DOI:
10.1039/C9RA03030A
(Paper)
RSC Adv., 2019,
9, 20463-20471
A novel, fast, high sensitivity biosensor for supporting therapeutic decisions and onset actions for chest pain cases†
Received
23rd April 2019
, Accepted 20th June 2019
First published on 1st July 2019
Abstract
In this work, a novel and promising organic nano linker (NL) was prepared via refluxing 5-aminoisophthalic acid and 1,2-phenylenediamine at 80 °C for 48 h. After that, this linker was reacted with manganese chloride to afford a novel manganese metal–organic framework (Mn–MOF). The produced materials were characterized using 1H-NMR, 13C-NMR, mass spectrometry, elemental analysis, UV, IR, FE-SEM, EDX, TEM, and thermal study. In addition to X-ray diffraction, XPS, magnetic properties and photoluminescence investigation for Mn–MOF. The study was extended to apply Mn–MOF as electroactive material for the preparation of a novel cardiac troponin I (cTn) potentiometric membrane biosensor. The biosensor, based on Mn–MOF with an optimized membrane composition, exhibits a fast, stable and linear-Nernstian response to cTn in the concentration range between 0.01 and 30.0 ng mL−1 with a pH range between 5.6 and 10.1 and a fast response time of 20 ± 5 s. The detection and quantification limits are 0.055 and, 0.168 ng mL−1, respectively. The lifetime of the electrode is between 3–12 week without a significant change in the membrane compositions and the performance characteristics based on the storage conditions. The electrode shows high selectivity towards cTn with respect to common interfering analytes. This approach of Mn–MOF-electrode could be addressed, facilitated and helped an emergency departments (EDs) decision-making in patients with chest pain and early myocardial infarction diagnosis. The future vision is converting the present approach to a small device with satisfactory results which will be used in term of point-of-care testing (POCT) for measuring the most important cardiac blood biomarkers.
Introduction
Acute coronary syndrome (ACS) is a clinical term used to describe a group of cases resulting from deficient blood flow to the heart muscle.1,2 These cases starting with chest pain and discomfort reach myocardial infarction (MI), and then cardiogenic shock.3 Symptoms can include chest pain including tightness and heaviness in the chest, discomfort in the arms and upper body, shortness of breath and other constitutional symptoms.1,2 ACS diagnosis is established on a medical history of the patient, physical inspection, electrocardiogram that evaluated the heart electrical activity and then the laboratories blood investigations that evaluated the attendance of biological biomarkers which resulting from cardiac cell injury.2 Sixteen million people worldwide die yearly by cardiovascular diseases especially stroke or heart attacks,4 Chest pain cases are among the top ten reasons that visit the emergency department (ED), approximately about a five percent of all ED visits5 expected prevalence of acute myocardial infarction (AMI) in chest pain patients in the ED.6
Cardiac troponin I (cTn) is a protein found in the myocardial muscle. It is the preferred marker for cardiac injury detection in AMI diagnosis.7 Cardiac troponin I levels increase within 3–6 hours after the onset of the symptoms, the highest values at 18–24 hours and stay elevated for 4–7 days.8 Due to the high positive predictive value (PPV) of troponin I for AMI, a positive result can contribute to early triage/risk stratification of at-risk patients indicates a high risk for AMI.9 However, a negative result does not exclude AMI and further testing is required (Fig. 1). Early detection of myocardial damage is indispensable. Community-based reports have shown that the overall mortality rate of cases with presumed AMI or ACS in the first month is nearly fifty percent, and about fifty percent of these cases within the first two hours the deaths occur.10 Several analytical techniques widely have been used for detection of cTn such as electrochemiluminescent,11 plasmonic biosensor,12 photoelectrochemical immunosensor,13 label-free immunosensor.14 In the ESI† file (Appendix A) approximately most of published methods and techniques which were used for cardiac troponin determination in last years.
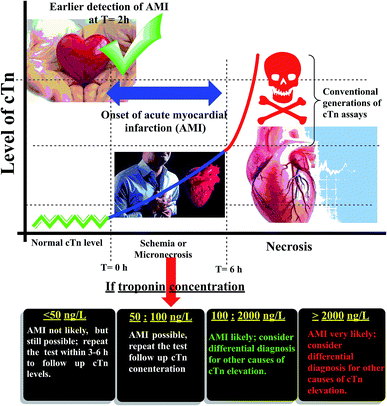 |
| | Fig. 1 Schematic diagram of the relation between cardiac troponin (cTn) level and diagnosis of myocardial infarction. | |
Accordingly, the potentiometric method based on using of a novel ion-selective electrodes (ISE) is the method of choice owing to its simplicity, ease of preparation, facile procedures, simple instrumentation, short analysis time, low cost, high selectivity, wide dynamic range, and wide linear concentration range, non-destructive approach for drug and proteins assay.15
Today, metal–organic frameworks (MOFs) are attracted enormous interest to a lot of researchers because of their fascinating structures and potentially useful properties.16,17 MOFs are hybrid molecules with a high molecular weight that are formed by the monomeric unit's repetition.18 These monomeric units are formed by a combination of metal ions and linkers (organic ligands) through coordination bonds.19 MOFs are substances that have a broad variety of physical and chemical properties and have a lot of promising application like; sensing/biosensing ability, catalysis, drug delivery, thin films, used as composite materials, and more.20–23 On the other hand, the linkers (ligands) which are used in MOFs synthesis must have specific functional groups as carboxylate, phosphate, pyridyl, etc. and must have various modes of coordination to metal ions, and have the ability to act as a hydrogen-bond acceptor and donor.24,25
In the present study, to the best of our knowledge, a novel nano linker was synthesized and used to fabricate a novel manganese metal–organic framework (Mn–MOF). Both produced materials are fully characterized. The target of this study was extended to fabricate of Mn–MOF electroactive membrane and used it as economic, fast, simple, sensitive, and a selective potentiometric biosensor for determination of cardiac troponin I (cTn) in whole blood, serum, and plasma samples. The present approach was used for cTn quantification based on using Mn–MOF/PVC matrix. Therefore, this approach could facilitate and helps EDs decision-making in patients with chest pain at the point of care and assistance in MI cases diagnosis. Assistance in the risk stratifies cation of patients with symptoms suggestive of ACS with respect to the relative risk of all-cause mortality and major adverse cardiac events. The goal of our approach is to measure troponin which always uses in conjunction with clinical presentation, history, and other diagnostic information.
Results and discussion
Characterization of organic nano linker (NL)
The organic nano linker (NL) was prepared according to the reaction scheme (Scheme S1†) by addition of Aip solution (ethanol–DMF, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) to a solution of Phen in ethanol in the presence of 1 mL glacial acetic acid. The summarized physical data were: melting point ∼300 °C. Anal. (%) Calcd for the NL of C47H74N8O20 (1071.15 g mol−1): C, 52.70; H, 6.96; N, 10.46. Found: C, 53.06; H, 6.79; N, 10.53. The yield was 92.0%. Electrospray ionization mass spectrum of NL: 1070.49 m/z. 1H-NMR (500 MHz, [D6, DMSO], 25 °C, TMS): δ = 1.04 ppm (t, 3H; CH3), 2.07 ppm (s, 6H; DMSO), 2.49 ppm (m, 6H; DMF), 3.43 ppm (q, 2H; CH2), 5.58 ppm (broad, H; NH2, OH), 7.3–7.6 ppm (m, H; phenyl, DMF), 12.3 ppm (broad, H; OH).13C-NMR (500 MHz, [D6] DMSO, 25 °C, TMS): δ = 18.58 ppm (CH3), 34.17 ppm (DMF), 39.5 ppm (DMSO-d6), 56.06 ppm (CH2), 117.5,118.08,132.07,149.12, and 167.4 ppm (CH-phenyl, DMF). IR spectrum (KBr, cm−1): 3658–3160 ν(NH2, NH and H2O); 2915 ν(CH); 1620 ν(C
:4 v/v) to a solution of Phen in ethanol in the presence of 1 mL glacial acetic acid. The summarized physical data were: melting point ∼300 °C. Anal. (%) Calcd for the NL of C47H74N8O20 (1071.15 g mol−1): C, 52.70; H, 6.96; N, 10.46. Found: C, 53.06; H, 6.79; N, 10.53. The yield was 92.0%. Electrospray ionization mass spectrum of NL: 1070.49 m/z. 1H-NMR (500 MHz, [D6, DMSO], 25 °C, TMS): δ = 1.04 ppm (t, 3H; CH3), 2.07 ppm (s, 6H; DMSO), 2.49 ppm (m, 6H; DMF), 3.43 ppm (q, 2H; CH2), 5.58 ppm (broad, H; NH2, OH), 7.3–7.6 ppm (m, H; phenyl, DMF), 12.3 ppm (broad, H; OH).13C-NMR (500 MHz, [D6] DMSO, 25 °C, TMS): δ = 18.58 ppm (CH3), 34.17 ppm (DMF), 39.5 ppm (DMSO-d6), 56.06 ppm (CH2), 117.5,118.08,132.07,149.12, and 167.4 ppm (CH-phenyl, DMF). IR spectrum (KBr, cm−1): 3658–3160 ν(NH2, NH and H2O); 2915 ν(CH); 1620 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1394–1294 ν(CC) aromatic; 1103–746 ν(CH) aromatic. UV-vis (solid): λmax = 230, 303, 386, and 630 nm. Thermal analysis: DTGmax = 75.13, 120.93, 290.25 and 367.58 °C. The proposed structure of NL was in a good agreement with the data obtained from microanalytical analysis and different spectroscopic tools. Elucidation of the structure in the following sections discussed in detail.
O); 1394–1294 ν(CC) aromatic; 1103–746 ν(CH) aromatic. UV-vis (solid): λmax = 230, 303, 386, and 630 nm. Thermal analysis: DTGmax = 75.13, 120.93, 290.25 and 367.58 °C. The proposed structure of NL was in a good agreement with the data obtained from microanalytical analysis and different spectroscopic tools. Elucidation of the structure in the following sections discussed in detail.
Mass spectrum. (Fig. S2 and S3†) show the mass spectrum and the suggested fragmentation of the NL, respectively. It can be seen that the molecular ion peak at 1071.15 m/z is consistent with the proposed structure molecular weight.
1H and 13C NMR spectra. The 1H and 13C NMR spectra of the NL were analyzed in DMSO-d6 at room temperature (Fig. S4 and S5†), respectively. The 1H NMR spectrum (Fig. S4†) shows three peaks for ethanol; a triplet peak at 1.04 ppm for (CH3), the quartet at 3.43 ppm for (CH2) and broad signal appears at 12.3 ppm for (OH). The signals of DMF were observed at 2.49 and 7.34 ppm. Abroad signal appears at 5.58 ppm for (NH2 and OH) the broadly and the shift occurs due to high hydrogen bonding of water molecules. The signals appear between 7.3 and 7.6 ppm are corresponding to phenyl protons. Whereas, 13C NMR spectrum (Fig. S5†), of the NL displays two signals at 18.58 and 56.06 ppm due to ethanol. Another two signals at 34.17 and 167.4 ppm shows due to DMF. The signals found in the region between 117.5 and 149.12 ppm are due to phenyl groups.
FT-IR spectrum. The FT-IR spectrum of NL is shown in (Fig. S6†), and the significant frequencies in the spectrum were investigated. The bands between 3658 and 3160 cm−1 are assigned to NH2, NH and water molecules. The band at 2915 cm−1 is assigned to sp3 CH. The band at 1620 cm−1 is assigned to the CO group. The bands between 1394 and 1294 cm−1 are due to aromatic CC, and bands between 1103 and 746 cm−1 are due to aromatic CH.
UV-vis spectrum. The electronic absorption spectra of solid starting ligands (Aip and Phen) and NL were measured in a range between 200–2200 nm, at room temperature. In addition, the bandgap spectrum represented in (Fig. S7a to d†). (Fig. S7a, b, and c†) showed the UV-vis spectrum, from this figures it was found that the NL exhibits absorption bands at 230, 303, 386 and 630 nm, which were assigned to intra ligand charge transfer (n–π* and π–π*). The data obtained in the region 800–2200 nm are consistent with the data obtained from the low and mid-region of FT-IR. The band gap of the linker and starting ligands using the plot of (αhv)2 versus photon energy (hv) are revealed in (Fig. S7d†). From this figure, it can see that the bandgap of Aip is about 2.98 and 3.92 eV and for Phen is 1.79 and 2.11 eV which is due to highly conjugated of Aip and Phen and the presence of NH2 groups. After the reaction of starting ligands to achieve the nano linker, the band gap for the nano linker (NL) was about 2.0, 2.96, and 3.64 eV, they provide more bonding sites with the organic linker, and the band gap of linker is very close to Aip as well as highly conjugated as Phen precursor.
Thermal analysis. The thermogravimetric analysis (TGA) and thermal stability study of NL were performed up to 1000 °C under nitrogen gas with a heating rate of 10 °C min−1. The thermal analysis (TGA-DTGA) plots (Fig. S8†) suggest that the NL pass through three stages of decompositions, underwent a weight loss due to the expulsion of ethanol, water, and DMF molecules in the temperature range between 46 to 168 °C with a weight loss of 18.48% (calculated: 17.9%). The linker collapse due to successive decomposition stages of Aip and Phen in the temperature range 220–377 °C (DTGmax = 71.18, 120.93, 290.25 and 367.58 °C, mass loss found 81.52, calculated: 82.1%).
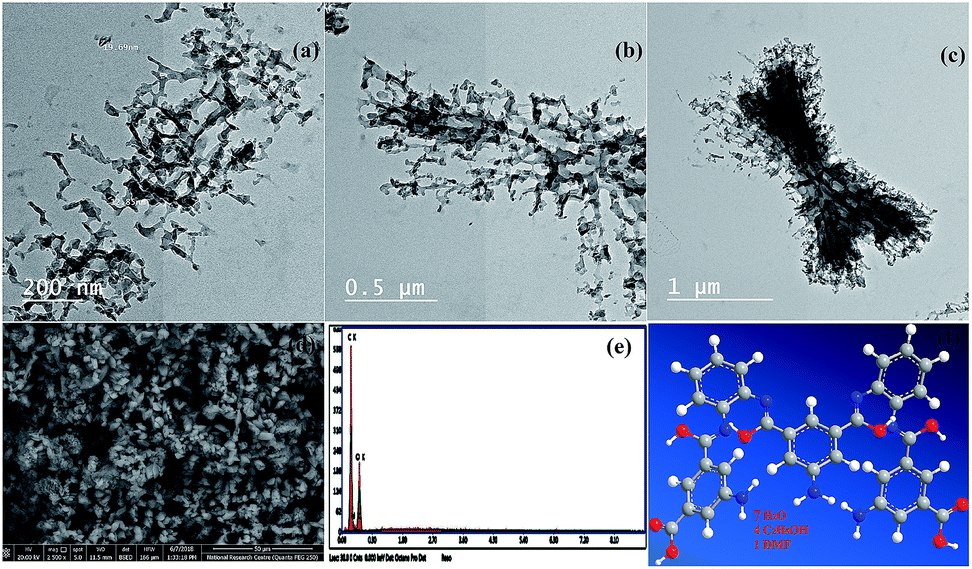
Transmission electron microscopy. Fig. 2a, b, and c show TEM images of the NL. The morphology of the NL looks like a network with uniformed disperse particles. The particles had an average size between 12 and 19 nm.
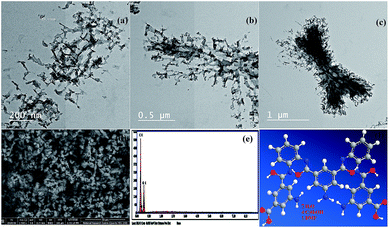 |
| | Fig. 2 (a, b, and c) Transmission electron microscopy images (TEM) of the nano linker (NL) at different magnification, (d) field-emission scanning electron microscopy image of NL, (e) energy-dispersive X-ray analysis with a single point EDX mapping analysis of NL, and (f) three-dimensional structural representation of the monomeric unit of NL. | |
FE-SEM combined with EDX spectroscopy. FE-SEM images of the NL are shown in Fig. 2d. The NL appeared to be like uniformed overlapped disperse particles. On the other hand, these images which are in good conformity with the TEM data. Furthermore, the single point EDX mapping analysis (Fig. 2e, Table S9†) of NL revealed the presence of C, N, and O as the building-block elements in the individual particles. EDX analysis revealed the good distribution of the elements C, N, and O along the cross-section (Fig. 2e). The EDX data is in a good agreement with the elemental analysis data: C, 52.70; N, 10.46; O, 29.87. Found: C, 52.75; N, 10.36; O, 36.89 (see Table S11†).Based on the physical and spectral data of the NL, we can deduce that the structure of NL is suggested as shown in Fig. 2f.
Characterization of Mn–MOF
The Mn–MOF was prepared according to the reaction scheme (Scheme S10†) by addition of MnCl2·4H2O solution to a flask containing nano linker which was prepared according to (Scheme S1†). The physical data were summarized as the following: melting point >300 °C. Anal. (%) Calcd for the Mn–MOF of C47H76Mn2N8O25 (1263.03 g mol−1): C, 44.70; H, 6.07; N, 8.87. Found: C, 44.41; H, 5.99; N, 8.93. The yield was 89.0%. 1H-NMR (500 MHz, [D6, DMSO], 25 °C, TMS): δ = 1.04 ppm (t, 3H; CH3), 2.07 ppm (s, 6H; DMSO), 2.49 ppm (m, 6H; DMF), 3.41 ppm (q, 2H; CH2), 4.35 ppm (broad, H; NH2, OH), 6.92–7.95 ppm (m, H; phenyl, DMF). IR spectrum (KBr, cm−1): 3700–3040 ν(NH2 and H2O); 2925 ν(CH); 1637 ν(CO); 1550–1234 ν(CC) aromatic; 1197–709 ν(CH) aromatic; 598 ν(Mn–O); 420 ν(Mn–N). UV-vis (solid): λmax = 225, 387, and 416 nm. Thermal analysis: DTGmax = 71.2, 211.51, 286.52, 524.02 and 798.22 °C. The proposed structure of Mn–MOF was in a good agreement with the data obtained from microanalytical analysis and different spectroscopic tools. Elucidation of the structure in detail is discussed in the following sections.
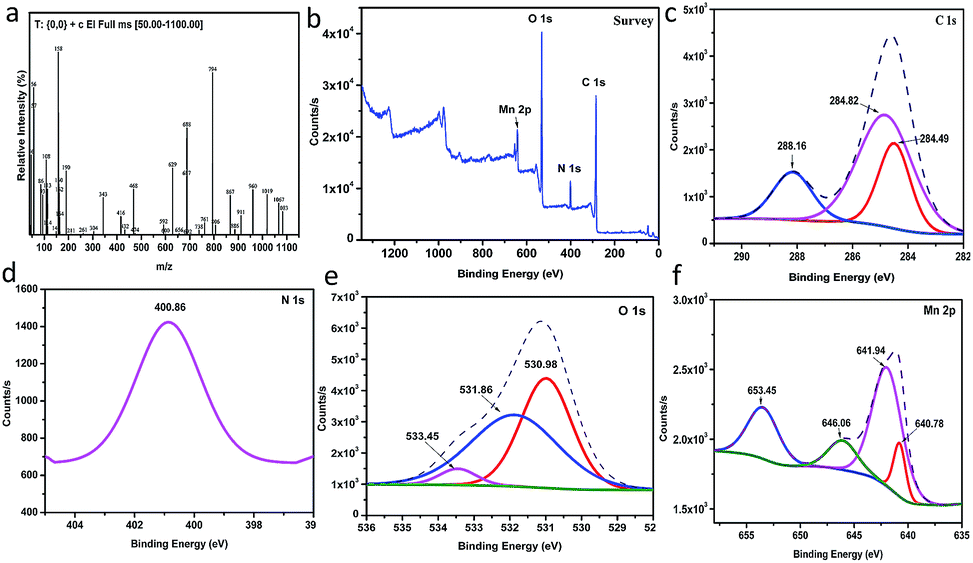
Mass spectrum. (Fig. 3a and S11†) show the mass spectrum and the suggested fragmentation of the Mn–MOF, respectively. It can be noticed that the molecular ion peak at 1263.03 m/z theoretically due to the limitation of the range 50–1100 m/z, but the subsequent fragmentations was inconsistent with the molecular weight of the proposed structure.
1H NMR spectrum. The 1H-NMR spectrum of the Mn–MOF was performed in DMSO-d6 at room temperature (Fig. S12†). It shows three peaks for ethanol; a triplet peak at 1.04 ppm for (CH3), the quartet at 3.41 ppm for (CH2), and at 4.35 ppm for (OH). The signals of DMF observed at 2.49 and 7.35 ppm. A signal appears at 4.35 ppm for (NH2 and OH) and the shift occurs is due to hydrogen bonding of water molecules. Moreover, the signals between 6.92 and 7.95 ppm are due to phenyl protons.
FT-IR spectrum. The FT-IR spectrum of NL and Mn–MOF is shown in Fig. S13† and the significant frequencies of the spectrum are investigated. The band between 3700 and 3040 cm−1 is attributed to NH2 and water molecules. The band at 2925 cm−1 is attributed to sp3 CH. The band appears at 1637 cm−1 is attributed to the CO group. Whereas, the bands appeared between 1550 and 1234 cm−1 are due to aromatic CC, and between 1197 and 709 cm−1 are due to aromatic CH. The band appeared at 598 cm−1 assigned to the coordination bonding between Mn and oxygen ν(Mn–O), the band at 420 cm−1 assigned to the covalent bonding between Mn and nitrogen (Mn ← N), these bands proved that the chelation between the NL and the metal through the CO and NH.
UV-vis spectrum. The electronic absorption spectra of the solid NL and Mn–MOF were measured in a range between 200 to 2200 nm, at room temperature in addition to bandgap spectrum represented in Fig. S14.† From (Fig. S14a, b, and c†) it found that the Mn–MOF exhibits absorption bands at 225, 303, 387 and 416 nm, which were was assigned to the ligand–metal charge transfers (LMCT) transition and to intra-ligand charge transfers (n–π* and π–π*). The band gap of linker and Mn–MOF were performed with the diffuse reflectance spectra (Fig. S14d†). As shown in (Fig. S14d†) it can notice that the band gap for the linker is about 2.0, 2.96, and 3.64 eV; whereas the band gap for Mn–MOF at 2.79 and 3.1 eV and it is located in the range of semiconducting materials as shown in (Fig. S14d†). Moreover, from the comparison between the band gap values, the Mn–MOF was reduced due to a highly conjugated of linker and presence of NH2 groups, which leads to an increase in the HOMO valance band energy and subsequently the Mn–MOF band gap reduced.26,27
XRD analysis. The XRD patterns of Mn–MOF were recorded in the 2θ range between 3.0°–70.0° as shown in Fig. S15.† The powder XRD patterns of Mn–MOF show that all the diffraction peaks match with JCPDS card no. 73–1826 for Mn2O3. And it also shows sharp peaks indicating that the crystalline phase Mn–MOF was obtained. Tables S16 and S17† summarized the XRD data, Millar indices, lattice parameter values, crystallite size and interplanar distances of Mn–MOF according to the Scherrer equation calculation.
XPS analysis. (Fig. 3b) shows the XPS analysis of the as-prepared Mn–MOF sample. From this figure, it can prove the existence of C, Mn, N, and O in the sample without any impurities. As depicted from (Fig. 3c), C 1s spectrum shows the presence of three XPS signals at 284.49, 284.82, and 288.16 eV which were attributed to C–C, C–N, and CO, respectively.28 N 1s shows a single peak at 400.86 eV which corresponded to quaternary N in sample29 (Fig. 3d). O 1s deconvoluted into three peaks at 530.98, 531.86, and 533.45 eV, which were attributed to O–Mn–O, C–O, and CO, respectively30 (Fig. 3e). Moreover, it was observed that four signals related to Mn 2p as shown in (Fig. 3f). The peaks detected at 641.94 and 653.45 eV, with a spin–orbital splitting of about 11.51 eV, were ascribed to Mn 2p3/2 and Mn 2p1/2, respectively, which proved the presence of Mn4+ in sample.31 In addition, the peak observed at 646.06 eV is characterized by the satellite signal of Mn2+ oxide.32 However, the signal at 640 eV was attributed to the presence of Mn3+ state in sample.33
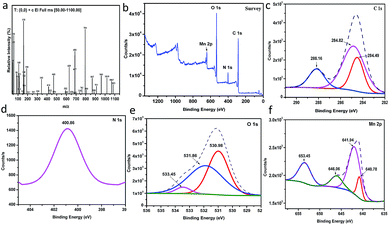 |
| | Fig. 3 (a) Mass spectrum of the manganese metal–organic framework (Mn–MOF), (b) to (f) The XPS analysis of the Mn–MOF [(b) survey, (c) C 1s, (d) N 1s, (e) O 1s, and (f) Mn 2p]. | |
Thermal analysis. The thermogravimetric analysis (TGA) and thermal stability study of Mn–MOF were performed up to 1000 °C under atmospheric nitrogen gas with a heating rate of ten °C min−1. The thermal analysis (TGA-DTGA) plots (Fig. S18†) suggest that the Mn–MOF pass through four stages of decompositions, underwent a weight loss due to the expulsion of ethanol, water, and DMF molecules in the temperature range between 58 to 211 °C with a weight loss of 11.33% (calculated: 11.4%). The Mn–MOF collapse due to followed two decomposition stages which related to the removal of phenyl rings in the temperature range of 252–798 °C (DTGmax = 286.52, 524.02 and 798.22 °C, mass loss found 81.52, calculated: 82.1%). The remaining residue is Mn2O3 (found 22.11, calculated: 22.46%). The obtained data were in a good agreement with XRD data.
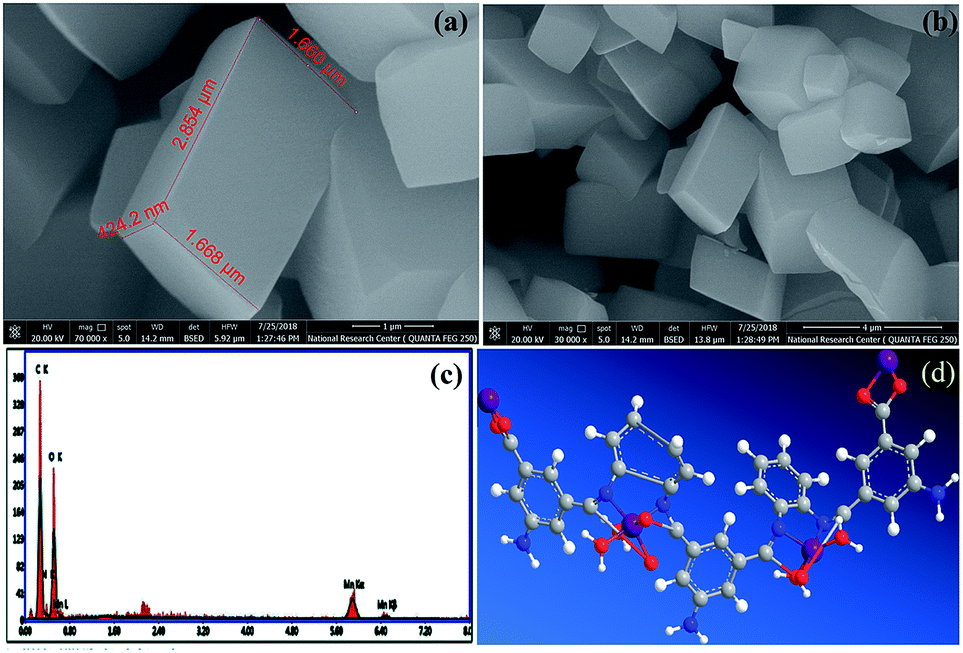
FE-SEM combined with EDX spectroscopy. FE-SEM images of the Mn–MOF are shown in (Fig. 4a, and b). The Mn–MOF appeared to be sharp rectangular bricks with an average size of 0.424 × 1.66 × 2.85 μm. Furthermore, the single point EDX mapping analysis (Fig. 4c, Table S19†) of Mn–MOF revealed the presence of C, N, O, and Mn as the building-block elements in the individual particles. The good distribution of the elements C, N, O, and Mn along the cross-section revealed by EDX analysis (Fig. 4c) also affirmed the formation of the Mn–MOF. The EDX data is in a good agreement with the elemental analysis data: C, 44.70; N, 8.87; O, 31.67; Mn, 8.70. Found: C, 44.97; N, 8.44; O, 37.85; and Mn, 8.74 (see Table S19†).
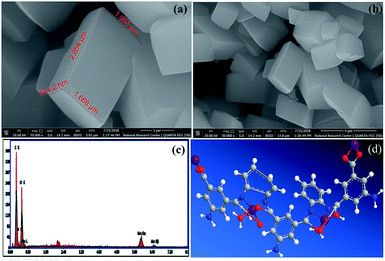 |
| | Fig. 4 (a and b) Field-emission scanning electron microscopy images of the manganese metal–organic framework (Mn–MOF) at different magnification, (c) energy-dispersive X-ray analysis with a single point EDX mapping analysis of Mn–MOF, and (d) three-dimensional structural representation of the monomeric unit of Mn–MOF. | |
Magnetic properties of Mn–MOF. Fig. S20† shows the magnetization curve obtained by VSM at room temperature (300 K) for the Mn MOF. From this curve, it can be seen that the saturation magnetization value (Ms) of the Mn–MOF was found to be 42.067 emu g−1 at 2 kOe with negligible coercivity and remanence. The magnetization curve features show a superparamagnetic (SPM) behavior of the Mn–MOF which can be assigned to an assembly of non-interacting enormous magnetic moments and their small crystallite size, which makes it easier for the Mn–MOF to be thermally activated to overcome the magnetic anisotropy.34On the basis of the physical and spectral data of the Mn–MOF discussed, we can deduce that the structure is as suggested in (Fig. 4d).
Photoluminescence properties of Mn–MOF. The photoluminescence of the Mn–MOF was recorded at room temperature with different excitation wavelengths as shown in (Fig. S21†). Mn–MOF were irradiated with UV radiation to investigate their capability to photoluminescence. Excitation spectra were first obtained to determine the optimum wavelength for photoluminescence excitation via our monitoring emission at the wavelength with the highest intensity. Form Fig. S21,† it was found that the Mn–MOF exhibit an emission peak at 422 nm when excited at 300 nm. It is most likely that the fluorescent behavior of the Mn–MOF is attributed to π–π* or π–n molecular orbital transitions within the aromatic rings of the nano linker, with redshifts caused by complexation with Mn (see Fig. S21†). The excitation and emission spectra of the Mn–MOF is represented in (Fig. S22†).
Application using of Mn–MOF as potentiometric membrane biosensors for troponin cardiac biomarker
In the present application, several potentiometric membranes were prepared based on Mn–MOF with different plasticizers as (o-NPOE, DOP, or DOS) according to the procedure of Thomas et al.35 The membrane composition consists of 2% Mn–MOF, 33% PVC powder and 65% o-NPOE, DOP, or DOS with 5–6 mL THF. The mixture mixed well and then poured into a Petri dish. The galvanic cell, as well as the electrodes, were prepared according to Thomas et al.35 and used for potentiometric measurements in the system according to the schematic diagram showed in (Fig. S23†). The preliminary experiments result in addition to the effect of experimental parameters and conditioning of the electrode were discussed as follows:
Effect of plasticizers. Electrodes with different plasticizers o-NPOE, DOP, and DOS were prepared using Mn–MOF as active membrane component. The response characteristics of these electrodes are given in Table S24.† The electrode with o-NPOE as plasticizer obviously has better response characteristics for cTn compared to the others. It seems that o-NPOE is favourable with a greater solubility of Mn–MOF in comparison with the other solvents.
Biosensor characteristics calibration curve and LOD. The performance characteristics of the electrode are summarized in Table 1 at pH in the range between 5.6 and 10.1. The sensor displays a linear and stable response in the range between 0.01 and 30.0 ng mL−1 for cTn with a slope of 59.05 ± 0.99 mV/decade, the limit of detection of 0.055 ng mL−1, the limit of quantitation of 0.168 ng mL−1 and correlation coefficient r2 equal 0.995. The least squares equations obtained from the calibration data are follows: E (mV) = (59.05 ± 0.99) log[cTn] + (170.65 ± 1.85). A calibration graph constructed by plotting the recorded potential as a function of the logarithm troponin concentration (log[cTn]) shown in (Fig. 5a). The resulting graphs were used for subsequent measurements of unknown cTn concentration. The lower values of LOD and LOQ, in addition to the wide linear range, are considered as evidence of the high sensitivity of the proposed approach. A comparison between the analytical performances of the present approach and other previous reports in the literatures for the determination of cTn is presented in (Table S25†). From the comparison and table data, it can be noted that the present approach introduces a wide linear range, lower LOD, and LOQ compared to the other methods.
Table 1 Response characteristics of the Mn–MOF-PVC membrane sensorsa
| Parameter |
Value |
| Average of five replicates. |
| Slope |
59.05 ± 0.99 |
| Intercept |
170.651 |
| Correlation coefficient (r2) |
0.995 |
| Standard deviation |
0.9923 |
| Linear range, ng mL−1 |
0.01–30.0 |
| Limit of detection (LOD), ng mL−1 |
0.055 |
| Limit of quantification (LOQ), ng mL−1 |
0.168 |
| Response time, s |
20 ± 5 |
| Working pH range |
5.6–10.1 |
| Lifetime of the electrode, week |
3.0–12.0 |
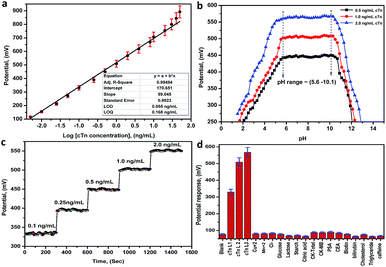 |
| | Fig. 5 (a) A Linear relationship (calibration graph) between the potential and the logarithm troponin concentration (log[cTn]), (b) the effect of pH on the potential response of the biosensor (Mn–MOF/PVC electrode), (c) the response time of the biosensor (Mn–MOF/PVC electrode), (d) the potential response histogram of the Mn–MOF/PVC electrode toward the (cTn) against different types of cancer biomarkers and interfering analytes. cTn cardiac troponin I, PSA prostate-specific antigen, CEA carcinoembryonic antigen, CK-total total creatine kinase, CK-MB creatine kinase-muscle/brain. | |
pH effect. The effect of pH on the potential response (performance response) of the biosensor was investigated by recording the potentials at three concentrations levels 0.5, 1.0 and 2.0 ng mL−1 cTn at a pH ranged from 1.5–13.0. From the potential-pH plot (Fig. 5b) it was observed a stable response not more than (±0.99 mV) in the electrode potential at pH ranged 5.6–10.1. Whereas, at pH < 5.6 and pH > 10.1 the potential revealed by the sensors was rapidly decreased. This can may be attributed to the increase in the acid nature as well as the basic nature leads to membrane decomposing.
Response time of the electrode. The response time of the sensors was defined as the time required for the sensor to reach the steady state potentiometric value. The response time recorded with the pH meter connected to the PC interface to give a change after each 1s for the optimal membranes conditions for the sensor. From obtained results, the best response time is 20 ± 5 s, and after new solutions are exposed to the electrodes the responses change rapidly and remain at a constant value (Fig. 5c).
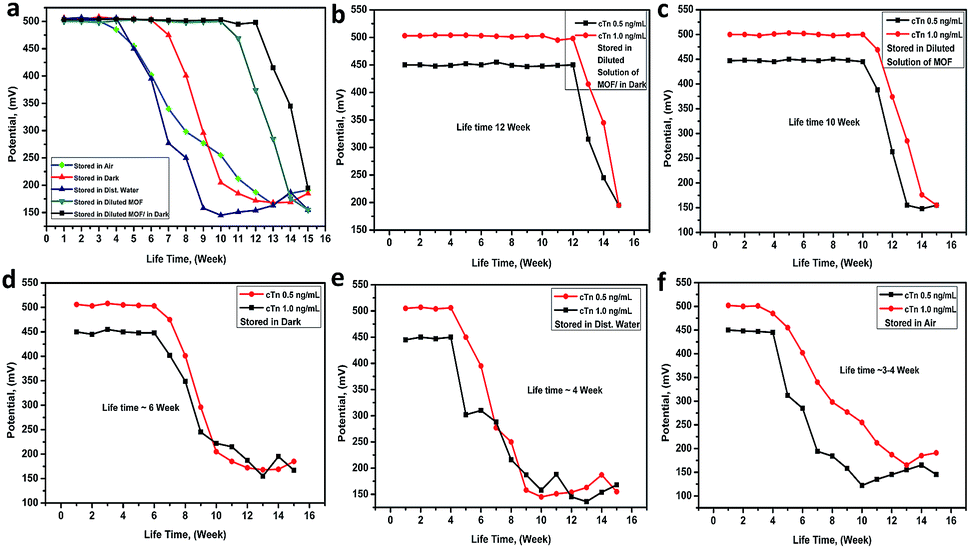
The lifetime of the electrode. The electrode lifetime was investigated under different storage conditions. From (Fig. 6a), it was found that the electrode lifetime is between 3.0 and 12.0 week without a significant change in the membrane compositions and performance characteristics when unused. The lifetime of the electrode was basically depended on the storage system. When the electrode stored in the dark and diluted solution of Mn–MOF the lifetime was around 12 weeks (Fig. 6b), while it stored in diluted solution of Mn–MOF the lifetime was slightly decreased to 10 week (Fig. 6c), whereas, while stored in dark the lifetime was around 6 weeks (Fig. 6d), whilst stored in distilled water the lifetime rapidly decreased to 4 weeks (Fig. 6e), and when stored in air the lifetime was around 3–4 week (Fig. 6f). Before used the electrode for measurements must be conditioned by soaking it in 1 × 10−5 M of Mn–MOF solution for 1 day.
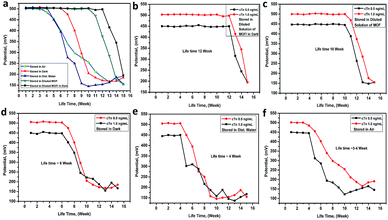 |
| | Fig. 6 The Mn–MOF/PVC electrode lifetime under deferent storage conditions. | |
The selectivity of the electrode. The potentiometric selectivity coefficient 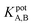 of an ISE is commonly used as a quantitative expression of the ability of the electrode to respond primarily to the analyte ion (cTn) at three different concentrations levels (cTn L1, 0.1 ng mL; cTn L2, 1.0 ng mL; and cTn L1, 2.0 ng mL−1) in the presence of interfering analytes as (Cu+2 1.0 M, Mn+2 1.0 M, Cl−1 1.0 M, glucose 100 mg dL−1, lactose 1.0 M, starch 1.0 M, citric acid 1.0 M, total creatine kinase (CK-total) 1.0 ng mL−1, creatine kinase-muscle/brain (CK-MB) 1.0 ng mL−1, prostate-specific antigen (PSA) 100 ng mL−1, carcinoembryonic antigen (CEA) 100 ng mL−1, biotin 200 ng mL−1, bilirubin 1.0 mg mL−1, cholesterol 10 mg mL−1, triglyceride 15 mg mL−1, and caffeine 10 μm mL−1). The influence of the presence of some different species on the potentiometric response of cTn sensor was investigated and presented in (Fig. 5d). The selectivity coefficients of the proposed biosensor were calculated in the presence of similar drugs, organic and inorganic substances using the separate solutions method according to IUPAC recommendations.36 The results are given in Table S26.† The results reveal reasonable selectivity to cTn over other interfering analytes.
of an ISE is commonly used as a quantitative expression of the ability of the electrode to respond primarily to the analyte ion (cTn) at three different concentrations levels (cTn L1, 0.1 ng mL; cTn L2, 1.0 ng mL; and cTn L1, 2.0 ng mL−1) in the presence of interfering analytes as (Cu+2 1.0 M, Mn+2 1.0 M, Cl−1 1.0 M, glucose 100 mg dL−1, lactose 1.0 M, starch 1.0 M, citric acid 1.0 M, total creatine kinase (CK-total) 1.0 ng mL−1, creatine kinase-muscle/brain (CK-MB) 1.0 ng mL−1, prostate-specific antigen (PSA) 100 ng mL−1, carcinoembryonic antigen (CEA) 100 ng mL−1, biotin 200 ng mL−1, bilirubin 1.0 mg mL−1, cholesterol 10 mg mL−1, triglyceride 15 mg mL−1, and caffeine 10 μm mL−1). The influence of the presence of some different species on the potentiometric response of cTn sensor was investigated and presented in (Fig. 5d). The selectivity coefficients of the proposed biosensor were calculated in the presence of similar drugs, organic and inorganic substances using the separate solutions method according to IUPAC recommendations.36 The results are given in Table S26.† The results reveal reasonable selectivity to cTn over other interfering analytes.
Accuracy, precision and recovery study. The accuracy, precision and recovery study of the present method for cTn quantification was investigated and the results summarized in (Table S27†). Our study was achieved by spiking of cTn standard at three concentrations levels (0.1, 2.0, and 20.0 ng mL−1) to serum samples. Each experiment was repeated five times within the same day and in different days, and then the proposed method was carried out to determine cTn in the serum samples. From the calculated results, it was found that, the average values of the standard deviation (SD) of cTn was (0.076 and 0.10), the average values of the coefficient of variation (CV) was (0.013 and 0.019), and the average values of the relative error percent (RE%) was (1.014 and 0.987) for intra and inter day investigation, respectively. The lower values of SD, CV and RE% demonstrating the high precision and accuracy of the present method. The recovery values are between (97.33 and 100.7%) and between (97.75 and 100.1%) intra and inter day investigation, respectively. The recovery values are around 100%, and this means that the present approach could be applied as troponin cardiac biomarker for detection of cardiac injury in the diagnosis of AMI in the EDs and in medical laboratories with sufficient accuracy and precision.
Analytical applications in real samples. The proposed method was tested by screening and quantification of the cTn concentration for fifteen patient's samples (whole blood, serum, and plasma samples) which are provided from emergency department National Heart Institute (NHI), Egypt. Five samples were used as its (whole blood) and the test is carried out immediately. Five samples were centrifuged immediately for 15 min at 4000 rpm min−1 to obtain on serum samples. Five samples were collected in tubes with different additives like EDTA, citrate, etc., and centrifuged for 15 min at 4000 rpm min−1 to obtain on plasma samples. The aim was to use different forms of blood to study the effect of blood matrix in for cardiac troponin I (cTn) determination at the same time. The results obtained from the present method was compared with that data obtained from two official methods (cardiac markers rapid tests37 as qualitative methods “test device containing membrane” and with enzyme-linked immunosorbent assay38 as quantitative method “ELISA kit”). The results obtained are summarized in (Table 2). The data demonstrated that one hundred percent agreement between observed results obtained and the results obtained using both the standard methods (qualitative and quantitative) and no significant differences between these methods. This is strong evidence for the effectiveness, applicability, accuracy, and precision of the proposed approach for detection of cTn. Moreover, this approach will be future primary support which helps EDs to address and facilitate the actions and decisions for the cases with chest pain and early diagnosis for the patients with ACS, and the onset of AMI in just a few seconds (25 s). In addition, the present approach can be converted to a small device in the nearest future and can be used in term of point-of-care testing (POCT) for measuring the most important cardiac blood biomarkers.
Table 2 Comparison between the results of the emergency departments samples which analyzed by the cardiac markers rapid tests, ELISA standard method and by Mn–MOF biosensor
| Sample |
Qualitative and Quantitative cTn |
Average potential readingc (mV) |
Corresponding average amount of cTn foundc, (ng mL−1) |
SD |
CV |
RE% |
| Rapid testa |
ELISAb (ng mL−1) |
| Cardiac markers rapid tests, the concentration of cTn to the healthy people from 0.0 to 0.2 ng mL−1 (cTn < 0.2 negative, cTn > 0.2 positive). The mean values were performed with official standard method (enzyme-linked immunosorbent assay for the quantitative determination of cTn). Each reading was repeated five times; SD, standard deviation; CV, coefficient of variation; RE%, relative error percent. |
| Whole blood sample |
Positive |
2.64 |
592.5 |
2.658 |
0.11 |
0.012 |
0.993 |
| Positive |
1.84 |
564.4 |
1.791 |
0.019 |
4 × 10−4 |
1.027 |
| Negative |
0.17 |
379.3 |
0.167 |
0.002 |
3 × 10−6 |
1.02 |
| Negative |
0.20 |
391.9 |
0.199 |
0.004 |
1 × 10−5 |
1.005 |
| Positive |
0.64 |
482.3 |
0.618 |
0.006 |
3 × 10−5 |
1.036 |
| Serum samples |
Negative |
0.19 |
387.9 |
0.187 |
0.006 |
3 × 10−5 |
0.983 |
| Positive |
3.00 |
602.4 |
2.904 |
0.053 |
0.003 |
0.968 |
| Positive |
2.77 |
596.2 |
2.703 |
0.039 |
0.001 |
0.976 |
| Negative |
0.16 |
374.5 |
0.159 |
0.002 |
4 × 10−6 |
0.991 |
| Positive |
0.97 |
514.6 |
0.943 |
0.012 |
1 × 10−4 |
0.972 |
| Plasma samples |
Positive |
1.13 |
526.5 |
1.111 |
0.034 |
0.001 |
0.983 |
| Negative |
0.19 |
387.9 |
0.186 |
0.003 |
8 × 10−6 |
0.978 |
| Positive |
2.15 |
576.5 |
2.112 |
0.044 |
0.002 |
0.983 |
| Negative |
0.2 |
391.9 |
0.198 |
0.003 |
6 × 10−6 |
0.991 |
| Positive |
0.89 |
507.9 |
0.865 |
0.011 |
1 × 10−4 |
0.972 |
Mechanism of the electrode response. The mechanism of the potentiometric response of the Mn–MOF-PVC membrane sensor is based on the ions exchange equilibrium and analyte extraction process at the membrane interface. Commonly the membrane contained on hydrophobically trapped, and mobile sites in the plasticized PVC.39 This membrane with charge sites is named sited membrane. Ions with the opposite sign in the membrane are counter-ions, whereas the ions with the same sign as the site are not present in significant quantities are known as coions. The sited membrane is selective to counter ion. That only counter-ions exchange into the membrane and therefore have some mobility in the membrane bulk. The photoluminescence spectrum of Mn–MOF (0.001 M) in distilled water and in the presence of buffered cTn solution was studied (Fig. S28†). The change of the PL spectra of the Mn–MOF when contacting a cTn containing solution might shed some light on the response mechanism of the electrode. Fig. S28† shows the PL spectra of Mn–MOF with and without interaction with cTn. By comparing the spectra of Mn–MOF and Mn–MOF–cTn, it can be noted that the photoluminescence intensity of Mn–MOF was high quenched after addition of cTn. This can serve as a more or less experimental indication that coordination really occurred between cTn and Mn–MOF. The mechanism of fluorescence quenching can be explained as follows: the observed chemical sensing behavior of Mn–MOF toward cTn may be attributed to the high affinity of cTn for the amine group lone electron pair of the Mn–MOF.40 The interaction mechanism may be also related to metal–ligand charge transfer due to Mn–MOF having low-lying π* orbitals localized in amine groups in the aromatic ring. In addition, according to chelation theory, when the lipophilicity of the Mn–MOF increased, the polarity of the metal ion decreased.16 This may be due to the possible π-electron delocalization within the chelate ring and a partial share of the positive charge with the donor groups, which subsequently increased the probability of chelation to the biological target.16 Moreover, this interaction may also occur between the aromatic chromophore of the Mn–MOF and the active site of cTn. Moreover, the interaction of Mn–MOF with cTn is normally due to covalent bonding.19
Future perspectives and socio-economic impact. The point-of-care testing (POCT) systems and near patient testing devices market in Europe has an annual turnover of more than 3.5 billion Euros, with the home care sector accounting for a major market share. Resource-limited countries will increase the use of cost-effective POCT. Therefore, the efforts to develop inexpensive POCT method and device solutions should be strengthened. In our country, Egypt will try to solve some of the economic problems related to buying or importing many tools and devices like the tools for cardiac biomarker. This will save a lot of foreign currency. We will try to apply new nanotechnology and new idea to overcome this big economic issue. In ESI† (Appendix C) a plan to a system with it we can convert our approach to the future device.
Experimental
Materials
See the ESI† file (Appendix B) for details on materials.
Instruments
See the ESI† file (Appendix B) for details on equipment.
Procedure
Synthesis of the nano linker (NL). A Schiff base of a twenty milliliters of Aip (3.0 mmol, 1.0872 g) in ethanol/dimethylformamide DMF (16:4 v/v) was added dropwise to a flask containing a dissolved Phen (2.0 mmol, 0.432 g) in 15 mL ethanol in presence of 1 mL glacial acetic acid. Then the system was refluxed at 80 °C for 48 h (Scheme S1†). The color of the solution was changed from a light yellow to a green suspension and then a yellowish orange precipitate is formed. This precipitate was filtered off, washed, and then dried under a vacuum.
Synthesis of the Mn–MOF. The organic linker prepared according to (Scheme S1†) was added dropwise to 15 mL distilled water containing a dissolved MnCl2·4H2O (2.0 mmol, 0.7916 g), and then the system was refluxed at 80 °C for 48 h (Scheme S10†). The color of the solution was changed from yellowish orange to a pink precipitate. The precipitate was filtered off, washed, and then dried under a vacuum.
Membrane composition. The Mn–MOF/PVC membrane electrode was prepared according to the procedure of Thomas et al.35 The optimized membrane composition consists of 2% active materials (Mn–MOF), 65% plasticized solvent mediator (o-NPOE, DOP, or DOS) and 33% powdered PVC and 5–6 mL of the tetrahydrofuran (THF), this mixture was mixed well. After mixing well the mixture was poured into a three-centimeter diameter Petri dish. At room temperature, the Petri dish was covered with a filter paper to allow the solvent to be evaporated slowly.
Electrode preparation and system set up. The potentiometric electrode was prepared as follows: Mn–MOF-PVC membrane was sectioned with a cork borer ten milliliters diameter and attached to PVC tubing three centimeters length, eight milliliters diameter with THF. The PVC tubing was filled with internal reference solution (1 × 10−4 M of potassium chloride). One milliliter Ag/AgC1 internal reference wire was immersed in the internal solution which was subsequently connected to a shielded cable. The prepared electrode was used for the potentiometric measurements in the system as described in the instrumental section. The following a schematic diagram of an electrochemical cell for potentiometric measurements and illustrate in the (Scheme S23†). The electrode is conditioned in 1 × 10−5 M of Mn–MOF for one day before used for successive uses and optimum stored in 1 × 10−5 M of Mn–MOF in dark place when un-use, and to clean the sensors after measuring the samples, occurs by soaking them in a beaker containing distilled water with stirring for 5 min.
Determination of cTn in real samples. The patient samples should be treated according to standard precautions, potentially infectious, and handled appropriately. Venous whole blood stored in lithium or sodium heparin tubes without separating gel is acceptable and the samples are stable for eight hours at room temperature. Collected blood by jugular venipuncture in tubes with different additives like EDTA, citrate, and sodium fluoride is acceptable. These samples centrifuged immediately for 15 min at 4000 rpm min−1. The obtained serum or plasma samples were stored at −20 °C until assayed.
Conclusions
In this article, a novel nano linker (NL) and manganese metal–organic frameworks (Mn–MOF) were successfully synthesized and fully characterized. The physicochemical characterization proved the proposed structures. The potentiometric application was extended to fabrication of Mn–MOF as a fast, direct, simple, economic, sensitive, and selective a novel cardiac troponin I (cTn) potentiometric membrane biosensor over a wide stable and linear-Nernstian response to cTn concentration range of 0.01–30 ng mL−1 with a pH range from 5.6 to 10.1 and a fast response time of 20 ± 5 s, the lifetime of electrode was 3 to 12 week based on the storage conditions, with a limit of detection (LOD) of 0.055 ng mL−1 and a limit of quantitation (LOQ) of 0.168 ng mL−1 without significant interference. These limits are lower than those previously reported with other different approaches. Subsequently, the Mn–MOF electrode was used successfully for cTn detection and quantification at clinically relevant concentrations. The statistical evaluations of the proposed approach in comparison with the standard official method indicate that this method is accurate and precise. The proposed approach can be used by healthcare professionals, ED staff, and in medical laboratories for the detection and quantification of troponin cardiac blood biomarker in different types of human blood samples with satisfactory results. This approach will aid in the risk stratify cation of patients with symptoms suggestive of ACS with respect to the relative risk of all-cause mortality and major adverse cardiac events.
Ethical statement
The human serum samples used in this study were provided by the Family Medical Laboratory, Ministry of Health, Cairo, Egypt. Informed consent was obtained. The studies were approved by the appropriate ethics committee (Ministry of Health, Egypt) and were performed in accordance with ethical standards. We did not use the samples in any research involving human participants or research involving physical interventions on study participants or involving the processing of personal data but conducted the research according to the method described in the article.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- I. Biyik, N. Isiksacan, S. Opan, F. Nihan, T. Caglar, M. Erturk, S. Yazan, M. R. Yildirim, O. F. Baycan, B. Ozalp, H. Karakurt and I. F. Akturk, Am. J. Cardiol., 2018, 121, e35 CrossRef.
- A. Kumar and C. P. Cannon, Mayo Clin. Proc., 2009, 84, 917–938 CrossRef PubMed.
- B. M. Scirica, J. Am. Coll. Cardiol., 2010, 55, 1403–1415 CrossRef CAS PubMed.
- R. Lozano, M. Naghavi, K. Foreman and S. Lim, Lancet, 2010, 380, 2095–2128 CrossRef.
- N. Bandstein, R. Ljung, M. Johansson and M. J. Holzmann, J. Am. Coll. Cardiol., 2014, 63, 2569–2578 CrossRef CAS PubMed.
- C. Mueller, C. Patrono, M. Valgimigli, J. Collet and M. Roffi, Eur. Heart J., 2015, 37, e15–e21 Search PubMed.
- K. Thygesen, J. Mair, H. Katus, M. Plebani, P. Venge, P. Collinson, B. Lindahl, E. Giannitsis, Y. Hasin, M. Galvani, M. Tubaro, J. S. Alpert, L. M. Biasucci, W. Koenig, C. Mueller, K. Huber, C. Hamm and A. S. Jaffe, Eur. Heart J., 2010, 31, 2197–2206 CrossRef CAS PubMed.
- D. A. Morrow, C. P. Cannon, R. L. Jesse, L. K. Newby, J. Ravkilde, A. B. Storrow, A. H. B. Wu and R. H. Christenson, Circulation, 2007, 115, 356–375 CrossRef PubMed.
- E. M. Antman, M. J. Tanasijevic, B. Thompson, M. Schactman, C. H. Mccabe, C. P. Cannon, G. A. Fischer, A. Y. Fung, C. Thompson, D. Wybenga and E. Braunwald, N. Engl. J. Med., 1996, 335, 1342–1349 CrossRef CAS PubMed.
- F. Van de Werf, J. Bax, A. Betriu, C. Blomstrom-Lundqvist, F. Crea, V. Falk, G. Filippatos, K. Fox, K. Huber, A. Kastrati, A. Rosengren, P. G. Steg, M. Tubaro, F. Verheugt, F. Weidinger and M. Weis, Eur. Heart J., 2008, 29, 2909–2945 CrossRef CAS PubMed.
- M. Tang, Z. Zhou, L. Shangguan, F. Zhao and S. Liu, Talanta, 2018, 180, 47–53 CrossRef CAS PubMed.
- X. Han, H. S. Kojori, R. M. Leblanc and S. J. Kim, Anal. Chem., 2018, 90, 7795–7799 CrossRef CAS PubMed.
- J. Chen, L. Kong, X. Sun, J. Feng, Z. Chen, D. Fan and Q. Wei, Biosens. Bioelectron., 2018, 117, 340–346 CrossRef CAS PubMed.
- W. Zhou, K. Li, Y. Wei, P. Hao, M. Chi and Y. Liu, Biosens. Bioelectron., 2018, 106, 99–104 CrossRef CAS PubMed.
- M. A. Ahamed, A. B. Farag, M. H. Ibrahim, A. M. Kamel, M. M. Abd-Elzaher and S. M. Sheta, Sens. Lett., 2017, 15, 632–638 CrossRef.
- S. M. Sheta, S. M. El-Sheikh and M. M. Abd-Elzaher, Dalton Trans., 2018, 47, 4847–4855 RSC.
- A. H. Valekar, B. S. Batule, M. I. Kim, K. H. Cho, D. Y. Hong, U. H. Lee, J. S. Chang, H. G. Park and Y. K. Hwang, Biosens. Bioelectron., 2018, 100, 161–168 CrossRef CAS PubMed.
- S. M. Sheta, S. M. El-Sheikh, M. M. Abd-Elzaher and A. R. Wassel, Appl. Organomet. Chem., 2019, 33, e4777 CrossRef.
- S. M. Sheta, S. M. El-sheikh and M. M. Abd-elzaher, Anal. Bioanal. Chem., 2019, 411, 1339–1349 CrossRef CAS PubMed.
- G. Gauglitz, Anal. Bioanal. Chem., 2017, 409, 1–2 CrossRef CAS PubMed.
- F. Jeremias, V. Lozan, S. K. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC.
- X. Zhang, Z. Zhan, X. Liang, C. Chen, X. Liu, Y. Jia and M. Hu, Dalton Trans., 2018, 47, 3272–3282 RSC.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Dalton Trans., 2012, 41, 14003–14027 RSC.
- X. Lian, T. Miao, X. Xu, C. Zhang, B. Yan, B. Lian, X. Miao, T. Xu, X. Zhang and C. Yan, Biosens. Bioelectron., 2017, 97, 299–304 CrossRef CAS PubMed.
- S. Zhu, X. Lin, P. Ran, Q. Xia, C. Yang, J. Ma and Y. Fu, Biosens. Bioelectron., 2017, 91, 436–440 CrossRef CAS PubMed.
- J. Li, T. Musho, T. Li, J. Wu, X. Xiao, B. Zhang, Z. Hu and J. Zhou, RSC Adv., 2019, 6, 13914–13919 RSC.
- J. Li and T. Musho, J. Electrochem. Soc., 2019, 166, 3029–3034 CrossRef.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. Zhang, J. Am. Chem. Soc., 2016, 138, 6924–6927 CrossRef CAS PubMed.
- T. Feng, J. M. Wang, S. T. Gao, C. Feng, N. Z. Shang, C. Wang and X. L. Li, Appl. Surf. Sci., 2019, 469, 431–436 CrossRef CAS.
- Y. Liu, J. Li, W. Li, Y. Li, Q. Chen and Y. Liu, Int. J. Hydrogen Energy, 2015, 40, 9225–9234 CrossRef CAS.
- F. Zheng, D. Zhu, X. Shi and Q. Chen, J. Mater. Chem. A, 2015, 3, 2815–2824 RSC.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS PubMed.
- M. Kundu, C. C. A. Ng, D. Y. Petrovykh and L. Liu, Chem. Commun., 2013, 49, 8459–8461 RSC.
- M. E. Guillermo and C. Eugenio, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- A. Craggs, G. J. Moody and J. D. R. Thomas, J. Chem. Educ., 1974, 51, 541–544 CrossRef CAS.
- International Union of Pure and Applied Chemistry, Pure Appl. Chem., 2000, 72(10), 1851–2082 Search PubMed.
- Cardiac markers rapid tests, http://www.rapidtest.com/index.php?i=Cardiac-Rapid-tests%26id=225%26cat=23, (Accessed March, 2019).
- Enzyme-linked immunosorbent assay, https://www.thermofisher.com/elisa/product/Cardiac-Troponin-I-TNNI3-Human-ELISA-Kit/EHTNNI3, (Accessed March, 2019).
- International Union of Pure and Applied Chemistry, Pure Appl. Chem., 1994, 66, 2527–2536 Search PubMed.
- Y. Wu, W. Wu, L. Zou, J. Feng, C. Gu, B. Li, S. R. Batten, R. Yadav and A. Kumar, Inorg. Chem. Commun., 2016, 70, 160–163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03030a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Said M. El-Sheikh
*a,
Said M. El-Sheikh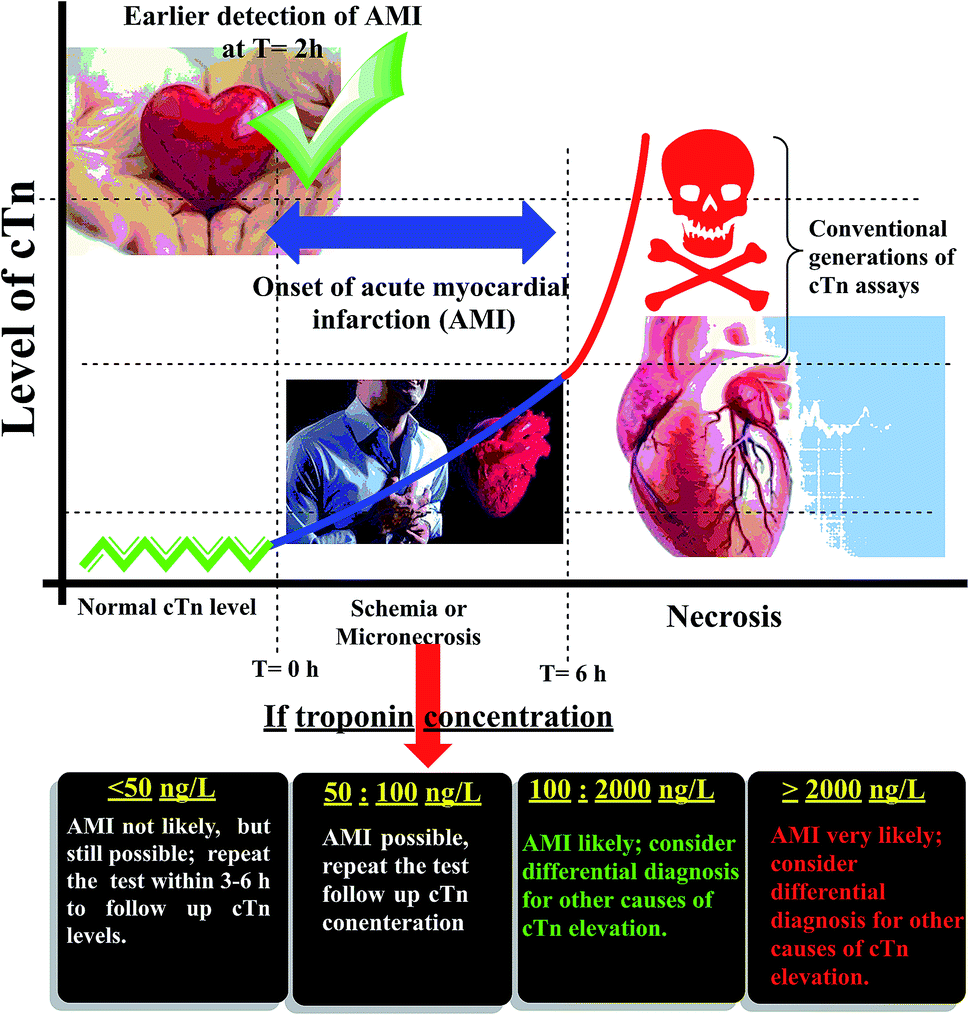

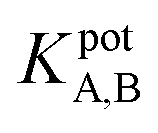 of an ISE is commonly used as a quantitative expression of the ability of the electrode to respond primarily to the analyte ion (cTn) at three different concentrations levels (cTn L1, 0.1 ng mL; cTn L2, 1.0 ng mL; and cTn L1, 2.0 ng mL−1) in the presence of interfering analytes as (Cu+2 1.0 M, Mn+2 1.0 M, Cl−1 1.0 M, glucose 100 mg dL−1, lactose 1.0 M, starch 1.0 M, citric acid 1.0 M, total creatine kinase (CK-total) 1.0 ng mL−1, creatine kinase-muscle/brain (CK-MB) 1.0 ng mL−1, prostate-specific antigen (PSA) 100 ng mL−1, carcinoembryonic antigen (CEA) 100 ng mL−1, biotin 200 ng mL−1, bilirubin 1.0 mg mL−1, cholesterol 10 mg mL−1, triglyceride 15 mg mL−1, and caffeine 10 μm mL−1). The influence of the presence of some different species on the potentiometric response of cTn sensor was investigated and presented in (Fig. 5d). The selectivity coefficients of the proposed biosensor were calculated in the presence of similar drugs, organic and inorganic substances using the separate solutions method according to IUPAC recommendations.36 The results are given in Table S26.† The results reveal reasonable selectivity to cTn over other interfering analytes.
of an ISE is commonly used as a quantitative expression of the ability of the electrode to respond primarily to the analyte ion (cTn) at three different concentrations levels (cTn L1, 0.1 ng mL; cTn L2, 1.0 ng mL; and cTn L1, 2.0 ng mL−1) in the presence of interfering analytes as (Cu+2 1.0 M, Mn+2 1.0 M, Cl−1 1.0 M, glucose 100 mg dL−1, lactose 1.0 M, starch 1.0 M, citric acid 1.0 M, total creatine kinase (CK-total) 1.0 ng mL−1, creatine kinase-muscle/brain (CK-MB) 1.0 ng mL−1, prostate-specific antigen (PSA) 100 ng mL−1, carcinoembryonic antigen (CEA) 100 ng mL−1, biotin 200 ng mL−1, bilirubin 1.0 mg mL−1, cholesterol 10 mg mL−1, triglyceride 15 mg mL−1, and caffeine 10 μm mL−1). The influence of the presence of some different species on the potentiometric response of cTn sensor was investigated and presented in (Fig. 5d). The selectivity coefficients of the proposed biosensor were calculated in the presence of similar drugs, organic and inorganic substances using the separate solutions method according to IUPAC recommendations.36 The results are given in Table S26.† The results reveal reasonable selectivity to cTn over other interfering analytes.