larrea, C. F. Sosa-Ferreyra, R. F. Mercado-Curiel, J. Manríquez and E. Bustos, J. Nanomater., 2014, 2014, 1–19 CrossRef.
- M. Ghaffari, G. Dehghan, F. Abedi-Gaballu, S. Kashanian, B. Baradaran, J. E. N. Dolatabadi and D. Losic, Eur. J. Pharm. Sci., 2018, 122, 311–330 CrossRef CAS PubMed.
- L. M. Bronstein and Z. B. Shifrina, Chem. Rev., 2011, 111, 5301–5344 CrossRef CAS PubMed.
- L. Palmerston Mendes, J. Pan and V. Torchilin, Molecules, 2017, 22, 1401 CrossRef PubMed.
- S. S. Santos, R. V. Gonzaga, J. V. Silva, D. F. Savino, D. Prieto, J. M. Shikay, R. S. Silva, L. H. A. Paulo, E. I. Ferreira and J. Giarolla, Can. J. Chem., 2017, 95, 907–916 CrossRef CAS.
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274–5300 CrossRef CAS PubMed.
- T. Kuanga, D. Fub, L. Changb, Z. Yangb, Z. Chenc, L. Jine, F. Chenb and X. Peng, Curr. Org. Chem., 2016, 20, 1820–1826 CrossRef.
- T. Okuda, A. Sugiyama, T. Niidome and H. Aoyagi, Biomaterials, 2004, 25, 537–544 CrossRef CAS PubMed.
- A. Kwok, G. A. Eggimann, M. Heitz, J. L. Reymond, F. Hollfelder and T. Darbre, ChemBioChem, 2016, 17, 2223–2229 CrossRef CAS PubMed.
- C. Liu, X. Liu, P. Rocchi, F. Qu, J. L. Iovanna and L. Peng, Bioconjugate Chem., 2014, 25, 521–532 CrossRef CAS PubMed.
- J. B. Kim, J. S. Choi, K. Nam, M. Lee, J.-S. Park and J.-K. Lee, J. Controlled Release, 2006, 114, 110–117 CrossRef CAS PubMed.
- T. Kim, C. Z. Bai, K. Nam and J. Park, J. Controlled Release, 2009, 136, 132–139 CrossRef CAS PubMed.
- H. Hirose, T. Takeuchi, H. Osakada, S. Pujals, S. Katayama, I. Nakase, S. Kobayashi, T. Haraguchi and S. Futaki, Mol. Ther., 2012, 20, 984–993 CrossRef CAS PubMed.
- K. Luo, C. Li, L. Li, W. She, G. Wang and Z. Gu, Biomaterials, 2012, 33, 4917–4927 CrossRef CAS PubMed.
- T. Kim, J. Baek, C. Zhe Bai and J.-S. Park, Biomaterials, 2007, 28, 2061–2067 CrossRef CAS.
- X. Liu, C. Liu, J. Zhou, C. Chen, F. Qu, J. J. Rossi, P. Rocchi and L. Peng, Nanoscale, 2015, 7, 3867–3875 RSC.
- A. Kwok, G. A. Eggimann, J.-L. L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- S. Falkovich, D. Markelov, I. Neelov and A. Darinskii, J. Chem. Phys., 2013, 139, 064903 CrossRef CAS PubMed.
- G. H. Sorland, in Dynamic Pulsed-Field-Gradient NMR, Springer-Verlag Berlin, Heidelberger Platz 3, D-14197 Berlin, Germany, 2014, vol. 110, pp. 169–190 Search PubMed.
- A. Jerschow and N. Müller, J. Magn. Reson., Ser. A, 1996, 123, 222–225 CrossRef.
- N. N. Sheveleva, D. A. Markelov, M. A. Vovk, M. E. Mikhailova, I. I. Tarasenko, I. M. Neelov and E. Lähderanta, Sci. Rep., 2018, 8, 1–7 CrossRef CAS PubMed.
- D. A. Markelov, S. G. Falkovich, I. M. Neelov, M. Y. Ilyash, V. V. Matveev, E. Lähderanta, P. Ingman and A. A. Darinskii, Phys. Chem. Chem. Phys., 2015, 17, 3214–3226 RSC.
- A. Abragam, The principles of nuclear magnetism, Clarendon Press; Oxford University Press, Oxford [Oxfordshire], New York, 1983 Search PubMed.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- I. Solomon, Phys. Rev., 1955, 99, 559–565 CrossRef CAS.
- V. I. Chizhik, Y. S. Chernyshev, A. V. Donets, V. V. Frolov, A. V. Komolkin and M. G. Shelyapina, Magnetic Resonance and Its Applications, Springer, Cham, 2014 Search PubMed.
- M. H. Levitt, Spin dynamics: basics of NMR, Chichester, England, Hoboken, NJ, 2nd edn, 2008 Search PubMed.
- Y. Y. Gotlib and D. A. Markelov, Polym. Sci., Ser. A, 2004, 46, 815–832 Search PubMed.
- O. V Shavykin, I. M. Neelov and A. A. Darinskii, Phys. Chem. Chem. Phys., 2016, 18, 24307–24317 RSC.
- M. Dolgushev, S. Schnell and D. A. Markelov, Appl. Magn. Reson., 2017, 48, 657–671 CrossRef.
- D. A. Markelov, F. Fürstenberg and M. Dolgushev, Polymer, 2018, 144, 65–71 CrossRef CAS.
- Y. Y. Gotlib and D. A. Markelov, Polym. Sci., Ser. A, 2007, 49, 1137–1154 CrossRef.
- D. A. Markelov, S. V. Lyulin, Y. Y. Gotlib, A. V. Lyulin, V. V. Matveev, E. Lahderanta and A. A. Darinskii, J. Chem. Phys., 2009, 130, 1–9 CrossRef PubMed.
- I. Neelov, S. Falkovich, D. Markelov, E. Paci, A. Darinskii and H. Tenhu, in Dendrimers in Biomedical Applications, The Royal Society of Chemistry, 2013, pp. 99–114 Search PubMed.
- I. M. Neelov, D. A. Markelov, S. G. Falkovich, M. Y. Ilyash, B. M. Okrugin and A. A. Darinskii, Polym. Sci., Ser. C, 2013, 55, 154–161 CrossRef CAS.
- D. A. Markelov, M. Dolgushev, Y. Y. Gotlib and A. Blumen, J. Chem. Phys., 2014, 140, 244904 CrossRef PubMed.
- M. Dolgushev, D. A. Markelov, F. Fürstenberg and T. Guérin, Phys. Rev. E, 2016, 94, 012502 CrossRef.
- D. A. Markelov, A. N. Shishkin, V. V Matveev, A. V Penkova, E. Lähderanta and V. I. Chizhik, Macromolecules, 2016, 49, 9247–9257 CrossRef CAS.
- J. Grimm and M. Dolgushev, Phys. Chem. Chem. Phys., 2016, 18, 19050–19061 RSC.
- D. A. Markelov, M. Dolgushev and E. Lähderanta, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2017, vol. 91, pp. 1–66 Search PubMed.
- L. F. Pinto, R. Riguera and E. Fernandez-Megia, J. Am. Chem. Soc., 2013, 135, 11513–11516 CrossRef CAS.
- L. F. Pinto, J. Correa, M. Martin-Pastor, R. Riguera and E. Fernandez-Megia, J. Am. Chem. Soc., 2013, 135, 1972–1977 CrossRef CAS PubMed.
- J. Vondrášek, P. E. Mason, J. Heyda, K. D. Collins and P. Jungwirth, J. Phys. Chem. B, 2009, 113, 9041–9045 CrossRef PubMed.
- J. Gao, C. Tang, M. A. Elsawy, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2017, 18, 826–834 CrossRef CAS PubMed.
- D. Lee, J. Lee and C. Seok, Phys. Chem. Chem. Phys., 2013, 15, 5844–5853 RSC.
- C. P. Schneider, D. Shukla and B. L. Trout, J. Phys. Chem. B, 2011, 115, 7447–7458 CrossRef CAS PubMed.
- D. A. Markelov, V. V. Matveev, P. Ingman, M. N. Nikolaeva, A. V. Penkova, E. Lahderanta, N. I. Boiko and V. I. Chizhik, Sci. Rep., 2016, 6, 24270 CrossRef PubMed.
- V. V Matveev, D. A. Markelov, S. V Dvinskikh, A. N. Shishkin, K. V Tyutyukin, A. V Penkova, E. A. Tatarinova, G. M. Ignat’eva and S. A. Milenin, Sci. Rep., 2017, 7, 13710 CrossRef PubMed.
- D. A. Markelov, V. V. Matveev, P. Ingman, M. N. Nikolaeva, E. Lähderanta, V. A. Shevelev and N. I. Boiko, J. Phys. Chem. B, 2010, 114, 4159–4165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02461a |
| This journal is © The Royal Society of Chemistry 2019 |