
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisproportionation-induced solid-state fluorescence in 6,13-dihydropentacenes†
Tomoyuki Tajima *a,
Rai Sandaa,
Katsuya Nishiharaa,
Hitoshi Shiraia,
Yasuhiro Okudab,
Akihiro Oritab and
Yutaka Takaguchi*a
*a,
Rai Sandaa,
Katsuya Nishiharaa,
Hitoshi Shiraia,
Yasuhiro Okudab,
Akihiro Oritab and
Yutaka Takaguchi*a
aGraduate School of Environmental and Life Science, Okayama University, 3-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan. E-mail: tajimat@cc.okayama-u.ac.jp; yutaka@cc.okayama-u.ac.jp
bDepartment of Applied Chemistry and Biotechnology, Okayama University of Science, 1-1 Ridai-cho, Kita-ku, Okayama 700-0005, Japan
First published on 31st May 2019
Abstract
The thermally and photolytically induced disproportionation of 6,13-dihydropentacene derivatives into tetrahydropentacenes and pentacenes results in unique solid-state fluorescence. The fluorescence thereby depends on the molecular structure and the molecular arrangement in the solid state.
Disproportionation reactions are arguably one of the most important classes of organic reactions, especially in the context of coal liquefaction and aromatization, as well as Cannizzaro and biochemical reactions. However, reports on disproportionation reactions of aromatic hydrocarbons remain scarce. Stein and co-workers have reported the disproportionation of 9,10-dihydroanthracene (>350 °C; liquid phase pyrolysis) into tetrahydroanthracene and anthracene.1 Pentacene has also been reported to undergo disproportionation at 320 °C in a horizontal vapor phase deposition furnace to produce 6,13-dihydropentacene and polycondensed aromatic hydrocarbons.2 It has been postulated that H-atom-transfer processes play a crucial role in such disproportion reactions. Indeed, H-atom transfer from 9,10-dihydroantharacene,3,4 xanthene,4 fluorene,5 acridan,6 and other H donors (D–H) with weak C–H bonds7 to unsaturated H acceptors has been widely explored.
On the other hand, the molecular design of π-conjugated systems is crucial for the development of new functional materials. Nevertheless, reports on π-conjugated systems exhibiting disproportionation-induced photoluminescence changes are relatively rare. Against this background, we decided to explore the utility of disproportionation reactions for the development of fluorescent chromic materials. Herein, we report the synthesis and disproportion of 6,13-dihydropentacene derivatives, as well as a detailed investigation into the fluorescence properties of the obtained compounds in the solid state.
The synthesis of N,N′-bisalkyl-6,13-dihydropentacene[2,3:9,10]biscarboxyimides 3a–c is outlined in Scheme 1. We have previously reported the synthesis of key starting material 6,13-dihydropentacene-2,3,9,10-tetracarboxylic acid (1).8 Tetracarboxylic acid 1 was converted into the corresponding anhydride (2) in good yield using a modification of the procedure reported by Qian and co-workers.9 N,N′-Bisalkyl-6,13-dihydropentacene[2,3:9,10]biscarboxyimides 3a–c were then obtained in 20–64% yield by stirring anhydride 2 under reflux in the presence of the corresponding amines. The structure of the obtained dihydropentacene bisimides was confirmed by 1H and 13C NMR spectroscopy, IR spectroscopy, FAB mass spectrometry, and elemental analysis. The 1H NMR spectrum of 3a in CDCl3 shows a singlet at 4.36 ppm, which was assigned to the central ring of dihydropentacene. In the aromatic region, two singlets appear at 8.03 and 8.23 ppm, which were attributed to the naphthalene ring. The 13C NMR of 3a displays 13 signals, including five aromatic carbon atoms (123.5, 128.0 × 2, 134.4, 138.4, and 168.3 ppm) for the naphthalene ring, one carbonyl carbon atom (186.3 ppm), and one sp3-hybridized carbon atom of the central ring (38.5 ppm). In general, the spectra of 3a–c are very similar.
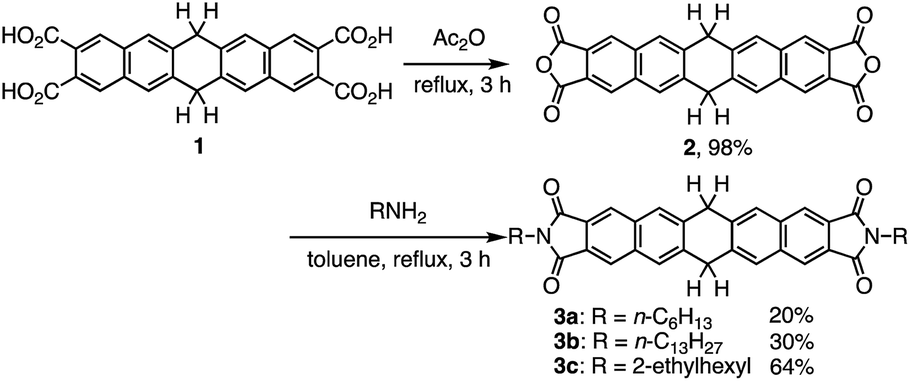 | ||
| Scheme 1 Synthesis of N,N′-bisalkyl-6,13-dihydropentacene[2,3:9,10]bis-carboxyimides 3a–c. | ||
Subsequently, we examined the disproportionation reaction of dihydropentacene 3a in the solid state (Scheme 2). Interestingly, upon heating solid 3a from room temperature to 230 °C, the color of the solid changed from colorless to green. To elucidate the origin of this green material, we investigated the absorption features of a thin film of 3a before and after heating (250 °C; 3 h). In the difference spectrum of 3a (Fig. 1), the positive absorption bands at 523, 560, and 611 nm were attributed to an expansion of the π-conjugated system. A comparison with the absorption spectrum of independently synthesized N,N′-bispentylpentacene[2,3:9,10]biscarboxyimide (4a) revealed very similar features. The formation of pentacene derivative 4a was also confirmed by 1H NMR spectroscopy (Fig. S1†). After heating (250 °C; 40 min), the main signals were assigned to the starting material (3a), and the new signals suggested a conversion <7%. The 1H NMR spectrum in CDCl3 showed three singlets at 8.50, 8.94, and 9.12 ppm, which were assigned to the protons attached to the pentacene core, respectively. Moreover, three singlets for aromatic protons (8.06, 8.22, and 8.39 ppm) and two singlets for methylene protons (4.27 and 4.81 ppm) were observed for tetrahydropentacene derivative 5a. Upon exposure to air, the signals of 4a disappeared and two singlets at 8.56 and 9.14 ppm appeared, which were assigned to pentacene quinone 7a, i.e., the oxidation product of pentacene 4a (Fig. S2†).8a,10 An integral 5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7a ratio of 1:1 was estimated. These results strongly indicate that the thermal reaction of dihydropentacene 3a affords pentacene 4a and tetrahydropentacene 5a (Scheme 2).
:7a ratio of 1:1 was estimated. These results strongly indicate that the thermal reaction of dihydropentacene 3a affords pentacene 4a and tetrahydropentacene 5a (Scheme 2).
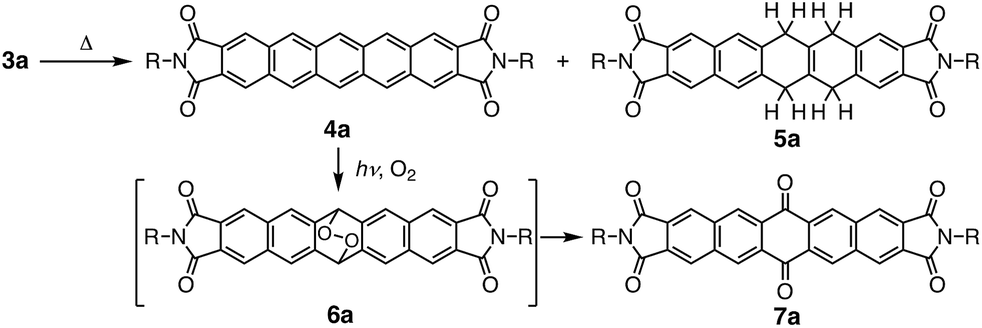 | ||
| Scheme 2 Disproportionation reaction of dihydropentacene 3a and decomposition of pentacene derivative 4a. | ||
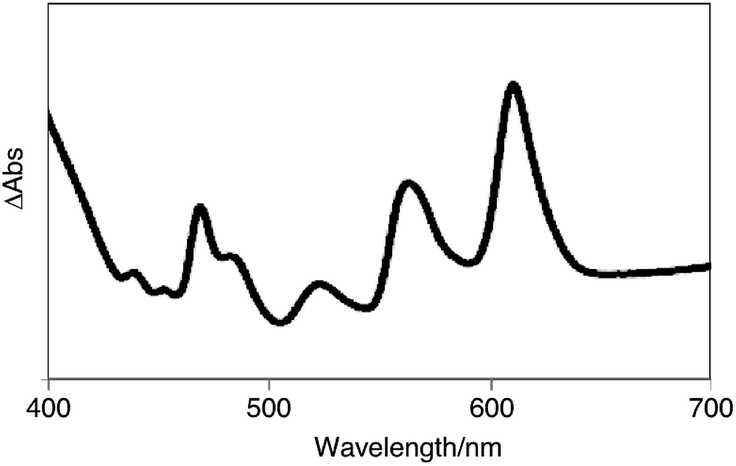 | ||
| Fig. 1 The absorbance difference spectrum of a thin film of 3a before and after the heating. | ||
Moreover, we discovered that N,N′-bisalky-6,13-dihydropentacene[2,3:9,10]bis-carboxyimide 3a exhibits solid-state fluorescence (Fig. 2); under illumination with UV light (λex = 365 nm), bright blue fluorescence was observed. Interestingly, upon heating 3a in solid state from room temperature to 230 °C, the color of the apparent fluorescence gradually changed from bright blue to yellow and then red (Fig. 2a). Such phosphorchromaticity was observed for both film and crystalline samples (Fig. 2b), and the original color was not recovered even after cooling to room temperature. The differential scanning calorimetry (DSC) cooling and heating curves of 3a showed no peaks between room temperature and 270 °C (Fig. S3†). In the case of 3b and 3c, similar fluorescence color changes were observed, while the crystal appearance changed from transparent to opaque. Polarized optical microscopy images revealed rough and cracked surfaces of the crystals of 3b and 3c after heating (Fig. S4†).
 | ||
| Fig. 2 (a) Apparent fluorescence color change of a 3a film with the increasing temperature. (b) Apparent fluorescence color change of crystals of 3a. | ||
We also observed significant changes in the fluorescence spectra of a thin film of 3a with increasing temperature (Fig. S5†). The intensity of the fluorescence of the naphthaleneimide chromophore (300–550 nm) decreased with increasing temperature, while the intensity of the new fluorescence band at 638 nm, which was assigned to a pentacene chromophore, increased. The decaying emission of the naphthaleneimide skeleton may be explained in terms of an energy transfer from 3a to 4a.11
Fig. S6† shows the concentration-dependent emission spectra of 3a in CHCl3. At a concentration of 1.9 × 10−4 M, a monomer emission band from the naphthaleneimide moiety was observed at 390 and 400 nm (λex = 280 nm). Upon increasing the concentration of 3a beyond 7.7 × 10−4 M, the fluorescence intensity of the naphthaleneimide moieties decreased and two new excimer emission bands emerged at approximately 440 and 460 nm with growing intensity. These results indicate that the solid-state fluorescence at 440 nm does not correspond to monomer emission, but to aggregate emission.
In order to determine the molecular conformation in the crystals and rationalize the observed reactivity, a single-crystal X-ray diffraction analysis was carried out (Fig. 3). Depending on the solvent, two different crystalline structures were identified in the case of 3a. A structural analysis of single crystals of 3a–c grown from benzonitrile solutions revealed a planar central six-membered ring of the dihydropentacenes (Fig. 3a, and S8†). The bond angle sums of the central six-membered rings are close to 720°. Numerous hitherto reported crystal structures are characterized by flattened boat conformations for dihydroanthracene and dihydropentacene derivatives, whereas the preferred conformation of dihydroaromatics should be a planar according to a theoretical study.12 In fact, there are a few crystallographic reports on dihydroanthacene13 and dihydropentacene14 skeletons with a planar geometry. In contrast, the structural analysis for crystals of 3a grown from a THF solution revealed a V-shaped dihydropentacene skeleton with a bent central six-membered ring 3a(v).
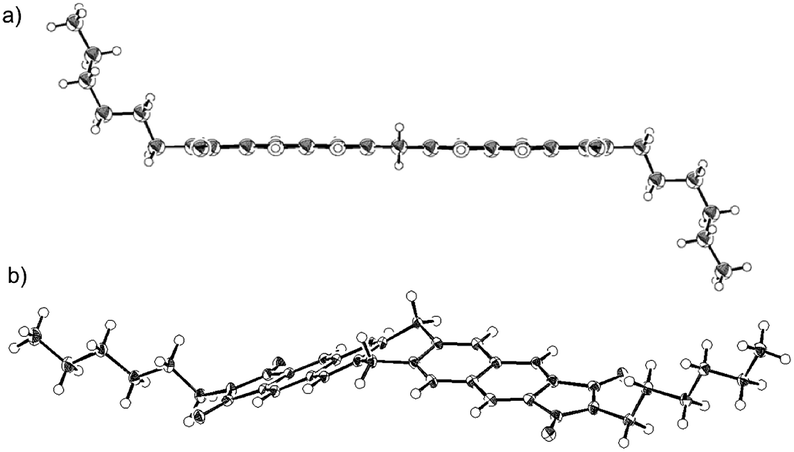 | ||
| Fig. 3 ORTEP drawings of (a) 3a(planer) and (b) 3a(v) (thermal ellipsoids at 50% probability). | ||
The energy difference between the planar and V-shaped conformers was examined using density functional theory (DFT) calculations on model compound 3′, which bears H atoms instead of alkyl chains. The V-shaped conformer 3′(v) is by 4.0 kcal mol−1 thermodynamically more stable than the planar conformer 3′(planar). These results suggest a small energy difference between the two conformers. Consequently, the solid-state structure of 3a is most likely determined by packing forces in the crystal.
Interestingly, the phosphorchromaticity changes in the solid state were also triggered by photoirradiation (>300 nm); however, the reaction was limited to the surface of the specimen. The phosphorchromaticity switching rate was sensitive to the substituents on the dihydropentacene. Upon photoirradiation of solids 3a(planar) and 3b (λex > 300 nm; 1 h), the apparent fluorescence color gradually changed from bright blue to red. In the case of 3(v) and 3c, the apparent fluorescence color did not change, not even after two days of photoirradiation. To clarify the different reactivity of the planar and V-shape conformers of 3a, further DFT calculations were carried out. Fig. 4 shows the relevant Kohn–Sham molecular orbitals for the optimized structures of 3′(planar) and 3′(v). Both HOMOs are clearly localized on the C–H bonds of the central six-membered ring and the two naphthalene units. Interestingly, the LUMO of 3′(planar) is notably less localized around the C–H bond of the central six-membered ring than the LUMO of 3′(v). These results indicate that the excited state of 3a(planar) favors the elimination of the C–H protons of the central six-membered ring compared to the case of 3a(v).
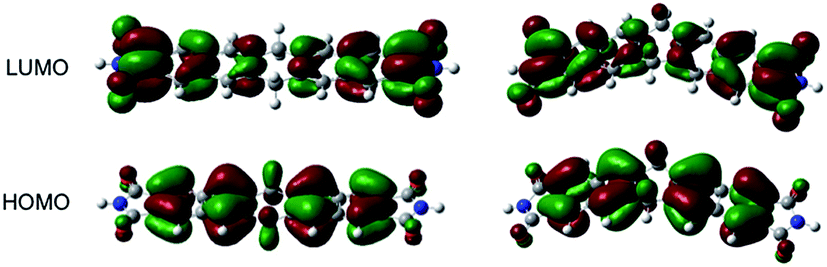 | ||
| Fig. 4 Molecular Kohn–Sham orbitals of 3′(v) and 3′(planer), calculated at the M06-2X/6-31G* level of theory. | ||
To evaluate the differences in photoreactivity depending on the substituents, the molecular arrangements in the crystals were investigated. None of the crystals contained any solvent in the lattice. The distances between the H (from the C–H bonds of the central six-membered ring) and C (from the naphthalene ring of the nearest-neighboring molecule atom) atoms were ca. 3.0 Å (3a), 2.8 Å (3b), and 3.3 Å (3c). The long distance in the crystals of 3c should thus be unfavorable for hydrogen-atom transfer from the C–H bond of the central six-membered ring to the naphthalene ring of the neighboring molecule.
Conclusions
N,N′-Bisalkyl-6,13-dihydropentacene[2,3:9,10]biscarboxyimides 3a–c undergo disproportionation reactions in the solid state. The molecular structure and conformation in the crystals strongly influence the disproportionation reaction. Upon heating or photoirradiation of 3 in the solid state, the fluorescence color gradually changed from bright blue to red. The solid-state fluorescence behavior15 induced by disproportionation may serve as an example for guided molecular engineering, providing fascinating possibilities to tune materials for sensing, as well as optical and thermal recording applications.Experimental section
Instruments
Unless otherwise stated, all experiments were performed under an atmosphere of argon. Solvents were dried by standard methods and distilled prior to use. CDCl3 was purchased from Cambridge Isotopes. NMR spectra were recorded on a JEOL JNM AL-300 or a Varian NMR System 600 spectrometer. The 1H and 13C NMR shifts are given in ppm relative to tetramethylsilane (TMS; δ = 0 ppm) and referenced internally with respect to the residual proton (CDCl3: δ = 7.26 ppm), or the 13C NMR resonance of the solvent (CDCl3: δ = 77 ppm). UV-visible spectra were measured on a Shimadzu UV-3150 spectrometer. Emission and excitation spectra were recorded on a Jasco FP-6500 spectrometer, while IR spectra were obtained on a Thermo Nicolet IR Affinity-1 spectrophotometer. Thermogravimetric analysis (TGA) was performed on a Shimadzu DTG-60 in air. Wet column chromatography (WCC) was performed using Wakogel C-200. The melting points were determined on a MEL-TEMP micro melting point apparatus and are uncorrected. Reagents were purchased from Wako Pure Chemical Industries Ltd., Tokyo Chemical Industries Co., Ltd., Kanto Kagaku Co., Ltd., Nacalai Tesque Co., Ltd., and Aldrich Chemical Co. Pentacene-2,3,9,10-tetracarboxylic acid (1) was prepared according to reported procedures.8Synthesis of 2 and 3a–c
000), 305 (18000), 365 (9000) nm; IR (KBr) 2956, 2922, 2828, 1770, 1708, 1392 cm−1. Anal. calcd for C52H66N2O4: C, 79.79; H, 8.50; N, 3.58; found: C, 79.53; H, 8.56; N, 3.55.X-ray data collection
Single crystals of 3a–c were grown by slow evaporation of saturated solutions of 3a–c in benzonitrile at room temperature. Single crystals of 3a(v) were grown by slow evaporation of a saturated solution of 3a(v) in THF at room temperature. The crystals were coated with hydrocarbon oil, mounted on glass fibers, and placed under a cold stream of N2 in the diffractometer. The intensity data for 3a–c were collected on a Rigaku VariMax diffractometer with a Saturn CCD detector using graphite-monochromated Mo-Kα radiation (λ = 0.71071 Å) up to 2θmax = 50° at 103 K. All structures were solved by Patterson methods (SHELXS-97)16 and refined by full-matrix least-squares procedures on F2 for all reflections (SHELXL-97)17. All hydrogen atoms were located following AFIX instructions. All other atoms were refined anisotropically. CCDC-1911683 (3a(planar)), CCDC-1911685 (3a(v)), CCDC-1911684 (3b), and CCDC-1911686 (3c) contain the supplementary crystallographic data for this paper.3a(planar): C38H38N2O4, MW = 586.70, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), a = 4.755(15) Å, b = 6.057(19) Å, c = 27.46(9) Å, α = 86.36(9)°, β = 89.245(10)°, γ = 84.00(6), V = 785(4) Å3, Z = 1, Dcalcd = 1.241 g cm−3, μ = 0.080 mm−1, R1(I > 2σ(I)) = 0.0616, wR2 (all data) = 0.2120, T = 103(2) K, GOF = 1.051.
(#2), a = 4.755(15) Å, b = 6.057(19) Å, c = 27.46(9) Å, α = 86.36(9)°, β = 89.245(10)°, γ = 84.00(6), V = 785(4) Å3, Z = 1, Dcalcd = 1.241 g cm−3, μ = 0.080 mm−1, R1(I > 2σ(I)) = 0.0616, wR2 (all data) = 0.2120, T = 103(2) K, GOF = 1.051.
3a(v): C80H84N4O9, MW = 1245.51, triclinic, space group P (#2), a = 4.483(3) Å, b = 16.529(1) Å, c = 21.775(13) Å, α = 85.587(10)°, β = 88.775(17)°, γ = 87.293(12), V = 1074.5(11) Å3, Z = 1, Dcalcd = 1.287 g cm−3, μ = 0.074 mm−1, R1(I > 2σ(I)) = 0.045, wR2 (all data) = 0.1036, T = 103(2) K, GOF = 1.031.
3b: C52H66N4O4, MW = 783.07, triclinic, space group P (#2), a = 4.638(2) Å, b = 9.316(5) Å, c = 24.909(14) Å, α = 91.5765(12)°, β = 91.622(12)°, γ = 92.166(15), V = 1074.5(11) Å3, Z = 1, Dcalcd = 1.210 g cm−3, μ = 0.075 mm−1, R1(I > 2σ(I)) = 0.0491, wR2 (all data) = 0.1352, T = 103(2) K, GOF = 1.073.
3c: C42H46N4O4, MW = 642.81, triclinic, space group P (#2), a = 7.451(5) Å, b = 8.965(5) Å, c = 14.802(12) Å, α = 78.46(5)°, β = 84.78(5)°, γ = 63.33(4), V = 865.7(11) Å3, Z = 1, Dcalcd = 1.233 g cm−3, μ = 0.079 mm−1, R1(I > 2σ(I)) = 0.0476, wR2 (all data) = 0.1264, T = 103(2) K, GOF = 1.059.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was partially supported by JSPS KAKENHI Grants 15H03519 (Y. T.), 16K05895 (T. T.), 18H04430 in Middle Molecular Strategy (A. O.) and 18K05134 (A. O.), 19K15574 (Y. O.), Okayama Foundation of Science and Technology (Y. O.), Promotion and Mutual Aid Corporation for Private Schools of Japan (Y. O.) and OUS Research Project (OUS-RP-19-4 to A. O. and Y. O.). The authors (A. O. and Y. O.) thank to Research Instruments Center, Okayama University (JNM-ECS400 and JNM-ECZ400).Notes and references
- S. E. Stein, L. L. Griffith, R. Billmers and R. H. Chen, J. Org. Chem., 1987, 52, 1582 CrossRef CAS.
- L. B. Roberson, J. Kowalik, L. M. Tolbert, C. Kloc, R. Zeis, X. Chi, R. Fleming and C. Wilkins, J. Am. Chem. Soc., 2005, 127, 3069 CrossRef CAS PubMed.
- (a) M. Gerst and C. Rüchardt, Tetrahedron Lett., 1993, 34, 7733 CrossRef CAS; (b) F. Schaffer, H.-D. Beckhaus, H.-J. Rieger and C. Rüchardt, Chem. Ber., 1994, 127, 557 CrossRef CAS; (c) M. Gerst, H.-D. Beckhaus, C. Rüchardt, E. E. B. Campbell and R. Tellgmann, Tetrahedron Lett., 1993, 34, 7729 CrossRef CAS; (d) C. Rüchardt, M. Gerst, J. Ebenhoch, H.-D. Beckhaus, E. R. B. Campbell, R. Tellgmann, H. Schwarz, T. Weiske and S. Pitter, Angew. Chem., Int. Ed. Engl., 1993, 32, 584 CrossRef; (e) M. Gerst, H.-D. Beckhaus, C. Rüchardt, E. R. B. Campbell and R. Tellgmann, Tetrahedron Lett., 1993, 34, 7729 CrossRef CAS; (f) N. Naghizada, G. H. C. Prado and A. de Klerk, Energy Fuels, 2017, 31, 6800 CrossRef CAS.
- (a) C. Rüchardt, M. Gerst and M. Nölke, Angew. Chem., Int. Ed. Engl., 1992, 31, 1523 CrossRef; (b) M. Coellen and C. Rüchardt, Chem.–Eur. J., 1995, 1, 564 CrossRef CAS; (c) C. Höfler and C. Rüchardt, Liebigs Ann., 1996, 183 Search PubMed.
- K. Rakus, S. P. Verevkin, J. Schätzer, H.-D. Beckhaus and C. Rüchardt, Chem. Ber., 1994, 127, 1095 CrossRef CAS.
- H. Friebolin and C. Rüchardt, Liebigs Ann., 1995, 1339 CrossRef CAS.
- (a) C. Rüchardt, M. Gerst and J. Ebenhoch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1406 CrossRef; (b) J. A. Franz and D. M. Camaioni, J. Org. Chem., 1984, 49, 3563 CrossRef CAS; (c) M. Gerst and C. Rüchardt, Chem. Ber., 1993, 126, 1039 CrossRef CAS; (d) F. Payan and A. de Klerk, Energy Fuels, 2018, 32, 9340 CrossRef CAS.
- (a) T. Tajima, A. Yamakawa, K. Fukuda, Y. Hayashi, M. Nakano and Y. Takaguchi, Chem. Lett., 2012, 41, 2702 CrossRef; (b) H. Shirai, T. Tajima, K. Kubo, T. Nishihama, H. Miyake and T. Takaguchi, Bull. Chem. Soc. Jpn., 2016, 89, 437 CrossRef CAS; (c) K. Kubo, T. Tajima, H. Shirai, T. Nishihama and T. Takaguchi, ChemistrySelect, 2017, 8, 2452 CrossRef.
- K. Baathulaa, Y. Xu and X. Qian, J. Photochem. Photobiol., A, 2010, 216, 24 CrossRef CAS.
- H. Yamada, Y. Yamashita, M. Kikuchi, H. Watanabe, T. Okujima, H. Uno, T. Ogawa, K. Ohara and N. Ono, Chem.–Eur. J., 2005, 11, 6212 CrossRef CAS PubMed.
- B. Manna, R. Ghosh and D. K. Palit, J. Phys. Chem. C, 2016, 120, 7299 CrossRef CAS.
- K. B. Lipkowitz and P. W. Rabideau, J. Org. Chem., 1982, 47, 1002 CrossRef CAS.
- J. Shi, D. Wu, Y. Ding, D. Wu, H. Hu and G. Lu, Tetrahedron, 2012, 68, 2770 CrossRef CAS.
- (a) D. Lehnherr, A. H. Murray, R. McDonald and R. R. Tykwinski, Angew. Chem., Int. Ed., 2010, 49, 6190 CrossRef CAS PubMed; (b) W. Liu, Y. L. Tnay, K. P. Gan, Z. Liu, W. H. Tyan and K. Narasaka, Helv. Chim. Acta, 2012, 95, 1953 CrossRef CAS.
- C. Yuan, S. Saito, C. Camacho, S. Irle, I. Hisaki and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 8842 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELX-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1911683–1911686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03297e |
| This journal is © The Royal Society of Chemistry 2019 |