DOI:
10.1039/C9RA03082D
(Paper)
RSC Adv., 2019,
9, 17560-17570
Syntheses, structures, and magnetic properties of mixed-ligand complexes based on 3,6-bis(benzimidazol-1-yl)pyridazine†
Received
25th April 2019
, Accepted 27th May 2019
First published on 4th June 2019
Abstract
Six new metal–organic coordination polymers (CPs) [Ni(L)(2,5-TDC)(H2O)]n (1), [Ni(L)(1,3-BDC)(H2O)]n (2), [Ni(L)(1,4-BDC)(H2O)]n (3), [Mn(L)(2,5-TDC)(H2O)]n (4), [Mn(L)(2,6-PYDC)(H2O)]n (5) and [Mn(L)(1,4-NDC)]n (6) were achieved by reactions of the corresponding metal salt with mixed organic ligands (L = 3,6-bis(benzimidazol-1-yl)pyridazine, 2,5-H2TDC = thiophene-2,5-dicarboxylic acid, 1,3-H2BDC = isophthalic acid, 1,4-H2BDC = terephthalic acid, 2,6-H2PYDC = pyridine-2,6-dicarboxylic acid, 1,4-H2NDC = naphthalene-1,4-dicarboxylic acid) under solvothermal condition. CPs 1–6 were characterized by single-crystal X-ray diffraction, IR, TG, XRD and elemental analyses. Their structures range from the intricate 3D CPs 1, 3, 4 and 6 to the 2D coordination polymer 2 and the infinite 1D chain 5. The CPs 1–4 and 6 underlying networks were classified from the topological viewpoint, disclosing the distinct sql (in 1), pcu (in 3 and 6), new topology (in 2), and dia (in 4) topological nets. Moreover, analysis of thermal stability shows that they had good thermal stability. Finally, magnetic properties of CPs 1–6 have been studied, the results showed that complex 2 had ferromagnetic coupling and complexes 1, 3–6 were antiferromagnetic.
Introduction
Metal–organic coordination polymers (CPs), as a new class of inorganic–organic hybrid materials, have received extensive attention due to their structural diversity, interesting properties as well as potential application in some fields such as gas storage/separation, magnetism, catalysis and chemical sensing.1–4 Particularly, the most important aspect is molecular magnets, in which the interesting magnetic properties such as SMMs (single molecule magnets)5 and SCMs (single chain magnets)6 are closely related to the molecular topological structures. So this prompts researchers to study magneto–structural correlation for the purpose of deepening the understanding and improving the magnetic properties. However, the magnetic properties of the CPs can be fine-tuned by suitable modification of their chemical composition (varying the metal or metal cluster nodes and of the organic linkers). This in turn affects the coordination environments around the metal centers, and of the magnetic exchange pathways.7–10 The inclusion of N-donor ligands along with the ancillary carboxylate ligands is an interesting modification of the synthetic strategy. The coordination complementarity or competition between carboxylate and N-donating ligands can significantly diversify the structure and magnetic features of the resulting CPs.11,12 Interestingly, experimental results have shown that the same building precursors may lead to completely different structural arrangements.13 It is proposed that the different structures might be caused by the subtle competitive relationship among various different coordination bonds.14 In this work, we have used benzimidazole ligands and different organic carboxylic acids to obtain six new complexes under the solvothermal condition with the Ni(II)/Mn(II) metal salt. Namely, [Ni(L)(2,5-TDC)(H2O)]n (1), [Ni(L)(1,3-BDC)(H2O)]n (2), [Ni(L)(1,4-BDC)(H2O)]n (3), [Mn(L)(2,5-TDC)(H2O)]n (4), [Mn(L)(2,6-PYDC)(H2O)]n (5) and [Mn(L)(1,4-NDC)]n (6). The analysis of structure displays that their molecular structures are also diverse, including different kinds of 1-D chains, 2-D networks, and 3-D frameworks. In addition, the magnetic properties of complexes 1–6 have been investigated and discussed.
Experimental
Materials and physical measurements
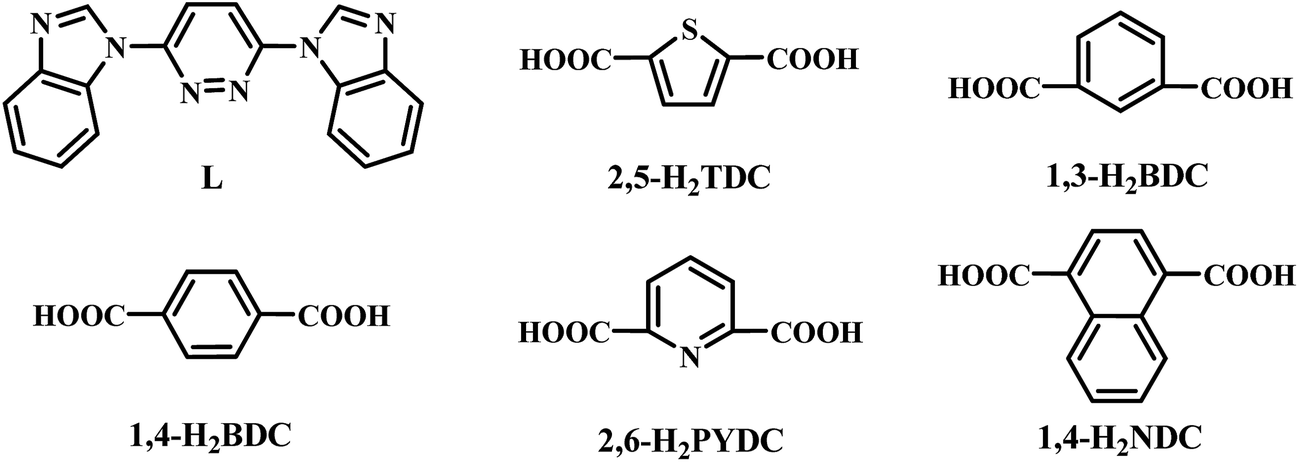
Starting materials were analytical grade and used as commercially obtained without further purification (Scheme 1). The ligand of L (Scheme 1) was synthesized by an analogous method described in the literature.15,16 Elemental analyses (C, H, and N) were performed by PerkinElmer 240 analyzer. FT-IR spectra were recorded by a Thermo Scientific Nicolet 6000 FT-IR spectrophotometer with KBr pellets in the 400–4000 cm−1 region. Powder X-ray diffraction (PXRD) data were recorded on a Rigaku D/Max-2500 diffractometer. Network topologies for all crystal structures were analyzed by using TOPOS4.0 software package.17 Thermogravimetric analyses were carried out by the Japan's neo-confucianism standard TG-DTA analyzer under a flow of Ar at a heating rate of 10 °C min−1. Magnetic measurements of the samples were performed on a SQUID MPMS-XL-7 magnetometer.
 |
| | Scheme 1 The L ligand and organic acids used in this work. | |
Synthesis of [Ni(L)(2,5-TDC)(H2O)]n (1). A mixture of Ni(AcO)2·4H2O (24.8 mg, 0.1 mmol), 2,5-H2TDC (17.2 mg, 0.1 mmol), and L (31.2 mg, 0.1 mmol) in 8 mL of solution (VH2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VCH3CN = 2:1) was sealed in a teflon-lined autoclave (25 mL) and heated at 160 °C for 12 h, and then cooled down to room temperature. Green stripe crystals of 1 were collected and washed with water. Yield: ∼42% based on L ligand. Elemental analysis (%): anal. calcd for C24H16N6O5SNi: C, 51.54; H, 2.88; N, 15.03. Found: C, 51.45; H, 2.73; N, 15.12. IR (cm−1, KBr pellets): 3354 w, 3132 w, 3104 w, 3080 w, 2903 w, 1607 w, 1565 m, 1527 s, 1478 m, 1458 s, 1438 s, 1399 s, 1372 s, 1304 m, 1150 m, 1032 m, 773 s, 738 s, 686 m, 626 m.
:VCH3CN = 2:1) was sealed in a teflon-lined autoclave (25 mL) and heated at 160 °C for 12 h, and then cooled down to room temperature. Green stripe crystals of 1 were collected and washed with water. Yield: ∼42% based on L ligand. Elemental analysis (%): anal. calcd for C24H16N6O5SNi: C, 51.54; H, 2.88; N, 15.03. Found: C, 51.45; H, 2.73; N, 15.12. IR (cm−1, KBr pellets): 3354 w, 3132 w, 3104 w, 3080 w, 2903 w, 1607 w, 1565 m, 1527 s, 1478 m, 1458 s, 1438 s, 1399 s, 1372 s, 1304 m, 1150 m, 1032 m, 773 s, 738 s, 686 m, 626 m.
Synthesis of [Ni(L)(1,3-BDC)(H2O)]n (2). A mixture of Ni(NO3)2·6H2O (29.0 mg, 0.1 mmol), 1,3-H2BDC (16.6 mg, 0.1 mmol), and L (31.2 mg, 0.1 mmol) in 8 mL of solution (VH2O:VDMF = 1:1) was sealed in a teflon-lined autoclave (25 mL) and heated at 140 °C for 12 h, and then cooled down to room temperature. Green stripe crystals of 2 were collected and washed with water. Yield: ∼46% based on L ligand. Elemental analysis (%): anal. calcd for C26H17N6O5Ni: C, 56.45; H, 3.23; N, 15.13. Found: C, 56.56; H, 3.10; N, 15.22. IR (cm−1, KBr pellets): 3439 w, 3123 w, 3087 w, 3029 w, 1611 m, 1564 m, 1529 s, 1460 s, 1441 s, 1404 m, 1381 s, 1314 m, 1248 m, 1149 m, 1030 m, 744 s, 720 s, 620 m.
Synthesis of [Ni(L)(1,4-BDC)(H2O)]n (3). A mixture of Ni(AcO)2·4H2O (24.8 mg, 0.1 mmol), 1,4-H2BDC (16.6 mg, 0.1 mmol), and L (31.2 mg, 0.1 mmol) in 6 mL of solution (VH2O:VCH3OH = 1:1) was sealed in a teflon-lined autoclave (25 mL) and heated at 130 °C for 12 h, and then cooled down to room temperature. Green stripe crystals of 3 were collected and washed with water. Yield: ∼35% based on L ligand. Elemental analysis (%): anal. calcd for C26H18N6O5Ni: C, 56.45; H, 3.28; N, 15.19. Found: C, 56.56; H, 3.10; N, 15.22. IR (cm−1, KBr pellets): 3271 m, 3139 w, 3076 w, 2824 w, 1605 m, 1567 s, 1520 s, 1506 s, 1478 m, 1456 s, 1440 s, 1392 s, 1362 m, 1300 m, 1248 m, 1187 w, 1149 m, 1034 m, 760 m, 747 s.
Synthesis of [Mn(L)(2,5-TDC)(H2O)]n (4). Preparation of complex 4 was similar to that of 1, except that Mn(AcO)2·4H2O (24.5 mg, 0.1 mmol) was used instead of Ni(AcO)2·4H2O. Orange prisms crystals of 4 were collected and washed with methanol. Yield: ∼38% based on L ligand. Elemental analysis (%): anal. calcd for C24H16N6O5SMn: C 51.89, H 2.90, N 15.13. Found (%): C 51.76, H 2.75, N 15.02. IR (cm−1, KBr pellets): 3376 m, 3117 m, 1963 w, 1686 w, 1566 s, 1525 s, 1457 s, 1375 s, 1209 s, 1031 m, 997 m, 824 m, 764 m, 747 m.
Synthesis of [Mn(L)(2,6-PYDC)(H2O)]n (5). A mixture of Mn(BF4)2·H2O (15.9 mg, 0.1 mmol), 2,6-H2PYDC (16.7 mg, 0.1 mmol), NaOH (8 mg, 0.20 mmol) and L (31.2 mg, 0.1 mmol) in 8 mL of water was sealed in a teflon-lined autoclave (25 mL) and heated at 160 °C for 12 h, and then cooled down to room temperature. Orange stripe crystals of 5 was collected and washed with methanol. Yield: ∼40% based on L ligand. Elemental analysis (%): anal. calcd for C25H17N7O5Mn: C, 54.56; H, 3.11; N, 17.81. Found: C, 54.47; H, 3.03; N, 17.70. IR (cm−1, KBr pellets): 3334 m, 3150 m, 3082 m, 2963 m, 2003 w, 1609 s, 1456 s, 1435 s, 1392 s, 1363 s, 1209 s, 724 s, 749 s.
Synthesis of [Mn(L)(1,4-NDC)]n (6). A mixture of Mn(BF4)2·H2O (15.9 mg, 0.1 mmol), 1,4-H2NDC (21.6 mg, 0.1 mmol), NaOH (40 mg, 1 mmol) and L (31.2 mg, 0.1 mmol) in 8 mL of water was sealed in a teflon-lined autoclave (25 mL) and heated at 140 °C for 12 h, and then cooled down to room temperature. Orange stripe crystals of 6 were collected and washed with methanol. Yield: ∼42% based on L ligand. Elemental analysis (%): anal. calcd for C30H18N6O4Mn: C, 61.97; H, 3.12; N, 14.45. Found: C, 61.85; H, 3.03; N, 14.28. IR (cm−1, KBr pellets): 3642 w, 3149 w, 3060 w, 1945 w, 1589 m, 1552 s, 1505 s, 1458 s, 1435 s, 1361 s, 1241 m, 1203 m, 829 m, 783 m, 740 m.
X-ray crystallography
The crystallographic data of complexes 1–6 were collected on a Bruker SMART Apex II diffractometer equipped with graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å) at 298 K using a multi-scan technique. All the structures were solved by the direct method of SHELXS-2014 and refined by full-matrix least-squares on F2 with anisotropic displacement using SHELXTL software package.18 The disordered segments were subjected to geometric restraints during the refinements. Non-hydrogen atoms were refined with anisotropic displacement parameters during the final cycles. All hydrogen atoms were located in calculated positions and refined isotropically, except the hydrogen atoms of water molecules were fixed in a difference Fourier map and refined isotropically. Crystallographic data and structure refinement details for structural analyses are summarized in Table 1 and the selected bond lengths and angles are given in Table S1 of ESI.†
Table 1 Crystal data and structure refinement summary for complexes 1–6
| |
1 |
2 |
3 |
4 |
5 |
6 |
| R = Σ(‖F0| − |FC‖)/Σ|F0|. wR = [Σw(|F0|2 − |FC|2)2/Σw(F02)]1/2. |
| Formula |
C24H16N6O5SNi |
C26H17N6O5Ni |
C26H18N6O5Ni |
C24H16N6O5SMn |
C25H17N7O5Mn |
C30H18N6O4Mn |
| Formula wt |
559.2 |
552.16 |
553.17 |
555.43 |
550.4 |
581.44 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
Cc |
C2/c |
Pn |
Cc |
P-1 |
P21/n |
| T (K) |
170.15 |
100.00 |
100.00 |
173.15 |
100.00 |
173.15 |
| a (Å) |
13.039(5) |
16.6905(7) |
11.7121(4) |
19.829(3) |
9.3365(7) |
11.035(3) |
| b (Å) |
18.606(7) |
11.6251(3) |
6.1520(2) |
5.8486(10) |
11.9034(11) |
9.024(2) |
| c (Å) |
10.943(6) |
26.5469(13) |
16.1276(4) |
20.750(3) |
11.9507(11) |
24.497(6) |
| α (°) |
90 |
90 |
90 |
90 |
66.724(9) |
90 |
| β (°) |
121.201(6) |
110.743(5) |
91.235(3) |
109.388(4) |
68.005 |
97.215(6) |
| γ (°) |
90 |
90 |
90 |
90 |
77.863 |
90 |
| V (Å3) |
2270.8(17) |
4817.0(4) |
1161.77(6) |
2270.0(6) |
1128.2(2) |
2420.2(10) |
| Z |
4 |
8 |
2 |
4 |
2 |
4 |
| D (mg m−3) |
1.636 |
1.523 |
1.581 |
1.625 |
1.62 |
1.596 |
| μ (mm−1) |
0.998 |
0.857 |
0.888 |
0.726 |
0.641 |
0.599 |
| F (000) |
1144 |
2264 |
568 |
1132 |
562 |
1188 |
| Crystal size (mm3) |
0.23 × 0.19 × 0.15 |
0.15 × 0.13 × 0.11 |
0.13 × 0.12 × 0.11 |
0.25 × 0.18 × 0.11 |
0.18 × 0.15 × 0.12 |
0.23 × 0.19 × 0.17 |
| Theta rang (deg) |
2.129–26.698 |
3.282–29.555 |
2.127–29.619 |
1.040–27.583 |
3.525–29.506 |
1.676–27.473 |
| Goof (F2) |
1.059 |
1.077 |
1.028 |
1.049 |
1.042 |
1.007 |
| Measured refins |
7171 |
22593 |
14734 |
16528 |
10282 |
21200 |
| Obsd refins |
3970 |
5961 |
5313 |
4747 |
5286 |
5518 |
| Rint |
0.0331 |
0.0303 |
0.0424 |
0.0426 |
0.0278 |
0.0941 |
| Ra/wRb |
0.0353/0.0999 |
0.0363/0.0820 |
0.0361/0.0687 |
0.0320/0.0775 |
0.0403/0.0982 |
0.0502/0.1143 |
CCDC 1873981–1873986 contains the supplementary crystallographic data for complexes [Ni(L)(2,5-TDC)(H2O)]n (1), [Ni(L)(1,3-BDC)(H2O)]n (2), [Ni(L)(1,4-BDC)(H2O)]n (3), [Mn(L)(2,5-TDC)(H2O)]n (4), [Mn(L)(2,6-PYDC)(H2O)]n (5) and [Mn(L)(1,4-NDC)]n (6), respectively.
Results and discussion
Description of the crystal structure
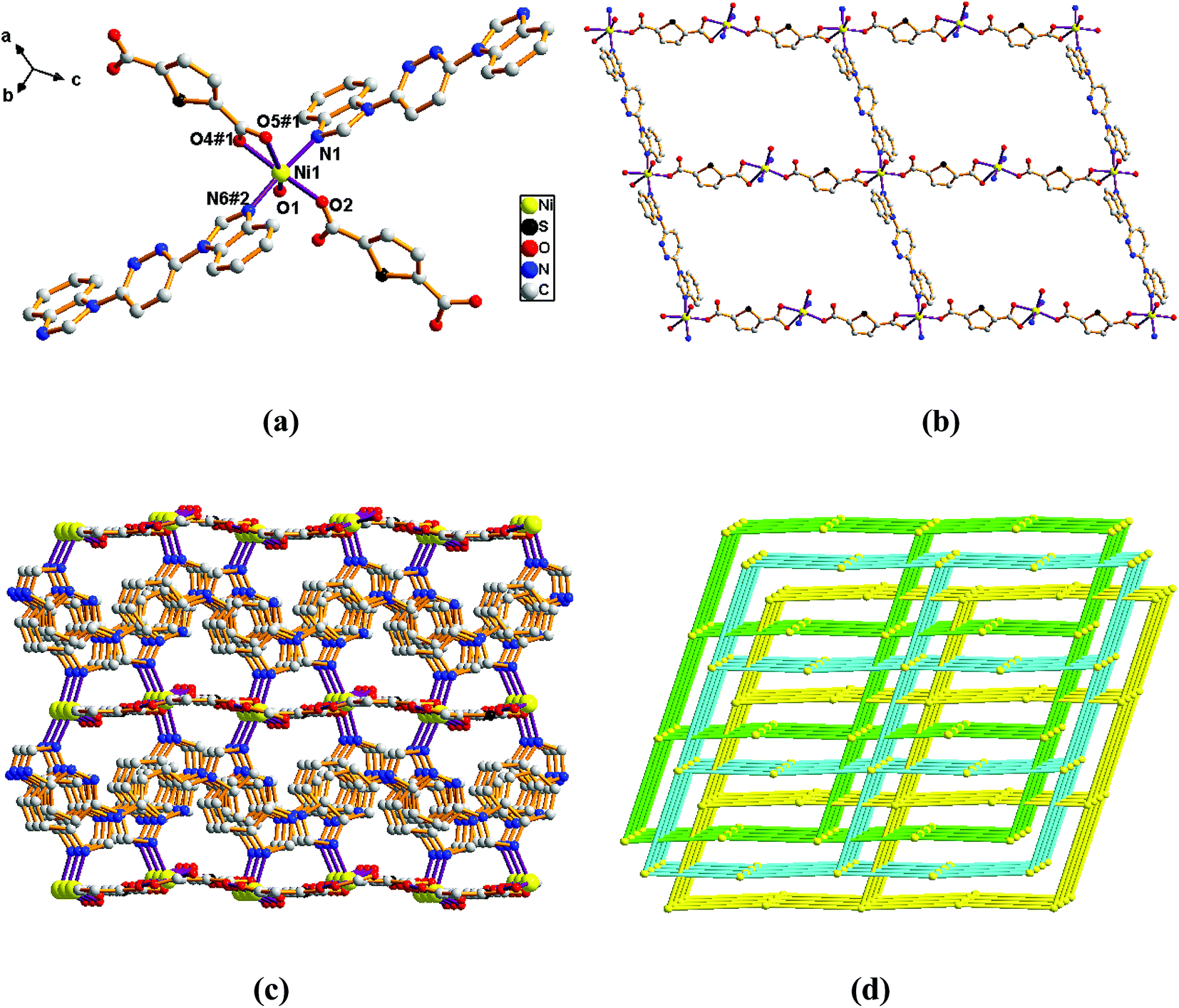
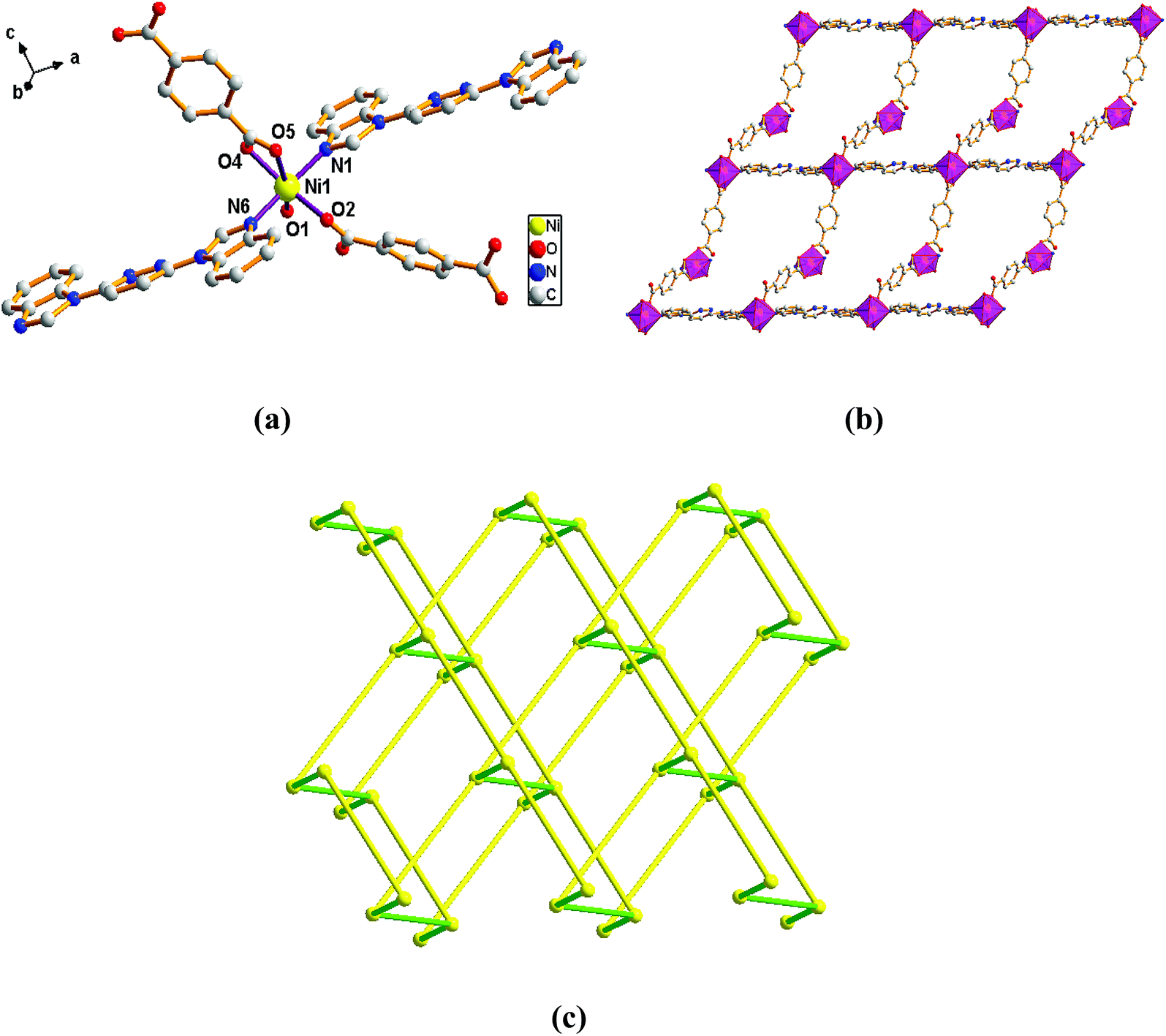
Structure of [Ni(L)(2,5-TDC)(H2O)]n (1). Single-crystal X-ray diffraction analysis reveals that complex 1 is based on a dense 3-fold interpenetrating three-dimensional (3D) framework and crystallizes in the monoclinic space group Cc. The construction unit of the 3D framework consists of one Ni ion, one L ligand, one 2,5-TDC2− anion and one water molecule of coordination. The Ni1 ion is six-coordinated and the coordination geometry around the individual Ni center is a distorted octahedron which the equatorial plane are occupied by O1, O2, O4#1 and O5#1 donor atoms, and the axial sites are occupied by N1 and N6#2 atoms with an N1–Ni1–N6#2 angle of 171.74°. The O1, O2, O4#1 and O5#1 donor atoms come from two different 2,5-TDC2− ligands and one water molecule of coordination, and the N1 and N6#2 sites originate from the bis-bidentate bridging ligand (Fig. 1a). The lengths of Ni–N bond are 2.077(5) and 2.080(5) Å, while the lengths of Ni–O bond is in the range of 2.038(4)–2.264(4) Å.
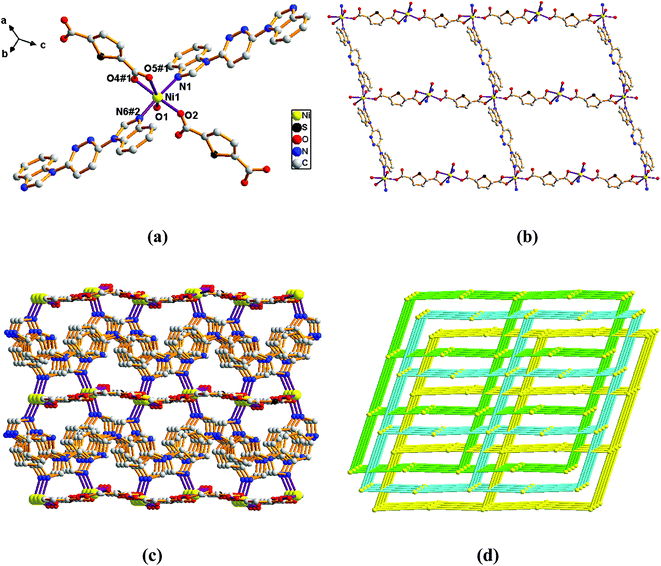 |
| | Fig. 1 View of (a) the coordination environment of Ni(II) atom in complex 1; (b) the 2D plane structure of 1; (c) the 3D framework structure of 1; (d) the 4-connected net topology with the Schläfli symbol of (66). | |
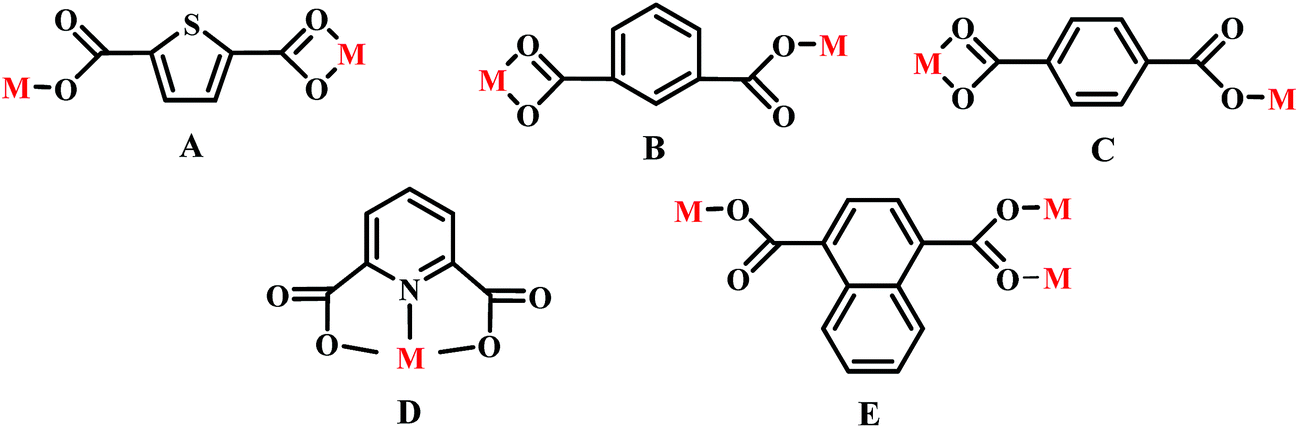
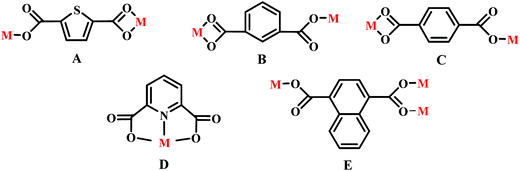
Adjacent Ni centers were connected by carboxylate ligands to form a 1D chain with the Ni⋯Ni distance of 10.4767 Å, then adjacent 1D chains are further extended by other L ligands to give the 2D plane network structure (Fig. 1b). The 2,5-TDC2− ligand adopts (κ2)-(κ1)-μ2 coordination mode (Scheme 2A) and the L ligand adopts bidentate bridging coordination. The 2D layers are further extended by residual carboxylate groups and L ligands to give the 3D Ni–organic framework (Fig. 1c), the Ni center is connected by organic ligands to four neighboring Ni centers. Thus, the Ni centers can be considered as a 4-connected node and the ligands can be considered as linkers. Topological analysis reveals that 1 is a uninodal 4-connected network with the point symbol of {66}-sql topology (Fig. 1d).
 |
| | Scheme 2 The coordination mode of organic acids in this work. | |
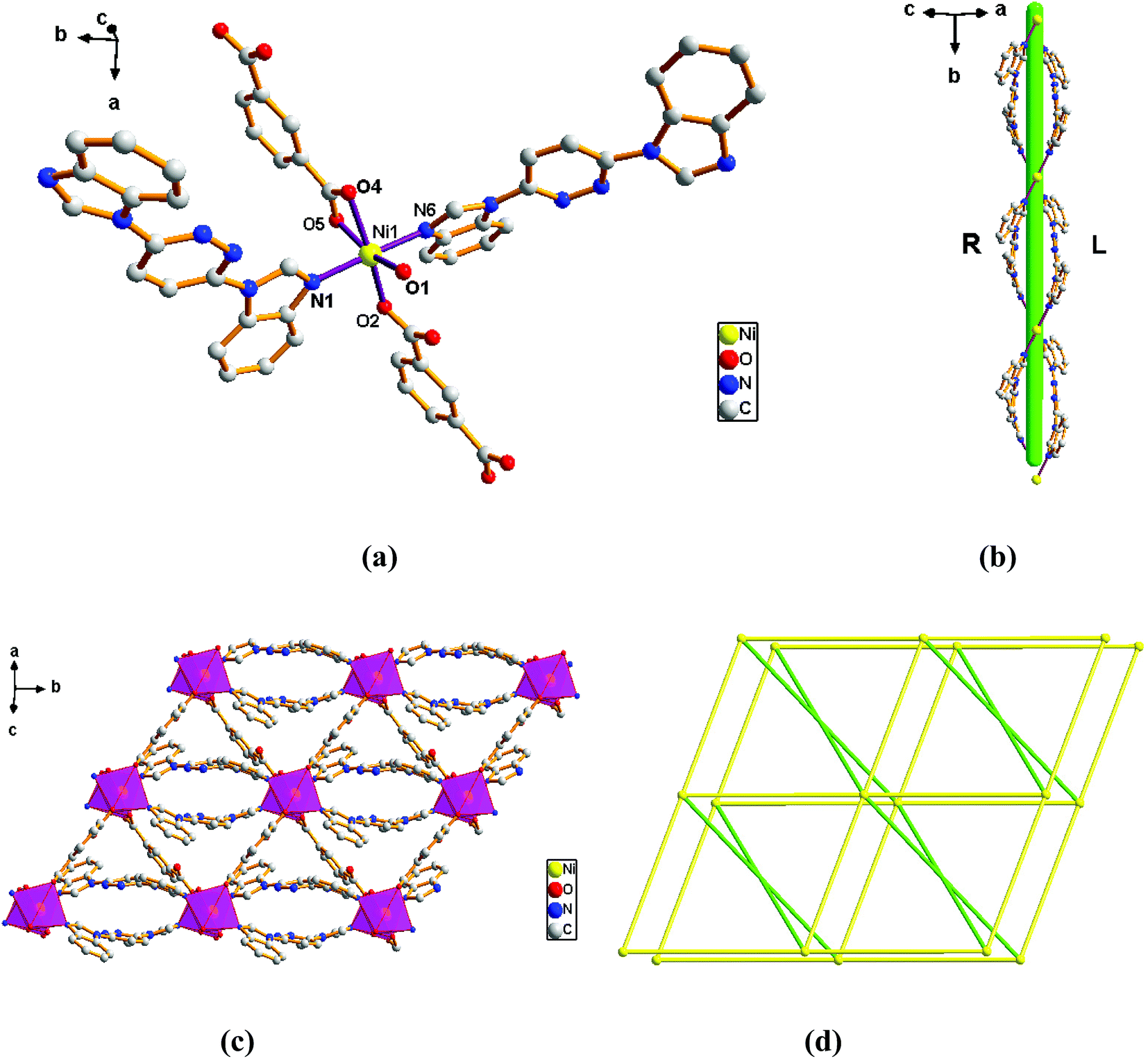
Structure of [Ni(L)(1,3-BDC)(H2O)]n (2). Complex 2 crystallizes in the monoclinic C2/c space group in which one Ni ion, one L ligand, one 1,3-BDC2− anion and one molecule of coordination water constitute the construction unit. The Ni1 ion is six-coordinated that ligated by four O atoms (O1, O2, O4 and O5) originating from two different 1,3-BDC2− ligands and one water molecule of coordination in the equatorial plane position and two N atoms (N1 and N6) form two different L ligands at axial positions (Fig. 2a) with an N1–Ni1–N6 angle of 176.92°. Analysis of the coordination geometry around the Ni center showed that the ion resides in a distorted octahedral environment. The lengths of Ni–N bond are 2.0879(15) and 2.1053(15) Å, while the lengths of Ni–O bond is in the range of 1.9934(12)–2.1362(12) Å.
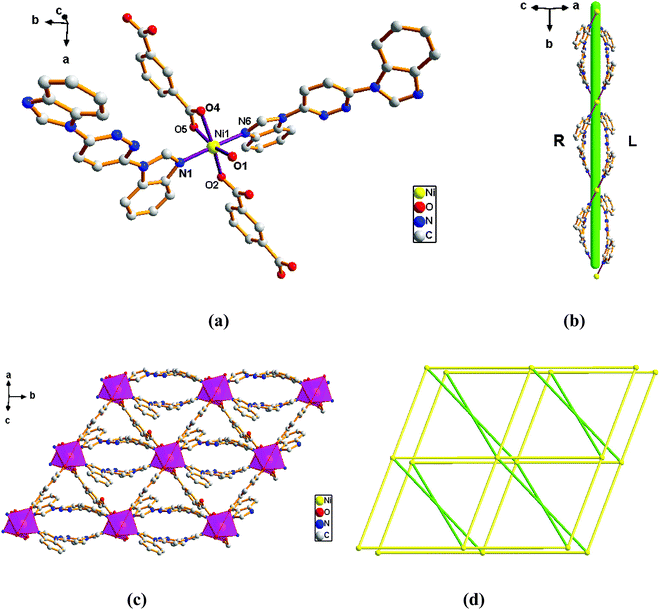 |
| | Fig. 2 View of (a) the coordination environment of Ni(II) atom in complex 2; (b) the 1D chain of 2; (c) the 2D plane of 2; (d) the 6-connected net topology with the Schläfli symbol of (412·63). | |
The outstanding structural feature of complex 2 is the presence of two types helical chains (Fig. 2b), the Ni1 ion is bridged by two N atoms from L ligands to form an infinitely extended double helical chain of left-handed and right-handed along the b-axis. These helical chains are then connected via other 1,3-BDC2− ligands in a (κ2)-(κ1)-μ2 coordination mode (Scheme 2B) to generate an infinitely extended 2D network structure. Furthermore, structural analysis showed that 2D Ni–organic network of two-fold interpenetration structure (Fig. 2c). For a better understanding of the intricate framework, a topological analysis of complex 2 was performed. In the 2D network structure, we can define the Ni ion as four-connected nodes with all crystallographically independent L ligands and 1,3-BDC2− anion as linkers. Therefore, the two-fold interpenetration structure of 2 can be represented as a 6-connected new topology with the point symbol of (412·63) (Fig. 2d).
Structure of [Ni(L)(1,4-BDC)(H2O)]n (3). When 1,4-H2BDC was introduced in place of 1,3-H2BDC, complex 3 was obtained. Single-crystal X-ray diffraction analysis reveals that complex 3 is based on a dense 3D framework and crystallizes in the monoclinic space group Pn. The construction unit of the 3D framework consists of one Ni ion, one L ligand, one 1,4-BDC2− anion and one water molecule of coordination. The Ni1 ion is six-coordinated and the coordination geometry around the individual Ni center is a distorted octahedron in which the equatorial plane is occupied by O1, O2, O4 and O5 donor atoms, and the axial sites are occupied by N1 and N6 atoms with an N1–Ni1–N6 angle of 174.33°. The O1, O2, O4 and O5 donor atoms come from two different 1,4-BDC2− ligands and one water molecule of coordination, and N1 and N6 sites originate from the bis-bidentate bridging ligand (Fig. 3a). The lengths of Ni–N bond are 2.062(3) and 2.084(3) Å, while the lengths of Ni–O bond is in the range of 2.026(2)–2.158(2) Å.
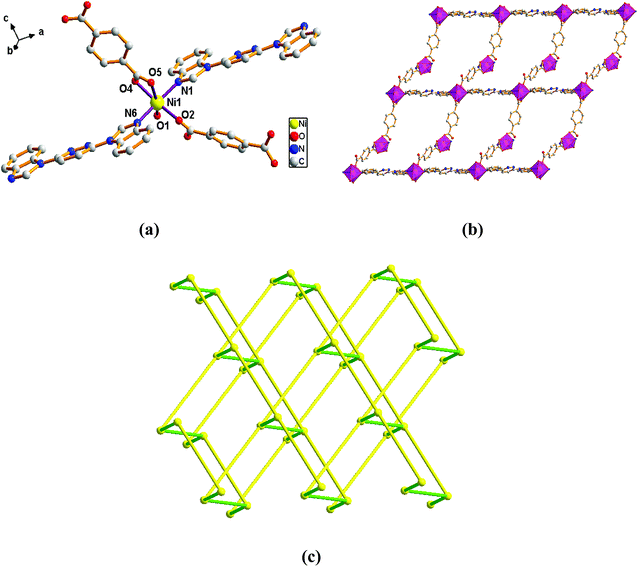 |
| | Fig. 3 View of (a) the coordination environment of Ni(II) atom in complex 3; (b) the 2D plane of 2; (c) the 4-connected net topology with the Schläfli symbol of (66). | |
Adjacent Ni centers were connected by organic ligands to form an undulating 2D plane structure (Fig. 3b). The 1,4-H2BDC ligand adopts a (κ2)-(κ1)-μ2 coordination mode (Scheme 2C) and the L ligand adopt bidentate bridging coordination. The 2D layers are further extended by residual L ligands to give the 3D Ni–organic framework (Fig. S1a†), the Ni center is connected by organic ligands to four neighboring centers. Thus, Ni centers can be considered as a 4-connected node with all crystallographically independent L ligands and 1,4-BDC2− anion as linkers. Topological analysis reveals that 3 is a 3D framework of uninodal 4-connected with the point symbol of {66} topology (Fig. 3c).
Structure of [Mn(L)(2,5-TDC)(H2O)]n (4). Compared with complex 1, the complex 4 was obtained when Mn ion was introduced in place of the Ni ion. Single-crystal X-ray diffraction analysis reveals that complex 4 is based on a dense 3D diamondoid framework and crystallizes in the monoclinic space group Cc. The construction unit of the 3D framework consists of one Mn(II) ion, one L ligand, one 2,5-TDC2− anion and one water molecule of coordination. The individual Mn1 ion is six-coordinated, and coordination geometry around the individual Mn(II) centers is a distorted octahedron in which the equatorial plane is occupied by O1, O2, O3 and O5 donor atoms, and the axial sites are occupied by N1 and N6 atoms with an N1–Ni1–N6 angle of 174.42°. The O1, O2, O3 and O5 donor atoms come from two different 2,5-TDC2− ligands and one water molecule of coordination, and N1 and N6 sites originate from the bis-bidentate bridging ligand (Fig. 4a). The lengths of Mn–N bond are 2.264(4) and 2.274(4) Å, while the lengths of Mn–O bond is in the range of 2.087(3)–2.290(3) Å.
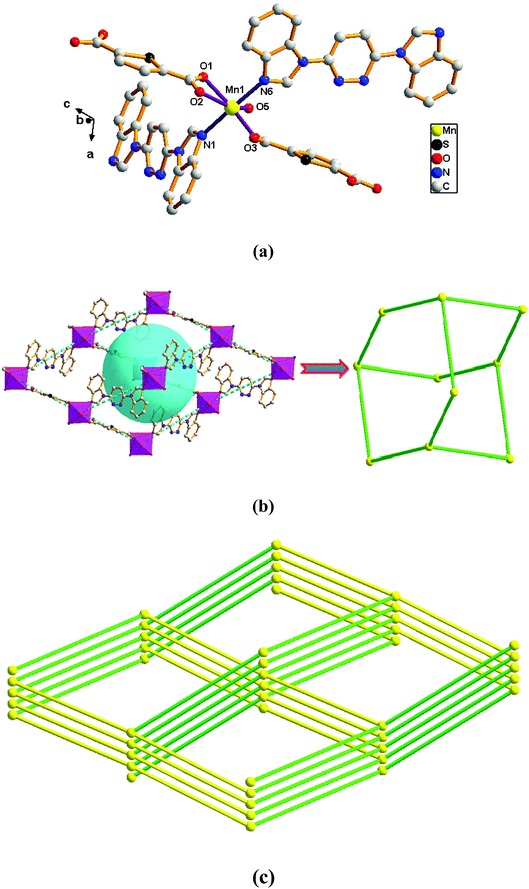 |
| | Fig. 4 View of (a) the coordination environment of Mn(II) atom in complex 4; (b) the 2D adamantane structure of 4; (c) the 4-connected net topology with the Schläfli symbol of (66). | |
Adjacent Mn(II) centers were connected by organic ligands to form a 2D plane network structure (Fig. 4b). The 2,5-TDC2− ligands adopts a (κ2)-(κ1)-μ2 coordination mode (Scheme 2A) and the L ligand adopts bidentate bridging coordination. The 2D layers are further extended by residual carboxylate groups and L ligands to give 3D Mn–organic framework contained 1D channels (Fig. S1b†). The structure of complex 4 is quite different from complex 1. It is worth noting that the diamondoid framework is observed in complex 4, the Mn centers are connected by organic ligands to four neighboring centers. Therefore, the Mn center can be considered as a 4-connected node and ligands as linkers. Topological analysis reveals that complex 4 is a 3D diamondoid framework of uninodal 4-connected with the point symbol of {66}-dia topology (Fig. 4c).
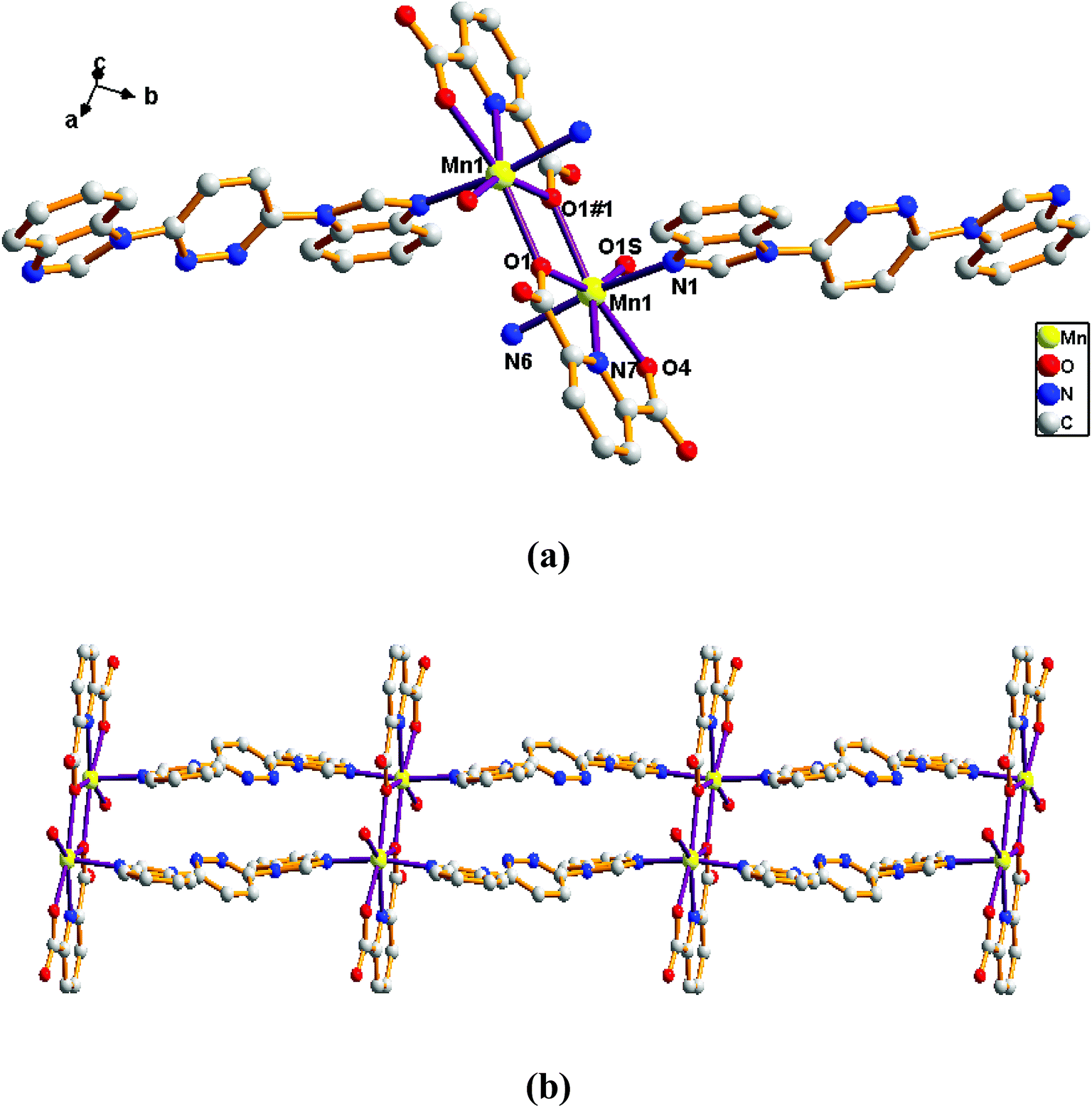
Structure of [Mn(L)(2,6-PYDC)(H2O)]n (5). Comparison with complex 4, the complex 5 was collected when 2,6-H2PYDC was introduced in place of 2,5-H2TDC. Complex 5 is based on an infinitely extended 1D chain framework and crystallizes in the triclinic space group P-1. The construction unit consists of one Mn ion, one L ligand, one 2,6-PYDC2− anion and one water molecule of coordination. The individual Mn1 ion is seven-coordinated, and coordination geometry around the Mn centers is a distorted pentagonal bipyramid in which the equatorial plane is occupied by O1, O1#1, O1s, O4, and N7 donor atoms, and the axial sites are occupied by N1 and N6 atoms with an N1–Mn–N6 angle of 169.57°. The O1, O1#1, O1s, O4, and N7 donor atoms come from two different 2,6-PYDC2− ligands and one water molecule of coordination, and N1 and N6 donor atoms from the bis-bidentate bridging ligand (Fig. 5a). The lengths of Mn–N bond are 2.2466(18), 2.2856(16) and 2.3306(17) Å, while the lengths of Mn–O bond is in the range of 2.1503(15)–2.3921(15) Å.
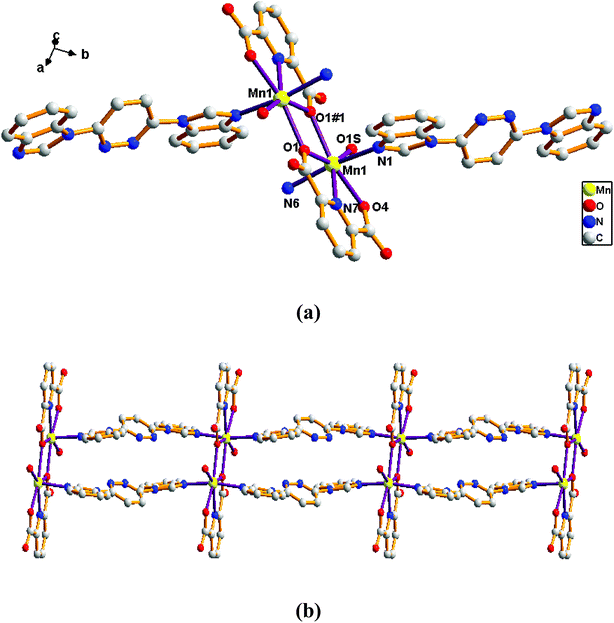 |
| | Fig. 5 View of (a) the coordination environment of Mn(II) atom in complex 5; (b) the 1D chain of 5. | |
Adjacent Mn(II) centers were connected by carboxylate ligands to form a binuclear [Mn2(CO2)2N2] building unit with Mn⋯Mn distance of 3.851 Å. It is worth noting that the availability of an inversion center in the binuclear metal node (Fig. 5a). Then these binuclear building units are connected via L ligands to generate an infinitely extended 1D chain (Fig. 5b). In complex 5, the carboxylate groups in 2,6-PYDC2- adopt a (κ1)-(κ1)-μ1 coordination mode (Scheme 2D) and the L ligand adopt bidentate bridging coordination.
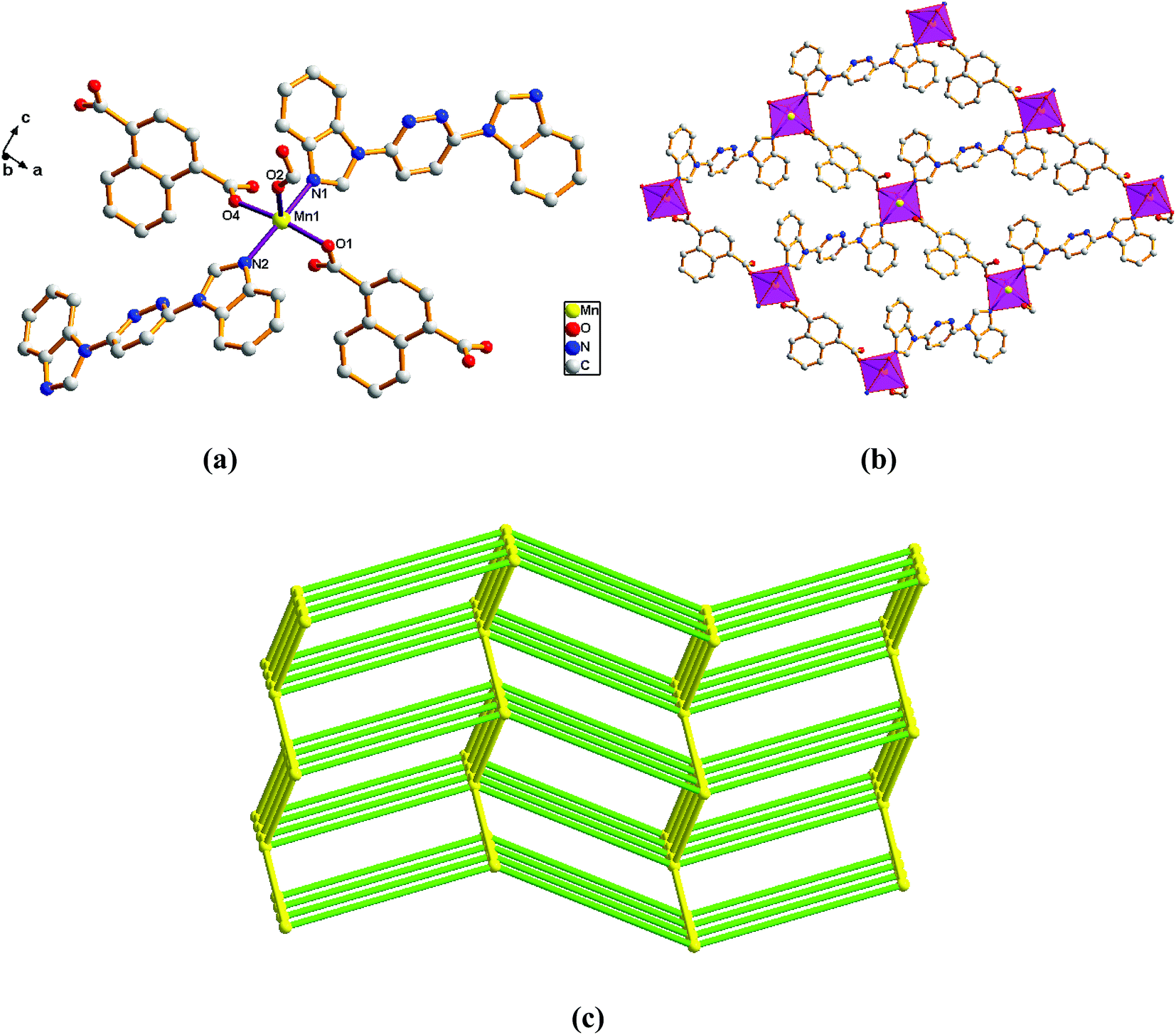
Structure of [Mn(L)(1,4-NDC)]n (6). Comparison with complex 5, the complex 6 was collected when 1,4-H2NDC was introduced in place of 2,6-H2PYDC. Single-crystal X-ray diffraction analysis reveals that complex 6 is based on a dense three-dimensional framework and crystallizes in the monoclinic space group P21/n. The construction unit of the 3D framework consists of one Mn(II) ion, one L ligand, and one 1,4-TDC2− anion. The individual Mn1 ion is five-coordinated, and coordination geometry around the individual Mn(II) centers is a distorted trigonal bipyramid in which the equatorial plane is occupied by O1, O2, and O4 donor atoms, and the axial sites are occupied by N1 and N2 atoms with an N1–Mn1–N2 angle of 167.99°. The O1, O2, and O4 donor atoms come from three different 1,4-NDC2− ligands, and N1 and N2 sites originate from the bis-bidentate bridging ligand (Fig. 6a). The lengths of Mn–N bond are 2.242(2) and 2.263(2) Å, while the lengths of Mn–O bond is in the range of 2.138(19)–2.152(2) Å.
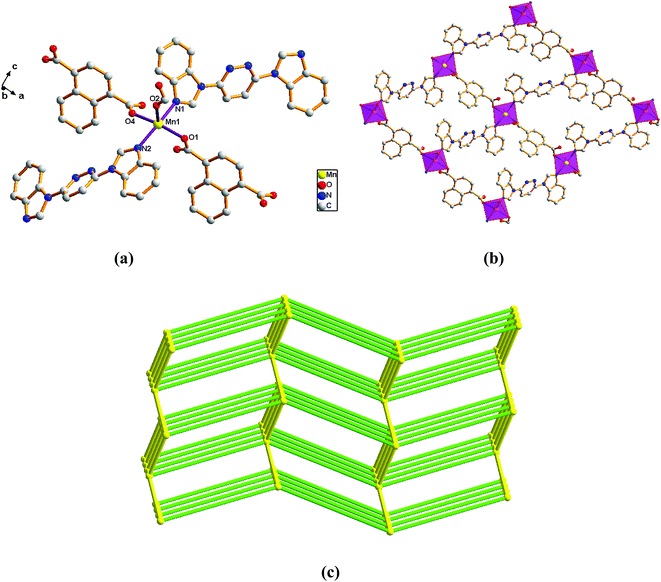 |
| | Fig. 6 View of (a) the coordination environment of Mn(II) atom in complex 6; (b) the 2D plane of 6; (c) the 6-connected net topology with the Schläfli symbol of (412·63). | |
Adjacent Mn(II) centers were connected by carboxylate ligands and L ligands to form a 2D (4,4) grid plane network structure (Fig. 6b), and the rectangular window of the 2D motif has a dimension of ca. 11.035 × 14.514 Å2. The 1,4-NDC2− ligands adopts a (κ1)-(κ1-κ1)-μ3 coordination mode (Scheme 2E) and the L ligand adopts bidentate bridging coordination. The 2D layers are further extended by residual carboxylate groups and L ligands to give 3D Mn–organic framework with 1D channels (Fig. S1c†), the Mn center is connected to five neighboring centers through four 1,4-TDC2− anions and two L ligands. Therefore, the Mn(II) centers can be considered as a 6-connected node and all ligands as linkers. Topological analysis reveals that it is a 3D framework of uninodal 6-connected with the point symbol of {412·63} topology (Fig. 6c).
Comparison of the structures 1–6
Crystallographic analyses revealed that CPs 1–6 shows varied structures from the 1D chain (5), 2D network (2) to 3D frameworks (1, 3, 4 and 6). Complexes 1, 3 and 4 are 4-connected topological types, which 1 display a 3-fold interpenetrating 3D framework structure, the 3 is a 3D framework structure and 4 displays a 3D diamondoid structure. Complexes 2 and 6 are 6-connected topological types, which 2 has a new 2D topological structure of helical chain structure and 6 displays a 3D framework structure. Complex 5 is a 1D chain structure. In addition, the carboxylate ligands exhibit varied coordination modes. The results herein further confirm that the metal center, anions, solvent system, temperature and varied coordination modes of the carboxylate ligand exert significant influence on the structural diversity of the frameworks, which can effectively regulate the structure of the CPs.
Spectroscopic and thermal analysis
PXRD analysis. The PXRD patterns of complexes 1–6 are presented in Fig. S2.† The diffraction peaks of both the simulated and experimental patterns match in the key positions, indicating the phase purity of the complexes. The differences in the intensity may be contribute to the preferred orientation of the crystal samples.
TG analysis. Thermogravimetric analyses (TGA) were performed to check the thermal stability of the frameworks under nitrogen atmosphere, and the obtained plots are presented in Fig. S3.† The results show that the initial mass losses of 1–6 are in accordance with the solvent losses calculated from the elemental analysis data. The TGA analysis revealed that 1–4 were stable up to 200 °C. For 1, 2 and 4 its weight loss of 48.57% (1), 50.80% (2) or 44.5% (4) in the range of 200–550 °C (1), 200–750 °C (2) or 200–650 °C (4) corresponded to the collapse of the framework. For 3, the first weight loss of 12.04% in the range of 200–300 °C corresponded to the loss of water molecules and partial framework decomposition and a further weight loss was observed at about 400 °C, corresponding to the collapse of the framework. For 5, the first weight loss of 5.33% in the range of 100–170 °C corresponded to the loss of water molecules and partial framework decomposition and a further weight loss was observed at about 280 °C, corresponding to the collapse of the framework. For 6, the TGA analysis revealed that 6 were stable up to 375 °C. The weight loss of 50% in the range of 375–550 °C corresponded to the loss of collapse of the framework.
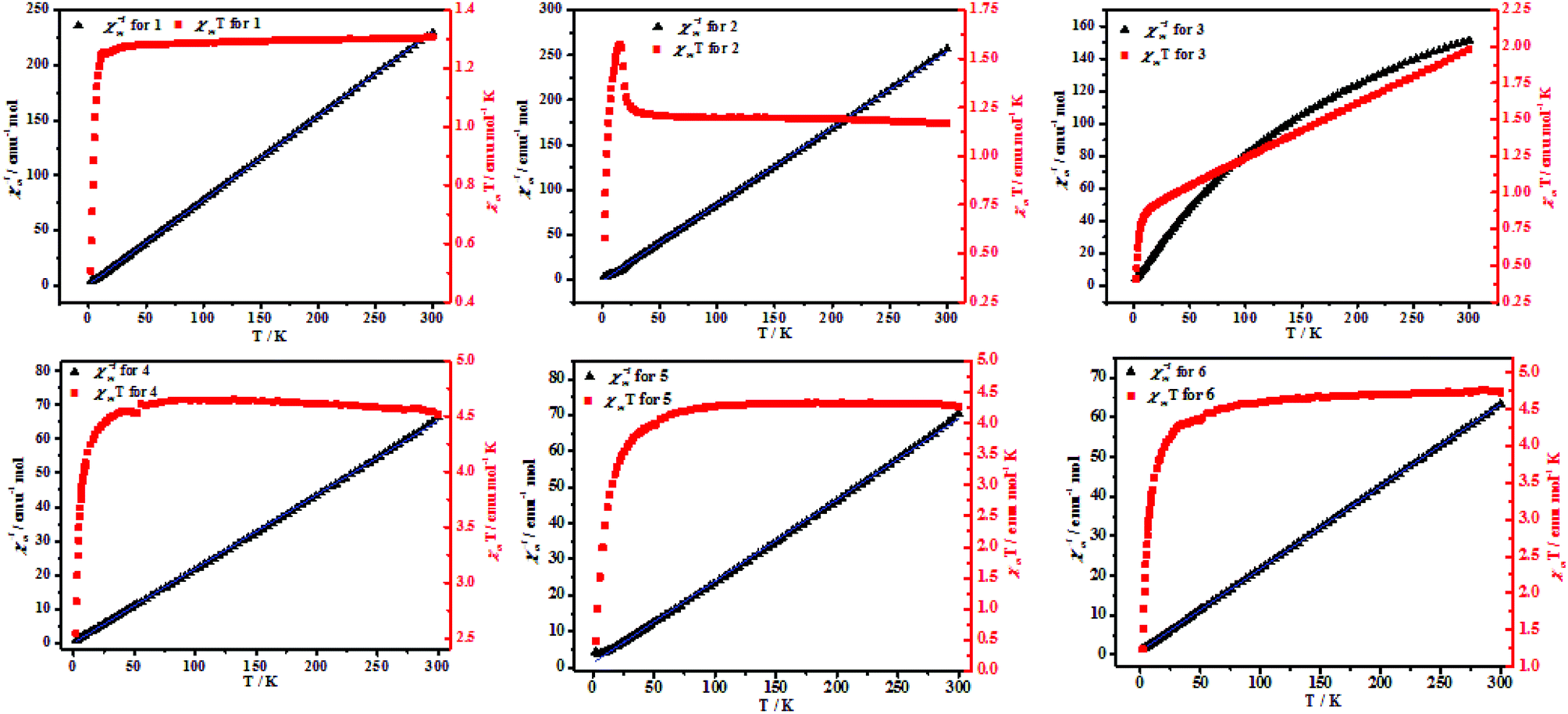
Magnetic properties. Variable-temperature magnetic susceptibility of complexes 1–6 studies were carried out on a SQUID MPMS-XL-7 with an applied magnetic field of 1000 Oe and in the temperature range of 2–300 K. Magnetic data as plots of χMT and χm−1 versus T are depicted in Fig. 7. The results showed that complex 2 was ferromagnetic coupling and complexes 1, 3–6 were antiferromagnetic. For 2, at room temperature, the product χMT is slightly higher than expected for one isolated Ni(II) ions with g = 1.1 (this would correspond to 1.17 emu mol−1 K), indicating the presence of predominantly ferromagnetic interactions in 2. They increase upon cooling with a more pronounced slope as the temperature becomes closer to zero to reach maxima at 1.62 emu mol−1 K, near 15 K, and below 15 K, it decreases quickly to 0.62 emu mol−1 K at 3 K, which might be mainly due to the presence of zero-field splitting (ZFS) for Ni(II) ions.19 Fitting of the χm−1–T above 25 K using the Curie–Weiss law χm = C/(T − θ) gives the curie constants C = 1.17 emu mol−1 K and the weiss constants θ = 1.65 K, which support the presence of overall ferromagnetic interactions in the complex 2. The carboxylate ligands that link the double helical chains to form 2D net will provide a pathway for weak antiferromagnetic coupling.20 For 1, 3–6, the magnetic coupling through this ligands in this bridging mode is very weak and usually antiferromagnetic. The maximum χMT value of complexes 1, 3–6 at 300 K are closer to 1.31, 1.95, 4.45, 4.15, and 4.55 emu mol−1 K, respectively, which is larger than the spin-only value for magnetically isolated systems, as expected for a Mn(II) system with a significant contribution from the unquenched orbital momentum in an octahedral ligand field.21 The curve indicates the typical antiferromagnetic and spin–orbit coupling interactions present in the complexes 1, 3–6. With lowering of temperature, the χMT values are smoothly decreases when the temperature closer to 2 K and obtain a minimum χMT values (Table 1), indicating that antiferromagnetic interactions occurred. The magnetic susceptibility χm−1 versus T obeys the Curie–Weiss law [χm = C (T − θ)] gives a C and θ value at the corresponding temperature range (Table 1), respectively. The negative θ further confirms antiferromagnetic interactions present in the complexes 1, 3–6. It is worth noting that the spin–orbit coupling is contributing for the negative weiss constant (Table 2).22
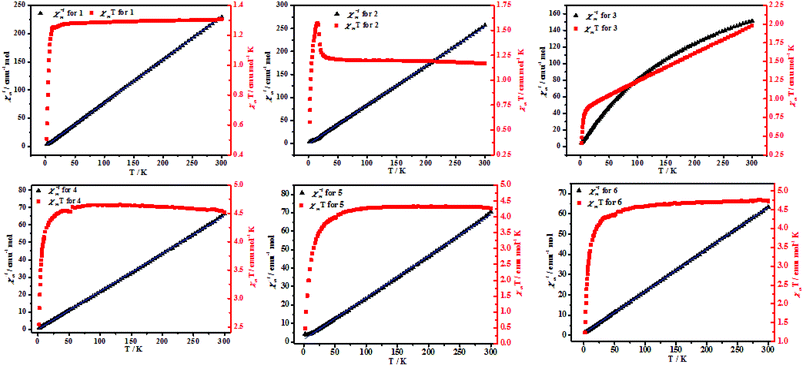 |
| | Fig. 7 Temperature-dependent magnetic susceptibilities of 4–6 in the temperature range of 2–300 K under an applied magnetic field of 1000 O. | |
Table 2 Magnetic properties date of complexes 1–6
| |
1 |
2 |
3 |
4 |
5 |
6 |
| T (K) |
25–300 |
25–300 |
25–300 |
60–300 |
50–300 |
70–300 |
| C (emu mol−1 K) |
1.3 |
1.17 |
1.94 |
4.59 |
4.42 |
4.81 |
| θ (K) |
−1.26 |
1.65 |
−34.64 |
−0.28 |
−5.74 |
−4.52 |
| χMT value at 300 K |
1.31 |
1.17 |
1.95 |
4.45 |
4.15 |
4.55 |
| χMT value at 2 K |
0.51 |
0.62 |
0.41 |
2.52 |
0.31 |
1.56 |
Conclusion
In conclusion, six new CPs with a 3,6-bis(benzimidazol-1-yl)pyridazine ligand and different dicarboxylate ligands as the auxiliary ligands have been structurally, magnetically and catalytically characterized in this work. Results analysis manifests that complexes 1–6 have high thermal stability. Complex 2 has a new topological structure and complex 4 has a diamond structure. The polycarboxylates plays an important role in the assembly of the final complexes structures. Moreover, complex 2 was ferromagnetic coupling and complexes 1, 3–6 were antiferromagnetic.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21361026).
References
-
(a) C.-W. Kung, K. Otake, C.-T. Buru, S. Goswami, Y.-X. Cui, J.-T. Hupp, A.-M. Spokoyny and O.-K. Farha, J. Am. Chem. Soc., 2018, 140, 3871–3875 CrossRef CAS PubMed;
(b) P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 13464–13481 CrossRef PubMed;
(c) J.-T. Dovgan, M.-J. Polinski, B.-Q. Mercado and E.-M. Villa, Cryst. Growth Des., 2018, 18, 5332–5341 CrossRef CAS;
(d) S.-K. Seth, CrystEngComm, 2013, 15, 1772–1781 RSC;
(e) A. Hossain, S.-K. Seth, A. Bauzá, S. Mukhopadhyay and A. Frontera, Polymers, 2018, 10, 182–196 CrossRef PubMed.
-
(a) Z.-Z. Xue, D. Zhang, J. Pan, S.-D. Han, J.-H. Li and G.-M. Wang, Dalton Trans., 2017, 46, 13952–13956 RSC;
(b) N. Tannert, S.-J. Ernst, C. Jansen, H.-J. Bart, S.-K. Henningerc and C. Janiak, J. Mater. Chem. A, 2018, 6, 17706–17712 RSC.
-
(a) F. Haque, A. Halder and D. Ghoshal, Cryst. Growth Des., 2018, 18, 5231–5244 CrossRef CAS;
(b) D. Mondal, M.-C. Majee, S. Kundu, M. Mörtel, G. Abbas, A. Endo, M.-M. Khusniyarov and M. Chaudhury, Inorg. Chem., 2018, 57, 1004–1016 CrossRef CAS PubMed.
-
(a) Y.-H. Tang, F. Wang and J. Zhang, Dalton Trans., 2018, 47, 4032–4035 RSC;
(b) C. Huang, Y.-Y. Zhang, H.-Y. Yang, D.-D. Wang, L.-W. Mi, Z.-C. Shao, M.-J. Liu and H.-W. Hou, Cryst. Growth Des., 2018, 18, 5674–5681 CrossRef CAS;
(c) S.-M. Tang, J. Zhou, H.-H. Zou, X. Liu and L. Zhang, Inorg. Chem., 2018, 57, 1242 CrossRef CAS PubMed;
(d) D. Zhao, X.-H. Liu, J.-H. Guo, H.-J. Xu, Y. Zhao, Y. Lu and W.-Y. Sun, Inorg. Chem., 2018, 57, 2695–2704 CrossRef CAS PubMed;
(e) Y.-S. Kang, Y. Lu, K. Chen, Y. Zhao, P. Wang and W.-Y. Sun, Coord. Chem. Rev., 2019, 378, 262–280 CrossRef CAS.
-
(a) P.-P. Cen, S. Zhang, X.-Y. Liu, W.-M. Song, Y.-Q. Zhang, G. Xie and S.-P. Chen, Inorg. Chem., 2017, 56, 3644–3656 CrossRef CAS PubMed;
(b) Y.-S. Ding, N.-F. Chilton, R. E.-P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed;
(c) F. El-Khatib, B. Cahier, F. Shao, M. Lopez-Jorda, R. Guillot, E. Riviere, H. Hafez, Z. Saad, J.-J. Girerd, N. Guihery and T. Mallah, Inorg. Chem., 2017, 56, 4601–4608 CrossRef PubMed.
-
(a) M. Chen, H. Zhao, E.-C. Sanudo, C.-S. Liu and M. Du, Inorg. Chem., 2016, 55, 3715–3717 CrossRef CAS PubMed;
(b) Y.-Z. Zhang, H.-H. Zhao, E. Funck and K.-R. Dunbar, Angew. Chem., Int. Ed., 2015, 54, 5583–5587 CrossRef CAS PubMed.
-
(a) S. Goswami, G. Leitus, B.-K. Tripuramallu and I. Goldberg, Cryst. Growth Des., 2017, 17, 4393–4404 CrossRef CAS;
(b) S. Ganguly, S. Mukherjee and P. Dastidar, Cryst. Growth Des., 2016, 16, 5247–5259 CrossRef CAS;
(c) T.-P. Hu, B. Zheng, X. Wang and X. Hao, CrystEngComm, 2015, 17, 9348–9356 RSC.
- X.-X. Wang, X.-Q. Wang, X.-Y. Niu and T.-P. Hu, CrystEngComm, 2016, 18, 7471–7477 RSC.
- H.-B. Zhou, K. Wu, C. Chen, R.-Y. Dong, Y.-S. Liu and X.-P. Shen, Eur. J. Inorg. Chem., 2017, 3946–3952 CrossRef CAS.
- S. Sujit, H. Susanta, K. Parimal, D. Supriya, R. Gopalan, S.-E. Carolina and M. Sasankasekhar, Inorg. Chem., 2011, 50, 7257–7267 CrossRef PubMed.
-
(a) J.-L. Dong, K.-H. He, D.-Z. Wang, Y.-H. Zhang and D.-H. Wang, J. Solid State Chem., 2018, 263, 164–171 CrossRef CAS;
(b) Z.-Y. Li, J.-W. Dai, M. Damjanovic, T. Shiga, J.-H. Wang, J. Zhao, H. Oshio, M. Yamashita and X.-H. Bu, Angew. Chem., Int. Ed., 2019, 58, 4339–4344 CrossRef CAS PubMed.
-
(a) B. Xia, Y. Zhou, Q.-L. Wang, X.-F. Xu, Y.-Z. Tong, X.-H. Bu and J.-R. Li, Dalton Trans., 2018, 47, 15888–15896 RSC;
(b) S.-D. Han, X.-H. Miao, S.-J. Liu and X.-H. Bu, Dalton Trans., 2015, 44, 560–567 RSC.
-
(a) B. Aguila, Q. Sun, X.-L. Wang, E. O'Rourke, A.-M. Al-Enizi, A. Nafady and S.-Q. Ma, Angew. Chem., Int. Ed., 2018, 57, 10107–10111 CrossRef CAS PubMed;
(b) X.-L. Yang, M.-H. Xie, C. Zou, Y.-B. He, B.-L. Chen, M. O'Keeffe and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 10638–10645 CrossRef CAS PubMed.
-
(a) S.-B. Zhou, X.-F. Wang, C.-C. Du, D.-Z. Wang and D.-Z. Jia, CrystEngComm, 2017, 19, 3124–3137 RSC;
(b) K. Fan, S.-S. Bao, W.-X. Nie, C.-H. Liao and L.-M. Zheng, Inorg. Chem., 2018, 57, 1079–1089 CrossRef CAS PubMed.
- X.-F. Wang, C.-C. Du, S.-B. Zhou and D.-Z. Wang, J. Mol. Struct., 2017, 1128, 103–110 CrossRef CAS.
- J.-P. Li, J.-Z. Fan and D.-Z. Wang, J. Solid State Chem., 2016, 239, 251–258 CrossRef CAS.
- V.-A. Blatov, and D.-M. Proserpio, TOPOS4.0, program package for multipurpose crystallochemical analysis, University of Milano, Italy, 2009 Search PubMed.
- G.-M. Sheldrick, SHELXS-2014, Program for the Crystal Structure Solution, University of Göttingen, Germany, 2014 Search PubMed.
- A.-R. Paital, W.-T. Wong, G. Aromí and D. Ray, Inorg. Chem., 2007, 46, 5727–5733 CrossRef CAS PubMed.
- B.-W. Hu, J.-P. Zhao, J. Tao, X.-J. Sun, Q. Yang, X.-F. Zhang and X.-H. Bu, Cryst. Growth Des., 2010, 10, 2829–2831 CrossRef CAS.
-
(a) C.-C. Du, X. F. Wang, S.-B. Zhou, D.-Z. Wang and D.-Z. Jia, CrystEngComm, 2017, 19, 6758–6777 RSC;
(b) J. Zhao, W.-W. Dong, Y.-P. Wu, Y.-N. Wang, C. Wang, D.-S. Li and Q.-C. Zhang, J. Mater. Chem. A, 2015, 3, 6962–6969 RSC.
- D.-Z. Wang, J.-Z. Fan, D.-Z. Jia and C.-C. Du, CrystEngComm, 2016, 18, 6708–6723 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1873981–1873986. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03082d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab