
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and dynamics of γ-secretase with presenilin 2 compared to presenilin 1†
Budheswar Dehurya,
Ning Tanga,
Tom L. Blundellb and
Kasper P. Kepp *a
*a
aDepartment of Chemistry, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark. E-mail: kpj@kemi.dtu.dk; Tel: +045 45252409
bDepartment of Biochemistry, University of Cambridge, Cambridge, CB2 1GA, UK
First published on 4th July 2019
Abstract
Severe early-onset familial Alzheimer's disease (FAD) is caused by more than 200 different mutations in the genes coding for presenilin, the catalytic subunit of the 4-subunit protease complex γ-secretase, which cleaves the C99 fragment of the amyloid precursor protein (APP) to produce Aβ peptides. γ-Secretase exists with either of two homologues, PS1 and PS2. All cryo-electron microscopic structures and computational work has so far focused on γ-secretase with PS1, yet PS2 mutations also cause FAD. A central question is thus whether there are structural and dynamic differences between PS1 and PS2. To address this question, we use the cryo-electron microscopic data for PS1 to develop the first structural and dynamic model of PS2-γ-secretase in the catalytically relevant mature membrane-bound state at ambient temperature, equilibrated by three independent 500 ns molecular dynamics simulations. We find that the characteristic nicastrin extra-cellular domain breathing mode and major movements in the cytosolic loop between TM6 and TM7 occur in both PS2- and PS1-γ-secretase. The overall structures and conformational states are similar, suggesting similar catalytic activities. However, at the sequence level, charge-controlled membrane-anchoring is extracellular for PS1 and intracellular for PS2, which suggests different subcellular locations. The tilt angles of the TM2, TM6, TM7 and TM9 helices differ in the two forms of γ-secretase, suggesting that the two proteins have somewhat different substrate processing and channel sizes. Our MD simulations consistently indicated that PS2 retains several water molecules near the catalytic site at the bilayer, as required for catalysis. The possible reasons for the differences of PS1 and PS2 are discussed in relation to their location and function.
Introduction
Alzheimer's disease (AD) is the most prevalent chronic neurodegenerative disorders, characterized by progressive loss of memory, cognitive impairment, and personality change; more than 30 million people globally suffer from the disease.1 The onset and progression of AD is thought to relate to the formation of aggregated β-amyloid (Aβ) peptides.2–4 Aβ is formed by proteolytic cleavage of the amyloid precursor protein (APP), a type I membrane protein. Initially, β-secretase cleaves APP to produce a 99-amino-acid membrane-bound C-terminal fragment (C99).5,6 The C99 transmembrane domain is then cleaved by the intramembrane-cleaving aspartyl protease γ-secretase, releasing the APP intracellular domain (AICD) and Aβ.7 The processing of C99 starts at the cytoplasmic border at residues 48 or 49 (ε-sites) and progresses stepwise by three or four amino acids towards the N-terminus.8–11 Depending on the initial ε-site, Aβ peptides of different lengths are generated, most notably Aβ40 and Aβ42. Aβ42 is highly aggregation-prone, neurotoxic, and a major component of senile plaques, and thus considered a possible pathogenic culprit of AD.12–17More than 200 mutations in the genes coding for the proteins presenilin 1 and 2 (PS1 and PS2) cause particularly severe disease inherited within families, known as early-onset familial AD (FAD).18,19 PS1/PS2 functions as the catalytic subunit of γ-secretase and is thus directly responsible for producing Aβ, but many other substrates are also cleaved by the enzyme complex, including importantly Notch.20,21 FAD-causing PS1/PS2 mutations tend to decrease the activity of γ-secretase and at the same time consistently increase the produced Aβ42/Aβ40 ratio.22–25 The step-wise C99 cleavage mechanism8,26 explains the variable cleavage products produced, and the recently published structure of C83 bound to the protein is consistent with this mechanism.27 It is also not known how the pathogenic FAD mutations cause disease and produce longer Aβ peptides, although the increased Aβ42/Aβ40 caused by mutation correlates with clinical severity and thus implies a relationship to the disease.25,28–30
Because of the importance of γ-secretase and PS1/PS2 to AD, we need to understand why humans use two types of presenilins. PS2 mutations tend to be clinically milder than PS1 mutations and associate with atypical dementia, dementia with Lewy bodies, and frontotemporal dementia.22 Unlike mutations in PS1, PS2 variants display incomplete penetrance, and the mean age at symptom onset has been stated to be at least 10 years higher.22 From the data set of Ryman et al.,22 we obtain an average age of onset of 43 and 58 years for PS1 and PS2 carriers, respectively. From our compiled larger data set,29 we estimate values of 44 and 62 years. Although the number of confirmed pathogenic PS2 mutations is small, this strongly suggests that PS2 variants are less disruptive than PS1 variants.
PS1 (467 aa; 46 kDa) and PS2 (448 aa; 55 kDa) share 66.3% sequence identity (with a sequence similarity of 71.9%) at the amino acid level; both harbor the two conserved catalytic aspartic acid residues, Asp-263 and Asp-366 (PS2 numbering), and are involved in a variety of biological processes.31–34 PS2 is located predominantly within the endoplasmic reticulum (ER) and early Golgi apparatus and is primarily expressed in neurons, whereas PS1 associates mainly with cell membranes.35 To reach a mature active state, both proteins are proteolytically autocleaved to generate two chains, the N-terminal and C-terminal fragments (NTF and CTF).7,36,37
PS1 is considered more important to γ-secretase-associated C99 and Notch cleavage than PS2, because knockout of PS1 in mice results in an embryonic lethal phenotype and a significant decrease in Aβ levels.38 Loss of function responsible for this phenotype has been suggested in two forms, one that emphasizes the important physiological role of Aβ,39 and another that emphasizes loss of PS function in relation to other functions.40,41 Knockout of PS2 alone does not change Aβ levels and is viable. Independent of their role in γ-secretase activity, presenilins also function as passive Ca2+ leak channels in the endoplasmic reticulum,42 and accordingly mutations in both presenilins affect intracellular calcium homeostasis.43–46 Comparative studies of PS1- and PS2-γ-secretase activities indicate higher measured activity of PS1-γ-secretase, but the specific activities of PS1 and PS2 are probably similar once accounting for the PS-constitution in the complex, which is more difficult for PS2.47,48 Different γ-secretases containing either PS1 or PS2 associate with distinct sub-cellular compartments,35 and some PS2 mutations dramatically change the Aβ42/Aβ40 ratios.48,49
Recent topologies of γ-secretase obtained from cryo-electron microscopy (cryo-EM)50–52 have enabled the study of the structure and dynamics of γ-secretase at the atomic scale using molecular dynamics (MD) simulations.32,53–57 Six cryo-EM structures of human γ-secretase without substrate bound are reported in the Protein Data Bank (PDB) with PDB codes 4UI2, 5A63, 5FN2, 5FN3, 5FN4, and 5FN5,50,51,58 and two new structures are reported with models of C99 (C83) (ref. 27) and Notch.59 These structures provide the overall subunit topology but miss many important details (loops and parts of helices) and notably the membrane environment at ambient temperature where these complexes feature in vivo. They all contain PS1 together with the three other subunits nicastrin (NCT), anterior pharynx-defective 1A (APH-1A), and presenilin enhancer 2 (PEN-2).34 NCT represents a type-1 transmembrane glycoprotein formed by a single TM in its C-terminal region and a large N-terminal extracellular domain. This domain is presumed to play a key role in selectively controlling entry of bulky substrates into the active site of PS1.60
All-atom and coarse-grained MD simulations have provided precise structural and dynamical insight into disease-related human proteins and their mutants,61–65 including PS1 and γ-secretase.32,54,56,57,66 Although necessarily building on (and reproducing) the experimental cryo-EM data, the computational PS1 models have greater atomic precision (they are at infinite atomic resolution). These models reveal the impact of the membrane (which is not seen in the cryo-EM but substantially affects protein conformation), the auto-cleavage of the large hydrophilic loop (not in the cryo-EM, but required for activity), and detailed TM helix movements that are elusive in cryo-EM; they directly probe the conformational dynamics of PS1-γ-secretase contributing to substrate processing and Aβ production.53,55,57 The three plausible different conformational states of γ-secretase, i.e. open, semi-open, and closed states, are governed by distinct tilt angles of the gate helices 2 and 6 and large movements of the NCT extracellular domain (ECD) and the hydrophilic loops that affect the compactness of the binding site, the associated affinity, retention time, and extent of trimming of C99 and the resulting Aβ42/Aβ40 ratio, viz. the Fit-Stay-Trim (FIST) mechanism of γ-secretase cleavage.57,67,68
Despite PS2 being associated with FAD, almost all work has so far focused on PS1, and neither experimental nor theoretical structures are available for PS2. We asked the question whether PS2 structure and dynamics inside γ-secretase are different from those of PS1-γ-secretase. In the absence of experimental structures of PS2, we assumed that the general structure and topology, due to the high sequence identity, are similar to that for PS1. We merged a three-dimensional homology model of PS2 based on our previous all-atom PS1 model,56,57 with the cryo-EM structure of the human γ-secretase (5A63 and 5FN2) as template, with all missing parts added, to produce a complete (except the elusive N-terminal part of PS1/2) atomic model of PS2-γ-secretase fully consistent with currently known structural information. To understand the structure and dynamics of complete PS2-γ-secretase, we employed all-atom MD simulations of the full mature auto-cleaved multimeric structure of the enzyme complex in a realistic membrane model at ambient temperature where thermal disorder effects on the membrane-protein system can be assessed as a complement to the low-temperature cryo-EM data.
Computational methods
Model of all-atom PS2 subunit
Despite the improvements in resolution of structures obtained from cryo-EM, a three-dimensional structure of PS2 has not been determined yet. We developed our model based on the amino acid sequence of PS2 from the UniProtKB database (ID: P49810). BLASTp and DELTA-BLAST search against protein structures in the PDB suggested the cryo-EM structures of γ-secretase (5A63 and 5FN2) as the most suitable templates. The cryo-EM structures of γ-secretase miss important regions such as the C-terminal residue segments of length 11 and 21 in nicastrin and APH1, whereas the PEN2 structure is completely available. The catalytic subunit PS1 lacks several features, most notably the large cytosolic hydrophilic loop region whose autocleavage is required to obtain the mature active state. Thus, the activity driven by maturation is arguably not deducible from the static cryo-EM structures. TM2 situated close to the catalytic site and the extracellular hydrophilic loop 1 are also absent in the highest-resolution structure (5A63).51We used a multi-template strategy to ensure the best possible coverage and avoid bias in our model built from Modeller version 9.20,69 by considering PS1 from 5FN2 and 5A63 and our previously established fully MD-equilibrated all-atom PS1 model. This model was constructed on the basis of a range of homology modeling programs; it includes HL2 and accounts for the high-resolution features of all PS helices TM1–TM9, which is not the case for individual cryo-EM structures.56,57 PS2 has a substantially shorter HL2, making its modeling and equilibration more straightforward than HL2 of PS1. The final PS2 structure spanned from Glu-77 to Ile-448, as templates are unavailable for the N-terminal loop. Since template-based methods may produce errors when the template proteins are not similar enough to the target protein, we further employed ab initio structure refinement as implemented in Galaxy Refine.70 The model structure with the smallest Cα-RMSD compared to the structural homologs was used and further validated by SAVES version 5.0, ProSA-Web,71 Molprobity,72 and ProQ73 model validation servers. Based on the model validation statistics (compared to experimental PS1 in Table S1†), our model was an appropriate basis for construction of the complete multi-subunit model of PS2-γ-secretase.
Development of complete mature PS2-γ-secretase model
The developed model is essentially complete as it includes all atoms of 5A63 and 5FN2, except the elusive 1–70 residue N-terminal of PS1 which has no template and is far from the catalytic site. Thus, the model is as complete as that of our previous model of PS1-γ-secretase.57 The model of PS2-γ-secretase was developed upon structural superposition and coordinate transfer of each subunit using PyMOL version 2.0. Our final complete model displayed a Cα-RMSD of 0.194 Å, 0.172 Å, and 0.123 Å respectively vs. that of 5A63, 5FN2 and our previously reported complete model of PS1-γ-secretase, respectively, showing that our models respect all the significant features of the experimental cryo-EM data while at the same time adding many new features to them. Upon MD simulation, this model is importantly directly comparable to our previously simulated model of PS1-γ-secretase.57We produced the mature model of PS2 from the optimized non-mature structure by cleaving the peptide bond between Ala-297 and Met-298 followed by addition of one oxygen atom and two hydrogen atoms to the CO and NH groups of Ala-297 and Met-298, respectively, using BIOVIA Discovery Studio Visualizer version 4.5. Cleavage of the peptide bond resulted in an N-terminal fragment (NTF: 77–297) and a C-terminal fragment (298–448) as a model of mature PS2. This state is additionally trimmed by ∼10 residues in vivo leaving a mixture of matured isoforms whose relative prevalence is unknown and will certainly affect loop dynamics much less than the initial autocleavage. This loop maturation produces two chains with major disorder required for catalytic activity, which are completely absent in the cryo-EM structures, but fortunately straightforwardly explored by MD. Bad contacts were removed using BIOVIA DSV before merging the mature model with the membrane-solvent system.
All-atom MD simulations of PS2-γ-secretase within the membrane
γ-Secretase is a membrane protein complex, but the available experimental structures do not describe the protein in the context of the cell membrane. Hence, to produce a realistic structural model of the protein complex in the membrane, the complete model was oriented using the PPM (position protein in membrane) server.74 This membrane-aligned model was then embedded into a membrane–solvent bilayer system using the membrane builder tool of the CHARMM-GUI web server.75 A homogeneous POPC lipid (302 lipids) bilayer was generated around the protein in a periodic rectangular simulation box by maintaining a water thickness of 20 Å (46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 105 water molecules) above and below the protein complex. Then the system was supplemented with salt ions to a formal concentration of 0.15 M NaCl (146 Na+ and 126 Cl− ions) using Monte Carlo randomization. The model was subjected to extensive quality control against experimental facts as discussed in the results and discussion section.
105 water molecules) above and below the protein complex. Then the system was supplemented with salt ions to a formal concentration of 0.15 M NaCl (146 Na+ and 126 Cl− ions) using Monte Carlo randomization. The model was subjected to extensive quality control against experimental facts as discussed in the results and discussion section.
All-atom MD simulations were performed using GROMACS gpu version 2018.2.76 We used the recently developed CHARMM36m force field, which was developed for good structure balance between secondary and disordered structure77 with TIP3P78 as an explicit water model. Langevin dynamics with a collision frequency of 1 ps−1 was applied at a temperature of 303.15 K, and system pressure was kept at 1 bar using the Berendsen barostat and a relaxation time of 0.5 ps. By applying the SHAKE algorithm the system was propagated with 2.0 femtosecond time steps. The Verlet cut-off scheme was used to treat non-bonded interactions with a large cut-off for Coulomb and Lennard–Jones interactions of 0.12 nm to ensure accurate long-range energetics as conformational changes in the protein occur on long length scales.57 Long-range electrostatic interactions were estimated using the Particle-Mesh Ewald (PME) method.79 The Lennard–Jones potential was truncated using a shift function between 0.9 and 1.2 nm. Full electrostatic interactions were evaluated between 0 and 1.2 nm using the Particle Mesh Ewald (PME) approach. The neighbor lists were updated every 20 steps and all bonds involving hydrogen atoms were constrained using the Linear Constraint Solver (LINCS) algorithm. Steepest descent minimization of the protein-lipid-solvent system was followed by six-step position-restrained equilibration for 500 ps to relax the atomic positions, with two steps in a canonical (NVT) ensemble and four steps within the isothermal–isobaric (NPT) ensemble. Finally, 500 ns of production simulations were carried out with a 2 fs time step with an NPT ensemble using the Nose–Hoover thermostat and Parrinello–Rahman barostat at 303.15 K and 1 atm, respectively.
In terms of simulation time and strategy, the helix tilts and loop movements have characteristic time scales of 1–100 ns (ref. 80 and 81) and the latter are well sampled only beyond 100 ns. In addition, we know from previous work that the first 200 ns are required to reach dynamic stability of the ensemble at physiologically relevant temperature.57 The relative helix movements of PS2 define the active site space, substrate binding affinity, the extent of cleavage, and thus the Aβ42/Aβ40 ratio, and are thus the central modes of the system from a catalytic and medicinal perspective.57 To get these dynamics accurate, we require all-atom descriptions (including helix hydrogens) with a structure-balanced force field and explicit water in a complete membrane-water system to ensure best possible realism at the tradeoff of very long (microsecond) simulations routinely done using coarse-grained models. Confirming the expected timescale of these modes,81 we see many helix tilt events and resulting broad tilt angle distributions, thus making 500 ns a necessary and sufficient time scale of simulation. This shows that we have an optimal balance between model realism and sampling requirements. To account for the stochastics of the simulations, we performed three independent simulations of the same system starting with different initial velocity seeds. All computations were performed using the high-performance computing facility at the Technical University of Denmark. In order to ensure adequate comparison with PS1-γ-secretase we also performed two seeded MD simulations of 500 ns each for PS1-γ-secretase using the exact same parameters as described above for PS2-γ-secretase.
Analysis of MD trajectories
The trajectories of the three simulations were analyzed using built-in GROMACS tools. Membrane properties including deuterium order parameters of the acyl chains, the density of the membrane environment, the area per lipid head group, and the bilayer thickness were computed using GridMATMD82 and FATSLiM.83 From the trajectories, coordinates were saved every 20 ps, yielding 25000 structural snapshots from each simulation. The root-mean-square deviation (RMSD) for backbone heavy atoms (i.e. N, Cα and carbonyl C atoms), root-mean-square fluctuation (RMSF) for Cα atoms, and radius of gyration (Rg) were computed using the gmx rmsdist, gmx rmsf and gmx gyrate tools, respectively. Visualization of MD trajectories was conducted using VMD version 1.9.3,84 and structural interpretations were performed with PyMOL and BIOVIA DSV.
Principal component analysis
Principal component analysis (PCA) was employed to identify the large-scale collective motions of atoms. We performed PCA of the last 300 ns of each simulation, to avoid artifacts from the first equilibration process up to 200 ns. A covariance matrix was constructed to capture the degree of collinearity of atomic motions for each pair of atoms using the gmx covar tool. Subsequently, each matrix was diagonalized to generate a diagonal matrix of eigenvalues using gmx anaeig. The eigenvectors (EV) define collective motions, and the values of the vectors indicate how much the corresponding atoms participate in the motion. The associated eigenvalue reflects the total motility associated with an eigenvector. Usually most of the motion in the system is described by relatively few EVs.Clustering and free energy landscape (FEL) analysis
To analyze the most frequently visited conformations of PS2-γ-secretase, we performed ensemble-average clustering analysis85 using the gmx cluster tool with a cut-off of 0.2 nm. The last 300 ns of each simulation were used in the clustering analysis, as these parts represent stable conformational ensembles. In addition, the Jarvis-Patrick algorithm (https://pypi.org/project/jarvispatrick/) was also used for comparison. The RMSD cutoff used to determine the number of nearest neighbors for the Jarvis-Patrick algorithm was 0.2 nm, and the algorithm was run for 10000 iterations. Snapshots that had at least three identical nearest neighbors were assigned to the same cluster. We performed FEL analysis of the last 300 ns of each system using the gmx sham module of GROMACS and OriginPro 2019. The FEL represents a mapping of all possible conformations adopted by the protein during simulation, together with their corresponding free energy estimated from the logarithm of their relative persistence in time. The FEL was based on the radius of gyration (Rg, PC1) and the root-mean-square deviation (PC2) that reflects conformational variability and was conducted for both PS2-γ-secretase and PS2 alone. We extracted a representative snapshot using get_timestamp.py script from the low-energy region of the FEL plot and compared with the top ranked cluster structure.
Results and discussion
Sequence differences and 9 TM topology of PS1 and PS2
It is evident from the pair-wise sequence alignment (Fig. S1†) that PS1 and PS2 share least similarity at their N-terminals and the large cytosolic hydrophilic loop regions connecting TM6 and TM7. Furthermore, in PS1 we observe a strongly negatively charged DEEED motif (residues 66–70) at the N-terminal, just before the conserved residues begin, signifying the extracellular membrane interface. This segment is very different and much less charged in PS2 (PPGLE). We speculate that this difference will facilitate the fixation of PS1 in a cell membrane with this motif functioning as an anchor on the extracellular side of the membrane, whereas PS2 lacks this feature, possibly because it is not required to be oriented towards the extracellular environment. PS2 is known to localize more towards the intracellular compartments.35 We also note that the cytosolic C-terminal of the NTF of PS1/PS2 is not strongly anchored by charge, whereas the N-terminal of the CTF is in both proteins (EDPEER and EEEEER in PS1 and PS2, respectively). We suspect that these differences are functionally important and will affect the relative conformational flexibility of TM1 and TM7. The ∼67% identity indicates that the proteins share the same 9 TM topology with the conserved catalytic residues numbered Asp-263 (TM6) and Asp-366 (TM7) in PS2 (257 and 385 in PS1).Quality control of PS2 model
Due to the gaps in the alignment, all single-template approaches failed to deliver a good-quality protein model. Therefore, a multiple-template approach was employed as described in the Methods section, yielding a PS2 model with very good scores with respect to the template structures in all validation servers as listed in Table S1†. The Φ and Ψ angles in the Ramachandran plot were in more than 98% cases associated with the allowed regions indicating a very good model †. The non-bonded interactions analyzed using ERRAT had 80.4% matches with those of highly refined structures. Similarly, the MolProbity analysis revealed the absence of bad backbone bonds and angles and Cβ deviations. The ProSA-webserver calculated the overall quality score (z-score) of the model to be −3.45, which is within the range of experimentally determined crystal structures of the same size.71 Other validation servers also presented high validation scores compared to the template cryo-EM structures (Table S1†). The hydrophilic loop 1 (residues Glu-114–Ile-173, HL1) connecting the TM1 and TM2 towards the extracellular space was found to superpose well, but the long hydrophilic loop 2 (Glu-279–Glu-355, HL2) between TM6 and TM7 differed somewhat in terms of orientation. The large loop also harbors an α-helical segment (307–324), a feature observed in our previous model of PS1.56 This would be difficult to observe in the cryo-EM structures but is likely a real feature supported by NMR and other studies.86 Inspection of TM9 reveals two helical segments (TM9a and TM9b) caused by a kink at the C-terminal, which reorients the CTF of PS2 towards APH-1A. Our simulations thus indicate that TM9 is less stable and can engage in conformational changes, consistent with its role in initial substrate binding to PS1.87,88Pair-wise structural superimposition of the Cα atoms of the template structures and modeled structure revealed an RMSD of 0.70, 0.97, and 0.84 Å for 5A63, 5FN2 and the homology model of PS1, testifying to the establishment of an accurate PS2 model (Fig. 1). The side chains of the catalytic residues superpose well with those of the high-resolution cryo-EM structures of PS1, with the distance between the Cα atoms of Asp-263 and Asp-366 being ∼10.2 Å, slightly less than the 10.6 Å measured for 5A63.
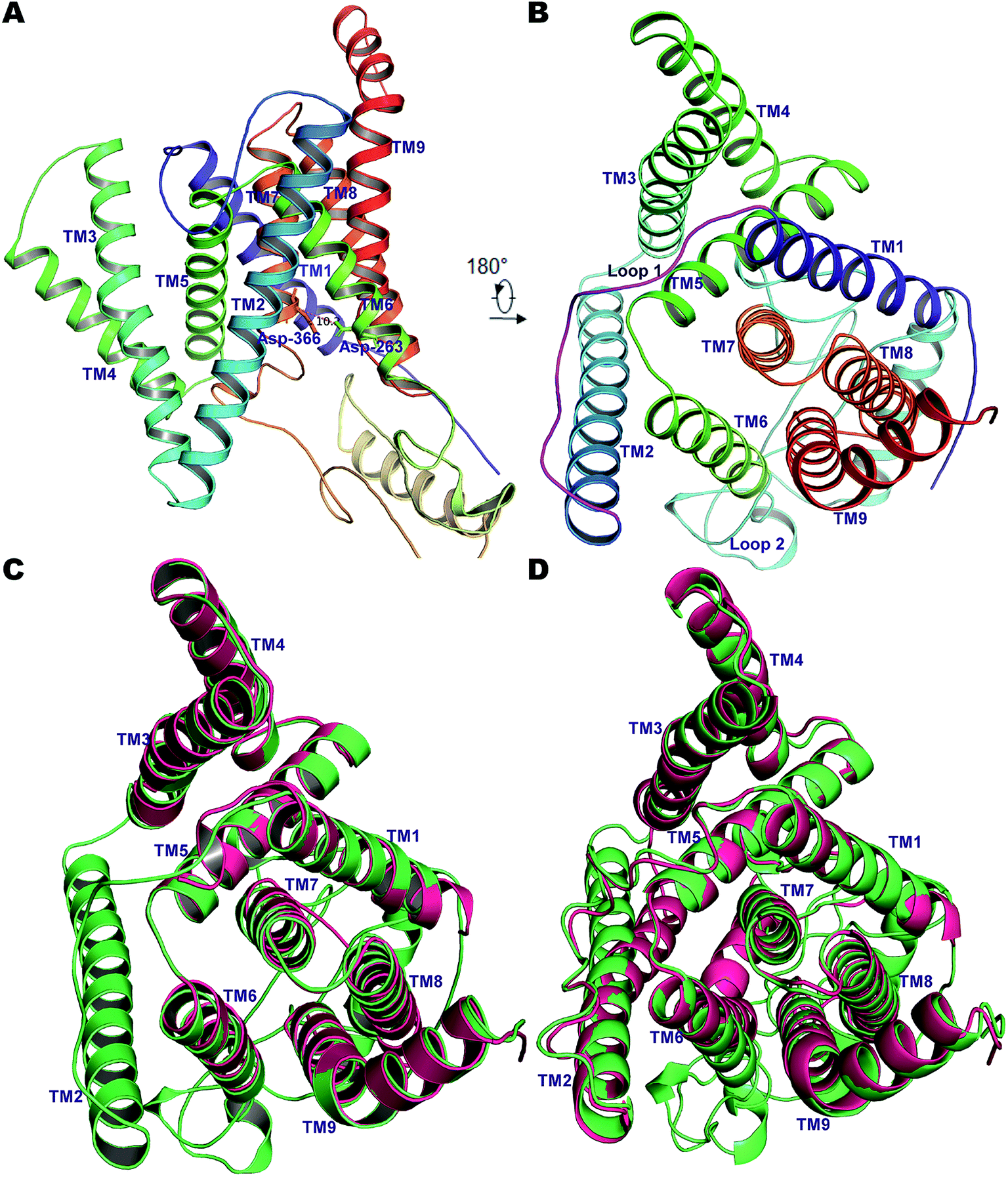 | ||
| Fig. 1 (A) The 9 TM architecture of PS2 modelled using cryo-EM structures of PS1-γ-secretase (PDB ID: 5A63, 5FN3) and our previously established homology model of PS1.56 (B) The rotated top view of the homology model of PS2. The small loop connecting TM1 and TM2 and the large cytosolic loop connecting TM6 and TM7 are colored magenta and cyan, respectively. (C) Structural superimposition of PS2 (green) with cryo-EM structure of PS1 from 5A63 (magenta/pink). (D) Structural superposition of modelled PS2 (green) and the cryo-EM structure 5FN2 (magenta/pink). The images were generated using PyMOL. | ||
Our complete structure of PS2-γ-secretase (Fig. S3†) upon structural superposition has an RMSD of 0.19 and 0.17 Å, respectively, vs. 5A63 and 5FN2 (and 0.12 Å vs. our all-atom PS1 γ-secretase model). The final, membrane-bound mature form of full PS2-γ-secretase model (including hydrogen atoms at pH 7) (Fig. 2A) was subject to three independently seeded all-atom MD simulation in the complete membrane-solvent system shown in Fig. 2B, simulated for 0.5 μs in each of the three simulations.
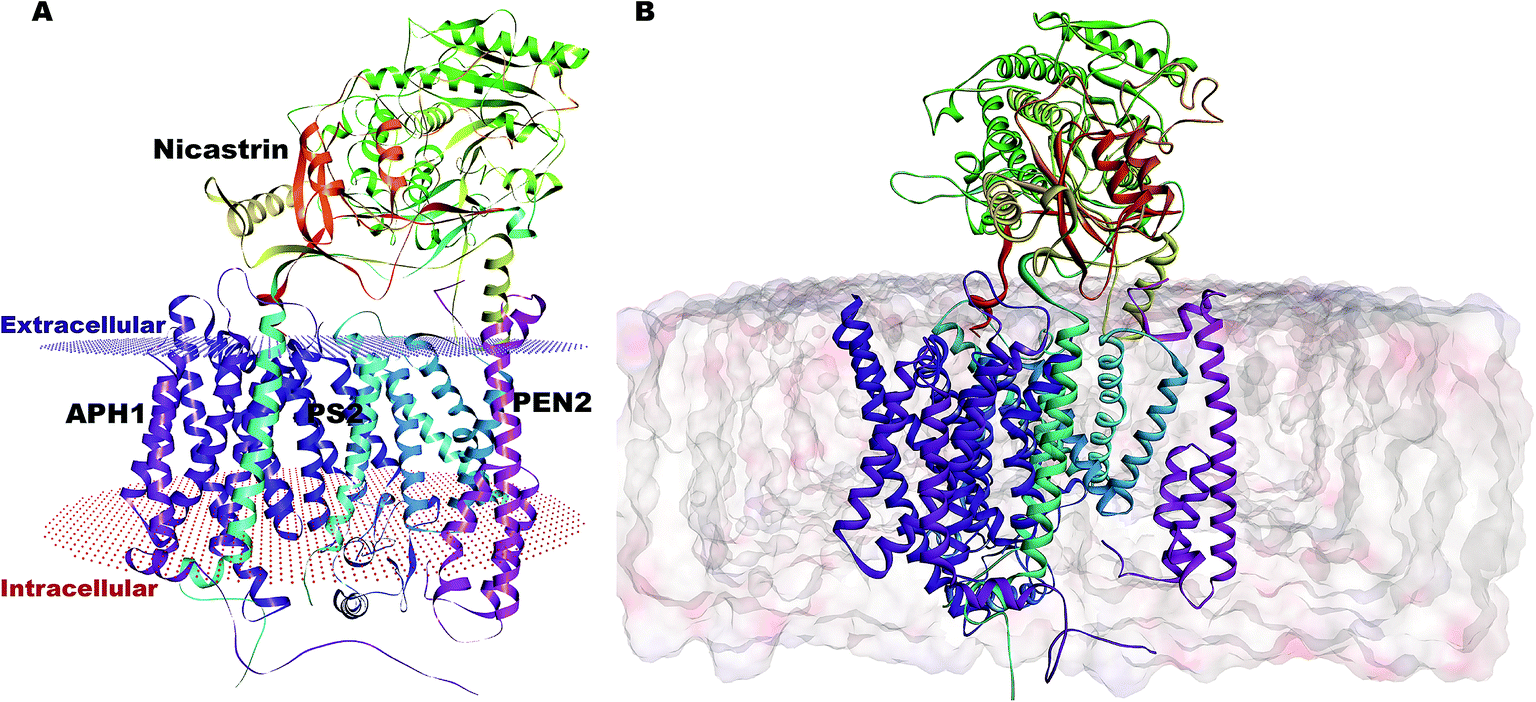 | ||
| Fig. 2 (A) Membrane-aligned schematic view of PS2-γ-secretase. (B) Complete mature PS2-γ-secretase in the POPC lipid bilayer system with bilayer structure shown. | ||
Quality control of the full membrane-γ-secretase model
The lipid composition of the membrane is likely to affect the conformation and proteolytic activity of γ-secretase, as this process occurs in the middle of the membrane,89,90 which makes our study an important complement to the cryo-EM data measured without the presence of a membrane. At ambient temperature, the thermal disorder of the complete membrane-protein system is much larger than at cryo-EM temperatures. The interplay between the structure and function of membrane proteins and membrane lipid dynamics is well known.32,53 The lipid membrane thickness affects the cleavage pattern of C99 with thicker membranes favoring production of Aβ40 and thinner membranes favoring Aβ42 production.91 Increasing the number of carbons in the lipid chain can increase the activity of γ-secretase and reduce the Aβ42/Aβ40 ratio.92 These findings follow the FIST model56,57,67 asserting that γ-secretase contains an open and a semi-open state, and that membrane packing increases fitting, retention time, and trimming of C99, leading to shorter Aβ peptides. Pathogenic PS1 and PS2 mutations disfavor the membrane-packed protein state and favor the open state with shorter retention time and reduced trimming leading to longer Aβ42/Aβ40 ratios, which correlate with disease severity.29 In contrast, γ-secretase modulators favor the more compact semi-open conformation by contributing their binding affinity to the stability and lifetime of the ternary complex (modulator-C99-γ-secretase), reducing the Aβ42/Aβ40 ratio.67The properties of a lipid bilayer are adequately described by the acyl chain order parameters, the area per lipid, and the bilayer thickness, and were thus computed as shown in Fig. S4 and S5.† The deuterium order parameter is central to quantify the structural orientation and flexibility of lipids in bilayers. The calculated deuterium order parameters per atom for carbon tails of the POPC membrane (sn1 and sn2 chains) and the splitting of the values near the head group are in excellent agreement with typical experimental values,93 as expected from the previous validation of CHARMM36m.94 The density of the head-group, tail-group and phosphate across the POPC lipid bilayer (along the z-axis) attained very typical values testifying to the realism and structural integrity of our membrane-protein model, with no undesired conformational changes occurring in the membrane (Fig. S4†). The density profile of headgroups exhibits a slight increase on the lower-leaflet. It is likely that polarization of the upper portions of the acyl chains by the water-headgroup interface may cause this effect, as NCT protects the membrane on the extracellular side. This asymmetry requires an all-atom description of the dynamics of both protein and membrane to be uncovered and thus does not show up in cruder (coarse-grained) models. The average area per lipid calculated using FATSLIM ranged from 59.1 Å2 to 60.4 Å2 for the three simulations (Fig. S5†). The surface area per lipid for POPC was slightly lower than seen in some other MD studies (64–65 Å2);95 such ∼5 Å2 variations are expected and are most likely due to differences in the water-lipid force field interaction parameters, as seen for POPS and phosphatidylcholine simulations.96
In order to characterize the POPC membrane integrity, we computed its thickness over the total simulation time (Fig. S5†). A thicker bilayer region in simulations 2 and 3 relative to simulation 1 testifies to the dynamics of the membrane-protein system, although overall structural integrity is maintained throughout the simulations. These variations in membrane thickness show that the membrane dynamics may affect the conformational states of γ-secretase, and thereby, as found previously, also its activity.91 Our results again fit well with the FIST model that emphasizes the overall compactness of the conformation state rather than local interactions with catalytic residues in determining the cleavage pattern.
To assess further the quality of our simulated structures, the overall Cα-RMSD between the cryo-EM structure of PS1-γ-secretase (5FN2) and PS-2-γ-secretase is below ∼3.5 Å, whereas the average Cα-RMSD (3.32 Å, 3.30 Å and 3.1 Å respectively) of our three representative PS2-γ-secretase structures are within the thermal fluctuation range (Fig. S6A†). Similarly, the structural superimposition of the MD simulated representative of PS2 with 5FN2 PS1 revealed Cα-RMSD of 2.78 Å, 1.69 Å and 1.97 Å, where TMs superpose well with each other except for TM2. The TM2, implicated in substrate recruitment displayed notable changes in orientation in C-terminal region, indicates its plasticity and disorder nature like that of PS1 (Fig. S6B†). In other words, as evidenced from Fig. S6†, the representative snapshots from the simulation respect all the constraints of the experimental cry-EM structures, which are mainly due to the TM helices, but in addition contain the missing membrane environment and the loop residues that are invisible in the cryo-EM data.
Essential dynamics of PS2-γ-secretase
In the analysis we emphasize the functionally important movement of TM helices in PS2 and the dynamics of the loops, known to have characteristic life times of typically 10–100 ns and thus accessible to MD simulations after equilibration at the 100 ns timescale.80,81 In PS1-γ-secretase these movements control the space near the catalytic aspartates and thus plausibly the retention time and trimming of C99;57 the maturation of the HL2 is directly required for activity and produces the multi-state nature of the protein, not seen in the immature form.56 To ensure that we capture these dynamics well, we performed three independent 500 ns simulations. For all simulations, the backbone RMSD relative to the initial positions (Fig. 3A) display a stable profile after 100–200 ns indicating a general equilibrium state at this time, and thus collecting statistics for the last 300 ns of each of the triplicate simulations is meaningful.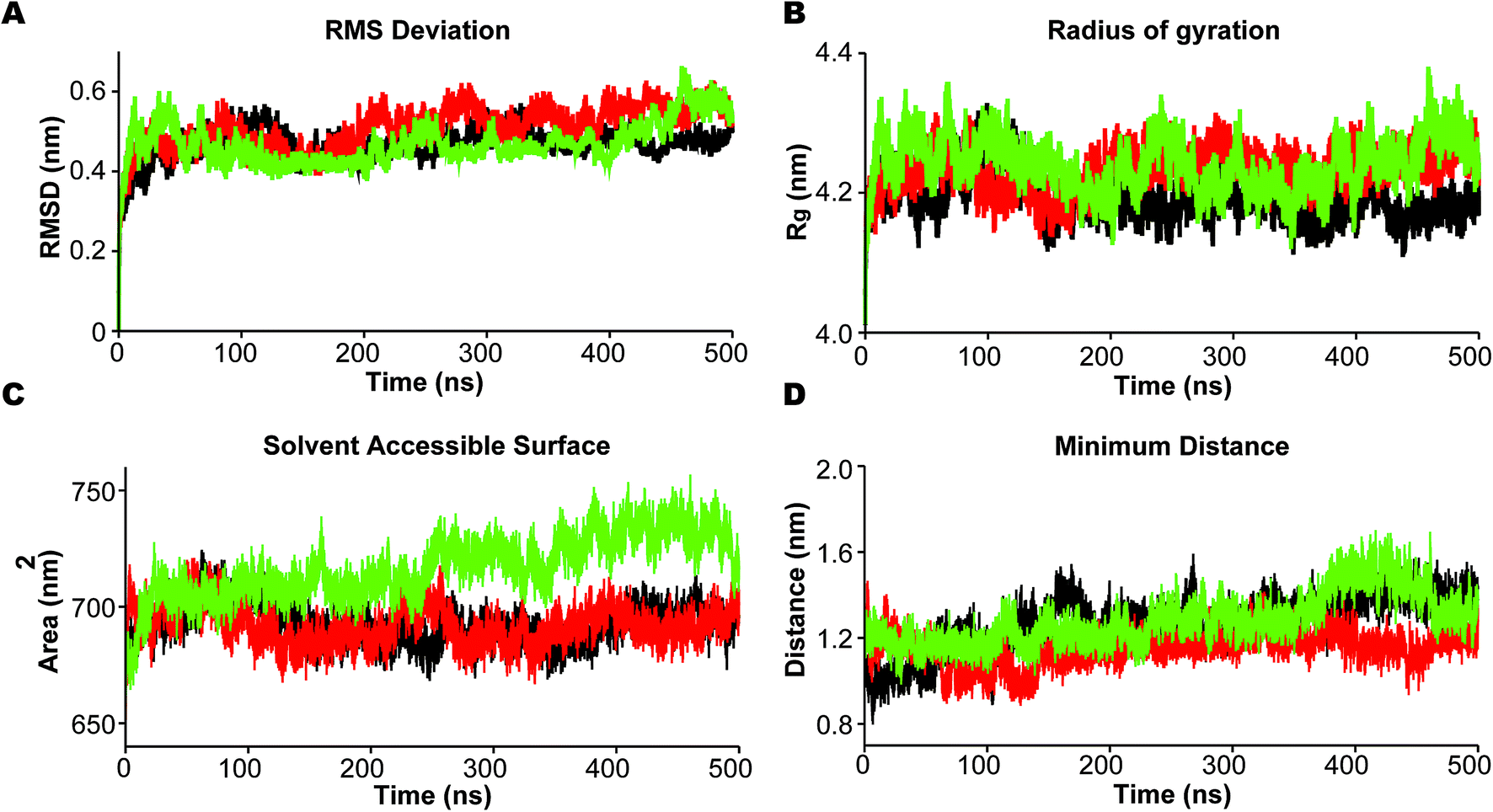 | ||
| Fig. 3 Dynamic stability of mature PS2-γ-secretase during three 500 ns MD simulations in the membrane. (A) The root-mean squared deviation (RMSD) of the backbone atoms of PS2-γ-secretase. (B) The compactness of the trajectories estimated by the mean of radius of gyration (Rg) over 500 ns time. (C) The solvent accessible surface area (SASA) of the three PS2-γ-secretase systems. (D) The minimum Cα-distance of the catalytic Asp-263 and Asp-366. Black lines represent simulation 1, red lines represent simulation 2, and green represents simulation 3. | ||
Rg varies within the narrow range of 4.15–4.35 nm (Fig. 3B). The solvent accessible surface area (SASA) was also relatively constant after 200 ns indicating small changes in overall fold structure during simulation (Fig. 3C); however, a very interesting increase in exposure was seen for simulation 3 (green), relating to a loose conformation state to be discussed further below. This increased exposure is probably catalytically important as it also increases the average distance between the two catalytic aspartates (Fig. 3D), with the exposed green simulation showing the largest catalytic pocket. Finally, as a test of the integrity of the overall complex, the RMSDs of the individual subunits of PS2-γ-secretase i.e., NCT, PS2, APH1, and PEN2, displayed stable trends after 200 ns, indicating that each subunit remained stable within the lipid bilayer system (Fig. S7†).
The corresponding Cα-root mean square fluctuations (RMSF) of all the subunits are shown in Fig. 4. The NCT movements near residue 600 (Fig. 4A) have been consistently seen in all previous simulations and relate to a large motion of the ECD. Importantly, we clearly see that the catalytic subunit PS2 (Fig. 4B) is much more dynamic than other subunits (Fig. 4A, C and D). HL2 connecting TM6 and TM7 is very flexible, and this is largely due to maturation that has produced the NTF and CTF as shown before for PS1, and it correlates with motions in TM2, TM6, and TM7.56 Variations in these helices are expected to affect Aβ42 production.56,92,97 HL1 connecting the TM1 and TM2 is less flexible than HL2 (Fig. 4B). Substrate binding to HL1 is indispensable for both ε- and γ-cleavage, whereas binding to the C-terminal of PS1 hampers cleavage.98 During the simulations, the RMSF of the catalytic aspartates is 0.17–0.28 nm (average 0.22 nm) and 0.12–0.15 nm (average 0.14 nm), respectively, for the Asp-263 and Asp-366, suggesting that the latter catalytic aspartate is much more restricted in its movement, which could be of catalytic relevance.
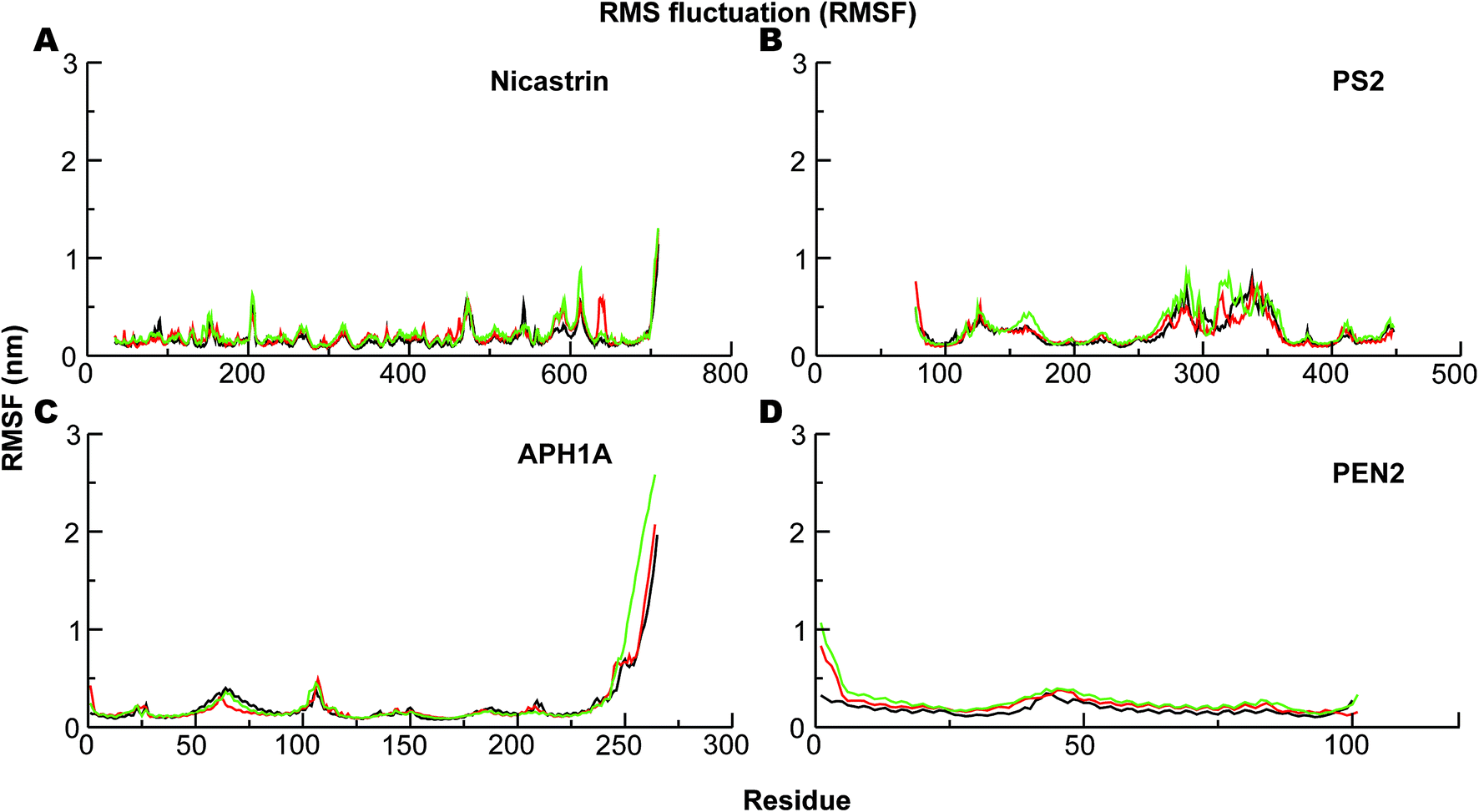 | ||
| Fig. 4 The Cα-root-mean-squared fluctuations (RMSF) of the four subunits of PS2-γ-secretase obtained using the last 300 ns trajectories from all-atom MD simulation in lipid bilayer. (A) The RMSF profile of NCT. (B) Cα-RMSF profile of PS2. (C) RMSF of the APH1A subunit of PS2 γ-secretase. (D) RMSF analysis of PEN2 subunit during last 300 ns of MD. Black lines represent simulation 1, red lines represent simulation 2, and green represents simulation 3. | ||
In order to understand the dynamics of mature PS2 within γ-secretase in the membrane in more complete detail, PCA was performed on the three systems.57 The global motions described by the first two EVs of all three simulations differ, with the third simulation behaving particularly differently (Fig. S8†). All three trajectories reach distinct minima with small energy barriers. The eigenvalues of simulation 3 indicate large conformational changes from a predominantly loose state to a more compact state. Porcupine plots of PC1 and PC2 (Fig. 5) quantify the features of the top-two EVs of the three ensembles. As expected, HL2 of PS2 and PSEN-2 displayed movements, as does the C-terminal part of APH1 (Fig. 5 and S7†). PEN2 plays a crucial role in endo-proteolysis and stabilization of the resulting NTF and CTF.99–101 TM3 and TM7 of PS2 also display tilt movements that are small compared to loop movements but catalytically important (see below). NCT movements are important as a gatekeeper of substrate entry into the PS1 catalytic site.55,102 Large movements of NCT–ECD relative to the membrane are also evident in PS2 (Fig. 5), providing a rationale for the gatekeeping dynamics also in PS2, not surprising considering the structural homology of the extracellular parts of the proteins.
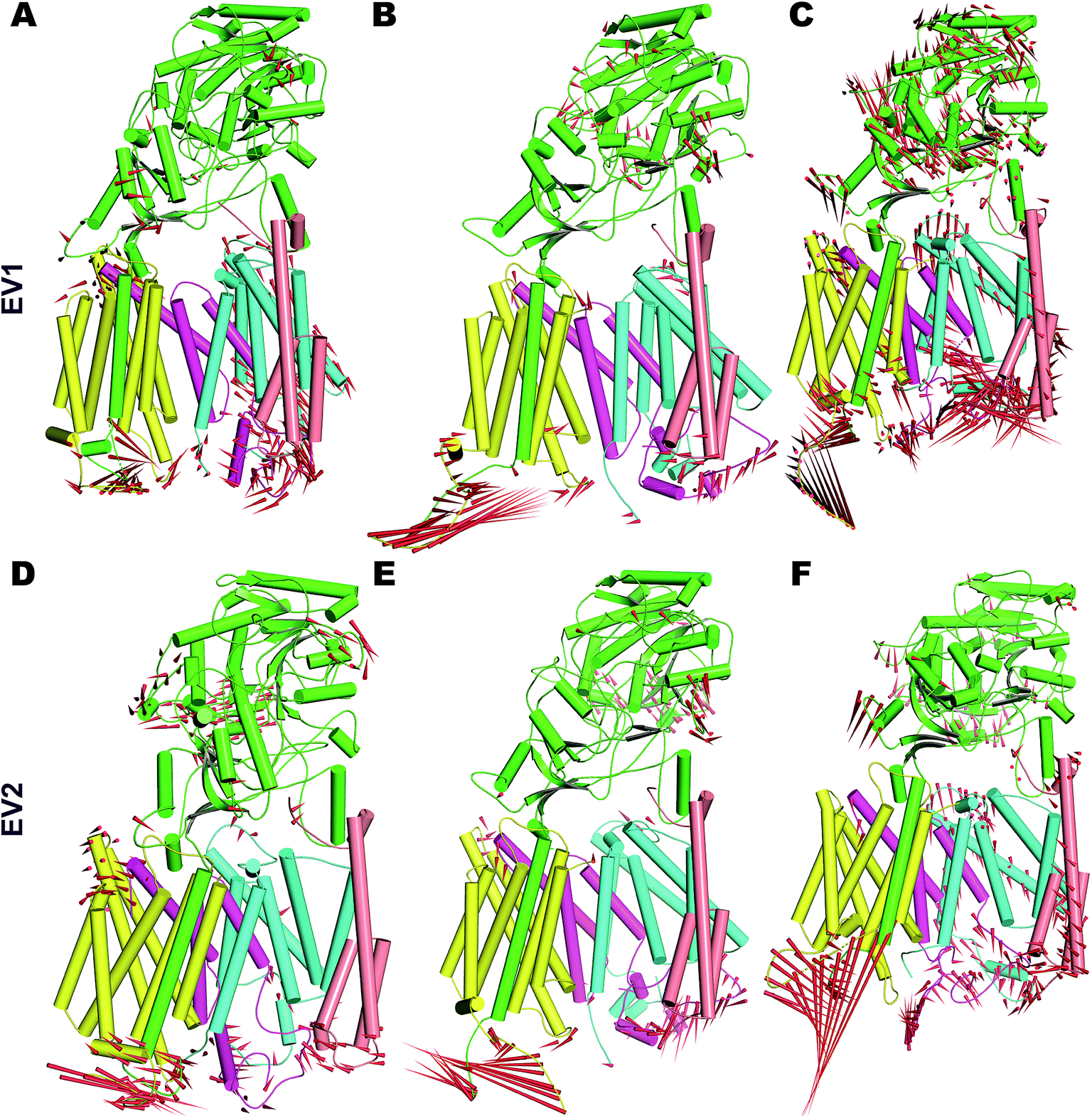 | ||
| Fig. 5 Porcupine plot depicting the dominant motions in PS2-γ-secretase obtained from the first two eigenvectors (EV1 and EV2) by PCA. (A) The movement of EV1 of simulation 1 (B). EV1 of simulation 2. (C) EV1 of simulation 3. (D) EV2 of simulation 1 (E) EV2 of simulation 2. (F) EV2 of simulation 3. Red needles show the direction (arrows) and amplitude of motion (the length). The different subunits are colored (Nicastrin: green; PS2-NTF: cyan; PS2-CTF: magenta; APH1: mellow; PEN2: tint wheat). | ||
Free energy landscape of conformation states of PS2-γ-secretase
The FELs sampled by equilibrated trajectory of the last 300 ns are shown in Fig. 6 for the full PS2-γ-secretase complexes for the three simulations (Fig. 6A, B and 6C) and for the corresponding PS2 subunit alone (Fig. 6D, E and F). The overall shapes of the FELs indicate that the main motions are similar in the simulated ensembles. However, simulation 3 had a distinctly different lowest minima compared to the other two simulations. Comparing these FELs to those for PS2 alone (Fig. 6D–F) suggests that the difference is not primarily due to the PS2 subunit, but largely to NCT. However, a substantially more variable free energy landscape is evident for Simulation 3 (the loose state) and largely due to PS2 dynamic variability (Fig. 6D). These findings are supported by PCA analysis (Fig. S9 and S10†) and quantify at high atomic precision a similar multistate tendency of PS2-γ-secretase as seen for PS1-γ-secretase involving NCT,103 but additionally an important PS2 dynamics that produce catalytically important differences, discussed below. Fluorescence lifetime imaging microscopy indicates that γ-secretase undergoes major conformational changes in the cell membrane,103–105 and our all-atom simulations in the membrane (which is absent in the cryo-EM structures) complement with the atomic contributions of the membrane to the NCT conformation states.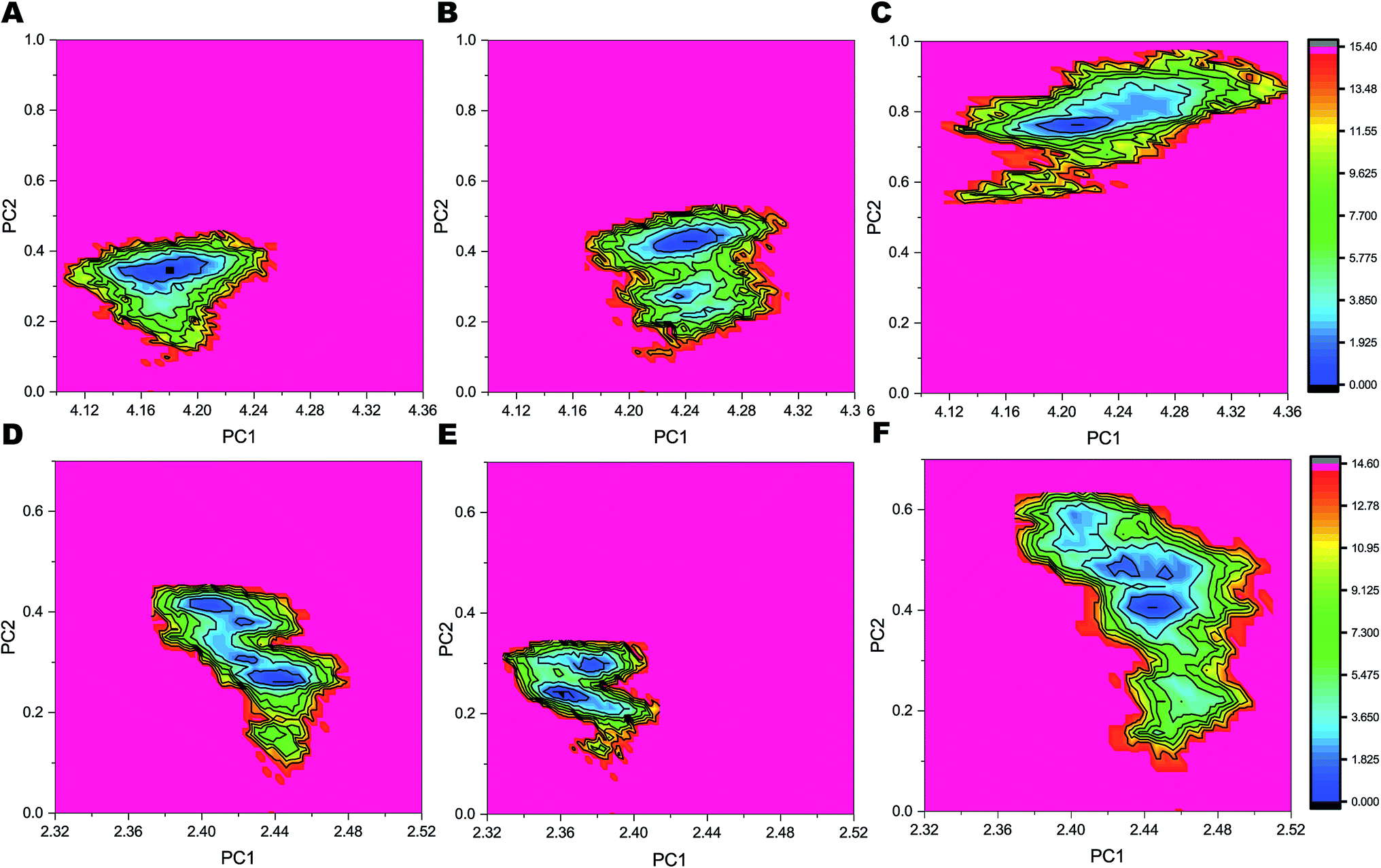 | ||
| Fig. 6 Free energy landscapes of the three simulations of PS2-γ-secretase and PS2 alone in the membrane as a function PC1 and PC2. The colored scale to the right defines the relative free energy, with dark blue being deep minima. (A) FEL profile of complete PS2-γ-secretase obtained from Simulation 1. (B) FEL profile of PS2-γ-secretase obtained from Simulation 2. (C) FEL profile of PS2-γ-secretase obtained from Simulation 3. (D) FEL profile of the PS2 subunit extracted from the full complex from Simulation 1. (E) FEL profile of PS2 from Simulation 2. (F) FEL profile of PS2 from Simulation 3. | ||
To analyze the most persistent conformations of PS2 γ-secretase, we considered the representative structures obtained from clustering analysis of each of the three simulations (Fig. S11†). The representative structure of a cluster has the smallest average RMSD distance to all other snapshot (time point) structures of the cluster. In simulation 1, cluster 1 included 63.2% of all conformations and cluster 2 included 21.8% of all conformations out of a total of 43 clusters. From simulation 2, 83 clusters were obtained with cluster 1 comprising 69.1% and cluster 2 comprising 17.1% of all conformations. Simulation 3 yielded 175 clusters; cluster 1 included 61.7% and cluster 2 19.8% of all conformations. The alternative Jarvis–Patrick algorithm produced the representative structures in Fig. S12.† As summarized in Fig. 7A–C, the top-ranked cluster contained >80% of the snapshots of the three systems. We aligned the TM helices of each representative cluster based on the Cα-atoms (Fig. 7D). The resulting RMSD values (2.17, 2.31 and 2.74 Å) show that the representative modeled ensemble structures are in very good agreement with experimental cryo-EM data of 5FN2 (resolution of 4.2 Å). The horse-shoe shaped architecture of PS2-γ-secretase and the TM helices of the four subunits are displayed in Fig. 7D. The size of the catalytic pocket, which determines the fit of the substrates, is of particular interest and can be compared directly with the cryo-EM structures of γ-secretase (5A63) where the membrane is absent.51 The Cα-distance between the catalytic Asp-263 and Asp-366 in the two-top ranked cluster structures was computed as 13.5 Å (cluster 1, simulation 1), 12.5 Å (cluster 2, simulation 1), 12.1 Å (cluster 1, simulation 2), 12.4 Å (cluster 2, simulation 2), 13.9 Å (cluster 1, simulation 3), and 13.8 Å (cluster 2, simulation 3). The intramembrane proteases may have an in-built “sloppy cleavage” yielding a broad profile of cleavage products, and a variable catalytic pocket size could cause this (Fig. 3D). While NCT movements define conformations controlling substrate entry, the distance between the catalytic aspartates (ranging from ∼0.8 to 1.6 nm with an average distance of 1.32 ± 0.08, 1.17 ± 0.05 and 1.34 ± 0.10 nm, respectively for the three simulations) defines these catalytically relevant states of PS2-γ-secretase (Fig. S13A†). Similarly, in case of PS1 γ-secretase (Fig. S13B†) the average distance ranges from 1.19 ± 0.09 nm (simulation 1) to 1.02 ± 0.03 nm (simulation 2); thus PS2 is somewhat more open than PS1 and implying a slightly different biding affinity and specific activity for a given substrate.
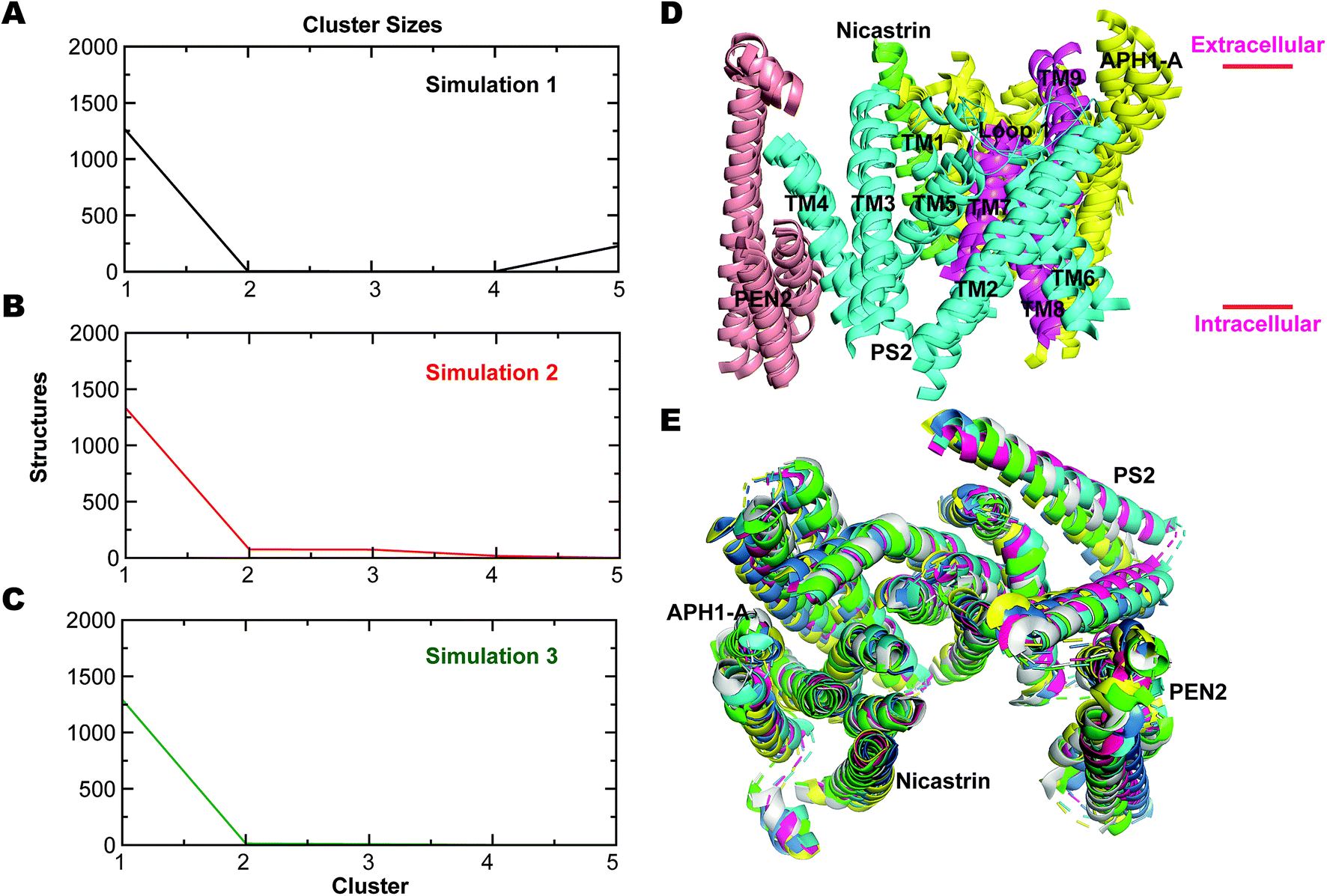 | ||
| Fig. 7 Jarvis-Patrick clustering analysis of trajectories of PS2 γ-secretase using 1500 structures as input. (A) Distribution of cluster sizes from simulation 1. (B) Same for simulation 2. (C) Same for simulation 3. (D) Structural superimposition of all helices of the four subunits of PS2 γ-secretase (the top-ranked cluster obtained using the Jarvis-Patrick algorithm) exhibiting very similar structural diversity (Nicastrin: green, PS2: NTF: cyan, CTF: magenta, APH1-A: yellow and PEN2: tint grey). (E) Superimposed top view of the horse-shoe-shaped architecture of PS2-γ-secretase with its four discrete subunit components (green: simulation 1, blue: simulation 2, pink: simulation 3, yellow: representative low energy state conformation (from FEL) of simulation 1, cyan: low energy state conformation (from FEL) of simulation 2, and grey: low energy state conformation (from FEL) of simulation 3). | ||
The nicastrin ECD is comprised of two discrete lobes, the large lobe and the small lobe (Fig. S12 and S14†). The independent motions of these lobes relative to PS2 were evaluated from the average distances and the angle between amino acids located in the large lobe, the small lobe, and the active site of PS2 as summarized in Fig. S14.† The distance between the catalytic Asp263 (PS2), Val328 (large lobe) and Leu121 (small lobe) displayed distinct variations (see Table S2†). The distance between V328 and Asp263 displayed short-lived fluctuations but more distinct global movement than the Leu121-Asp263 distance. The angle formed by Val328, Leu121, and Asp263 changed distinctly with movements of PS2 relative to the large lobe, but not so much the small lobe, implying that the large lobe-PS2 relative movement is decoupled from the movement of the small lobe. The small distance between the small lobe and the PS2 active site related to minor fluctuations in the nicastrin ECD.
The cluster analysis displayed three different states in this regard: compact, semi-compact, and loose (Fig. S15†), mainly differing by the relative positions of TM6 and TM7 harboring the catalytic aspartates and the PEN2/PS1 positioning. These states are distinct from the major conformation states of PS1-γ-secretase defined by the NCT movement that we also see.103 The additional conformation states that we identify relate specifically to the size of the catalytic PS2 pocket, and thus to substrate retention and trimming, as described by the FIST model, where the “fist” represents the PS1/PS2 subunit, and a stronger grab by the compact state increases stay time and trimming of the substrate to shorter Aβ.57,67
TM helix tilts and catalytic pocket size is dynamically correlated in PS2-γ-secretase
To specifically understand the PS2 dynamics and its impact on the catalytic pocket, we calculated the helical properties of each TM of PS2-γ-secretase (Fig. S16†). A well-oriented stable helix was observed for all the TMs, except TM9 (Fig. S16†), which displayed loss of helicity for one residue. The conformational change in TM9 helix (due the presence of kink at the C-terminal end) potentially contributes to the larger gate opening driven by the movements of TM2 and TM6, which is consistent with results from cysteine-scanning mutagenesis.87,88 TM9 of PS1 was previously found to be highly mobile.87 The flexibility of TM9 is attributed to the highly conserved PAL motif (Pro-414-Ala-415-Leu-416 using PS2 numbering). The strong association between the cytoplasmic end of TM9 and TM6 has been speculated to guide the substrate into the catalytic pocket, since mutation of Pro-433 in the PAL motif of PS1 results in structural rearrangement within the catalytic site.87As the most important helix dynamics in our ensembles in the membrane, we observed variation in tilt angles in TM6 and TM7 (with average tilt of 8.2 ± 2.5°, 8.9 ± 1.9°, and 16.6 ± 7.9° for TM6 and 12.3 ± 4.0°, 10.2 ± 2.1° and 14.4 ± 5.7° for TM7) for the three simulations of PS2-γ-secretase (Fig. 8A). In order to explore the correlation between the TM orientation and catalytic pocket size, we analyzed the last 15000 snapshot structures for each simulation by projecting the Asp–Asp distance on the helix tilt angle of all TMs of PS1 and PS2 (Fig. 8, S17 and S18†). This analysis shows that the concerted TM6 and TM7 tilting correlates strongly with the Asp–Asp distance and thus, the size of the catalytic pocket (Fig. 8B).
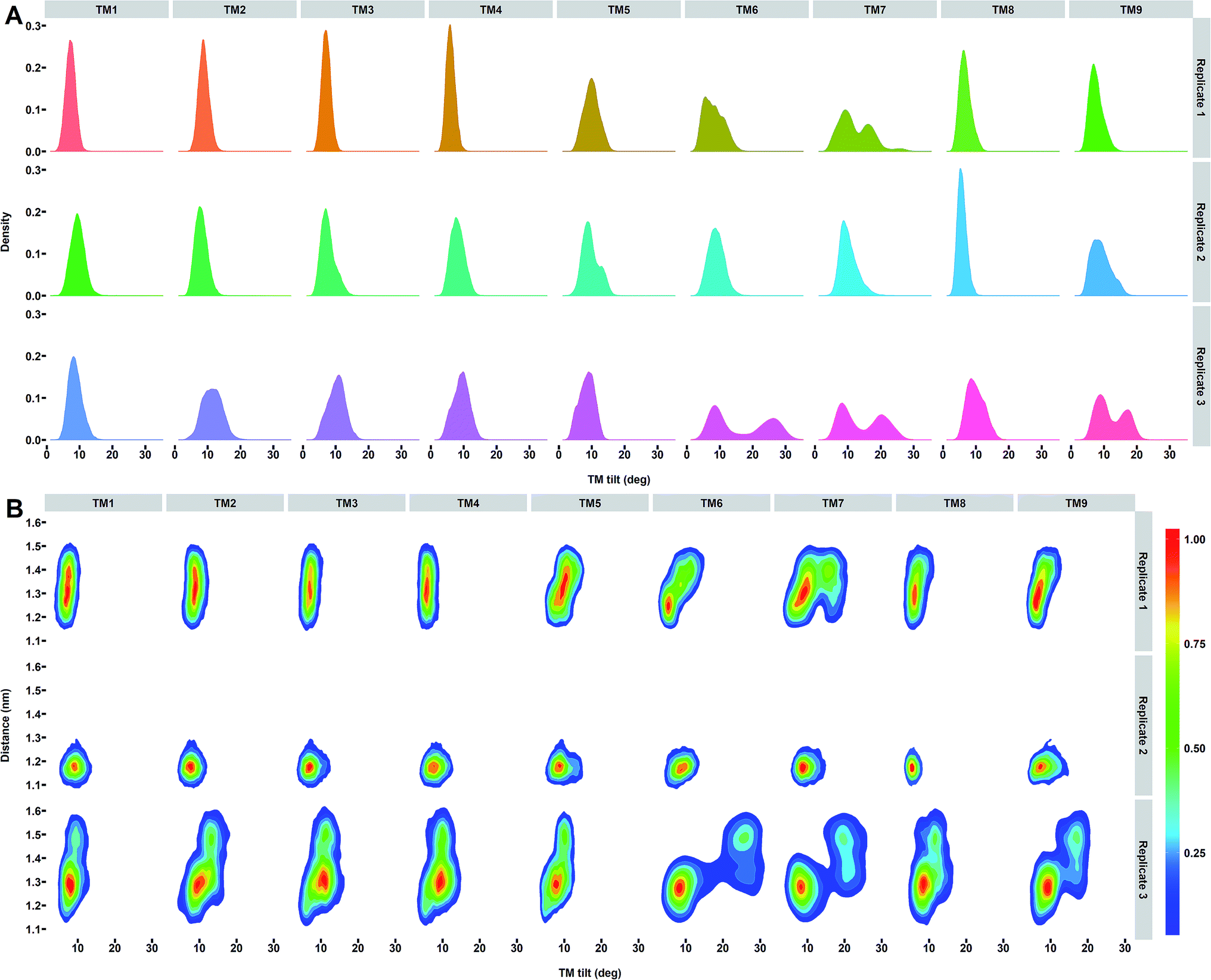 | ||
| Fig. 8 (A) Distribution of tilt angles for each TM helix of PS2 γ-secretase. (B) Distribution of snapshots projected onto the distance between the catalytic Asp-263 and Asp-366 and tilt angles of each TM helix of PS2 for the three independent simulations. The colored scale defines the relative populations of each state. | ||
Several studies of PS1-γ-secretase have indicated a concerted motion of TMs regulating the transition between active and inactive states,55–57,106 as directly rationalized by the Asp–Asp distances. The identified changes in tilt angles of TM2, TM6, TM7, and TM9 are consistent with but also explain the results of various experimental data.103,107,108 In particular, the high disorder of TM2, TM6, and TM7 in the cryo-EM data without membrane51 is consistent with our membrane simulations at ambient temperature and are seen to be due to two-state behavior of the helix tilt angles of TM6 and TM7 (Fig. 8A). Whereas our simulated PS1 (ref. 57) also shows two-state behavior for TM2, PS2 does not. TM2-TM6 arguably act as gate doors for substrate entry,51,56,108 with door opening controlled by correlated helix tilts that affect the catalytic pocket size and thus probably substrate affinity, retention, and trimming, as implied by the FIST model.57,67 Fig. 8A reveals that whereas TM2 motion is dampened in PS2 relative to PS1, TM9 tilting is more prevalent, possibly affecting the sloppiness of cleavage in the two homologues.
We hypothesized that the dynamics of HL1 may affect the TM dynamics of PS2. To investigate this, we plotted the average tilt angles of the TMs of PS2 against the average Cα distance to HL1 for all snapshots of the three simulations (Fig. S19†). The two-state behavior of TM3, TM6, TM7 and TM9 indicates their participation in a controlled mechanism that drives active site space. HL1 is also very flexible and the distance to TM6, TM7, and TM9 is strongly correlated with the TM tilt of these PS2 helices in the loose conformation state represented by simulation 3 (Fig. S19†).
Intramembrane proteolysis requires an adequate number of water molecules to perform the actual nucleophilic attack on the peptide bond and stabilize putative transition states, and possibly to help unwinding the substrate TM helix. In order to investigate if any water can afford such roles, we computed the average number of hydrogen bonds between the catalytic residues and water molecules from the last 300 ns trajectories of each simulation, averaged over all 15000 sampling frames (Fig. S20†). We find a consistent number (average of 11) of hydrogen bonds in the ensembles indicating that PS2 catalytic residues have access to adequate amount of water despite being in a lipid bilayer environment. Since two catalytic aspartates of PS2 are at the bottom of the cavity on the intracellular side, they are somewhat exposed to the aqueous environment (Fig. S21A†). One snapshot portraying the typical distribution of water molecules within 4 Å of Asp-263 and Asp-366 of PS2 is depicted in Fig. S21B.† Our simulations thus provide an all-atom rationale for the access of catalytic water to this intramembrane protease and shows that our overall membrane-protein water system accounts for this important feature; high-resolution bulk water molecules (and membrane) are not available in the cryo-EM data, thus making MD simulations an important complement to understand the water dynamics of the total system.
The TMs of PS1 and PS2 differ in length, but their overall structures are similar to each other. The catalytic residues located on the convex side of the TM horseshoe in PS1 and PS2 superpose well with each other. The structural conservation of residues in the catalytic pocket (Ile-259, Tyr-262, Val-267, Gly-363, Phe-369), the GxGD motif (Gly-363-Leu-364-Gly-365-Asp-366), and the PAL motif (Pro-414-Ala-415-Leu-416) motif suggests that PS2 exhibits similar cleavage activity as PS1-γ-secretase,51 as also seen.47 However, the shift of two-state behavior from TM2 to TM9 may change the details of substrate cleavage and its specificity to substrates. Furthermore, the mature HL2 of the two homologs adopts different conformation which we suggest could lead to differential preferences for various substrates, but these implications need to be tested further in future experiments.
Conclusions
γ-Secretase harboring either PS1 or PS2 catalytic subunits is directed toward different sub-cellular compartments, and the distinct lipid environments and substrates encountered may suggest differences in the two types of γ-secretase. PS2 plays a pivotal role in various physiological functions associated with AD including calcium homeostasis, innate immunity, and notch signaling, and mutations in both PS1 and PS2 can lead to AD. Accordingly, a structural-dynamic comparison of PS1 and PS2 is of substantial interest.We describe here the first structure of PS2-γ-secretase developed from available experimental and computational data for PS1-γ-secretase. The model is essentially complete, except the elusive N-terminal part of PS2, and respects all experimental constraints from the cryo-EM data, bot complements these data with all-atom dynamics in a complete explicit membrane-mimicking POPC lipid bilayer system surrounded by explicit water at ambient temperature. We gauge the similarities and differences between the homologs in terms of structure and molecular dynamics using a range of structural and dynamical comparisons.
Both PS1 and PS2 share the 9 TM architecture also after simulation in the membrane, with structures in very good agreement with the experimental cryo-EM data. Sequence-wise, PS1 and PS2 differ substantially at the N-terminus and in the large cytosolic hydrophilic loop connecting TM6 and TM7, which we suggest drives their different locations within the cell. Global conformational analysis reveals significant variation in the helix tilt angles, which are well-sampled on the time scale, as seen from the broad distributions of Fig. 8. The relatively fast correlated helix tilts of TM2, TM3, TM6, TM7, and TM9 control the distance between the catalytic aspartates, and are thus the most important modes in terms of substrate binding, retention and cleavage (the FIST model). As these are well-sampled, maximal realism using explicit all-atom hydrogens and structure-balanced force fields should be prioritized vs. very long simulations with less realistic models. TM6 and TM7 has two-state behavior in both homologs, enforced by two very similar free energy minima in the membrane-protein-water system. The different helix tilts lead to different space between the catalytic aspartates. In PS2, TM9 specifically has two-state behavior not seen in PS1, whereas PS2 has less two-state behavior of TM2 than observed for PS1.56,57 This variability is enforced by maturation of HL2, required for activity. The shift in dynamic variability from TM2 to TM9 in PS2 vs. PS1 may affect substrate specificity. The similar structure in the catalytic pocket implies similar specific activities of the enzymes, although PS2 is slightly more open.
Like other intramembrane-cleaving proteases such as site-2 protease109 and rhomboids,110 our simulations reveal the presence of water-rich areas within the bilayer of PS2 close to the catalytic aspartates, thus providing a structural-dynamic basis for the assess of nucleophilic catalytic water molecules to the intramembrane active site; such water molecules cannot be accurately inferred from the cryo-EM data at current resolution.
Our simulations consistently suggest that correlated motions of several TM helices drive the transitions between various conformations states of both PS1- and PS2-γ-secretase and thereby determine the Asp–Asp distance and size of the catalytic pocket. Thus, our simulations provide an atomic basis for the membrane-protein dynamics at ambient temperature, which is partly indicated by the disorder in the cryo-EM data, but adds substantial detail and clarifications to these data, specifically the previously unknown PS1-PS2 differences. One important question is why fewer mutations in PS2 cause FAD as compared to PS1. Since the proteins are expected to have similar catalytic proficiency, we propose that the difference in clinical importance relates to the different location, expression levels, and substrate specificity induced by the TM2/TM9 differences rather than specific activities per se.
Conflicts of interest
The authors declare that no competing financial interest exists.Acknowledgements
The authors acknowledge financial support for this work from the Novo Nordisk Foundation, grant NNF17OC0028860, and the Danish Council for Independent Research (DFF), grant 7016-00079B.References
- Alzheimer's Disease International, 2018, 1–48.
- C. L. Masters, D. C. Gajdusek and C. J. J. Gibbs, Brain, 1981, 104, 535–558 CrossRef CAS PubMed.
- C. L. Masters and D. J. Selkoe, Cold Spring Harbor Perspect. Med., 2012, 2, a006262 Search PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- E. H. Koo and R. Kopan, Nat. Med., 2004, 10, S26–S33 CrossRef PubMed.
- C. Haass, C. Kaether, G. Thinakaran and S. Sisodia, Cold Spring Harbor Perspect. Med., 2012, 2, a006270 Search PubMed.
- C. Kaether, C. Haass and H. Steiner, Neurodegener. Dis., 2006, 3, 275–283 CrossRef CAS PubMed.
- A. Fukumori, R. Fluhrer, H. Steiner and C. Haass, J. Neurosci., 2010, 30, 7853–7862 CrossRef CAS PubMed.
- H. Steiner, A. Fukumori, S. Tagami and M. Okochi, Cell Stress, 2018, 2, 292–310 CrossRef PubMed.
- M. S. Wolfe, Biol. Chem., 2012, 393, 899–905 CAS.
- R. Wolfenden, Bioorg. Med. Chem., 1999, 7, 647–652 CrossRef CAS PubMed.
- O. Quintero-Monzon, M. M. Martin, M. A. Fernandez, C. A. Cappello, A. J. Krzysiak, P. Osenkowski and M. S. Wolfe, Biochemistry, 2011, 50, 9023–9035 CrossRef CAS PubMed.
- A. Götz and C. Scharnagl, PLoS One, 2018, 13, 1–34 CrossRef PubMed.
- L. Chávez-Gutiérrez, L. Bammens, I. Benilova, A. Vandersteen, M. Benurwar, M. Borgers, S. Lismont, L. Zhou, S. Van Cleynenbreugel, H. Esselmann, J. Wiltfang, L. Serneels, E. Karran, H. Gijsen, J. Schymkowitz, F. Rousseau, K. Broersen and B. De Strooper, EMBO J., 2012, 31, 2261–2274 CrossRef PubMed.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- M. F. M. Sciacca, S. A. Kotler, J. R. Brender, J. Chen, D. K. Lee and A. Ramamoorthy, Biophys. J., 2012, 103, 702–710 CrossRef CAS PubMed.
- A. Ramamoorthy and M. H. Lim, Biophys. J., 2013, 105, 287–288 CrossRef CAS PubMed.
- N. S. Ryan, G. J. Biessels, L. Kim, J. M. Nicholas, P. A. Barber, P. Walsh, P. Gami, H. R. Morris, A. J. Bastos-Leite, J. M. Schott, J. Beck, S. Mead, L. Chavez-Gutierrez, B. de Strooper, M. N. Rossor, T. Revesz, T. Lashley and N. C. Fox, Neurobiol. Aging, 2015, 36, 3140–3151 CrossRef CAS PubMed.
- Y. F. Shea, L. W. Chu, A. O. K. Chan, J. Ha, Y. Li and Y. Q. Song, J. Formos. Med. Assoc., 2016, 115, 67–75 CrossRef CAS PubMed.
- R. Kopan and A. Goate, Genes Dev., 2000, 14, 2799–2806 CrossRef CAS PubMed.
- G. Periz and M. E. Fortini, J. Neurosci. Res., 2004, 77, 309–322 CrossRef CAS PubMed.
- D. C. Ryman, N. Acosta-Baena, P. S. Aisen, T. Bird, A. Danek, N. C. Fox, A. Goate, P. Frommelt, B. Ghetti, J. B. S. Langbaum, F. Lopera, R. Martins, C. L. Masters, R. P. Mayeux, E. McDade, S. Moreno, E. M. Reiman, J. M. Ringman, S. Salloway, P. R. Schofield, R. Sperling, P. N. Tariot, C. Xiong, J. C. Morris and R. J. Bateman, Neurology, 2014, 83, 253–260 CrossRef CAS PubMed.
- B. De Strooper, EMBO Rep., 2007, 8, 141–146 CrossRef CAS PubMed.
- M. Cacquevel, L. Aeschbach, J. Houacine and P. C. Fraering, PLoS One, 2012, 7, 1–13 CrossRef PubMed.
- L. Sun, R. Zhou, G. Yang and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2016, 114, E476–E485 CrossRef PubMed.
- D. Langosch and H. Steiner, Biol. Chem., 2017, 398, 441–453 CAS.
- R. Zhou, G. Yang, X. Guo, Q. Zhou, J. Lei and Y. Shi, Science, 2019, 363, eaaw0930 CrossRef PubMed.
- S. Veugelen, T. Saito, T. C. Saido, L. Chávez-Gutiérrez and B. De Strooper, Neuron, 2016, 90, 410–416 CrossRef CAS PubMed.
- N. Tang and K. P. Kepp, J. Alzheimer's Dis., 2018, 66, 939–945 CAS.
- A. K. Somavarapu and K. P. Kepp, J. Neurochem., 2016, 137, 101–111 CrossRef CAS PubMed.
- S. Stanga, C. Vrancx, B. Tasiaux, C. Marinangeli, H. Karlström and P. Kienlen-Campard, J. Cell. Mol. Med., 2018, 22, 823–833 CAS.
- R. Aguayo-Ortiz and L. Dominguez, Biochimie, 2018, 147, 130–135 CrossRef CAS PubMed.
- H. Acx, L. Serneels, E. Radaelli, S. Muyldermans, C. Vincke, E. Pepermans, U. Müller, L. Chávez-Gutiérrez and B. De Strooper, EMBO Mol. Med., 2017, 9, 1088–1099 CrossRef CAS PubMed.
- B. De Strooper, T. Iwatsubo and M. S. Wolfe, Cold Spring Harbor Perspect. Med., 2012, 2, a006304 Search PubMed.
- R. Sannerud, C. Esselens, P. Ejsmont, R. Mattera, L. Rochin, A. K. Tharkeshwar, G. De Baets, V. De Wever, R. Habets, V. Baert, W. Vermeire, C. Michiels, A. J. Groot, R. Wouters, K. Dillen, K. Vints, P. Baatsen, S. Munck, R. Derua, E. Waelkens, G. S. Basi, M. Mercken, M. Vooijs, M. Bollen, J. Schymkowitz, F. Rousseau, J. S. Bonifacino, G. Van Niel, B. De Strooper and W. Annaert, Cell, 2016, 166, 193–208 CrossRef CAS PubMed.
- I. Khan, S. Krishnaswamy, M. Sabale, D. Groth, L. Wijaya, M. Morici, I. Berger, C. Schaffitzel, P. E. Fraser, R. N. Martins and G. Verdile, Sci. Rep., 2018, 8, 1–15 CrossRef PubMed.
- T.-W. Kim, W. H. Pettingell, O. G. Hallmark, R. D. Moir, W. Wasco and R. E. Tanzi, J. Biol. Chem., 1997, 272, 11006–11010 CrossRef CAS PubMed.
- J. J. Palacino, B. E. Berechid, P. Alexander, C. Eckman, S. Younkin, J. S. Nye and B. Wolozin, J. Biol. Chem., 2000, 275, 215–222 CrossRef CAS PubMed.
- K. P. Kepp, Prog. Neurobiol., 2016, 143, 36–60 CrossRef CAS PubMed.
- C. A. Saura, S.-Y. Choi, V. Beglopoulos, S. Malkani, D. Zhang, B. S. Shankaranarayana Rao, S. Chattarji, R. J. 3rd Kelleher, E. R. Kandel, K. Duff, A. Kirkwood and J. Shen, Neuron, 2004, 42, 23–36 CrossRef CAS PubMed.
- J. Shen and R. J. Kelleher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 403–409 CrossRef CAS PubMed.
- H. Tu, O. Nelson, A. Bezprozvanny, Z. Wang, S.-F. Lee, Y.-H. Hao, L. Serneels, B. De Strooper, G. Yu and I. Bezprozvanny, Cell, 2006, 126, 981–993 CrossRef CAS PubMed.
- M. A. Leissring, T. R. Yamasaki, W. Wasco, J. D. Buxbaum, I. Parker and F. M. LaFerla, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8590–8593 CrossRef CAS PubMed.
- O. Nelson, C. Supnet, H. Liu and I. Bezprozvanny, J. Alzheimer's Dis., 2010, 21, 781–793 CAS.
- C. Supnet and I. Bezprozvanny, Cell Calcium, 2011, 50, 303–309 CrossRef CAS PubMed.
- C. Garwood, A. Faizullabhoy, S. B. Wharton, P. G. Ince, P. R. Heath, P. J. Shaw, L. Baxter, C. Gelsthorpe, G. Forster, F. E. Matthews, C. Brayne and J. E. Simpson, Neuropathol. Appl. Neurobiol., 2013, 39, 788–799 CrossRef CAS PubMed.
- Y. Yonemura, E. Futai, S. Yagishita, S. Suo, T. Tomita, T. Iwatsubo and S. Ishiura, J. Biol. Chem., 2011, 286, 44569–44575 CrossRef CAS PubMed.
- E. S. Walker, M. Martinez, A. L. Brunkan and A. Goate, J. Neurochem., 2005, 92, 294–301 CrossRef CAS PubMed.
- N. R. Graff-Radford, J. E. Crook, J. Lucas, B. F. Boeve, D. S. Knopman, R. J. Ivnik, G. E. Smith, L. H. Younkin, R. C. Petersen and S. G. Younkin, Arch. Neurol., 2007, 64, 354–362 CrossRef PubMed.
- G. Yang, R. Zhou and Y. Shi, Curr. Opin. Struct. Biol., 2017, 46, 55–64 CrossRef CAS PubMed.
- X. Bai, C. Yan, G. Yang, P. Lu, L. Sun, R. Zhou, S. H. W. Scheres and Y. Shi, Nature, 2015, 525, 212–218 CrossRef CAS PubMed.
- P. Lu, X. Bai, D. Ma, T. Xie, C. Yan, L. Sun, G. Yang, Y. Zhao, R. Zhou, S. H. W. Scheres and Y. Shi, Nature, 2014, 512, 166–170 CrossRef CAS PubMed.
- R. Aguayo-Ortiz, J. E. Straub and L. Dominguez, Phys. Chem. Chem. Phys., 2018, 20, 27294–27304 RSC.
- L. Dominguez, L. Foster, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5281–E5287 CrossRef CAS PubMed.
- R. Aguayo-Ortiz, C. Chávez-García, J. E. Straub and L. Dominguez, Chem. Sci., 2017, 8, 5576–5584 RSC.
- A. K. Somavarapu and K. P. Kepp, Neurobiol. Dis., 2016, 89, 147–156 CrossRef CAS PubMed.
- A. K. Somavarapu and K. P. Kepp, ACS Chem. Neurosci., 2017, 8, 2424–2436 CrossRef CAS PubMed.
- L. Sun, L. Zhao, G. Yang, C. Yan, R. Zhou, X. Zhou, T. Xie, Y. Zhao, S. Wu, X. Li and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6003–6008 CrossRef CAS PubMed.
- G. Yang, R. Zhou, Q. Zhou, X. Guo, C. Yan, M. Ke, J. Lei and Y. Shi, Nature, 2019, 565, 192–197 CrossRef CAS PubMed.
- S. Urban, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1112–1114 CrossRef CAS PubMed.
- J. D. Perlmutter, A. R. Braun and J. N. Sachs, J. Biol. Chem., 2009, 284, 7177–7189 CrossRef CAS PubMed.
- V. Rajendran, C. Gopalakrishnan and R. Sethumadhavan, J. Cell. Biochem., 2018, 119, 918–925 CrossRef CAS PubMed.
- V. Rajendran, Mol. BioSyst., 2016, 12, 2276–2287 RSC.
- L. Celik, J. D. D. Lund and B. Schiøtt, Biochemistry, 2007, 46, 1743–1758 CrossRef CAS PubMed.
- L. Thøgersen, B. Schiøtt, T. Vosegaard, N. C. Nielsen and E. Tajkhorshid, Biophys. J., 2008, 95, 4337–4347 CrossRef PubMed.
- M. Hitzenberger and M. Zacharias, ACS Chem. Neurosci., 2019 Search PubMed , acschemneuro.8b00725.
- N. Tang, A. K. Somavarapu and K. P. Kepp, ACS Omega, 2018, 3, 18078–18088 CrossRef CAS.
- M. Szaruga, B. Munteanu, S. Lismont, S. Veugelen, K. Horré, M. Mercken, T. C. Saido, N. S. Ryan, T. De Vos, S. N. Savvides, R. Gallardo, J. Schymkowitz, F. Rousseau, N. C. Fox, C. Hopf and B. De Strooper, Cell, 2017, 170, 443–456 CrossRef CAS PubMed.
- B. Webb and A. Sali, in Methods in Molecular Biology, Humana Press, New York, NY, 2017, vol. 1654, pp. 39–54 Search PubMed.
- L. Heo, H. Park and C. Seok, Nucleic Acids Res., 2013, 41, W384–W388 CrossRef PubMed.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, 407–410 CrossRef PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr. D, 2010, 66, 12–21 CrossRef CAS PubMed.
- B. Wallner and A. Elofsson, Protein Sci., 2003, 12, 1073–1086 CrossRef CAS PubMed.
- M. A. Lomize, I. D. Pogozheva, H. Joo, H. I. Mosberg and A. L. Lomize, Nucleic Acids Res., 2012, 40, D370–D376 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, vol. 1–2, pp. 19–25 Search PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2016, 14, 71–73 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- M. C. Zwier and L. T. Chong, Curr. Opin. Pharmacol., 2010, 10, 745–752 CrossRef CAS PubMed.
- S. A. Adcock and J. A. McCammon, Chem. Rev., 2006, 106, 1589–1615 CrossRef CAS PubMed.
- W. J. Allen, J. A. Lemkul and D. R. Bevan, J. Comput. Chem., 2009, 30, 1952–1958 CrossRef CAS PubMed.
- S. Buchoux, Bioinformatics, 2017, 33, 133–134 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. Van Gunsteren and A. E. Mark, Angew. Chem. Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- K. S. Knappenberger, G. Tian, X. Ye, C. Sobotka-Briner, S. V Ghanekar, B. D. Greenberg and C. W. Scott, Biochemistry, 2004, 43, 6208–6218 CrossRef CAS PubMed.
- A. Tolia, K. Horré and B. De Strooper, J. Biol. Chem., 2008, 283, 19793–19803 CrossRef CAS PubMed.
- C. Sato, S. Takagi, T. Tomita and T. Iwatsubo, J. Neurosci., 2008, 28, 6264–6271 CrossRef CAS PubMed.
- P. Osenkowski, W. Ye, R. Wang, M. S. Wolfe and D. J. Selkoe, J. Biol. Chem., 2008, 283, 22529–22540 CrossRef CAS PubMed.
- T. Hartmann, J. Kuchenbecker and M. O. W. Grimm, J. Neurochem., 2007, 103(Suppl), 159–170 CrossRef CAS PubMed.
- O. Holmes, S. Paturi, W. Ye, M. S. Wolfe and D. J. Selkoe, Biochemistry, 2012, 51, 3565–3575 CrossRef CAS PubMed.
- T. Iwatsubo, S. Takagi-Niidome, A. Tominaga, T. Cai and T. Tomita, J. Neurosci., 2016, 36, 1362–1372 CrossRef PubMed.
- N. Kučerka, M.-P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771 CrossRef PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- X. Zhuang, E. M. Dávila-Contreras, A. H. Beaven, W. Im and J. B. Klauda, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 3093–3104 CrossRef CAS PubMed.
- G. Shahane, W. Ding, M. Palaiokostas and M. Orsi, J. Mol. Model., 2019, 25, 76 CrossRef PubMed.
- X. Bai, E. Rajendra, G. Yang, Y. Shi and S. H. Scheres, Elife, 2015, 4, 551–560 CrossRef PubMed.
- S. Takagi-Niidome, T. Sasaki, S. Osawa, T. Sato, K. Morishima, T. Cai, T. Iwatsubo and T. Tomita, J. Neurosci., 2015, 35, 2646–2656 CrossRef CAS PubMed.
- C. Hu, J. Xu, L. Zeng, T. Li, M. Z. Cui and X. Xu, J. Alzheimer's Dis., 2017, 56, 1263–1269 CAS.
- L. Bammens, L. Chávez-Gutiérrez, A. Tolia, A. Zwijsen and B. De Strooper, J. Biol. Chem., 2011, 286, 12271–12282 CrossRef CAS PubMed.
- O. Holmes, S. Paturi, D. J. Selkoe and M. S. Wolfe, Biochemistry, 2014, 53, 4393–4406 CrossRef CAS PubMed.
- J. Y. Lee, Z. Feng, X. Q. Xie and I. Bahar, Biophys. J., 2017, 113, 2634–2649 CrossRef CAS PubMed.
- N. Elad, B. De Strooper, S. Lismont, W. Hagen, S. Veugelen, M. Arimon, K. Horré, O. Berezovska, C. Sachse and L. Chávez-Gutiérrez, J. Cell Sci., 2015, 128, 589–598 CrossRef CAS PubMed.
- Y. Li, S. H. J. Lu, C. J. Tsai, C. Bohm, S. Qamar, R. B. Dodd, W. Meadows, A. Jeon, A. McLeod, F. Chen, M. Arimon, O. Berezovska, B. T. Hyman, T. Tomita, T. Iwatsubo, C. M. Johnson, L. A. Farrer, G. Schmitt-Ulms, P. E. Fraser and P. H. St George-Hyslop, Structure, 2014, 22, 125–135 CrossRef CAS PubMed.
- C. C. Shelton, L. Zhu, D. Chau, L. Yang, R. Wang, H. Djaballah, H. Zheng and Y.-M. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20228–20233 CrossRef CAS PubMed.
- M. Hitzenberger and M. Zacharias, Front. Chem., 2019, 6, 640 CrossRef PubMed.
- A. Y. Kornilova, F. Bihel, C. Das and M. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3230–3235 CrossRef CAS PubMed.
- A. Fukumori and H. Steiner, EMBO J., 2016, e201694151 Search PubMed.
- L. Feng, H. Yan, Z. Wu, N. Yan, Z. Wang, P. D. Jeffrey and Y. Shi, Science, 2007, 318, 1608–1612 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang and Y. Ha, Nature, 2006, 444, 179–180 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information file contains additional data and analysis. The three representative snapshot structures from cluster analysis of all three simulations have been provided as pdb files. See DOI: 10.1039/c9ra02623a |
| This journal is © The Royal Society of Chemistry 2019 |