
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel polymeric derivatives of betulin with anticancer activity†
Daria Niewolika,
Katarzyna Krukiewiczad,
Barbara Bednarczyk-Cwynarb,
Piotr Ruszkowskic and
Katarzyna Jaszcz *a
*a
aDepartment of Physical Chemistry and Technology of Polymers, Silesian University of Technology, UL M Strzody 9, 44-100 Gliwice, Poland. E-mail: katarzyna.jaszcz@polsl.pl
bDepartment of Organic Chemistry, Poznan University of Medical Sciences, Poznan, Poland
cDepartment of Pharmacology, Poznan University of Medical Sciences, Poznan, Poland
dCentre for Research in Medical Devices, National University of Ireland, Galway, Ireland
First published on 4th July 2019
Abstract
In order to provide novel polymeric biomaterials for chemotherapeutic purposes, in this paper we described the synthesis and the characterization of the physicochemical properties of a betulin-based polyanhydride exhibiting anti-cancer effects. The polyanhydride was obtained by a melt polycondensation of a disuccinate betulin (3,28-di-O-succinyl betulin), and was thoroughly characterized through 1H NMR and 13C NMR spectroscopies, correlation spectroscopy, heteronuclear single quantum correlation, size exclusion chromatography, differential scanning calorimetry and FT-IR spectroscopy. It was confirmed, that the obtained polyanhydride undergoes hydrolytic degradation, releasing disuccinate betulin as a degradation product. Polyanhydride of a disuccinate betulin was tested for cytostatic activity against a wide range of cancer cell lines (HeLa, MCF-7, A-549, U-87MG, KB and HepG2), proving its efficiency in inhibiting the growth of selected cancer cells. To realize the concept of an easily administrated drug release system, polyanhydride was fabricated in a form of micro- (1–30 μm) and nanospheres (∼400 nm) by using an emulsion solvent evaporation method. The micro- and nanospheres were characterized by SEM.
1 Introduction
Nowadays, chemotherapeutic approaches are focused on the design of the biomaterial-based structures able to act locally, in order to enhance treatment and minimize the side effects of therapy.1,2 This new concept of a regional chemotherapy makes it necessary to design novel types of polymeric materials that exhibit cytostatic properties or that can act as the carriers for anti-cancer agents.Betulin(lup-20(29)-ene-3β-diol), a lupane derivative, belongs to the pentacyclic triterpenes and occurs commonly in nature. It is obtained on a large scale from the outer layer of the birch bark.3,4 Betulin and its derivatives have a broad spectrum of biological relevance, including anti-inflammatory, anti-viral and, in particular, anti-cancer activity, thus these compounds seems to be promising as new, potential therapeutic agents.3–8 The major problem limiting their potential pharmaceutical uses is the poor aqueous solubility of lupane triterpenes faced when trying to formulate pharmaceutical compounds from betulin. Nevertheless, this problem can be solved by providing betulin in a polymeric form, e.g. in a shape of micro- or nanoparticles, which, in turn, can be easily administrated by injection or inhalation. In literature, however, there are only several reports describing the fabrication of polymeric forms of betulin, including polyesters, polyurethanes and polyethylene oxide conjugates.9–12 These materials exhibit high porosity and are used mainly in non-biological applications, e.g. for gas adsorption studies. In these syntheses betulin, containing two hydroxyl groups, is used as a renewable diol. These hydroxyl groups, however, can be also easily transformed into carboxyl ones, e.g. in a reaction with succinic anhydride. Disuccinate betulin (DBB), formed in this way, was found to exhibit biological activity in terms of anti-cancer, anti-leishmania, hypolipidemic, fungicidal, bactericidal and antiviral effects, including Epstein–Barr virus and HIV.5,13–17 On the other hand, DBB containing two carboxylic groups is an excellent raw material to obtain polyanhydrides, a class of surface-degradable polymers. Due to their properties, such as easy tunable release kinetics and lack of toxicity, polyanhydrides are mainly used in medicine, both as drug carriers and biomaterials.18 For the best of our knowledge, so far there are no literature reports describing the synthesis of betulin-based polyanhydrides.
The aim of this work is to study the synthesis of a polyanhydride formed from disuccinate betulin and its fabrication into a form of micro- and nanoparticles. To fully characterize this polymer, we used nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, gel chromatography (GPC) and differential scanning calorimetry (DSC). Hydrolytic degradation of polyDBB in physiological conditions was also investigated. In order to determine the anti-cancer activity of disuccinate betulin and polyDBB, these materials were tested for cytotoxicity against a wide range of cancer cell lines, including HeLa, MCF-7, A-549, U-87MG, KB and HepG2. The micro- and nanospheres were further characterized using optical microscopy and scanning electron microscopy (SEM).
2 Experimental
2.1 Materials
Betulin > 95% (Natchem), succinic anhydride 99% (Sigma-Aldrich), pyridine (min 99.5%, Chempur), acetic anhydride (POCh S.A.), DMSO (Chempur), trichloroacetic acid (Chempur), acetic acid (Chempur), Tris buffer, sulforhodamine, poly(vinyl alcohol) (Mw = 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 88% hydrolyzed) (ACROS Organics), methylene chloride (Chempur) were used as supplied.
000 g mol−1, 88% hydrolyzed) (ACROS Organics), methylene chloride (Chempur) were used as supplied.
2.2 Methods
Infrared (FT-IR) spectra were recorded by means of a PerkinElmer Spectrum Two Spectrometer. NMR spectra were obtained using a Varian 600 MHz spectrometer in CDCl3 with TMS as an internal standard. Molecular weight of the polyanhydride was determined in methylene chloride by gel-permeation chromatography (GPC) using Agilent Technologies Infinity 1260 chromatograph equipped with a refractive index detector and calibrated with polystyrene standards. Thermal analyses were performed using a 822e DSC Mettler Toledo differential scanning calorimeter. Samples were tested in temperature range from −60 °C to 350 °C at a heating rate of 10 °C min−1. Shape and size of microspheres were estimated using optical microscope DELTA Optical ME 100 and PHAMIAS 2003 v.1.3 B software. The morphological characterization of micro- and nanospheres was carried out using a Phenom ProX scanning electron microscope (SEM).2.3 Synthesis of disuccinate betulin (DBB)
The synthesis of DBB was carried out, as follows. 4.43 g (10.0 mmol) of betulin was dissolved under heating in 100 mL of dried pyridine. 10.0 g (100 mmol) of succinic anhydride was added and the obtained solution was refluxed during 8 hours. The solution was cooled and poured into 500 mL of 5% HCl. The resulted precipitate was filtered off, washed with water to neutral pH and dried. The obtained amorphous powder was dissolved in ethanol under heating, boiled with active carbon and filtered. The product was precipitated with water, filtered off, washed again with water and dried. Yield: 92%, mp 106–110 °C, ESI-MS m/z 665,4 [M + Na]+ (calcd for DBB: 642, 87)IR: v = 2941 cm−1 (m, vC–H), 2844 cm−1 (w, vC–H), 1724–1706 cm−1 (s, vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1310–1155 cm−1 (vC–O); 1603 cm−1, 982 cm−1, 880 cm−1 (m, vCC).
O), 1310–1155 cm−1 (vC–O); 1603 cm−1, 982 cm−1, 880 cm−1 (m, vCC).
1H NMR (600 MHz, CDCl3, δ): 4.68 (1H, d, J = 1.7 Hz, C29–Ha); 4.59 (1H, d, J = 0.2 Hz, C29–Hb); 4.50 (1H, t, J = 7.9 Hz, C3–Hα); 4.30 (1H, d, J = 11.0 Hz, C28–Ha); 3.88 (1H, d, J = 11.0 Hz, C28–Hb); 2.72–2.64 (6H, m, HOOC–CH2–CH2–COO–, –OOC–CH2–CH2–COOH); 2.64–2.58 (2H, m, HOOC–CH2–CH2–COO–); 2.42 (1H, dt, J = 5.7 and 10.3 Hz, C19–H); 1.68 (3H, s, C30–H3) 1.95 (1H, m, C21–Ha); 1.81 (1H, d, C16–Ha); 1.74 (1H, t, C22–Ha); 1.69 (3H, s, C30–H3); 1.68–1.56 (7H, m, C15–Ha, C1–Ha, C12–Ha, C13–Ha, C2–Ha,b, C18–H); 1.51 (1H, m, C6–Ha); 1.45–1.35 (5H, m, C6–Hb, C11–Ha, C21–Hb, C7–Ha,b); 1.28 (1H, d, C9–H); 1.26–1.17 (2H, m, C11–Hb, C16–Hb); 1.13–1.04 (3H, m, C22–Hb, C12–Hb, C15–Hb); 1.03 (3H, s, C25–H3); 0.97 (3H, s, C27–H3); 0.94–0.92 (1H, m, C1–Hb); 0.86–0.82 (3× 3H, 3× s, C26–H3, C23–H3, C24–H3); 0.78 (1H, d, C5–H).
13C NMR (150 MHz, CDCl3, δ): 178.0 (Cq, C(O)OH); 172.37, 171.77 (Cq, C(O)O); 150.09 (Cq, C-20); 109.88 (CH2, C-29); 81.55 (CH, C-3); 63.19 (CH2, C-28), 55.39 (CH, C-5); 50.24 (CH, C-9); 48.76 (CH, C-18), 47.71 (CH, C-19); 46.42 (Cq, C-17); 42.69 (Cq, C-14); 40.87 (Cq, C-8); 38.34 (CH2, C-1); 37.82 (Cq, C-10); 37.58 (CH, C-13); 37.04 (Cq, C-4); 34.40 (CH2, C-22); 34.07 (CH2, C-7); 31.23 (CH2C(O)OH); 29.62 (CH2, C-21); 29.53 (CH2, C-16); 29.02 (CH2C(O)O); 27.88 (CH3, C-23); 27.00 (CH2, C-15); 25.13 (CH2, C-12); 23.58 (CH2, C-2); 20.80 (CH2, C-11); 19.08 (CH3, C-30); 18.15 (CH2, C-6); 16.51 (CH3, C-25); 16.14 (CH3, C-26); 16.02 (CH3, C-24); 14.80 (CH3, C-27).
DEPT: 6× CH3, 16× CH2, 6× CH.
2.4 Prepolymer and polymer synthesis
Polyanhydride was obtained by a two-step melt polycondensation of betulin disuccinate according to the procedure described earlier.19,20 Betulin disuccinate (5 g) was refluxed in acetic anhydride (1:10, w/v) under nitrogen flow for 40 min. After this time, the excess of acetic anhydride and acetic acid formed in the reaction were removed under vacuum. The remaining diacyl derivative of disuccinate betulin (prepolymer) was heated at 150 °C for 2 h with constant stirring under vacuum (0.1 mm Hg) and nitrogen. PolyDBB in a form of a solid, amorphous material was obtained with a yield of over 90%. The obtained polymer was stored in a freezer.
IR: v = 2941 cm−1 (m, vC–H), 2844 cm−1 (w, vC–H), 1827 cm−1 (m, vCO), 1724 cm−1 (s, vCO), 1034 cm−1 (vC–O).
1H NMR (600 MHz, CDCl3, δ): 4.69 (1H, d, C29–Ha); 4.59 (1H, d, C29–Hb), 4.50 (1H, t, C3–Hα); 4.30 (1H, d, C28–Ha); 3.89 (1H, d, C28–Hb); 2.84–2.74 (4H, m, –OCOOC–CH2–CH2–COO–); 2.74–2.62 (4H, m, –OCOOC–CH2–CH2–COO–); 2.42 (1H, td, C19–H); 2.18 (end groups); 1.96 (1H, m, C21–Ha); 1.82 (1H, d, C16–Ha); 1.76 (1H, t, C22–Ha); 1.68 (3H, s, C30–H3); 1.67–1.54 (7H, m, C15–Ha, C1–Ha, C12–Ha, C13–Ha, C2–Ha,b, C18–H); 1.51 (1H, m, C6–Ha); 1.45–1.35 (5H, m, C6–Hb, C11–Ha, C21–Hb, C7–Ha,b); 1.29 (1H, d, C9–H); 1.26–1.17 (2H, m, C11–Hb, C16–Hb); 1.13–1.04 (3H, m, C22–Hb, C12–Hb, C15–Hb); 1.02 (3H, s, C25–H3); 0.97 (3H, s, C27–H3); 0.94–0.92 (1H, m, C1–Hb); 0.88–0.80 (9H, s, C26–H3, C23–H3, C24–H3); 0.78 (1H, d, C5–H).
13C NMR (150 MHz, CDCl3, δ): 171.93 (Cq, C(O)O); 167.96 (Cq, C(O)OC(O)); 150.03 (Cq, C-20); 109.93 (CH2, C-29); 81.74 (CH, C-3); 63.36 (CH2, C-28), 55.40 (CH, C-5); 50.27 (CH, C-9); 48.81 (CH, C-18), 47.71 (CH, C-19); 46.43 (Cq, C-17); 42.71 (Cq, C-14); 40.91 (Cq, C-8); 38.36 (CH2, C-1); 37.86 (Cq, C-10); 37.61 (CH, C-13); 37.07 (Cq, C-4); 34.52 (CH2, C-22); 34.11 (CH2, C-7); 30.38–30.32 (CH2C(O)OC(O)); 29.72 (CH2, C-21); 29.57 (CH2, C-16); 28.62 (CH2C(O)O); 27.97 (CH3, C-23); 27.04 (CH2, C-15); 25.17 (CH2, C-12); 23.65 (CH2, C-2); 20.81 (CH2, C-11); 19.14 (CH3, C-30); 18.16 (CH2, C-6); 16.53 (CH3, C-25); 16.14 (CH3, C-26); 16.04 (CH3, C-24); 14.76 (CH3, C-27).
2.5 Hydrolytic degradation of polyDBB
Hydrolytic degradation experiments were performed in a phosphate buffer solution of pH 7.4 (PBS) or in distilled water (pH 5.5) at 37 °C. The hydrolytic degradation was monitored by both the mass loss of the test samples and the content of anhydride groups. PolyDBB powder samples (approximately 0.1 g) were placed in the weighed filters. Then, the filters with polyDBB were placed in glass vials containing 15 mL of PBS or distilled water, respectively. The vials were incubated at 37 °C for various time (from 1 h to 14 days). After incubation, the buffer or water were decanted, lyophilized and tested by 1H NMR to examine degradation products. The remaining samples were rinsed with distilled water, dried to constant weight in a vacuum oven, weighed to the nearest 0.0001 g and tested by 1H NMR to calculate the content of anhydride groups. The mass loss was defined as follows:where m1 represents the weight of the dry sample before degradation and m2 represents the weight of dry sample after degradation at different time intervals.
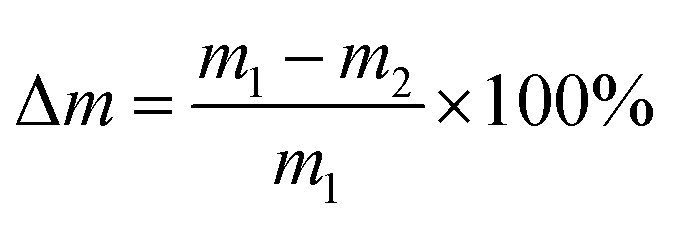
The ratio of anhydride groups to the sum of anhydride and ester groups in the polyanhydride (A/A + E) was calculated using the formula:
| A/A + E = ISAc/(ISAc + IE) |
2.6 Stability of polyDBB
In addition to the polyDBB hydrolytic degradation study, the degradation rate of the polyanhydride in air at room temperature was also investigated. For this purpose, polyDBB samples were placed in a glass vessel and left in the air. Every few days, samples were tested by 1H NMR to calculate the content of anhydride moieties to the sum of the anhydride and ester moieties. Calculations were carried out in the same manner as in the case of hydrolytic degradation.2.7 Cell cultures
KB, HeLa, MCF-7, and Hep-G2 cells were obtained from the European Collection of Cell Culture (ECACC) supplied by Sigma Aldrich. A-549, U-87, and HDF cells were purchased from the American Type Culture Collection (ATCC) through LGC Standards (Lomianki).KB, Hep-G2 and U-87 cells were cultured in EMEM medium while HeLa cells were grown in RPMI 1640 medium, A-549 cells in F-12K medium, MCF-7 cells in DMEM medium and HDF cells in Fibroblasts Growth Medium. Each medium was supplemented with 10% fetal bovine serum, 1% L-glutamine and 1% penicillin/streptomycin solution. All cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
2.8 Cytostatic activity of DBB and polyDBB
For the determination of the cytostatic activity of test compounds, the protein-staining sulforhodamine B (SRB) assay was employed. The SRB method has been assessed by the National Cancer Institute (USA) as suitable for the in vitro anti-tumour screening.22 The SRB assay estimates cell densities based on the measurement of cellular protein content. For the SRB assay, 100 μL of diluted cell suspension containing approximately 104 cells was added to the wells of 96-well plates. After 24 h, when a partial monolayer was formed, the supernatant was aspirated and 100 μL medium containing test compounds (DBB and polyDBB) at six different concentrations (0.1, 0.2, 1, 2, 10 and 20 μg mL−1) was added to the cells. Stock solutions of test compounds were prepared in DMSO and the concentration of DMSO in the assay did not exceed 0.1% which was found to be nontoxic to applied cell lines. After incubation for 72 h, 25 μL of 50% trichloroacetic acid was added to each well and the plates were incubated for 1 h at 4 °C. After that, the plates were washed with distilled water to remove traces of medium and were air-dried. Then, the dried plates were stained with 100 μL 0.4% SRB (prepared in 1% acetic acid) for 30 min at room temperature. Unbound dye was removed by rapid washing with 1% acetic acid and the plates were air-dried overnight. Finally, the protein-bound dye was dissolved in 100 μL of 10 mM unbuffered Tris and the absorbance was read at 490 nm.2.9 Microspheres and nanospheres preparation
In order to provide polyDBB in a form suitable for controlled drug release systems, attempts were made to obtain polymer micro- and nanospheres. Microspheres were prepared according to previous reports.19,21 In short, the polymer was dissolved in methylene chloride (20 mL, concentration 50 mg mL−1) and emulsified with 400 mL of aqueous solution (1.0% w/w) of poly(vinyl alcohol) (PVA 88% hydrolyzed, MW = 88000 g mol−1) using ULTRA-TURRAX T18 homogenizer, for 30 s. The speed of homogenizer was 3000 or 18000 rpm, respectively. The emulsion was then stirred with a magnetic stirrer at 1100 rpm at room temperature for 3 h to evaporate the organic solvent. After that, microspheres were collected by centrifugation at 5000 rpm for 5 min, washed 3 times with distilled water, lyophilized and stored in a freezer. Nanospheres were obtained by the same method as microspheres, using an ultrasonic homogenizer Omni Ruptor 250 (50% pulses, 50% power, 60 s).
Size and size distribution of particles were estimated using optical microscope, according to the procedure described earlier.21 The diameters of at least 100 particles were measured on each of the photomicrograph (magnification 100× and 400×). Then, number average diameters (Dn) and volume average diameters (Dv), standard deviation (S) and dispersity index (Dv/Dn) were calculated (Dn = ∑NiDi/∑Ni, Dv = ∑NiDi4/∑NiDi3, where Ni is the number of particles having diameter Di). The morphological characterization of microspheres was carried out using a Phenom ProX scanning electron microscope (SEM).
3 Results and discussion
3.1 PolyDBB synthesis and characterization
Betulin disuccinate (DBB) was synthesized by esterification of succinic anhydride and betulin. The reaction was carried out in dried pyridine, following a modified procedure described earlier by Sun et al.13 and Tian et al.23,24 DBB was isolated in high yields (92%), and its chemical structure was confirmed by IR and NMR spectroscopies and ESI-MS. The obtained results were consistent with the literature data provided earlier.13,23,24PolyDBB was obtained by melt a polycondensation of betulin disuccinate with the use of acetic anhydride (Fig. 1), and its physicochemical properties were thoroughly evaluated by a number of analytical and spectroscopic techniques.
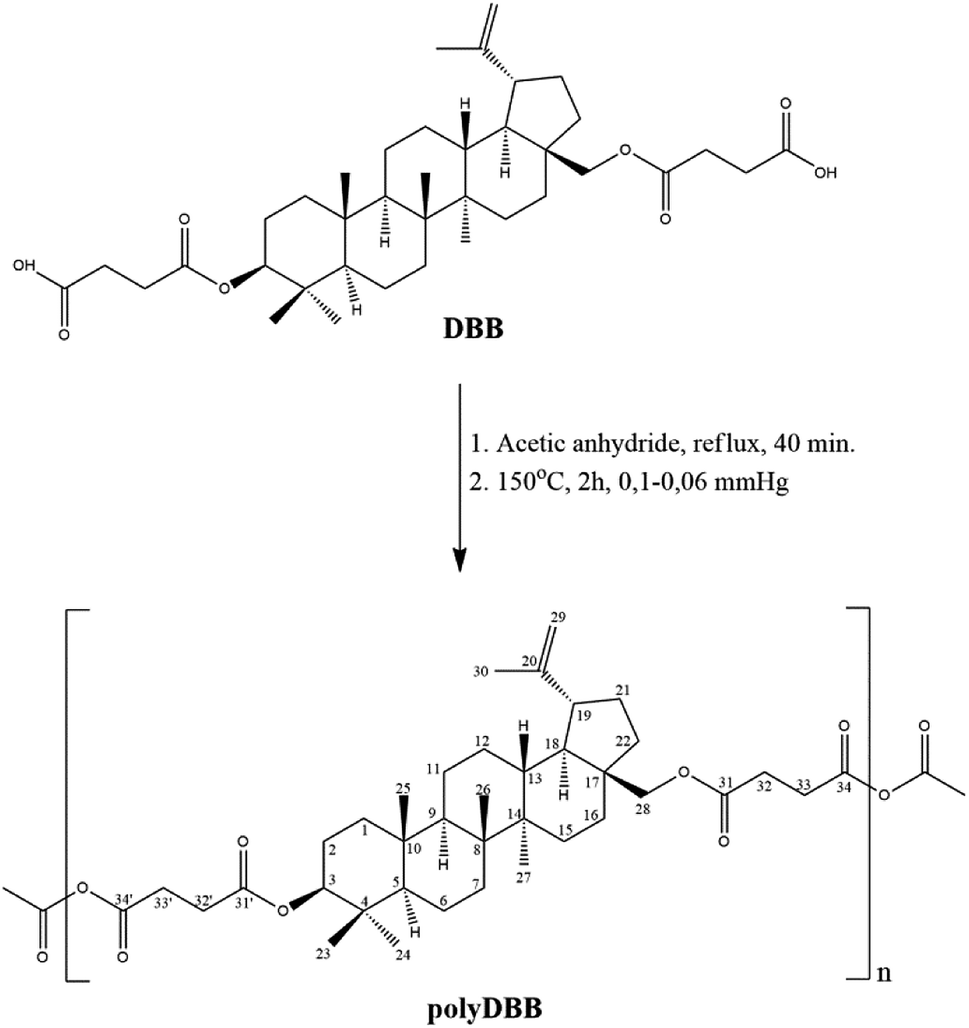 | ||
| Fig. 1 Reaction scheme of the synthesis of polyDBB. | ||
The polymer was found to be insoluble in water, ethanol, acetone, diethyl ether and hexane, but could be dissolved in chloroform, methylene chloride, toluene, THF and partially in DMSO. Thermal properties of obtained polymer were investigated using the DSC method. Comparative DSC thermograms of polyDBB before and after the heat treatment under a nitrogen atmosphere are shown on Fig. 2. The DSC analysis indicated that the polyDBB melts at a high temperature of 320 °C. The determined heat of melting was equal to 108 J g−1. In the first run of heating, exothermal transition with the peak at 238 °C, was also observed and attributed to the crystallization process. After being cooled down to room temperature, the melt of polyDBB did not crystallize in DSC experiments. In the second run of heating neither melting nor crystallization was observed. Melted polyDBB does not crystallize after cooling and remains glassy under ambient conditions, showing glass transition temperature (Tg) equal to 124 °C.
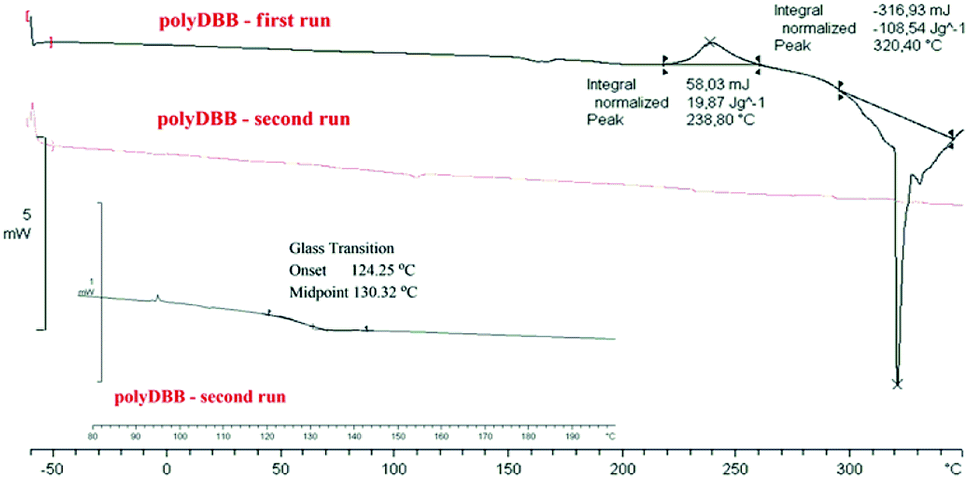 | ||
| Fig. 2 DSC thermograms of polyDBB. | ||
The molecular weight of polyDBB was calculated from 1H NMR and determined by GPC (Table 1). The GPC chromatogram of polyDBB made directly after synthesis has shown only one fraction with relatively low dispersity (Fig. 3). The Mn of the obtained product was approx. 8500 g mol−1, which corresponded to a polymer consisting of 13 repeating units. A good correlation was found between Mn determined by GPC and the one calculated from 1H NMR (Table 1). However, when the GPC analysis was made a few days after the synthesis, or the polymer solution for analysis was stored for too long, the appearance of the second fraction was observed in the GPC chromatograms. The molecular weight of the second fraction corresponded approximately to DBB which indicates a hydrolytic degradation of the polymer during its storage, which occurs with the release of free DBB.
| Mn (1H NMR) | Molecular weight (GPC) | Mw/Mn (GPC) | ||
|---|---|---|---|---|
| Mn | Mw | |||
| polyDBB | 8225 | 8496 | 24939 |
2.94 |
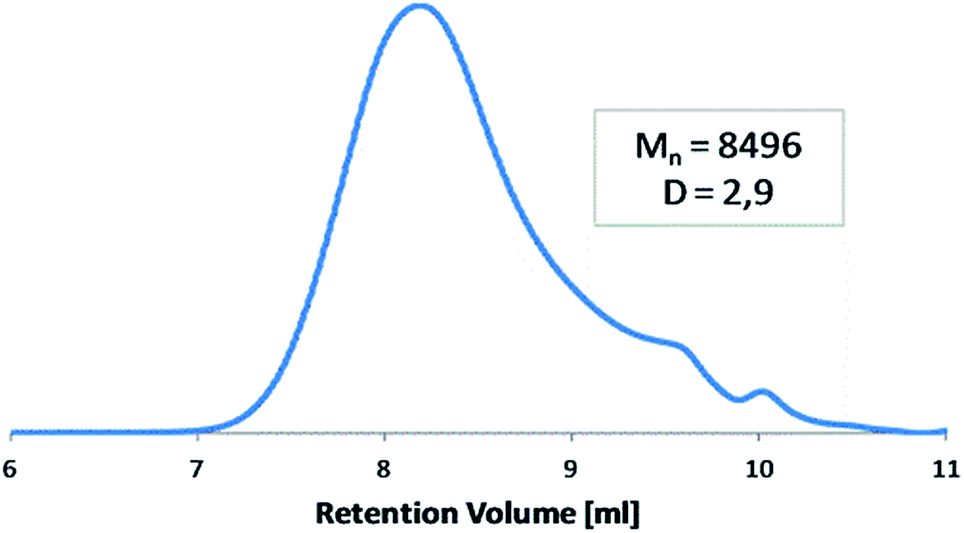 | ||
| Fig. 3 Representative GPC chromatogram of polyDBB. | ||
To determine the exact structure of polyDBB, IR (Fig. 4), 1H NMR (Fig. 5), 13C NMR (Fig. 6), HSQC (Fig. 7) and COSY spectra were made. A successful polyDBB synthesis was confirmed by the formation an anhydride bond. The presence of two peaks in the carbonyl region of the IR spectrum of polyDBB at 1724 cm−1 and 1827 cm−1 confirmed the formation of anhydride bonds.
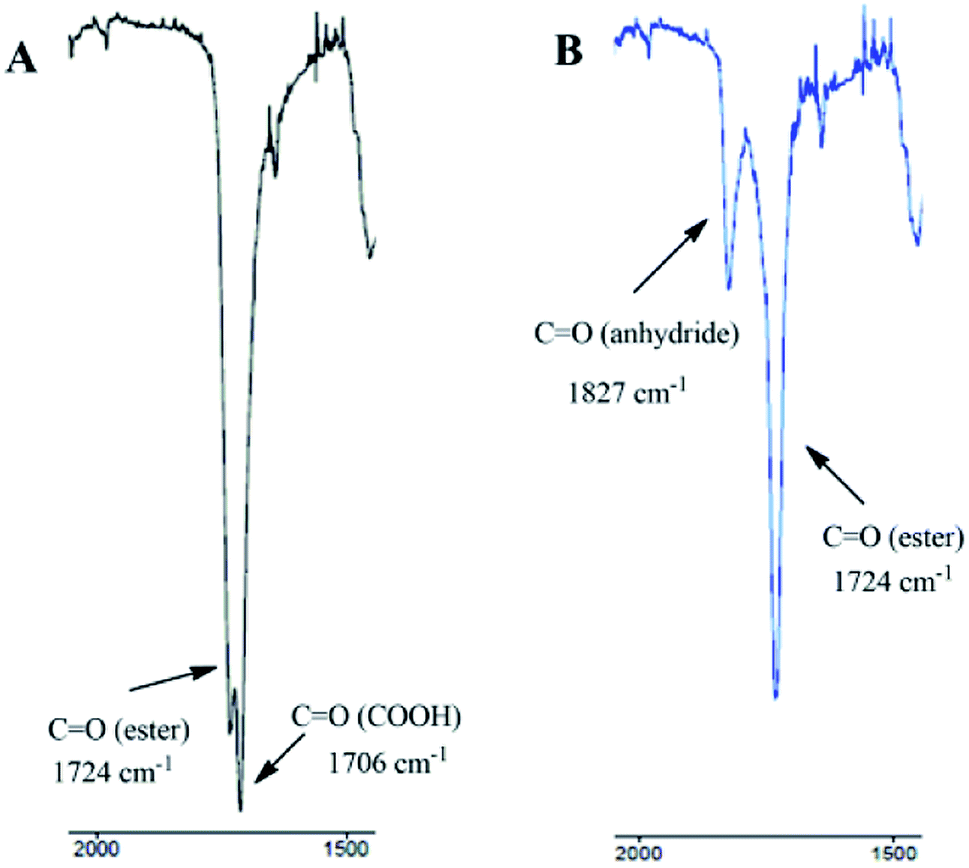 | ||
| Fig. 4 IR spectra (carbonyl region) of DBB (A) and polyDBB (B). | ||
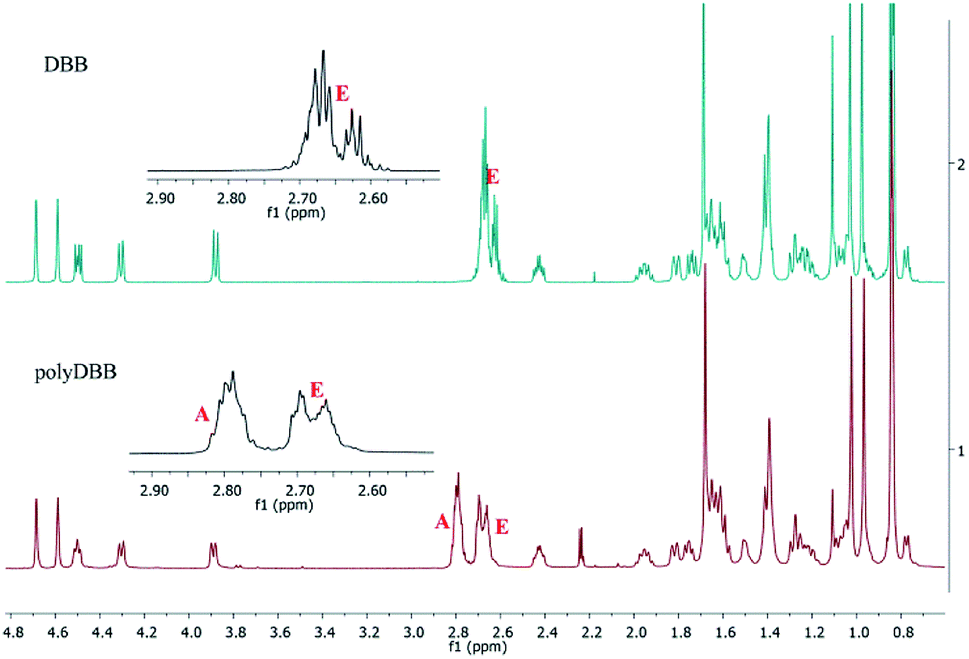 | ||
| Fig. 5 1H NMR spectra of DBB and polyDBB. | ||
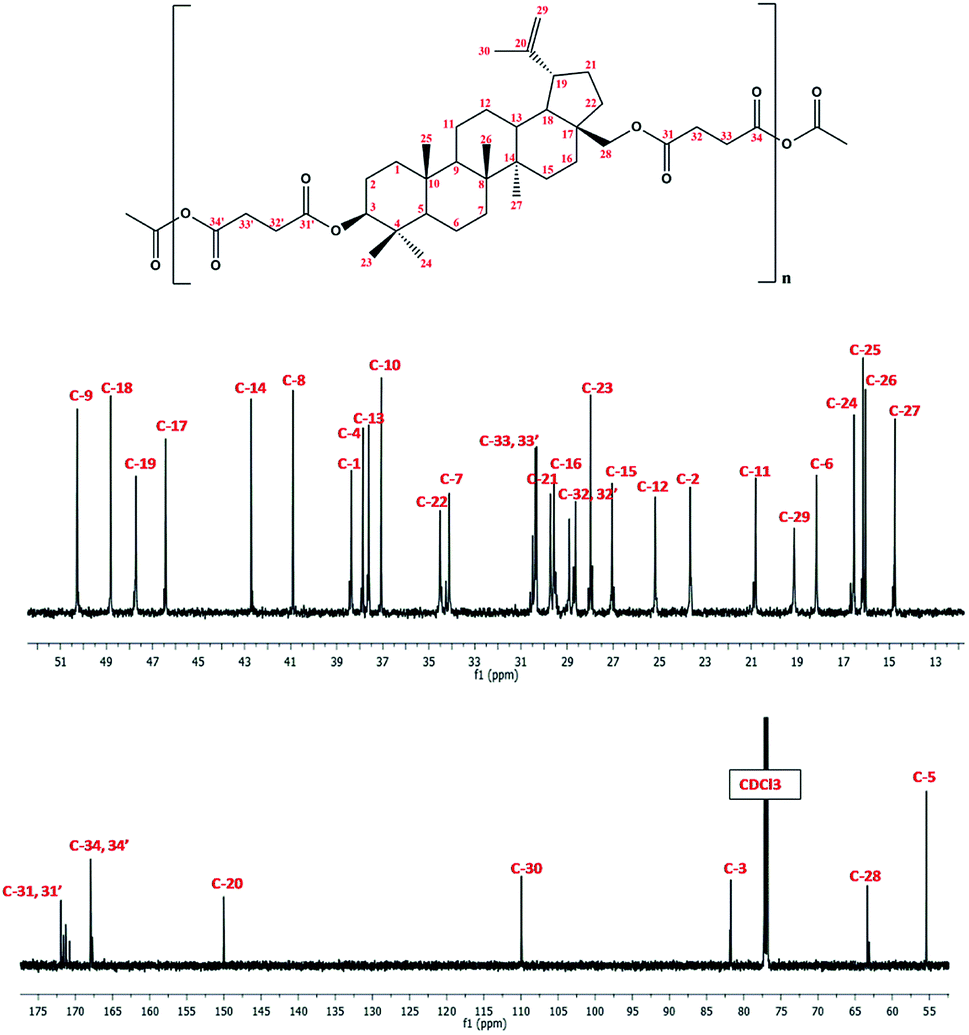 | ||
| Fig. 6 13C NMR spectrum of polyDBB. | ||
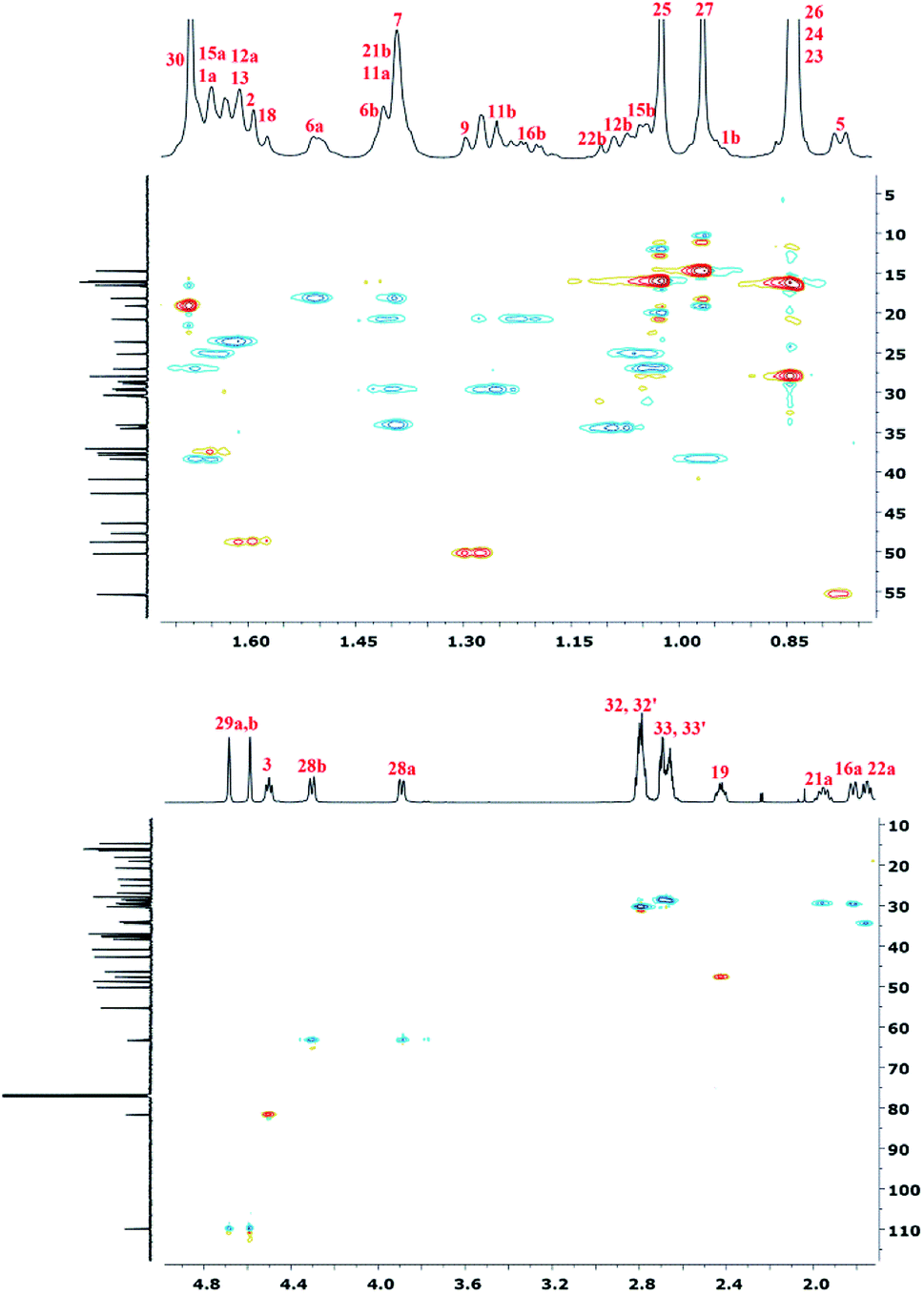 | ||
| Fig. 7 HSQC NMR spectrum of polyDBB. | ||
In 1H NMR spectra of polyDBB (Fig. 5), the signals at δ = 2.74–2.64 ppm and at δ = 2.82–2.77 ppm are observed, which could be assigned to methylene protons close to ester (E) and anhydride (A) groups, respectively. The presence of the signal at δ = 2.84–2.77 ppm in 1H NMR spectrum indicates the formation of the polymer. The molecular weight of polyDBB was calculated using the intensity of the signals of protons at δ = 4.69, 4.59, 4.50, 4.30, 3.89 ppm and the intensity of the end groups signals at δ = 2.18 ppm. The 13C NMR (Fig. 6) spectrum of polyDBB showed two different signals assigned to carbonyl carbon atoms in anhydride (δ = 167.96 ppm) and ester groups (δ = 171.93 ppm) as well as two different signals of methylene carbon atoms next to ester (δ = 28.62 ppm) and anhydride (δ = 30.38–30.32 ppm) groups, respectively. 13C NMR signals were assigned to the relevant polyDBB carbons, but due to the complex structure of polyDBB and the overlapping signals of protons in the rings of betulin, it was impossible to assign all 1H NMR signals to the corresponding protons.
To accurately describe the structure of polyDBB, a two-dimensional HSQC spectrum was made (Fig. 7) enabling to assign proton signals to the corresponding carbons and to determine all signals in the 1H NMR spectrum, except for the two methyl groups at carbons C-26 and C-25. This problem was overcome by collecting a COSY spectrum, which confirmed previous arrangements.
3.2 Hydrolytic degradation and stability of polyDBB
Hydrolytic degradation of polyDBB, which is of a significant importance for any drug delivery application, was carried out in PBS (pH 7.4) and in distilled water (pH 5.5) at 37 °C, in order to determine the disappearance of anhydride bonds and the weight loss. Additionally, the degradation products that have been dissolved in the supernatant were also tested. 1H NMR spectra of lyophilized residues after different degradation times revealed the presence of disuccinate betulin among the degradation products. This is typical for polyanhydrides, that easily undergo hydrolytic degradation to appropriate diacids.18 In case of polyDBB, this diacid is disuccinate betulin and hence it was the only product released during hydrolysis. What is more, no hydrolysis of ester bonds in disuccinate betulin was observed. In the 1H NMR spectra of the degradation products (Fig. 8), the signals at δ = 3.18 ppm, δ = 3.33 ppm and δ = 3.79 ppm assigned to methylene and methine protons next to hydroxyl groups were not observed.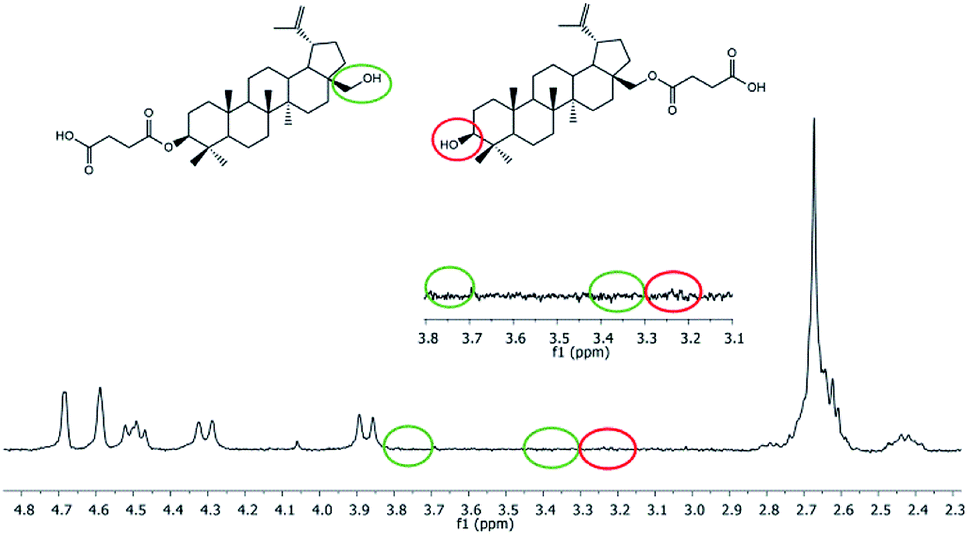 | ||
| Fig. 8 1H NMR spectrum of degradation products (after 14 days of degradation) of polyDBB. | ||
Similar polyanhydride prodrug system, was investigated and described by Uhrich et al., with salicylic acid contained in the backbone of the polymer. Salicylic acid release was depended on aliphatic diacid used as a comonomer in synthesis of the polyanhydride.25,26
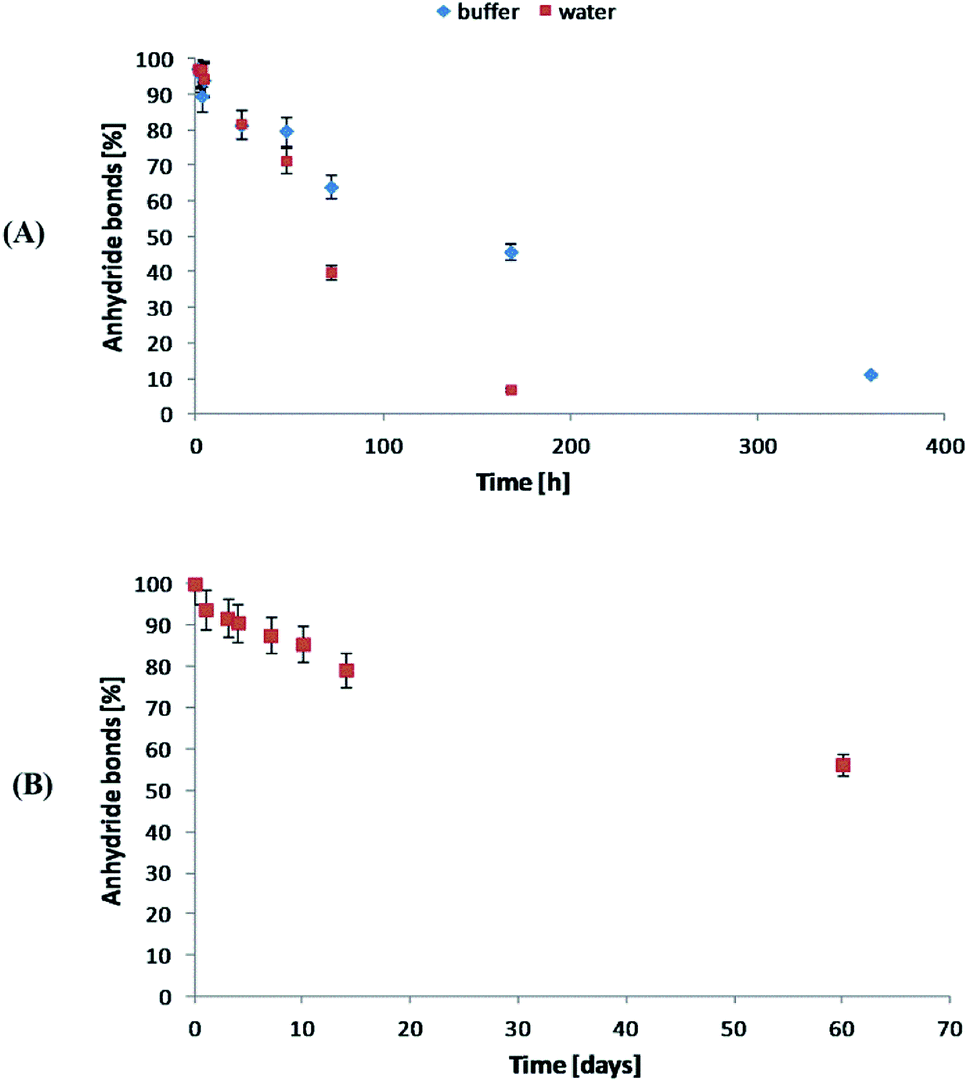
The degradation rate of polyanhydrides is strongly depended on the kind of diacid used in synthesis and varies from a few days to several years.18,25–27 The highly hydrophobic polyanhydrides exhibit surface-eroding properties.26,27 Due to the strong hydrophobicity of the polyDBB, it degrades relatively slowly (Fig. 9A). After 14 days under physiological conditions the complete disappearance of anhydride bonds was observed, indicating full degradation. The weight loss of sample, however, was only about 20% (w/w) after this time, which is supposed to be the effect of the poor solubility of degradation product (DBB) in water.
 | ||
| Fig. 9 Anhydride bonds loss during hydrolytic degradation of polyDBB in water and phosphate buffer (A) conducted at 37 °C and in the air (B) at 25 °C. | ||
The stability of polyDBB in air at room temperature was also investigated. As in the case of the hydrolytic degradation study, the amount of anhydride bonds was determined after certain time of keeping the sample in air (Fig. 9B). After 60 days under the testing conditions, more than 50% of anhydride bonds was still present in the sample, indicating good stability of polyDBB in the air.
3.3 Cytostatic activity of DBB and polyDBB
DBB and polyDBB were studied to determine their cytostatic activity against selected cancer cell lines. In these studies, cell lines representing cervix, breast, lung, liver, central nervous system and nasopharynx tumors were used to find concentrations causing inhibition of cell growth in culture by 50% (IC50). HDF cell lines were also used as non-proliferative cells, to compare results from human cancer cell lines and to establish selectivity between cancer and non-cancer cells. Cytarabine was used as the internal standard for methodological evaluation. Actinomycin D, a widely used anticancer agent, was used in this experiment to compare with our compounds and their activity. Actinomycin D has at least two mechanism of action: inhibition of topoisomerases (I and II), and DNA direct intercalation.The IC50 values obtained for DBB and polyDBB are listed in Table 2.
| Compound | Cytostatic activity IC50 [μg mL−1] | ||||||
|---|---|---|---|---|---|---|---|
| HeLa | MCF-7 | A-549 | U-87 MG | KB | HepG2 | HDF | |
| a N = 3 (t-test), p < 0.05.b Copolymer of DBB and PEG600 containing 40 wt% of PEG600 (Mn = 14166 g mol−1).c Cytarabine and actinomycin D were used as the standard. |
|||||||
| DBB | 8.25 ± 0.81 | 7.26 ± 0.79 | 7.09 ± 0.01 | 7.37 ± 0.26 | 7.17 ± 0.93 | 8.02 ± 0.04 | 14.80 ± 0.06 |
| PolyDBB | 16.23 ± 0.72 | 13.38 ± 0.06 | 16.19 ± 0.31 | 16.07 ± 0.02 | 17.81 ± 0.03 | 15.93 ± 0.12 | 27.13 ± 0.01 |
| PolyDBB_PEGb | 9.03 ± 0.51 | 9.79 ± 0.02 | 8.62 ± 0.19 | 9.07 ± 0.09 | 9.37 ± 0.02 | 9.84 ± 0.91 | 17.22 ± 0.88 |
| Cytarabinec | 1.40 ± 0.08 | — | 1.17 ± 0.21 | 1.03 ± 0.25 | 0.95 ± 0.02 | 1.49 ± 0.04 | 1.94 ± 0.01 |
| Actinomycinc | 1.13 ± 0.01 | — | 1.03 ± 0.83 | 0.92 ± 0.59 | 1.07 ± 0.05 | 1.19 ± 0.02 | 2.28 ± 0.11 |
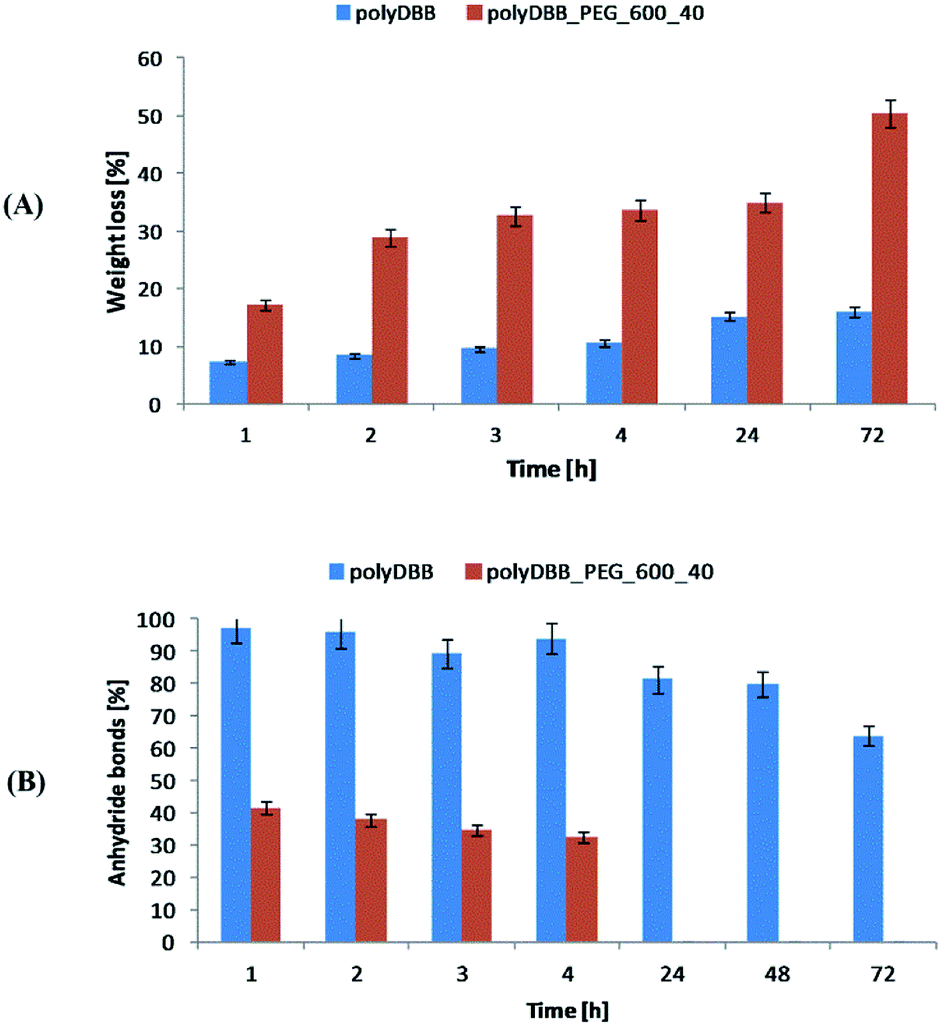
Cytostatic tests indicated the effectiveness of DBB and polyDBB in inhibition of growth of cancer cells (IC50 < 20 μg mL−1), with limited cytotoxicity towards normal cells. DBB showed moderate cytostatic activity against tested cancer cells (IC50 in the range of 7.09 to 8.25 μg mL−1). Biological activity of betulin disuccinate, was described in literature,5,13–17 and was further confirmed by our study. IC50 values calculated for polyDBB were higher than those determined for DBB, what might indicate milder cytostatic activity of the polyanhydride. Nevertheless, it must be emphasized that the anti-cancer activity of polyDBB depends on the degree of hydrolytic degradation of polymer and on the amount of DBB released. PolyDBB is a polymeric prodrug system in which the drug is chemically incorporated into the polymer backbone, with the actual active agent (DBB) becoming available as the polymer degrades. The cytostatic activity was determined after 72 h, at which polyDBB was degraded to elute DBB. During the 72 hour hydrolytic degradation the sample losses around 16% of their initial mass (Fig. 10A), releasing equivalent amount of DBB. What is important, DBB was the only degradation product that was found in the supernatant. Hence, polyDBB was shown to serve as a degradation-based delivery system for DBB, thus having long lasting cytostatic effect when compared to DBB. Cytostatic effects of the polymeric derivatives of betulin can be further modified by copolymerization of DBB with different diacids, allowing to optimize the degradation rate and tailor the period of cytotoxicity. For example, copolymerization of DBB with polyethylene glycol (PEG600) was found to accelerate the degradation process (Fig. 10), resulting in a significant increase in IC50 values determined after 72 h (Table 2).
 | ||
| Fig. 10 The comparison of hydrolytic degradation of polyDBB and polyDBB_PEG600_40 (A) mass loss and (B) anhydride bonds loss. | ||
Copolymer of DBB and PEG600 (polyDBB_PEG600_40), was obtained accordingly to the procedure described for polyDBB. 40 wt% of DBB was replaced by dicarboxylic derivatives of PEG600. Due to the increase in the hydrophilicity, polyDBB_PEG600_40 degraded faster (Fig. 10) than polyDBB losing over 50% of their initial weight and thus releasing more DBB after 72 h.
3.4 Microspheres and nanospheres from polyDBB
PolyDBB was applied for the preparation of micro- and nanospheres by means of an emulsion (O/W) solvent evaporation technique using poly(vinyl alcohol) as stabilizing agent. By changing the speed of homogenization, it was possible to form smooth microspheres with diameters of 20–30 μm (3000 rpm, Fig. 11A) and 1–3 μm (18000 rpm, Fig. 11B). When the ultrasounds were used, it was possible to form polyDBB nanospheres (Table 3). Two fractions of polymer nanospheres were obtained, those having the diameters of approx. 800–900 nm (obtained after the first centrifugation at 5000 rpm for 5 min) and of approx. 400–500 nm (obtained after the second centrifugation at 8000 rpm for 3 min) (Fig. 11C), both spherical in shape and having smooth surface. Nanospheres from the second fraction, however, had the tendency to agglomerate. This tendency, as well as the microsphere diameters, could be reduced by the copolymerization of DBB with PEG. The results will be published separately.
 | ||
| Fig. 11 SEM images of polyDBB microspheres obtained by using homogenization speeds of 3000 rpm (A) and 18000 rpm (B) and polyDBB nanospheres (C). | ||
| homogenizer rpm | Optical microscope | SEM | ||||
|---|---|---|---|---|---|---|
| Dn [μm] | S | Dv/Dn | Dn [μm] | S | Dv/Dn | |
| a The first fraction obtained after the first centrifugation (5000 rpm for 5 min).b The second fraction obtained after the second centrifugation (8000 rpm for 3 min). | ||||||
| 3000 | 23.81 | 4.56 | 1.11 | 27.25 | 10.93 | 1.29 |
| 18000 |
3.23 | 0.95 | 1.24 | 1.73 | 1.14 | 2.59 |
| Ultrasounda | — | — | — | 0.898 | 0.46 | 1.86 |
| Ultrasoundb | — | — | — | 0.417 | 0.55 | 3.85 |
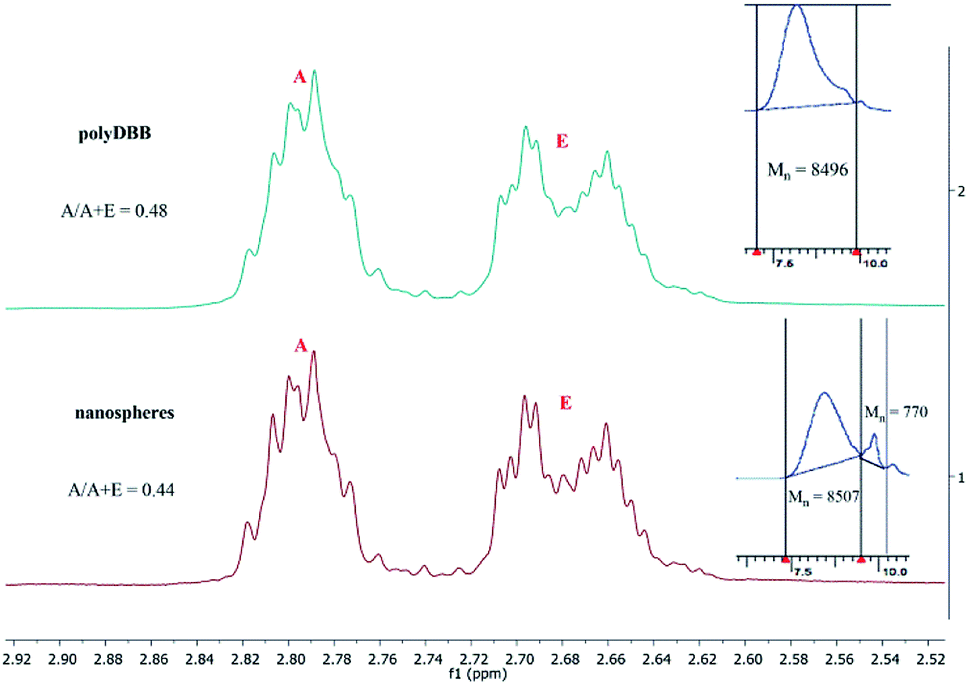
Because the polyDBB are hydrolytically unstable, it was necessary to quantify the degree of hydrolytic degradation that occurs during the micro- or nanospheres fabrication. For this purpose, the molecular weight (GPC) and anhydride bonds content (1H NMR) were determined for neat polymer (before microspheres fabrication) and polymer after microspheres preparation. The molecular weight loss, as well as the disappearance of anhydride bonds, were observed for all microspheres, but the degree of hydrolysis was the less significant the larger the microspheres were obtained. For the microspheres with diameters of 20–30 μm (3000 rpm), 20% reduction of Mn and 5% disappearance of anhydride bonds were observed, after microparticles formulation. The process of hydrolytic degradation of polymers was more significant when small microspheres (Dn = 1–3 μm) or nanospheres were produced. It is due to an increased surface area available for contact with water. Such dependence is characteristic for polyanhydrides, which undergo hydrolytic degradation at the surface. Fig. 9 shows 1H NMR spectra (in range of ester and anhydride groups) and GPC chromatograms of polyDBB before and after nanospheres preparation. The disappearance of anhydride bonds was less than 15%, but the almost 60% reduction of Mn was observed after nanospheres preparation. Mn of polyDBB after nanospheres preparation was equal to 3500 g mol−1, compared to the neat polymers with Mn = 8496 g mol−1. In the GPC chromatogram of polyDBB after nanospheres preparation (Fig. 12), the low molecular weight fraction with Mn = 770 g mol−1 was observed, besides the main fraction with Mn = 8507 g mol−1. The molecular weight of the main fraction is on the same level as the molecular weight of neat polymers, whereas the molecular weight of the second fraction corresponds approximately to DBB. This confirmed that polyDBB undergoes hydrolytic degradation from the end of the chain to release DBB as degradation products.
 | ||
| Fig. 12 1H NMR spectra (ester and anhydride groups region) and GPC chromatograms of polyDBB (A) before and (B) after nanospheres preparation. | ||
The possibility of making micro- and nanospheres from polyDBB indicates the potential applicability of this polymer as a controlled drug release system. As such, it possesses promisingly suitable and controllable degradation rate, making it a potentially suitable drug delivery system for DBB. Similar delivery systems of betulin or betulin derivatives have recently been investigated, and described in literature. Such systems were proposed in forms of e.g. nanoemulsion,28 nanoparticles,29 PLA nanovectors30 or micelles.31 It has been found that incorporating betulin substance into a nanosystem could significantly increase dissolution rate and/or its bioavailability.28–31 However, in all of the described nanosystems, betulin derivatives were physically entrapped in the carrier substance. Therefore, their release was mainly controlled by diffusion and could not be easily adjusted. In case of polyDBB, betulin disuccinate is chemically incorporated into the polymer backbone itself and becomes available as the polymer degrades. This design allows for a maximum amount of drug to be incorporated within the polymer structure, and its release to be controlled by the degree of degradation. Such systems are usually more stable, exhibit a smaller burst effect and exhibit better release characteristics.18,32,33
PolyDBB spheres may be also considered as vehicles for another biologically active compounds. The possibility of tailoring the size of the particles makes it possible to target for different forms of administration, including inhalation or intravenous injection (nanospheres), as well as intramuscular injection or implantation (microspheres).
4 Conclusions
In this work, a new polyanhydride, polyDBB, based on a betulin derivative was obtained and thoroughly characterized. Under physiological conditions (37 °C, pH = 7.4 or pH = 5.5), polyDBB was shown to undergo hydrolytic degradation to form betulin disuccinate, whose biological activity is known and confirmed. It was shown that polyanhydride can be fabricated in a form of micro- or nanospheres, being easily administrated by injection or inhalation. In this way, polyDBB serves as a promising degradation rate controlled drug delivery system for either DBB or other cytostatic agents.Conflicts of interest
The authors declare no conflict of interest.Notes and references
- S. Cai, T. R. Bagby and M. L. Forrest, Ther. Delivery, 2011, 2, 1467–1484 CrossRef PubMed.
- S. A. Stewart, J. Dominguez-Robles, R. F. Donnelly and E. Larraneta, Polymers, 2018, 10, 1379–1403 CrossRef PubMed.
- S. Alakurtti, T. Mäkelä, S. Koskimies and J. Yli-Kauhaluoma, Eur. J. Pharm. Sci., 2006, 29, 1–13 CrossRef CAS PubMed.
- S. Jager, H. Trojan, T. Kopp, M. N. Laszczyk and A. Scheffler, Molecules, 2009, 14, 2016–2031 CrossRef PubMed.
- J. Patočka, J. Appl. Biomed., 2003, 1, 7–12 CrossRef.
- A. Muhammad, R. M. Carlson, P. Krasutsky and M. Karim, J. Microbiol. Biotechnol., 2004, 14, 1086–1088 Search PubMed.
- C. M. Wang, K. L. Yeh, S. J. Tsai, Y. L. Jhan and C. H. Chou, Molecules, 2017, 22, 2119–2132 CrossRef PubMed.
- J. L. C. Sousa, C. S. R. Freire, A. J. D. Silvestre and A. M. S. Silva, Molecules, 2019, 24, 355 CrossRef PubMed.
- J. Jeromenok, W. Böhlmann, M. Antonietti and J. Weber, Macromol. Rapid Commun., 2011, 32, 1846–1851 CrossRef CAS PubMed.
- J. Zhao, H. Schlaad, S. Weidner and M. Antonietti, Polym. Chem., 2011, 1763–1768 Search PubMed.
- J. Jeromenok, W. Böhlmann, C. Jäger and J. Weber, ChemistryOpen, 2013, 2, 17–20 CrossRef CAS PubMed.
- M. Okada, K. Suzuki, Y. Mawatari and M. Tabata, Eur. Polym. J., 2019, 113, 12–17 CrossRef CAS.
- I. C. Sun, H. K. Wang, Y. Kashiwada, J. K. Shen, L. M. Cosentino, C. H. Chen, L. M. Yang and K. H. Lee, J. Med. Chem., 1998, 41, 4648–4657 CrossRef CAS PubMed.
- P. A. Krasutsky, US Pat., WO2002026761 A1, 2002.
- P. A. Krasutsky, US Pat., 20020119935 A1, 2002.
- S. Chowdhury, T. Mukherjee, S. R. Chowdhury, S. Gupta, S. Mukhopadhyay, P. Jaisankar and H. K. Majumder, Antimicrob. Agents Chemother., 2014, 58, 2186–2201 CrossRef PubMed.
- R. A. Pegova, et al., Medical Almanac, 2015, 3, 216 Search PubMed.
- A. J. Domb, N. Kumar, and A. Ezra. Biodegradable polymers in clinical use and clinical development. Wiley&Sons Inc., Hoboken, New Jersey, USA, 2011 Search PubMed.
- K. Jaszcz, Macromol. Symp., 2007, 254, 109–116 CrossRef CAS.
- K. Jaszcz, M. Śmiga-Matuszowicz and J. Łukaszczyk, React. Funct. Polym., 2008, 68, 351–360 CrossRef CAS.
- K. Jaszcz, Int. J. Polym. Mater. Polym. Biomater., 2014, 63, 97–106 CrossRef.
- P. Skehan, et al., J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- S. Q. Tian, K. Lu, G. Z. Fang, S. Y. Han and Q. Ai, Chem. Ind. Forest Prod., 2007, 27, 77–80 CAS.
- S. Q. Tian, K. Lu, G. Z. Fang, S. Y. Han and Q. Ai, J. Beijing For. Univ., 2009, 31, 108–111 CAS.
- A. Prudencio, R. C. Schmeltzer and K. Uhrich, Macromolecules, 2005, 38, 6895–6901 CrossRef CAS PubMed.
- K. Whitaker-Brothers and K. Uhrich, J. Biomed. Mater. Res., Part A, 2006, 76, 470–479 CrossRef PubMed.
- A. Göpferich and J. Tessmar, Adv. Drug Delivery Rev., 2002, 54, 911–926 CrossRef.
- C. A. Dehelean, S. Feflea, S. Ganta and M. Amiji, J. Biomed. Nanotechnol., 2011, 7, 317–324 CrossRef CAS PubMed.
- X. H. Zhao, et al., Drug Deliv., 2014, 21, 467–479 CrossRef CAS PubMed.
- R. Yadav, D. Kumar, A. Kumara and S. Kumar Yadav, Biotechnol. Lett., 2016, 38, 259–269 CrossRef CAS PubMed.
- K. Jelonek, et al., Indones. J. Pharm., 2019, 557, 43–52 CAS.
- Q. Sun, J. Wang, M. Radosz, and Y. Shen, Functional Polymers for Nanomedicine, in Functional Polymers for Nanomedicine, ed. Y. Shen, The Royal Society of Chemistry, 2013 Search PubMed.
- S. Dragojevic, J. Su Ryu and D. Raucher, Molecules, 2015, 20, 21750–21769 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03326b |
| This journal is © The Royal Society of Chemistry 2019 |