
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUPLC-MS-guided isolation of single ether linkage dimeric 2-(2-phenylethyl)chromones from Aquilaria sinensis†
Tongdong Kuang‡
ab,
Hui-Qin Chen‡ab,
Hao Wangab,
Fan-Dong Kongab,
Cai-Hong Caiab,
Wen-Hua Dongab,
Jing-Zhe Yuanab,
Wen-Li Mei*ab and
Hao-Fu Dai *ab
*ab
aHainan Engineering Research Center of Agarwood, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101, PR China. E-mail: meiwenli@itbb.org.cn; daihaofu@itbb.org.cn
bHainan Key Laboratory for Research and Development of Natural Products from Li Folk Medicine, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101, PR China
First published on 31st May 2019
Abstract
Eleven novel uncommon single ether linkage dimeric 2-(2-phenylethyl)chromones (aquilasinenones A–K) were isolated by a UPLC-MS guided method from artificial hole-induced agarwood originating from Aquilaria sinensis. Their structures were unambiguously deduced using HRESIMS data, detailed 1D and 2D NMR spectroscopic analysis, and experimental ECD spectra. All the compounds were tested for acetylcholinesterase (AChE) inhibitory activity by Ellman's colorimetric method, and compounds 9–11 displayed weak AChE inhibitory activity with the inhibition ranging from 15.6% to 16.8% at a concentration of 50 μg mL−1.
1. Introduction
Agarwood, widely used in perfumes, incenses, and traditional medicine, is an aromatic dark resin produced by the heartwood of the Aquilaria and Gyrinops species of the Thymelaeaceae family when suffering from a natural or artificial injury.1–3 Aquilaria sinensis (Lour.), which is distributed in southern China, is a peculiar plant resource of Chinese agarwood.42-(2-Phenylethyl)chromones are characterized as a group of uncommon chromones possessing a phenylethyl substituent at C-2 and have been reported to be one of the main diagnostic constituents of agarwood.5,6 Interestingly, 2-(2-phenylethyl)chromone dimers and trimers have been derived from agarwood recently; even though the first batch from Kalimantan agarwood was reported by Siu Kiyosawa's group,7–11 the process could not be traced until the analogues were rediscovered again in 2017.12–16 These distinctive structures originating from agarwood showed diverse linkages between the subunits via carbon–carbon bonds and/or ether bonds. However, not more than twenty structures featuring a single ether linkage between the 5,6,7,8-tetrahydro-2-(2-phenylethyl)chromone and 2-(2-phenylethyl)chromone units have been reported since the first one was found,3,16 and they exhibited various biological activities including acetylcholinesterase (AChE) inhibition, antineuroinflammatory action, and cytotoxicity in pharmacological researches.7–16
Previous phytochemical studies on the chemical constituents of artificial hole-induced agarwood originating from A. sinensis contributed to the identification of a series of sesquiterpenes17 and 2-(2-phenylethyl)chromone derivatives,18–20 including four 2-(2-phenylethyl)chromone dimers.13 Herein, the ongoing studies targeted the distinct characterization of the 5,6,7,8-tetrahydro-2-(2-phenylethyl)chromone and 2-(2-phenylethyl)chromone dimer derivatives guided by UPLC-MS, which led to the isolation and elucidation of eleven 2-(2-phenylethyl)chromone dimers with an ether linkage, as well as the identification of their absolute configurations. All the dimers were tested for acetylcholinesterase (AChE) inhibitory activity by Ellman's colorimetric method, and compounds 9–11 displayed weak AChE inhibitory activity with inhibitions in the range of 15.6–16.8% at a concentration of 50 μg mL−1.
2. Results and discussion
Aquilasinenone A (1) was obtained as a yellow oil. Its molecular formula was established as C37H36O13 based on the HRESIMS pseudomolecular ion peak at m/z 711.2057 [M + Na]+ (calcd for C37H36NaO13, 711.2048). The 1H NMR spectrum (Table 1) displayed three methoxy groups, two singlet olefinic protons, and two sets of ABX coupling aromatic systems, along with four methylenes and four oxygenated methines. The 13C NMR spectrum (Table 2) presented two carbonyl groups, twenty-four olefinic carbons, four oxygenated tertiary carbons, three methoxyl carbons, and four secondary carbons. The above data hinted a dimeric 2-(2-phenylethyl)chromone derivative comprising 5,6,7,8-tetrahydro-2-(2-phenylethyl)chromone (unit A) and 2-(2-phenylethyl)chromone (unit B) moieties.| Position | 1a | 2a | 3b | 4a | ||||
|---|---|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| a Measured in CD3OD.b Measured in DMSO-d6. | ||||||||
| 3/3′ | 6.13, s | 6.12, s | 6.12, s | 6.10, s | 6.12, s | 6.05, s | 6.14, s | 6.10, s |
| 5/5′ | 4.83, d (3.8) | 7.55, s | 4.82, d (3.7) | 7.54, s | 4.58, d (3.6) | 7.32, s | 4.82, d (3.8) | 7.56, s |
| 6/6′ | 4.14, dd (3.8, 2.3) | 4.14, dd (3.7, 2.1) | 3.87, dd (3.6, 2.3) | 4.14, dd (3.8, 2.3) | ||||
| 7/7′ | 4.48, dd (7.6, 2.3) | 4.47, dd (7.6, 2.1) | 4.30, dd (8.0, 2.3) | 4.47, dd (7.6, 2.3) | ||||
| 8/8′ | 5.63, d (7.6) | 7.60, s | 5.64, d (7.6) | 7.58, s | 5.39, d (8.0) | 7.55, s | 5.63, d (7.6) | 7.59, s |
| 2′′/2′′′ | 6.46, d (2.1) | 6.73, d (1.8) | 6.43, d (1.8) | 6.65, br s | 6.65, d (2.1) | 6.51, d (2.1) | 6.45, d (2.1) | 7.06, d (8.6) |
| 3′′/3′′′ | 7.76, d (8.6) | |||||||
| 5′′/5′′′ | 6.67, d (8.1) | 6.65, d (8.0) | 6.65, d (6.7) | 6.71, d (8.2) | 6.75, d (8.2) | 6.67, d (8.2) | 6.67, d (8.2) | 7.76, d (8.6) |
| 6′′/6′′′ | 6.32, dd (8.1, 2.1) | 6.59, dd (8.0, 1.8) | 6.29, dd (8.2, 1.8) | 6.55, dd (8.2, 1.8) | 6.55, d (8.2, 2.1) | 6.32, d (8.2, 2.1) | 6.31, dd (8.2, 2.1) | 7.06, d (8.6) |
| 7′′/7′′′ | 2.52, m | 2.93, m | 2.49, m | 2.88, m | 2.60, m | 2.82, m | 2.51, m | 2.94, m |
| 8′′/8′′′ | 2.64, m | 2.93, m | 2.62, m | 2.88, m | 2.60, m | 2.82, m | 2.64, m | 2.91, m |
| OCH3 | 3.75, s (4′′-OCH3) | 3.92, s (6′-OCH3) | 3.75, s (4′′-OCH3) | 3.90, s (6′-OCH3) | 3.69, s (4′′-OCH3) | 3.68, s (4′′′-OCH3) | 3.75, s (4′′-OCH3) | 3.93, s (6′-OCH3) |
| 3.74, s (3′′′-OCH3) | 3.74, s (4′′′-OCH3) | 3.71, s (4′′′-OCH3) | ||||||
| Position | 1a | 2a | 3b | 4a | ||||
|---|---|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| a Measured in CD3OD.b Measured in DMSO-d6. | ||||||||
| 2/2′ | 171.3, C | 171.0, C | 171.2, C | 171.0, C | 167.9, C | 167.9, C | 170.9, C | 171.2, C |
| 3/3′ | 114.5, CH | 110.1, CH | 114.5, CH | 110.1, CH | 113.2, CH | 108.7, CH | 114.5, CH | 110.1, CH |
| 4/4′ | 181.4, C | 179.8, C | 181.5, C | 179.8, C | 178.0, C | 176.1, C | 181.5, C | 179.8, C |
| 5/5′ | 66.3, CH | 105.8, CH | 66.4, CH | 105.8, CH | 64.4, CH | 108.1, CH | 66.3, CH | 105.8, CH |
| 6/6′ | 74.6, CH | 150.0, C | 74.6, CH | 150.0, C | 73.4, CH | 146.3, C | 74.6, CH | 150.0, C |
| 7/7′ | 70.7, CH | 156.2, C | 70.7, CH | 156.1, C | 69.0, CH | 153.4, C | 70.7, CH | 156.2, C |
| 8/8′ | 78.3, CH | 105.2, CH | 78.3, CH | 105.2, CH | 77.9, CH | 104.2, CH | 78.3, CH | 105.1, CH |
| 9/9′ | 162.0, C | 153.9, C | 162.0, C | 153.8, C | 159.5, C | 150.5, C | 162.0, C | 153.9, C |
| 10/10′ | 122.9, C | 118.6, C | 122.9, C | 118.5, C | 121.8, C | 117.7, C | 122.9, C | 118.5, C |
| 1′′/1′′′ | 133.8, C | 132.8, C | 133.8, C | 134.2, C | 132.5, C | 132.7, C | 133.8, C | 133.2, C |
| 2′′/2′′′ | 116.2, CH | 113.1, CH | 116.2, CH | 116.4, CH | 115.7, CH | 115.5, CH | 116.2, CH | 130.4, CH |
| 3′′/3′′′ | 147.6, C | 149.9, C | 147.6, C | 147.6, C | 146.3, C | 146.3, C | 147.6, C | 114.9, CH |
| 4′′/4′′′ | 147.6, C | 146.1, C | 147.6, C | 147.6, C | 146.0, C | 146.0, C | 147.6, C | 159.7, C |
| 5′′/5′′′ | 112.8, CH | 116.2, CH | 112.8, CH | 112.8, CH | 112.2, CH | 112.1, CH | 112.8, CH | 114.9, CH |
| 6′′/6′′′ | 120.2, CH | 121.8, CH | 120.3, CH | 120.5, CH | 118.7, CH | 118.5, CH | 120.2, CH | 130.4, CH |
| 7′′/7′′′ | 32.8, CH2 | 33.7, CH2 | 32.8, CH2 | 33.3, CH2 | 30.9, CH2 | 31.4, CH2 | 32.8, CH2 | 33.1, CH2 |
| 8′′/8′′′ | 36.6, CH2 | 37.3, CH2 | 36.5, CH2 | 37.0, CH2 | 34.6, CH2 | 35.2, CH2 | 36.6, CH2 | 37.2, CH2 |
| OCH3 | 56.4 (4′′-OCH3) | 56.7 (6′-OCH3) | 56.4 (4′′-OCH3) | 56.8 (6′-OCH3) | 55.6 (4′′-OCH3) | 55.6 (4′′′-OCH3) | 56.4 (4′′-OCH3) | 56.7 (6′-OCH3) |
| 56.3 (3′′′-OCH3) | 56.4 (4′′′-OCH3) | 55.6 (4′′′-OCH3) | ||||||
The structure of unit A was deduced to be the same as that of aquilarone A21 on the basis of 1D and 2D NMR data. The tetrahydrochromone fragment was concluded by the respective signals of a pyrone fragment [δC 171.3 (C-2), δC 114.5/δH 6.13 (1H, s, H-3), δC 181.4 (C-4), δC 162.0 (C-9), δC 122.9 (C-10)] and four consecutive oxymethines [δC 66.3/δH 4.83 (1H, d, J = 3.8 Hz, H-5), δC 74.6/δH 4.14 (1H, dd, J = 3.8, 2.3 Hz, H-6), δC 70.7/δH 4.48 (1H, dd, J = 7.6, 2.3 Hz, H-7), and δC 78.3/δH 5.63 (1H, d, J = 7.6 Hz, H-8)]. The phenylethyl moiety was deduced by the signal for a characteristic ABX coupling aromatic system [δC 133.8 (C-1′′), δC 116.2/δH 6.46 (1H, d, J = 2.1 Hz, H-2′′), δC 147.6 (C-3′′, 4′′), δC 112.8/δH 6.67 (1H, d, J = 8.1 Hz, H-5′′), δC 120.2/δH 6.32 (1H, dd, J = 8.1, 2.1 Hz, H-6′′)] and two methylene groups [δC 32.8/δH 2.52 (2H, m, H-7′′), δC 36.6/δH 2.64 (2H, m, H-8′′)]. The methoxyl group (δC 56.4/δH 3.75) attached to the aromatic carbon C-4′′ was confirmed by the HMBC correlation of OCH3/C-4′′, as well as the ROESY correlation from OCH3 to H-5′′.
Apart from the aforementioned NMR signals assigned to unit A, the remaining signals indicated a 2-(2-phenylethyl)chromone unit, which showed similarity to 6-methoxy-7-hydroxy-2-[2-(4-methoxyphenyl)ethyl]chromone22 with main differences in the substituents on the phenylethyl moiety. The NMR signals [δC 132.8 (C-1′′′), δC 113.1/δH (6.73, 1H, d, J = 1.8 Hz, H-2′′′), δC 149.9 (C-3′′′), δC 146.1 (C-4′′′), δC 116.2/δH (6.65, d, J = 8.0 Hz, H-5′′′), δC 121.8/δH (6.59, dd, J = 8.0, 1.8 Hz, H-6′′′)] revealed that an ABX aromatic coupling system existed, and the substituents 4′′′-OH and 3′′′-OCH3 were indicated by the downfield chemical shifts of C-3′′′ and C-4′′′, as well as the HMBC correlations from H-5′′′/OCH3 (δH 3.74) to C-3′′′ and from H-2′′′ to C-4′′′, and the ROESY correlation of 3′′′-OCH3/H-2′′′. Thus, unit B was assigned as 7-hydroxy-6-methoxy-2-[2-(4′-hydroxy-3′-methoxyphenyl)ethyl]chromone.
The presence of the ether linkage C8–O–C7′ between units A and B was determined by the key HMBC correlation from H-8 to C-7′, the ROESY cross-peak of H-8/H-8′, as well as through the analysis of the molecular formula. The relative configurations of the chiral centers C-5, C-6, C-7 and C-8 were deduced to be the same as those of crassin D,14 based on the similar 3JH–H coupling constants for H-5/H-6 (J = 3.8 Hz), H-6/H-7 (J = 2.3 Hz) and H-7/H-8 (J = 7.6 Hz). The ECD spectrum (Fig. 3) of 1 was opposite to that of crassin D, with a positive Cotton effect around λmax 280 nm and a negative Cotton effect around λmax 250 nm, indicating that the front chromophore existed in a clockwise arrangement relative to the rear chromophore, which suggested an R configuration for C-8. Thus, compound 1 was determined to have (5S,6S,7S,8R)-configuration and named as aquilasinenone A.
Aquilasinenone B (2) was obtained as a colorless oil. It possessed the molecular formula C37H36O13, as established by HRESIMS. The similarity of its 1H and 13C NMR spectroscopic data (Tables 1 and 2) to those of 1 revealed that they shared structural resemblance except for the substitutions of 3′′′-OH and 4′′′-OCH3 on unit B in 2. The assignment of 3′′′-OH and 4′′′-OCH3 was consistent with the HMBC correlation from 4′′′-OCH3 (δH 3.74) to C-4′′′ (δC 147.6), together with the ROESY correlation of 4′′′-OCH3/H-5′′′ (δH 6.71). The remaining substructures were identical to those of 1 based on the detailed analysis of their 2D NMR spectra (Fig. 2). The configuration of 2 was deduced to be the same as that of 1, based on the similar coupling constants of 3J5,6 (3.7 Hz), 3J6,7 (2.1 Hz) and 3J7,8 (7.6 Hz). Based on the similarity of the ECD curve of 2 to that of 1, the absolute configuration of 2 could be assigned to be 5S, 6S, 7S, and 8R, and it was given the trivial name of aquilasinenone B.
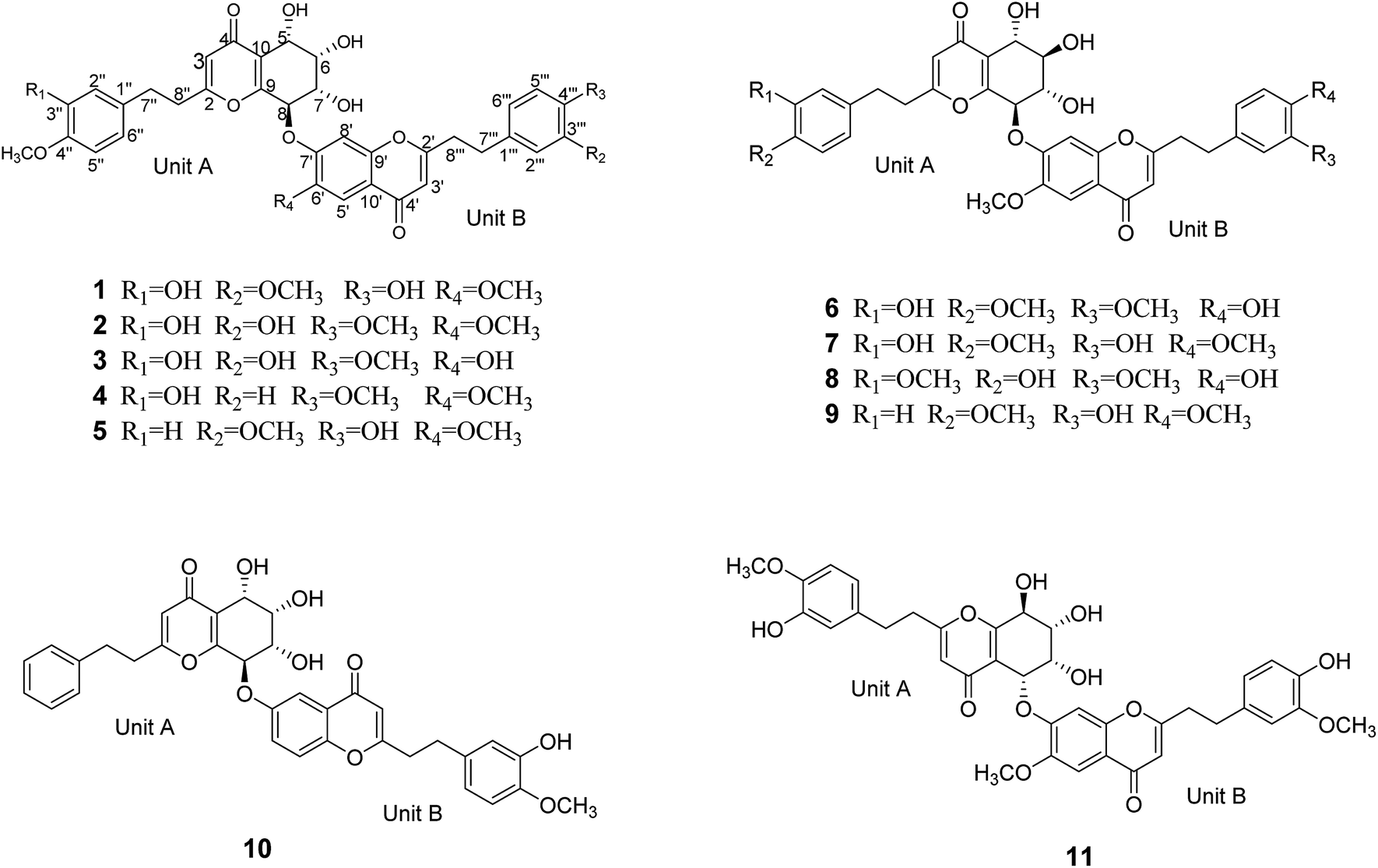 | ||
| Fig. 1 Structures of compounds 1–11. | ||
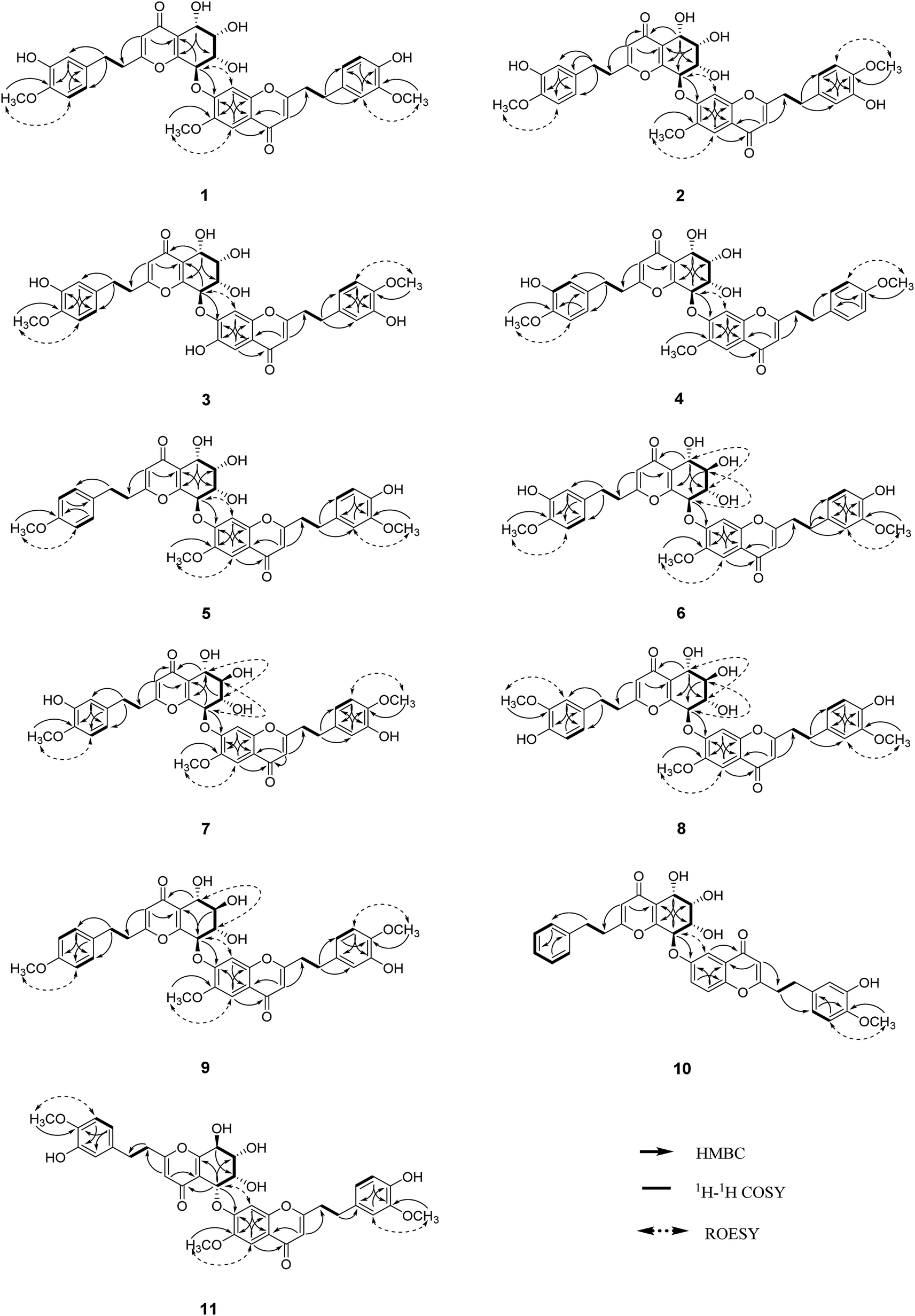 | ||
| Fig. 2 Key 2D NMR correlations of compounds 1–11. | ||
Aquilasinenone C (3) was obtained as a yellow oil. Its molecular formula was C36H34O13, as indicated by HRESIMS. Data comparison revealed that the structure of 3 closely resembled that of 2, except for the 6′-OH rather than 6′-OCH3 in 2, which was confirmed by the 14 amu difference between their molecular weights. The two methoxyl groups in the structure were located at C-4′′ and C-4′′′ according to the ROESY correlations of 4′′-OCH3 (δH 3.69)/H-5′′ (δH 6.75) and 4′′′-OCH3 (δH 3.68)/H-5′′′(δH 6.67), respectively. The downfield chemical shift of C-6′ (δC 146.3) revealed oxygenation at C-6′, and the molecular formula indicated that a hydroxyl group was located at C-6′. The relative configuration of 3 was deduced to be the same as that of 1 due to their similar 3JH–H coupling constants for H-5/H-6/H-7/H-8. The absorption in the ECD curve of 3 was close to zero, which indicated the existence of a racemate. Thus, compound 3 was identified as aquilasinenone C.
Aquilasinenone D (4) was obtained as a colorless oil, and its molecular formula was established as C37H36O12 based on the HRESIMS pseudomolecular ion peak at m/z 695.2095 [M + Na]+ (calcd for C37H36NaO12, 695.2099). The comparison of corresponding NMR data revealed that compound 4 was similar to that of 2, except for the substitutions on the phenylethyl part of the aromatic system in unit B. A typical AA′BB′ aromatic system in contrast to the ABX coupling system in 2 was deduced from the NMR data (Table 1), and the 4-methoxyl group was confirmed by the HMBC correlation from 4′′′-OCH3 (δH 3.71) to C-4′′′ (δC 159.7), as well as the ROESY correlation of 4′′′-OCH3 (δH 3.71)/H-3′′′ (δH 7.76). The presence of C8–O–C7′ linkage connecting units A and B was deduced according to the HMBC correlation from H-8 (δH 5.63) to the phenyl carbon C-7′ (δC 156.2), as well as the ROESY correlation of H-8/H-8′. The relative configuration of 4 was deduced to be the same as that of 1 due to their similar 3JH–H coupling constants for H-5/H-6/H-7/H-8. The absolute configuration of (5S,6S,7S,8R) was determined by analyzing its ECD data, which were the same as those of 1. Therefore, compound 4 was identified as aquilasinenone D.
Aquilasinenone E (5) was obtained as a colorless oil, and its molecular formula was established as C37H36O12 based on the HRESIMS pseudomolecular ion peak at m/z 695.2108 [M + Na]+ (calcd for C37H36NaO12, 695.2099). Analysis of the 1H and 13C NMR spectroscopic data (Tables 3 and 4) of 5 showed a close structural resemblance to that of 1. The major difference was the presence of one para-disubstituted methoxyl benzene moiety in unit A. These assignments were corroborated by the typical AA′BB′ aromatic system [132.7 (C-1′′), 6.82 (2H, d, J = 8.7 Hz, H-2′′, 6′′)/130.2 (C-2′′, 6′′), 6.69 (2H, d, J = 8.7 Hz, H-3′′, 5′′)/114.9 (C-3′′, 5′′), 159.7 (C-4′′)], as well as their HMBC correlation of 4′′-OCH3 (δH 3.70)/C-4′′ and ROESY correlation of 4′′-OCH3/H-3′′. The presence of C8–O–C7′ linkage connecting units A and B was deduced according to the HMBC correlation from the proton at δH 5.62 (H-8) to the phenyl carbon at δC 156.2 (C-7′), and the ROESY correlation of H-8/H-8′. The relative configurations of 5 were the same as those assigned for 1 on the basis of their 3JH–H coupling constants. A racemate existed with the (5S,6S,7S,8R)-configuration in excess, according to the slight positive Cotton effect around λmax 280 nm and the negative Cotton effect around λmax 250 nm in the ECD spectrum, which was similar to that of 1. Therefore, the structure was named as aquilasinenone E.
| Position | 5 | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| 3/3′ | 6.14, s | 6.14, s | 6.10, s | 6.11, s | 6.13, s | 6.12, s | 6.09, s | 6.11, s |
| 5/5′ | 4.82, d (3.7) | 7.56, s | 4.76, dd (7.2, 1.2) | 7.52, s | 4.77, dd (7.1, 1.5) | 7.56, s | 4.74, dd (7.1, 1.6) | 7.53, s |
| 6/6′ | 4.13, dd (3.7, 2.3) | 3.84, dd (9.9, 7.2) | 3.85, dd (9.9, 7.1) | 3.82, dd (9.8, 7.1) | ||||
| 7/7′ | 4.47, dd (7.6, 2.3) | 4.02, dd (9.9, 7.6) | 4.02, dd (9.9, 7.6) | 3.97, dd (9.8, 7.5) | ||||
| 8/8′ | 5.62, d (7.6) | 7.60, s | 5.63, d (7.6) | 7.60, s | 5.66, d (7.6) | 7.62, s | 5.63, dd (7.6, 1.5) | 7.60, s |
| 2′′/2′′′ | 6.82, d (8.7) | 6.74, d (1.9) | 6.43, d (1.9) | 6.70, d (1.6) | 6.44, d (2.1) | 6.66, d (2.1) | 6.48, d (1.9) | 6.70, d (1.9) |
| 3′′/3′′′ | 6.69, d (8.7) | |||||||
| 5′′/5′′′ | 6.69, d (8.7) | 6.65, d (8.0) | 6.65, d (8.2) | 6.63, d (8.0) | 6.67, d (8.2) | 6.74, d (8.20) | 6.57, d (8.0) | 6.62, d (8.0) |
| 6′′/6′′′ | 6.82, d (8.7) | 6.59, dd (8.0, 1.9) | 6.29, dd (8.2, 1.9) | 6.55, dd (8.0, 1.6) | 6.31, dd (8.2, 2.1) | 6.56, dd (8.2, 2.1) | 6.33, dd (8.0, 1.9) | 6.56, dd (8.0, 1.9) |
| 7′′/7′′′ | 2.57, m | 2.93, m | 2.47, m | 2.88, m | 2.50, m | 2.90, m | 2.53, m | 2.90, m |
| 8′′/8′′′ | 2.67, m | 2.93, m | 2.59, m | 2.88, m | 2.64, m | 2.90, m | 2.64, m | 2.90, m |
| OCH3 | 3.70, s (4′′-OCH3) | 3.92, s (6′-OCH3) | 3.72, s (4′′-OCH3) | 3.89, s (6′-OCH3) | 3.76, s (4′′-OCH3) | 3.94, s (6′-OCH3) | 3.68, s (3′′-OCH3) | 3.87, s (6′-OCH3) |
| 3.74, s (3′′′-OCH3) | 3.71, s (3′′′-OCH3) | 3.79, s (4′′′-OCH3) | 3.72, s (3′′′-OCH3) | |||||
| Position | 5 | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| 2/2′ | 171.3, C | 171.0, C | 171.3, C | 171.3, C | 171.3, C | 171.2, C | 171.4, C | 171.4, C |
| 3/3′ | 114.5, CH | 110.1, CH | 114.4, CH | 110.1, CH | 114.4, CH | 110.1, CH | 114.6, CH | 110.1, CH |
| 4/4′ | 181.4, C | 179.8, C | 182.0, C | 180.0, C | 182.0, C | 179.8, C | 182.0, C | 179.8, C |
| 5/5′ | 66.3, CH | 105.7, CH | 70.5, CH | 105.8, CH | 70.5, CH | 105.9, CH | 70.4, CH | 105.7, CH |
| 6/6′ | 74.6, CH | 150.0, C | 75.0, CH | 149.7, C | 75.1, CH | 149.8, C | 75.0, CH | 149.7, C |
| 7/7′ | 70.6, CH | 156.2, C | 73.5, CH | 155.8, C | 73.6, CH | 155.8, C | 73.6, CH | 155.8, C |
| 8/8′ | 78.3, CH | 105.1, CH | 79.2, CH | 105.0, C | 79.2, CH | 105.0, CH | 79.1, CH | 104.9, CH |
| 9/9′ | 162.0, C | 153.9, C | 159.9, C | 153.8, C | 160.0, C | 153.9, C | 160.0, C | 153.9, C |
| 10/10′ | 122.9, C | 118.5, C | 122.5, C | 118.5, C | 122.6, C | 118.6, C | 122.6, C | 118.5, C |
| 1′′/1′′′ | 132.7, C | 132.9, C | 133.7, C | 132.7, C | 133.8, C | 134.2, C | 132.3, C | 132.7, C |
| 2′′/2′′′ | 130.2, CH | 113.1, CH | 116.2, C | 113.1, CH | 116.2, CH | 116.4, CH | 112.8, CH | 113.1, CH |
| 3′′/3′′′ | 114.9, CH | 148.9, C | 147.5, C | 148.9, C | 147.6, C | 147.6, C | 149.0, C | 149.0, C |
| 4′′/4′′′ | 159.7, C | 146.1, C | 147.6, C | 146.0, C | 147.6, C | 147.6, C | 146.1, C | 146.1, C |
| 5′′/5′′′ | 114.9, CH | 116.2, CH | 116.2, CH | 112.7, CH | 112.9, CH | 112.8, CH | 116.2, CH | 116.2, CH |
| 6′′/6′′′ | 130.2, CH | 121.8, CH | 120.2, CH | 121.8, CH | 120.3, CH | 120.5, CH | 121.6, CH | 121.8, CH |
| 7′′/7′′′ | 32.7, CH2 | 33.7, CH2 | 32.7, CH2 | 33.6, CH2 | 32.8, CH2 | 33.4, CH2 | 33.1, CH2 | 33.7, CH2 |
| 8′′/8′′′ | 36.7, CH2 | 37.3, CH2 | 36.5, CH2 | 37.2, CH2 | 36.6, CH2 | 37.1, CH2 | 36.8, CH2 | 37.3, CH2 |
| OCH3 | 55.6 (4′′-OCH3) | 56.7 (6′-OCH3) | 56.4 (4′′-OCH3) | 56.8 (6′-OCH3) | 56.4 (4′′-OCH3) | 56.8 (6′-OCH3) | 56.3 (3′′-OCH3) | 56.7 (6′-OCH3) |
| 56.3 (3′′′-OCH3) | 56.3 (3′′′-OCH3) | 56.5 (4′′′-OCH3) | 56.4 (3′′′-OCH3) | |||||
Aquilasinenone F (6) was obtained as a yellow oil, and its molecular formula was established as C37H36O13 based on the HRESIMS pseudomolecular ion peak at m/z 711.2062 [M + Na]+ (calcd for C37H36NaO13, 711.2048). Upon a detailed comparison of the 1H and 13C NMR data of 6 with those of 1, it was found that they shared the same planar structures. The relative configurations of the four chiral centers in unit A were deduced by the analysis of the 3JH–H coupling constants and ROESY spectrum. The large J values of H-5/H-6 (7.2 Hz), H-6/H-7 (9.9 Hz) and H-7/H-8 (7.6 Hz) indicated the axial orientation of these protons, which was further supported by the ROESY correlations of H-5/H-7 and H-6/H-8. The positive long-wave Cotton effect around λmax 280 nm and the negative short-wave Cotton effect around λmax 250 nm (Fig. 4) in its ECD curve were the same as those reported for (5S,6R,7S,8R)-2-[2-(4-methoxyphenyl)ethyl]-5,6,7-trihydroxy-5,6,7,8-tetrahydro-8-{6-methoxy-2-[2-(3-methoxy-4-hydroxyphenyl)ethyl]chromonyl-7-oxy}chromone,13 which suggested a (5S,6R,7S,8R)-configuration for 6. It was named aquilasinenone F.
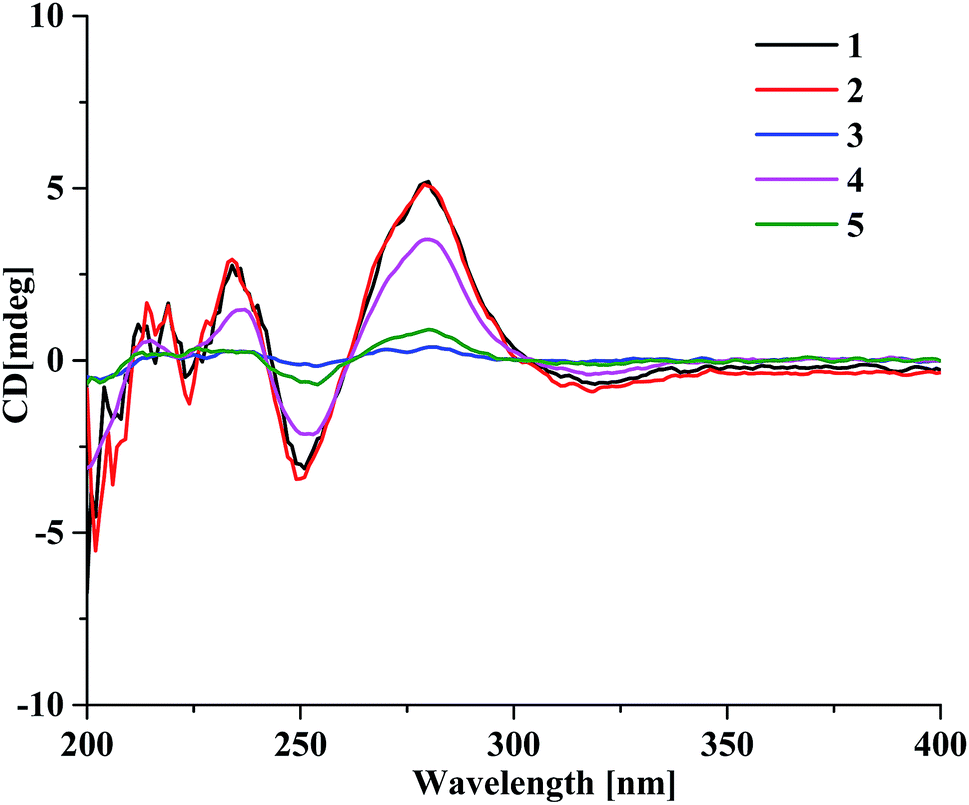 | ||
| Fig. 3 The ECD spectra of compounds 1–5. | ||
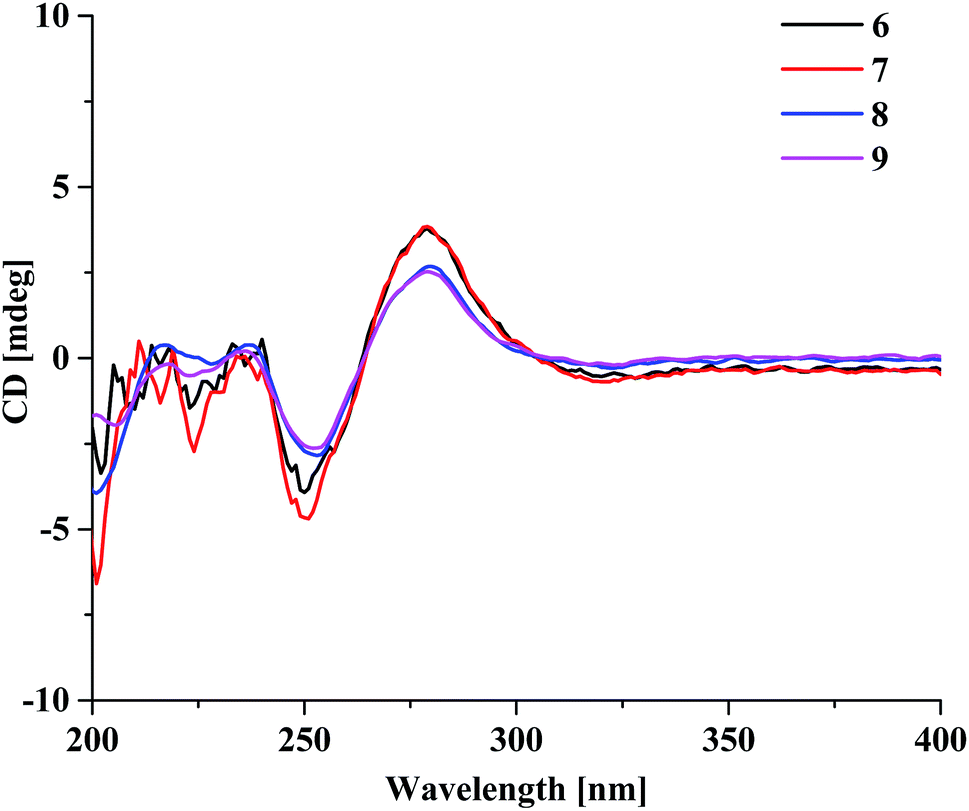 | ||
| Fig. 4 The ECD spectra of compounds 6–9. | ||
Aquilasinenone G (7) was obtained as a colorless oil, and its molecular formula was established as C37H36O13 based on the HRESIMS pseudomolecular ion peak at m/z 711.2121 [M + Na]+ (calcd for C37H36NaO13, 711.2048). On comparing the 1H and 13C NMR data of 7 with those of 2, it was found that they shared the same planar structures. However, the relative configuration of 7 was deduced to be the same as that of 6 due to their similar ROESY and 3JH–H coupling constant data for H-5/H-6/H-7/H-8. The ECD curve (Fig. 4) of 7 showed a similar Cotton effect as that of 6, indicating the same (5S,6R,7S,8R)-configuration. Thus, the structure of aquilasinenone G was established.
Aquilasinenone H (8) was obtained as a colorless oil, and its molecular formula was established as C37H36O13 based on the HRESIMS pseudomolecular ion peak at m/z 711.2046 [M + Na]+ (calcd for C37H36NaO13, 711.2048). Comparison of the 1D and 2D NMR spectroscopic data (Tables 3 and 4) of 8 showed a close structural resemblance to that of 6, and both shared the same relative configuration on the 4H-chromone part. The main differences were the substitutions for 4′′-OH and 3′′-OCH3 on the benzene moiety in unit A. These assignments were verified by the HMBC correlations of H-2′′ (δH 6.48)/C-4′′ (δC 146.1) and H-5′′ (δH 6.57)/C-1′′ (δC 132.3), and the ROESY correlation of 3′′-OCH3 (δH 3.68)/H-2′′. The absolute configuration of C-8 was deduced to be the same as that of 6 based on their similar ECD spectra. Thus, the structure of aquilasinenone H was established.
Aquilasinenone I (9) was obtained as a yellow oil, and its molecular formula was established as C37H36O12 based on the HRESIMS pseudomolecular ion peak at m/z 671.2145 [M − H]− (calcd for C37H35O12, 671.2134). A comparison of its 1D and 2D NMR spectroscopic data (Tables 5 and 6) with those of 7 revealed that they shared similar structures, with the main difference being the para-disubstituted methoxyl benzene moiety in unit A of 9. These assignments were verified by the HMBC correlation of 4′′-OCH3 (δH 3.70)/C-4′′ (δC 159.7), the ROESY correlation from 4′′-OCH3 to H-3′′/5′′ (δH 6.69), as well as the typical AA′BB′ aromatic coupling system [δC 132.8 (C-1′′), δC 130.1/δH 6.80 (2H, d, J = 8.6 Hz, H-2′′, 6′′), δC 114.9/δH 6.69 (2H, d, J = 8.6 Hz, H-3′′, 5′′), δC 159.7 (C-4′′)]. The relative configuration of 9 was deduced to be the same as that of 6, and the absolute configuration of C-8 was determined to be R via the analysis of its ECD data, indicating the same (5S,6R,7S,8R)-configuration. Therefore, compound 9 was identified as aquilasinenone I.
| Position | 9 | 10 | 11 | |||
|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| 3/3′ | 6.13, s | 6.13, s | 6.15, s | 6.15, s | 6.15, s | 6.11, s |
| 5/5′ | 4.77, d (7.2) | 7.55, s | 4.80, d (3.8) | 7.90, dd (4.0, 2.2) | 5.50, d (3.2) | 7.47, s |
| 6/6′ | 3.84, dd (9.9, 7.2) | 4.10, dd (3.8, 2.3) | 4.23, m | |||
| 7/7′ | 4.01, dd (9.9) | 4.38, dd (7.7, 2.3) | 7.62, d (1.7) | 4.14, dd (8.2, 2.3) | ||
| 8/8′ | 5.68, m | 7.64, s | 5.46, d (7.7) | 7.62, d (1.7) | 4.67, d (8.2) | 7.54, s |
| 2′′/2′′′ | 6.80, d (8.6) | 6.65, d (2.0) | 6.93, d (6.7) | 6.70, d (2.1) | 6.72, d (2.0) | 6.76, d (1.8) |
| 3′′/3′′′ | 6.69, d (8.6) | 7.13, t (7.4) | ||||
| 4′′/4′′′ | 7.10, t (7.4) | |||||
| 5′′/5′′′ | 6.69, d (8.6) | 6.72, d (8.2) | 7.13, t (7.4) | 6.76, d (8.2) | 6.83, d (8.2) | 6.69, d (1.8) |
| 6′′/6′′′ | 6.80, d (8.6) | 6.55, dd (8.2, 2.0) | 6.93, d (6.7) | 6.62, dd (8.2, 2.1) | 6.67, dd (8.2, 2.0) | 6.65, dd (8.0, 1.8) |
| 7′′/7′′′ | 2.56, m | 2.90, m | 2.61, m | 2.97, m | 2.93, m | 2.97, m |
| 8′′/8′′′ | 2.56, m | 2.90, m | 2.71, m | 2.97, m | 2.94, m | 3.01, m |
| OCH3 | 3.70, s (4′′-OCH3) | 3.76, s (4′′′-OCH3) | 3.77, s (4′′′-OCH3) | 3.75, s (4′′-OCH3) | 3.86, s (6′-OCH3) | |
| 3.74, s (3′′′-OCH3) | ||||||
| Position | 9 | 10 | 11 | |||
|---|---|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | Unit A | Unit B | |
| 2/2′ | 171.3, C | 171.3, C | 170.9, C | 171.8, C | 171.7, C | 171.1, C |
| 3/3′ | 114.1, CH | 110.1, CH | 114.5, CH | 110.1, CH | 114.2, CH | 110.2, CH |
| 4/4′ | 182.0, C | 179.8, C | 181.5, C | 180.2, C | 181.6, C | 179.9, C |
| 5/5′ | 70.5, CH | 105.7, CH | 66.3, CH | 110.4, CH | 74.4, CH | 105.6, CH |
| 6/6′ | 75.1, CH | 149.7, C | 74.5, CH | 158.6, C | 70.5, CH | 150.3, C |
| 7/7′ | 73.4, CH | 155.9, C | 70.8, CH | 126.2, CH | 72.5, CH | 155.0, C |
| 8/8′ | 78.7, CH | 104.9, CH | 78.2, CH | 120.9, CH | 69.7, CH | 104.7, CH |
| 9/9′ | 160.0, C | 153.9, C | 162.8, C | 153.4, C | 167.2, C | 154.1, C |
| 10/10′ | 122.6, C | 118.5, C | 122.7, C | 125.0, C | 118.7, C | 118.4, C |
| 1′′/1′′′ | 132.8, C | 134.1, C | 140.9, C | 134.1, C | 134.1, C | 132.8, C |
| 2′′/2′′′ | 130.1, CH | 116.4, CH | 129.2, CH | 116.4, CH | 116.4, CH | 113.1, CH |
| 3′′/3′′′ | 114.9, CH | 147.6, C | 129.5, CH | 147.6, C | 147.6, C | 148.9, C |
| 4′′/4′′′ | 159.7, C | 147.6, C | 127.4, CH | 147.6, C | 147.7, C | 146.1, C |
| 5′′/5′′′ | 114.9, CH | 112.8, CH | 129.5, CH | 112.8, CH | 112.9, CH | 116.3, CH |
| 6′′/6′′′ | 130.1, CH | 120.5, CH | 129.2, CH | 120.5, CH | 120.6, CH | 121.9, CH |
| 7′′/7′′′ | 32.6, CH2 | 33.4, CH2 | 33.4, CH2 | 33.6, CH2 | 33.2, CH2 | 33.8, CH2 |
| 8′′/8′′′ | 36.8, CH2 | 37.1, CH2 | 36.2, CH2 | 37.2, CH2 | 36.5, CH2 | 37.4, CH2 |
| OCH3 | 55.6 (4′′-OCH3) | 56.4 (4′′′-OCH3) | 56.4 (4′′′-OCH3) | 56.5 (4′′-OCH3) | 56.7 (6′-OCH3) | |
| 56.3 (3′′′-OCH3) | ||||||
Aquilasinenone J (10) was obtained as a yellow oil, and its molecular formula was established as C35H32O10 based on the HRESIMS pseudomolecular ion peak at m/z 635.1909 [M + Na]+ (calcd for C35H32NaO10, 635.1888). The comparison of the 1H and 13C NMR spectroscopic data of 10 and those of (5S,6R,7S,8R)-2-(2-phenylethyl)-5,6,7-trihydroxy-5,6,7,8-tetrahydro-8-[2-(2-phenylethyl)chromonyl-6-oxy]chromone16 indicated that they shared the same structure for unit A, and the difference being the replacement of the mono-substituted aromatic ring by a 1,3,4-trisubstituted aromatic ring in unit B in 10. These assignments were verified by the presence of an ABX aromatic coupling system [δC 134.1 (C-1′′′), δC 116.4/δH 6.70 (1H, d, J = 2.1 Hz, H-2′′′), δC 147.6 (C-3′′′, 4′′′), δC 112.8/δH 6.76 (1H, d, J = 8.2 Hz, H-5′′′), δC 120.5/δH 6.62 (1H, dd, J = 8.2, 2.1 Hz, H-6′′′)] together with the HMBC correlations of H-6′′′/C-4′′′and H-5′′′/C-3′′′, and the ROESY correlation of 4′′′-OCH3 (δH 3.77)/H-5′′′, which inferred the substitutions of 3′′′-OH and 4′′′-OCH3. The presence of C8–O–C6′ linkage connecting units A and B was deduced according to the HMBC correlation from the proton H-8 (δH 5.46) to the phenyl carbon C-6′ (δC 158.6) and the ROESY correlation of H-8/H-5′. The relative configuration of 10 was deduced to be the same as that of 1 due to their similar 3JH–H coupling constants for H-5/H-6/H-7/H-8. Thus, the structure of aquilasinenone J was established.
Aquilasinenone K (11) was obtained as a yellow oil, and its molecular formula was established as C37H36O13 based on the HRESIMS pseudomolecular ion peak at m/z 711.2106 [M + Na]+ (calcd for C37H36NaO13, 711.2048). A comparison of the 1D and 2D NMR spectroscopic data (Tables 5 and 6) of 11 showed that units A and B were the same as those in 1, respectively. Detailed analysis revealed that the major difference was the linkage between units A and B via C5–O–C7′ in 11. This assignment was verified by the HMBC correlations from H-5 (δH 5.50) to C-7′ (δC 155.0), as well as with the ROESY correlation of H-5/H-8′ (δH 7.54). Thus, the structure for aquilasinenone K was established as shown in Fig. 1.
For compounds 3 and 5, we tried to separate them with the available chiral column, but failed. Therefore, we have reported them as racemates. The ECD spectra of 10 and 11 were measured, but the curves could not be clearly presented; therefore, we reported their structures using relative configurations. All compounds were tested for AChE inhibitory activity; however, only compounds 9, 10 and 11 exhibited weak AChE inhibitory activity (Table 7).
3. Conclusion
In conclusion, eleven novel uncommon single ether linkage dimeric compounds (1–11) were isolated from the artificial hole-induced agarwood originating from Aquilaria sinensis. Herein, we reported the NMR and MS data of these compounds, as well as the absolute configurations of 1–9 using ECD spectra. Among them, compounds 9, 10 and 11 exhibited weak acetylcholinesterase (AChE) inhibitory activity.4. Experimental section
4.1 General
1H, 13C and 2D NMR spectra were recorded on a Bruker AV-500 and Bruker AVIII-600 spectrometer (Bruker), and the chemical shifts were referenced to the solvent residual peaks. HRESIMS spectra were measured with an API QSTAR Pulsar mass spectrometer (Bruker). UV spectra were obtained on a Shimadzu UV-2550 spectrometer (Beckman, America). IR absorptions were obtained on a Nicolet 380 FT-IR instrument (Thermo) using KBr pellets. Optical rotation was measured on a Rudolph Autopol III polarimeter. HPLC analysis was performed with an Agilent Technologies 1260 Infinity equipped with an Agilent DAD G1315D detector, and the separation columns were YMC-pack C18 columns (5 mm, 250 mm × 4.6 mm). Semi-preparative HPLC was performed on reversed-phase columns (YMC-packed C18, 5 mm, 250 mm × 10 mm). Silica gel (60–80, 200–300 mesh, Qingdao Marine Chemical Co. Ltd.), ODS gel (20–45 mm, Fuji Silysia Chemical Co. Ltd.), MCI gel (75–150 mm, Mitsubishi Chemical Co. Ltd.) and Sephadex LH-20 (Merck) were used for column chromatography. TLC was conducted on precoated silica gel G plates (Qingdao Marine Chemical Co. Ltd.), and the spots were detected by spraying them with 5% H2SO4 in EtOH followed by heating.4.2 Plant material
The artificial hole-induced agarwood of A. sinensis was harvested in November 2012 in the Xishuangbanna Dai autonomous prefecture of Yunnan province after four years of formation. The identification of the original plant as Aquilaria sinensis (Lour.) Gilg (Thymelaeaceae) was performed by Dr Jun Wang (Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Science). The voucher specimen (No. 20121108) was deposited at the Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Science, Haikou, China.4.3 Extraction and isolation
The artificial hole-induced agarwood of A. sinensis (4.7 kg, air-dried) was exhaustively extracted by refluxing with 95% EtOH (5 L × 3). The EtOH extract (510.0 g) was dissolved in H2O (2.5 L) and successively extracted by EtOAc (2.5 L × 3) and n-BuOH (2.5 L × 3). Subsequently, the EtOAc extract (310.0 g) was subjected to vacuum-liquid chromatography (VLC) using silica gel by employing a gradient of CHCl3/MeOH (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 50:1, 25:1, 15:1, 10:1, 5:1, 2:1, 1:1, 0:1, 8.0 L of each) to obtain nine fractions (Fr.1–9).
:0, 50:1, 25:1, 15:1, 10:1, 5:1, 2:1, 1:1, 0:1, 8.0 L of each) to obtain nine fractions (Fr.1–9).
Fr.5 and Fr.6 were found to be rich in bi-phenylethylchromone analogues according to their chromogenic reactions on TLC, UV absorptions, and UPLC-MS data analysis. Thus, a combined fraction (89.6 g) of Fr.5 and Fr.6 was purified using the MCI column by eluting with MeOH to remove pigments, and the residue was subjected to vacuum-liquid chromatography (VLC) and eluted with a stepwise gradient of petroleum ether (PE)/acetone (v/v, 2:1, 1:1, 1:5, 1:10, 1:20) to obtain six subfractions (Fr.A–F). Fr.D (31.4 g) was subjected to Sephadex LH-20 column (10 × 120 cm) and eluted with CHCl3/MeOH (v/v, 1:1) to give three fractions (Fr.D-1–3). Fr.D-2 (8.1 g) was applied to an ODS gel column and eluted with MeOH–H2O (v/v, 3:7, 2:3, 1:1, 11:9, 3:2, 13:7, 7:3, 4:1, 9:1, 1:0) to give nine fractions (Fr.D-2-1–9).
Fr.D-2-4 (642.8 mg) was eluted with a gradient of CHCl3/MeOH (v/v, 35:1, 30:1, 25:1) using silica gel to obtain six fractions (Fr.D-2-4-1–6). The following semi-preparative HPLC separation on a C18 column (CH3CN/H2O, v/v 25:75 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) afforded the pure compounds 6 (tR 21.5 min, 25.6 mg) and 7 (tR 24.1 min, 34.3 mg). Fr.D-2-4-3 (22.4 mg) was separated by a silica gel column with CHCl3/acetone/MeOH (v/v/v, 15:1:0.1) as the eluent to furnish three fractions (Fr.D-2-4-3-1–3). Fr.D-2-4-3-1 (19.7 mg) was further purified by semi-preparative HPLC on a C18 column (CH3CN/H2O, v/v 25:75 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to afford compound 8 (tR 17.7 min, 0.7 mg). Fr.D-2-4-5 (95.0 mg) was eluted with a gradient of PE/EtOAc (v/v, 1:2, 1:8) using silica gel to obtain two fractions (Fr.D-2-4-5-1–2). Fr.D-2-4-5-1 (47.4 mg) was followed up by semi-preparative HPLC on a C18 column (CH3CN/H2O, v/v 25:75 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to give compounds 1 (tR 19.3 min, 11.7 mg) and 2 (tR 21.7 min, 10.6 mg).
Fr.D-2-5 (696.4 mg) was separated using a silica gel column with CHCl3/MeOH (v/v, 25:1) as the eluent to give seven fractions (Fr.D-2-5-1–7). Fr.D-2-5-3 (187.2 mg) was subjected to a silica gel column eluting with a stepwise gradient of CHCl3/acetone/MeOH (v/v/v, 5:1:0.15, 5:1:0.2, 2:1:0.2, 2:1:2) to afford five fractions (Fr.D-2-5-3-1–5). Fr.D-2-5-3-2 (50.1 mg) was separated on a Sephadex LH-20 column by eluting with CHCl3/MeOH (v/v, 1:1) and then purified by semi-preparative HPLC on a C18 column (MeOH/H2O, v/v 55:45 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to afford compound 5 (tR 11.9 min, 2.0 mg). Fr.D-2-5-6 (132.3 mg) was separated using a Sephadex LH-20 column by eluting with CHCl3/MeOH (v/v, 1:1) to afford two fractions (Fr.D-2-5-6-1–2). Fr.D-2-5-6-1 (69.5 mg) was chromatographed on a silica gel column with CHCl3/acetone/MeOH (v/v/v, 2:1:0.2) as the eluent to give four fractions (Fr.D-2-5-6-1-1–4), and then, Fr.D-2-5-6-1-2 (19.7 mg) was followed up by semi-preparative HPLC on a C18 column (CH3CN/H2O, v/v 25:75 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to give compounds 4 (tR 10.0 min, 1.0 mg) and 11 (tR 26.0 min, 2.4 mg).
Fr.D-2-7 (1.2 g) was separated using a silica gel column with a stepwise gradient of CHCl3/MeOH (v/v, 50:1, 30:1, 25:1, 20:1) as the eluent to give nine fractions (Fr.D-2-7-1–9). Fr.D-2-7-2 (114.8 mg) was chromatographed on a silica gel column with CHCl3/acetone/MeOH (v/v/v, 10:1:0.1) as the eluent to give three fractions (Fr.D-2-7-2-1–3), and then, Fr.D-2-7-2-3 (85.1 mg) was further purified by silica gel with CHCl3/acetone/MeOH (v/v/v, 14:1:0.1) as the eluent to obtain three fractions (Fr.D-2-7-2-3-1–3). Furthermore, Fr.D-2-7-2-3-2 (33.4 mg) was purified by semi-preparative HPLC separation on an ODS column (CH3CN/H2O, v/v 32:68 as eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to obtain compound 9 (tR 15.4 min, 1.7 mg). Fr.D-2-7-4 (210.0 mg) was chromatographed on a silica gel column with CHCl3/acetone/MeOH (v/v/v, 10:1:0.1) as the eluent to give six fractions (Fr.D-2-7-4-1–6). Fr.D-2-7-4-5 (68.1 mg) was purified by semi-preparative HPLC separation on a C18 column (CH3CN/H2O, v/v 32:68 as the eluent; flow rate 4.0 mL min−1; UV detection at 254 nm) to obtain compound 10 (tR 18.5 min, 1.2 mg).
Compound 3 (tR 16.3 min, 4.3 mg) was isolated from the n-BuOH extract following the same procedures. The purities of the above compounds were analyzed by HPLC.
ε) 231 (4.56), 252 (4.34), 280 (4.20), 316 (3.99) nm; IR (KBr) νmax 3361, 2934, 2834, 1631, 1594, 1470, 1374, 1266, 1026, 856 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 711.2057 [M + Na]+ (calcd for C37H36NaO13, 711.2048).ε) 231 (4.69), 252 (4.49), 280 (4.36), 318 (3.99) nm; IR (KBr) νmax 3357, 2929, 2834, 1631, 1590, 1470, 1378, 1212, 1026, 856 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 711.2121 [M + Na]+ (calcd for C37H36NaO13, 711.2048).ε) 231 (4.54), 253 (4.39), 280 (4.23), 322 (2.98) nm; IR (KBr) νmax 3388, 3207, 2954, 1644, 1515, 1448, 1275, 1275, 1024, 762, 673 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 697.1884 [M + Na]+ (calcd for C36H34NaO13, 697.1892).ε) 224 (4.32), 256 (3.88), 278 (3.78), 314 (3.41) nm; IR (KBr) νmax 3331, 2922, 1635, 1600, 1511, 1470, 1431, 1383, 1211, 1179, 1078 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 695.2095 [M + Na]+ (calcd for C37H36NaO12, 695.2099).ε) 231 (4.78), 252 (4.66), 316 (4.26) nm; IR (KBr) νmax 3447, 3210, 2330, 1645, 1510, 1464, 1432, 1392, 1226, 1181, 1079, 1031 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 695.2108 [M + Na]+ (calcd for C37H36NaO12, 695.2099).ε) 231 (4.96), 252 (4.74), 280 (4.60), 318 (4.39) nm; IR (KBr) νmax 3365, 2934, 1635, 1590, 1470, 1378, 1212, 1034, 810 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 711.2062 [M + Na]+ (calcd for C37H36NaO13, 711.2048).ε) 231 (4.71), 252 (4.37), 280 (4.22), 316 (4.02) nm; IR (KBr) νmax 3363, 2921, 1631, 1586, 1428, 1378, 1212, 1026, 852 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 711.2121 [M + Na]+ (calcd for C37H36NaO13, 711.2048).ε) 228 (4.17), 254 (3.85), 280 (3.73), 314 (3.45) nm; IR (KBr) νmax 3441, 2921, 1600, 1515, 1470, 1383, 1270, 1210, 11123 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS: m/z 711.2046 [M + Na]+ (calcd for C37H36NaO13 711.2048).ε) 231 (4.45), 253 (4.25), 278 (4.09), 316 (3.92) nm; IR (KBr) νmax 3448, 2929, 1642, 1508, 1388, 1204 cm−1; 1H and 13C NMR data: Tables 5 and 6; HRESIMS m/z 671.2145 [M − H]− (calcd for C37H35O12, 671.2134).ε) 231 (4.31), 252 (4.17), 280 (3.86) nm; IR (KBr) νmax 3390, 2921, 1656, 1511, 1478, 1374, 1275, 1022, 839 cm−1; 1H and 13C NMR data: Tables 5 and 6; HRESIMS m/z 635.1909 [M + Na]+ (calcd for C35H32NaO10, 635.1888).ε) 230 (4.60), 280 (4.24), 318 (4.05) nm; IR (KBr) νmax 3413, 2930, 1629, 1513, 1438, 1390, 1269, 1131, 1085, 1026 cm−1; 1H and 13C NMR data: Tables 5 and 6; HRESIMS m/z 711.2106 [M + Na]+ (calcd for C37H36NaO13, 711.2048).4.4 Bioassay for AChE inhibitory activity
Compounds 1–11, each dissolved in DMSO, were tested for AChE inhibitory activity using a modified Ellman's colorimetric method.23 Briefly, 200 μL reaction mixture containing phosphate buffer (pH 8.0), 50 μg mL−1 test compound, and 0.02 U mL−1 acetylcholinesterase was incubated for 20 min at 30 °C. Afterward, 20 μL of 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellman's reagent) (2.48 mg mL−1) and 20 μL S-acetylthiocholine iodide (1.81 mg mL−1) were added, and the hydrolysis of acetylthiocholine was monitored at 405 nm for one hour. Tacrine (Sigma-Aldrich 99%) with a final concentration of 0.08 μg mL−1 served as the positive control, and DMSO with a final concentration of 0.1% served as the negative control. The percentage of inhibition was calculated by the equation: % inhibition = (E − S)/E × 100 (E is the activity of the enzyme without test compounds, and S is the activity of the enzyme with test compounds). The reagents for this reaction were purchased from Sigma Chemical. All the reactions were conducted in triplicates. The values are expressed as mean ± standard deviation (SD).Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The project was financially supported by the Innovative Research Team Grant of the Natural Science Foundation of Hainan Province (2017CXTD020), National Natural Science Foundation of China (31870668), and China Agriculture Research System (CARS-21).References
- R. Naef, Flavour Fragrance J., 2011, 26, 73–89 CrossRef CAS.
- C. T. Ma, T. Eom, E. Cho, B. Wu, T. R. Kim, K. B. Oh, S. B. Han, S. W. Kwon and J. H. Park, J. Nat. Prod., 2017, 80, 3043–3048 CrossRef CAS PubMed.
- M. Gao, X. M. Han, Y. Sun, H. J. Chen, Y. Yang, Y. Y. Liu, H. Meng, Z. H. Gao, Y. H. Xu, Z. Zhang and J. P. Han, RSC Adv., 2019, 9, 4113–4130 RSC.
- W. L. Mei, W. J. Zuo, D. L. Yang, W. H. Dong and H. F. Dai, Chin. J. Top. Crops, 2013, 34, 2513–2520 Search PubMed.
- H. Q. Chen, J. H. Wei, J. S. Yang, Z. Zhang, Y. Yang, Z. H. Gao, C. Sui and B. Gong, Chem. Biodivers., 2012, 9, 236–250 CrossRef CAS PubMed.
- S. R. Ibrahim and G. A. Mohamed, Nat. Prod. Res., 2015, 29, 1489–1520 CrossRef CAS PubMed.
- K. Iwagoe, T. Konishi, S. Kiyosawa, Y. Shimada, K. Miyahara and T. Kawasaki, Chem. Pharm. Bull., 1986, 34, 4889–4891 CrossRef CAS.
- K. Iwagoe, S. Kodama, T. Konishi, S. Kiyosawa, Y. Fujiwara and Y. Shimada, Chem. Pharm. Bull., 1987, 35, 4680–4682 CrossRef CAS.
- K. Iwagoe, T. Kakae, T. Konishi, S. Kiyosawa, Y. Fujiwara and Y. Shimada, Chem. Pharm. Bull., 1989, 37, 124–128 CrossRef CAS.
- T. Konishi, K. Iwagoe, S. Kiyosawa and Y. Fujiwara, Phytochemistry, 1989, 28, 3548–3550 CrossRef CAS.
- T. Konishi, K. Iwagoe, S. Kiyosawa and Y. Fujiwara, Chem. Pharm. Bull., 1991, 39, 1869–1870 CrossRef CAS.
- H. X. Huo, Z. X. Zhu, Y. L. Song, S. P. Shi, J. Sun, Y. F. Zhao, J. Zheng, J. K. Zjawiony, P. F. Tu and J. Li, J. Nat. Prod., 2017, 81, 543–553 CrossRef PubMed.
- P. Xiang, W. L. Mei, H. Q. Chen, F. D. Kong, H. Wang, G. Liao, L. M. Zhou and H. F. Dai, Fitoterapia, 2017, 120, 61–66 CrossRef CAS PubMed.
- Y. Yang, W. L. Mei, F. D. Kong, H. Q. Chen, W. Li, Z. B. Chen and H. F. Dai, Fitoterapia, 2017, 119, 20–37 CrossRef CAS PubMed.
- Y. Yang, H. Q. Chen, F. D. Kong, L. M. Zhou, W. Li, W. H. Dong, Z. B. Chen, W. L. Mei and H. F. Dai, Phytochemistry, 2018, 145, 207–213 CrossRef CAS PubMed.
- H. X. Huo, Y. F. Gu, Z. X. Zhu, Y. F. Zhang, X. N. Chen, P. W. Guan, S. P. Shi, Y. L. Song, Y. F. Zhao, P. F. Tu and J. Li, Phytochemistry, 2019, 158, 46–55 CrossRef CAS PubMed.
- W. Li, C. H. Cai, Z. K. Guo, H. Wang, W. J. Zuo, W. H. Dong, W. L. Mei and H. F. Dai, Fitoterapia, 2015, 100, 44–49 CrossRef CAS PubMed.
- W. Li, C. H. Cai, W. H. Dong, Z. K. Guo, H. Wang, W. L. Mei and H. F. Dai, Fitoterapia, 2014, 98, 117–123 CrossRef CAS PubMed.
- G. Liao, W. L. Mei, W. H. Dong, W. Li, H. Wang, F. D. Kong, C. J. Gai, X. Q. Song and H. F. Dai, Fitoterapia, 2016, 110, 38–43 CrossRef CAS PubMed.
- G. Liao, W. L. Mei, F. D. Kong, W. Li, J. Z. Yuan and H. F. Dai, Phytochemistry, 2017, 139, 98–108 CrossRef CAS PubMed.
- D. Chen, Z. R. Xu, X. Y. Chai, K. W. Zeng, Y. X. Jia, D. Bi, Z. Z. Ma and P. F. Tu, Eur. J. Org. Chem., 2012, 10, 5389–5397 CrossRef.
- B. Wu, S. W. Kwon, G. S. Hwang and J. H. Park, Helv. Chim. Acta, 2012, 95, 1657–1665 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, A. J. Valentino and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–90 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02597a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |