
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular docking, pharmacophore based virtual screening and molecular dynamics studies towards the identification of potential leads for the management of H. pylori†
Manoj G. Damalea,
Rajesh B. Patil*b,
Siddique Akber Ansaric,
Hamad M. Alkahtani c,
Abdulrahman A. Almehiziac,
Devanand B. Shinded,
Rohidas Arotee and
Jaiprakash Sangshetti*f
c,
Abdulrahman A. Almehiziac,
Devanand B. Shinded,
Rohidas Arotee and
Jaiprakash Sangshetti*f
aDepartment of Pharmaceutical Medicinal Chemistry, Srinath College of Pharmacy, Aurangabad, M.S. 431136, India
bSinhgad Technical Education Society's, Smt. Kashibai Navale College of Pharmacy, Kondhwa (Bk), Pune, India. E-mail: rajshama1@yahoo.com
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Po Box 2454, Riyadh 11451, Saudi Arabia
dShivaji University, Vidyanagar, Kolhapur 416004, MS, India
eDepartment of Molecular Genetics, School of Dentistry, Seoul National University, Seoul, Republic of Korea
fY. B. Chavan College of Pharmacy, Dr Rafiq Zakaria Campus, Rauza Baugh, Aurangabad, MS, India. E-mail: jnsangshetti@rediffmail.com
First published on 21st August 2019
Abstract
The enzyme pantothenate synthetase panC is one of the potential new antimicrobial drug targets, but it is poorly characterized in H. pylori. H. pylori infection can cause gastric cancer and the management of H. pylori infection is crucial in various gastric ulcers and gastric cancer. The current study describes the use of innovative drug discovery and design approaches like comparative metabolic pathway analysis (Metacyc), exploration of database of essential genes (DEG), homology modelling, pharmacophore based virtual screening, ADMET studies and molecular dynamics simulations in identifying potential lead compounds for the H. pylori specific panC. The top ranked virtual hits STOCK1N-60270, STOCK1N-63040, STOCK1N-44424 and STOCK1N-63231 can act as templates for synthesis of new H. pylori inhibitors and they hold a promise in the management of gastric cancers caused by H. pylori.
1 Introduction
The Helicobacter pylori infection in the patients with chronic gastritis and peptic ulcer can become the primary cause of gastric cancer.1–5 Gastric cancer is the fourth common malignancy worldwide causing over 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year. H. pylori is a microaerophilic, spiral-shaped Gram-negative bacterium which colonizes in the human stomach eventually causing duodenal and gastric ulcers. Broad spectrum antibacterials and antibiotics such as metronidazole, clarithromycin, levofloxacin, amoxicillin, tetracycline, furazolidone, and rifabutin are used in the treatment and management of H. pylori infection. Unfortunately, the efficacy of these antibiotics against H. pylori has weakened due to a strong resistance developed by H. pylori organism.6,7 Furthermore, many factors such as the strain of H. pylori, the host genetic factor like polymorphism in the interleukin-1, gender, and individual's habits like smoking and their diet may aggravate the H. pylori infection. It is established that the colonization of the H. pylori with the nitro sating bacteria in the achlorhydric stomach becomes the primary cause of gastric cancer.8 Therefore, eradication of the H. pylori infection and proper management and treatment of the duodenal and gastric ulcers is essential in the prevention of ensuing gastric cancer. Emergence of strong resistance is the main concern with most of the currently used broad spectrum antibacterials and antibiotics in H. pylori infection. Hence, design and development of the newer potential drug candidates effective against the newer targets specific for H. pylori may be advantageous. The enzyme pantothenate synthetase, encoded by the panC gene, catalyzes the biosynthesis of pantothenate (vitamin B5) from an adenosine triphosphate (ATP)-dependent condensation of the D-pantoate and the β-alanine in bacteria.9 The pantothenate is a key precursor of the coenzyme A and the acyl carrier protein. Many intracellular processes such as fatty acid metabolism, cell signaling, synthesis of the polypeptides and the non-ribosomal peptides are regulated by the coenzyme A and the acyl carrier protein. Interestingly, mammals lack the pantothenate synthetase and its biosynthetic pathway and derive the pantothenate from their diet.10 Thus, it is advantageous to target bacterium specific pantothenate synthetase in the development of potential inhibitors. Many experimental crystal structures of pantothenate synthetase are available which could be exploited in design and development of its potential inhibitors. But, most of these crystal structures and inhibitors are reported for the Mycobacterium tuberculosis. To best of our knowledge the H. pylori specific panC inhibitors has not been reported so far and may be due to the unavailability of the experimental crystal structure of the H. pylori specific pantothenate synthetase. The complete genome sequencing has identified the key proteins of H. pylori.11–13 The European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI)14 has assigned the accession number to gene of H. pylori panC as EBI-7515141 and the details of the interactions and protein features are available in UniProtKB database with ID P56061.15 In absence of the experimentally solved crystal structure, homology modeling is the most reliable method to construct the theoretical models of proteins under study.16 The homology modeling derived validated theoretical models of proteins can be used in molecular docking studies, which is the most popular technique to understand the binding mode and the affinities of ligands at the binding site of such proteins.17 In fact, such molecular modeling approaches has lead to identification of many potential anti-cancer agents.18–22 Furthermore, the best binding modes of the ligands with high affinity and the structural traits of such ligands can be used to build the pharmacophore.23,24 The features in such pharmacophore are usually exploited in the virtual screening which is an efficient and alternative approach to the High Throughput Screening to derive the promising hits.25–27 The deeper insights into the binding modes and energetic of such hits at the binding site of the modeled protein in simulated biological environment can be achieved through molecular dynamics simulations. The effects and risk of the promising hits can be accessed through the prediction of absorption, distribution, metabolism, elimination and toxicity (ADMET) parameters which aid in the design of drug like compounds.28,29 The fact that the inhibitors of the H. pylori specific pantothenate synthetase could be the best way to tackle the problem of poor prognosis of the H. pylori infection and the associated risk of the gastric cancer provoked us to investigate the computational aspects of this issue and provide the possible candidate molecules as inhibitors. Thus, the present work describes the homology modeling of the H. pylori specific pantothenate synthetase, the molecular docking studies with the known inhibitors, the pharmacopore development from the best docked poses of the top rank compounds, pharmacophore based virtual screening, the ADMET studies and the detail investigation of the possible mechanism of inhibition through the molecular dynamics simulations of the best hit molecules.
000 deaths per year. H. pylori is a microaerophilic, spiral-shaped Gram-negative bacterium which colonizes in the human stomach eventually causing duodenal and gastric ulcers. Broad spectrum antibacterials and antibiotics such as metronidazole, clarithromycin, levofloxacin, amoxicillin, tetracycline, furazolidone, and rifabutin are used in the treatment and management of H. pylori infection. Unfortunately, the efficacy of these antibiotics against H. pylori has weakened due to a strong resistance developed by H. pylori organism.6,7 Furthermore, many factors such as the strain of H. pylori, the host genetic factor like polymorphism in the interleukin-1, gender, and individual's habits like smoking and their diet may aggravate the H. pylori infection. It is established that the colonization of the H. pylori with the nitro sating bacteria in the achlorhydric stomach becomes the primary cause of gastric cancer.8 Therefore, eradication of the H. pylori infection and proper management and treatment of the duodenal and gastric ulcers is essential in the prevention of ensuing gastric cancer. Emergence of strong resistance is the main concern with most of the currently used broad spectrum antibacterials and antibiotics in H. pylori infection. Hence, design and development of the newer potential drug candidates effective against the newer targets specific for H. pylori may be advantageous. The enzyme pantothenate synthetase, encoded by the panC gene, catalyzes the biosynthesis of pantothenate (vitamin B5) from an adenosine triphosphate (ATP)-dependent condensation of the D-pantoate and the β-alanine in bacteria.9 The pantothenate is a key precursor of the coenzyme A and the acyl carrier protein. Many intracellular processes such as fatty acid metabolism, cell signaling, synthesis of the polypeptides and the non-ribosomal peptides are regulated by the coenzyme A and the acyl carrier protein. Interestingly, mammals lack the pantothenate synthetase and its biosynthetic pathway and derive the pantothenate from their diet.10 Thus, it is advantageous to target bacterium specific pantothenate synthetase in the development of potential inhibitors. Many experimental crystal structures of pantothenate synthetase are available which could be exploited in design and development of its potential inhibitors. But, most of these crystal structures and inhibitors are reported for the Mycobacterium tuberculosis. To best of our knowledge the H. pylori specific panC inhibitors has not been reported so far and may be due to the unavailability of the experimental crystal structure of the H. pylori specific pantothenate synthetase. The complete genome sequencing has identified the key proteins of H. pylori.11–13 The European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI)14 has assigned the accession number to gene of H. pylori panC as EBI-7515141 and the details of the interactions and protein features are available in UniProtKB database with ID P56061.15 In absence of the experimentally solved crystal structure, homology modeling is the most reliable method to construct the theoretical models of proteins under study.16 The homology modeling derived validated theoretical models of proteins can be used in molecular docking studies, which is the most popular technique to understand the binding mode and the affinities of ligands at the binding site of such proteins.17 In fact, such molecular modeling approaches has lead to identification of many potential anti-cancer agents.18–22 Furthermore, the best binding modes of the ligands with high affinity and the structural traits of such ligands can be used to build the pharmacophore.23,24 The features in such pharmacophore are usually exploited in the virtual screening which is an efficient and alternative approach to the High Throughput Screening to derive the promising hits.25–27 The deeper insights into the binding modes and energetic of such hits at the binding site of the modeled protein in simulated biological environment can be achieved through molecular dynamics simulations. The effects and risk of the promising hits can be accessed through the prediction of absorption, distribution, metabolism, elimination and toxicity (ADMET) parameters which aid in the design of drug like compounds.28,29 The fact that the inhibitors of the H. pylori specific pantothenate synthetase could be the best way to tackle the problem of poor prognosis of the H. pylori infection and the associated risk of the gastric cancer provoked us to investigate the computational aspects of this issue and provide the possible candidate molecules as inhibitors. Thus, the present work describes the homology modeling of the H. pylori specific pantothenate synthetase, the molecular docking studies with the known inhibitors, the pharmacopore development from the best docked poses of the top rank compounds, pharmacophore based virtual screening, the ADMET studies and the detail investigation of the possible mechanism of inhibition through the molecular dynamics simulations of the best hit molecules.
2 Materials and methods
2.1 Homology modeling
The H. pylori specific metabolic pathways were identified from the MetaCyc Metabolic Pathway Database (https://metacyc.org/).30–32 The key enzymes in these pathways were further indentified with the help of choke point finder tool available in this database. The database of essential genes (DEG) (http://www.essentialgene.org)33 was also explored to identify the essential genes and the corresponding metabolic enzymes in the Helicobacter pylori. The sequence of the key metabolic enzymes thus identified was retrieved from the Uniprot database (https://www.uniprot.org/).15,34 This protein sequence was subjected to the BLASTP search in order to identify the matching protein.35 These preliminary investigations lead to the identification of pantothenate synthetase, the unique protein specific for the H. pylori. The primary sequence of the panC specific for the H. pylori (Uniprot accession number P56061) was subjected to the similarity search in the Molecular Operating Environment (MOE).36 The similarity search was executed by transferring the sequence in the sequence query and with the search parameters set to the values for gap start −12, gap extend −2, E-value cutoff 10, E-value accept 0.5, Z iterations 100 and Z cutoff 6. Subsequently, the MOE homology model tool linked with Generalized Born/Volume Integral (GB/VI) scoring function and Amber99 force field was used to generate the homology models.37 MOE's site finder promodel module was employed to identify and validate the binding site. In this module the atoms on the protein's surface were identified, and the centers of spheres defined by combinations of four such points are marked. The clusters of such spheres defined the potential binding sites on the protein's surface, or voids within it. The likelihood of a hydrophilic contact was marked and this method identified 10 potential binding sites amongst which the first was used in the docking studies. Further validation of these models based on various computational approaches was carried out and the most suitable model was used in docking studies.2.2 Molecular docking
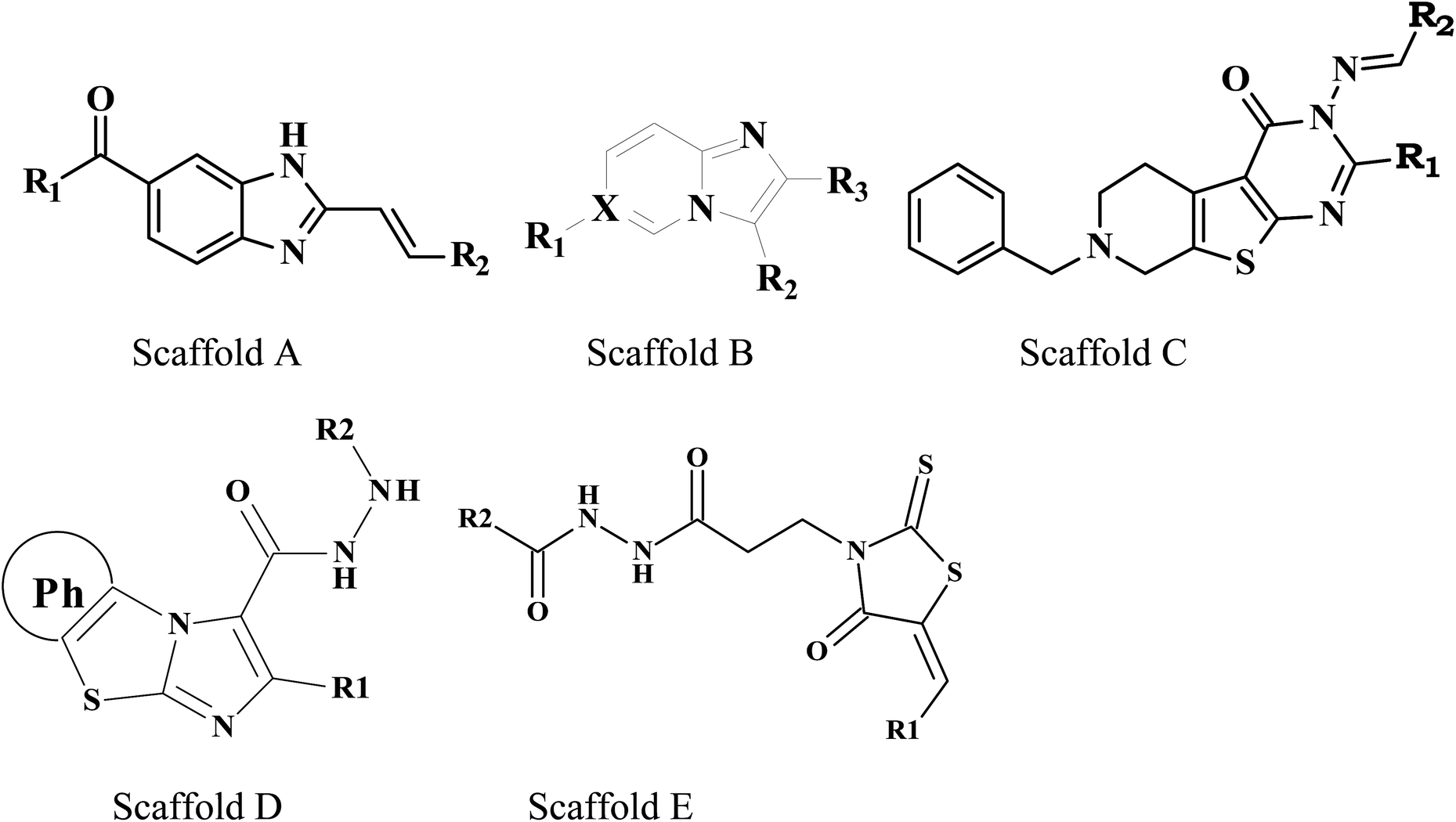
Molecular docking studies were carried out on MOE molecular modeling suite. The validated homology model structure was subjected to the structure preparation wizard to curate the inaccuracies in the protein structure using the LigX standard protocol. The promodel module was used for the active site prediction. One hundred fifteen already known M. tuberculosis PanC inhibitors38–42 were used for the docking studies (Table 1).
|
|||||
|---|---|---|---|---|---|
| Compound no. | Scaffold | R1 | R2 | R3 | X |
| 1 | A | 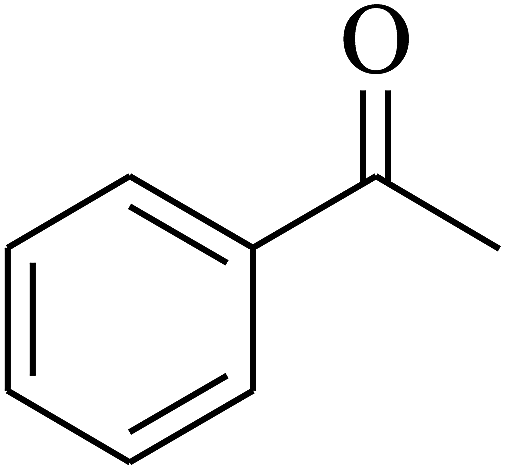 |
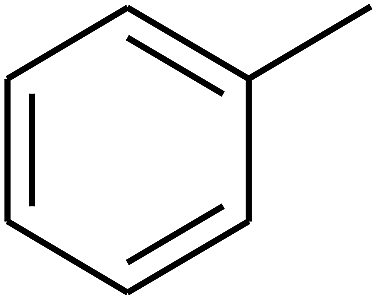 |
— | — |
| 2 | A | 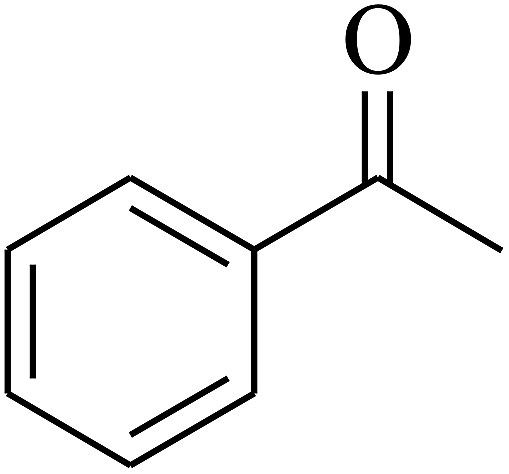 |
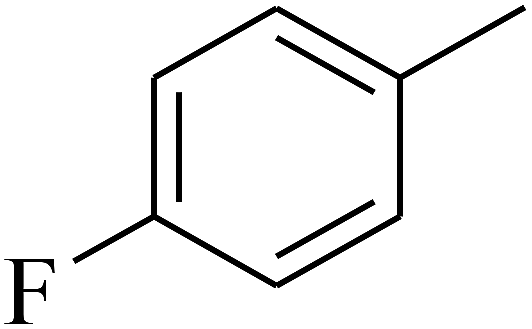 |
— | — |
| 3 | A | 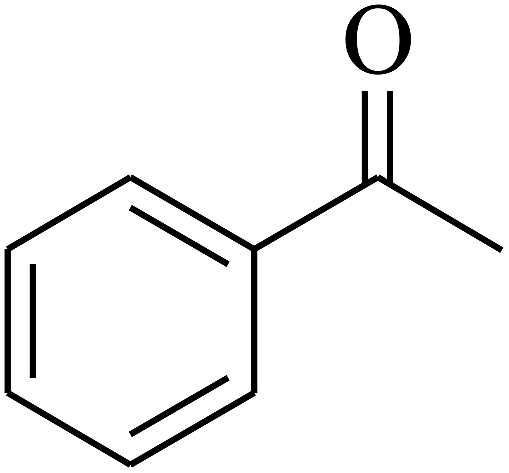 |
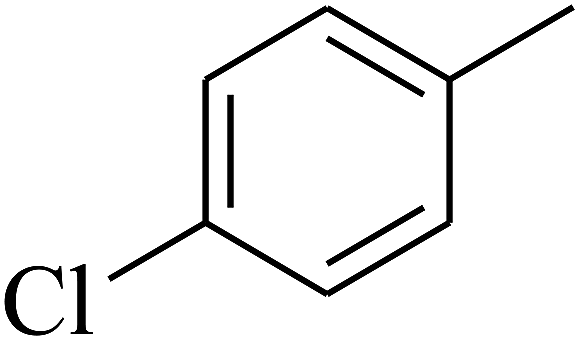 |
— | — |
| 4 | A | 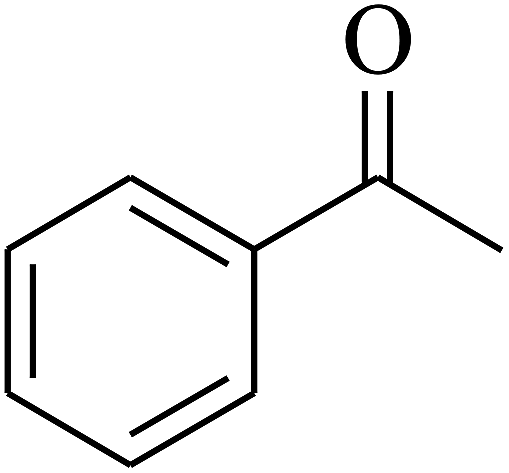 |
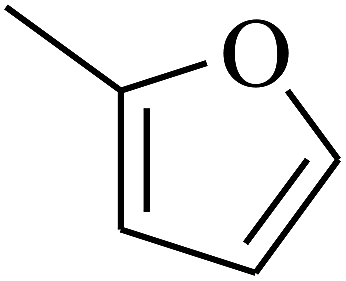 |
— | — |
| 5 | A | 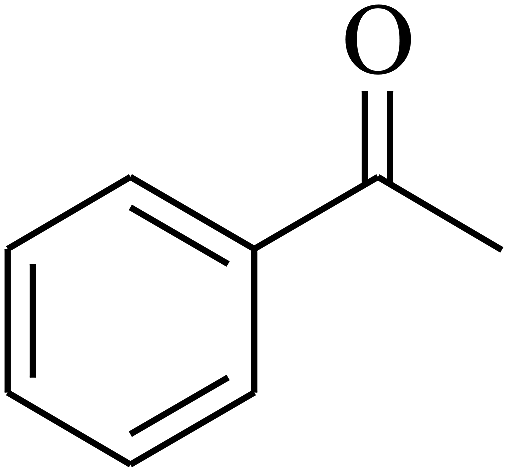 |
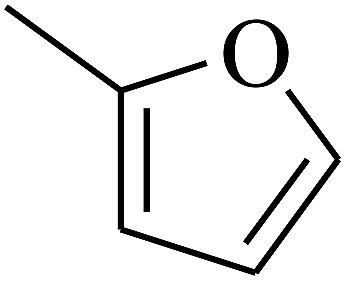 |
— | — |
| 6 | A | 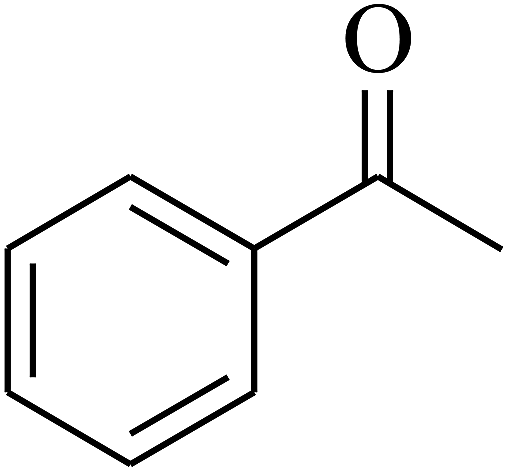 |
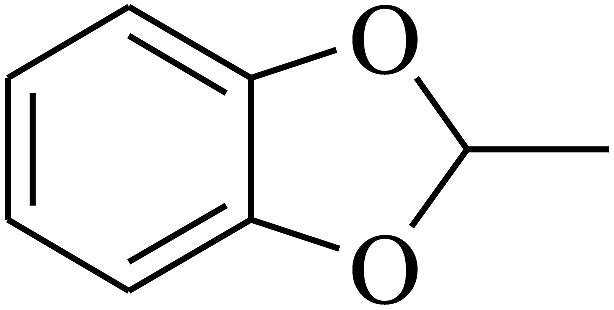 |
— | — |
| 7 | A | 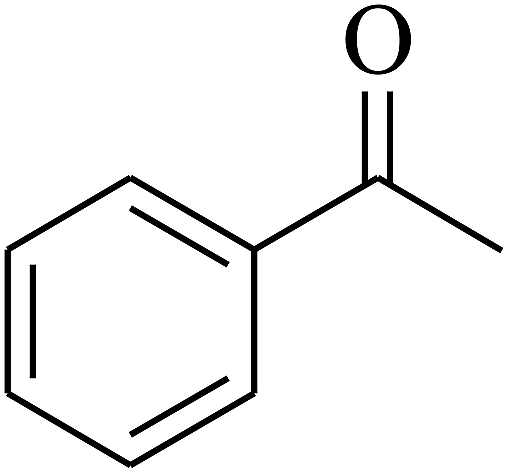 |
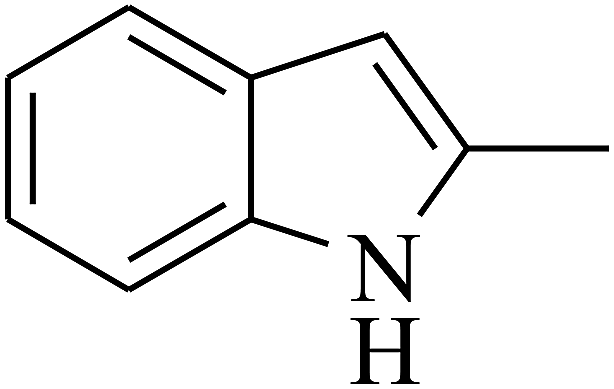 |
— | — |
| 8 | A | 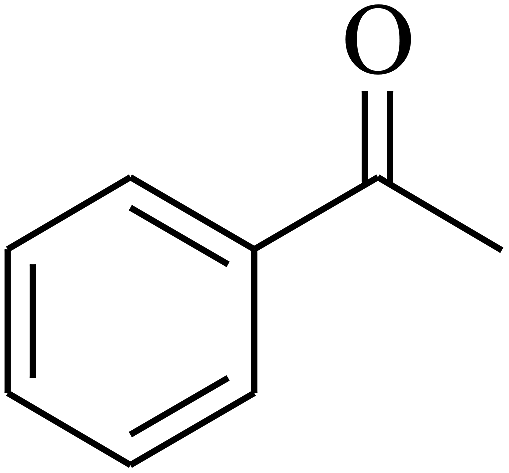 |
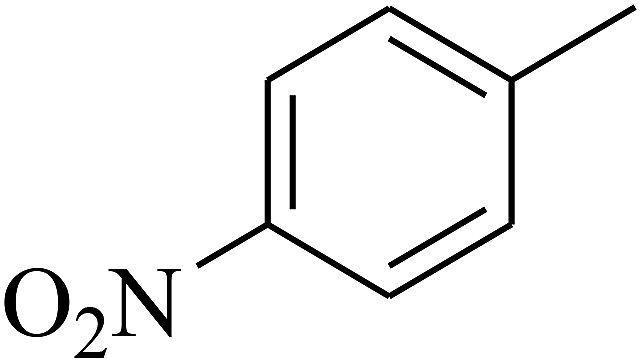 |
— | — |
| 9 | A | 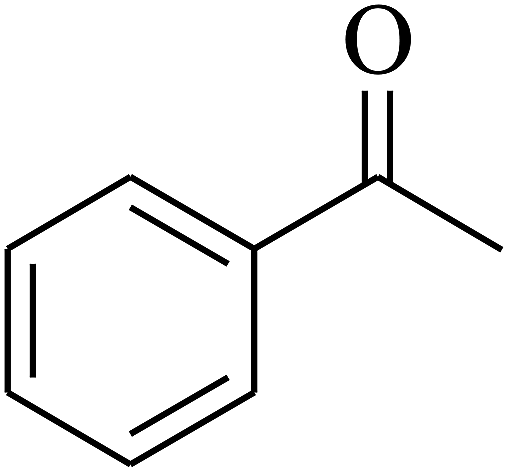 |
 |
— | — |
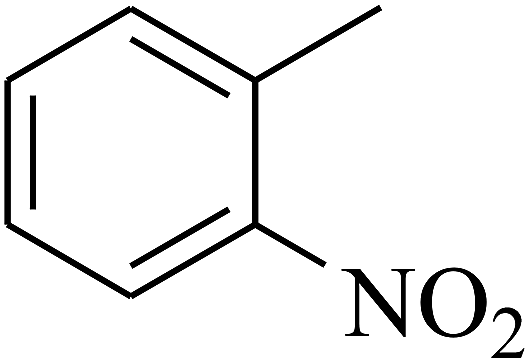
| 10 | A | –NO2 | 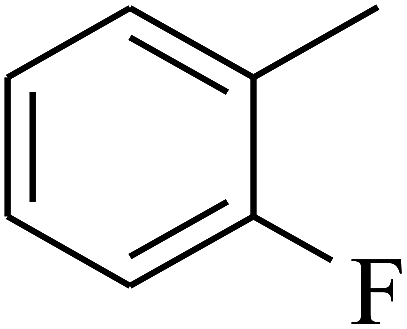 |
— | — |
| 11 | A | –H | –H | — | — |
| 12 | A | –NO2 | 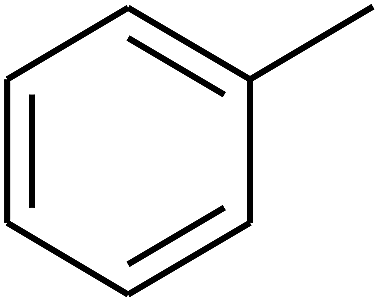 |
— | — |
| 13 | A | –NO2 | 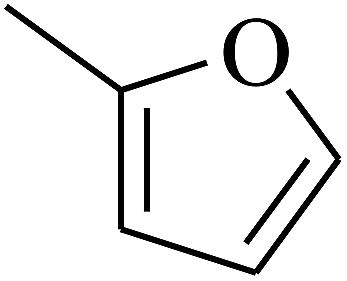 |
— | — |
| 14 | A | –NO2 | 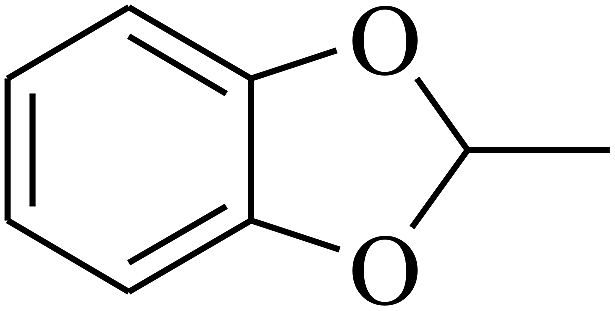 |
— | — |
| 15 | B | –H | (CH3)3CNH– | 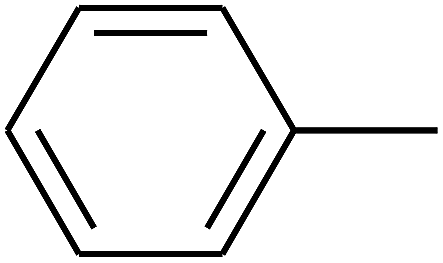 |
C |
| 16 | B | CH3 | (CH3)3CNH– | 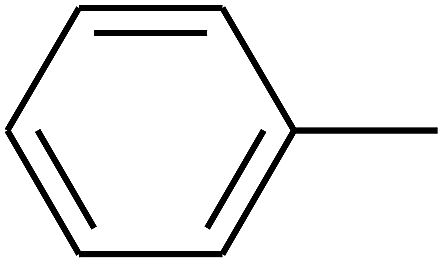 |
C |
| 17 | B | — | (CH3)3CNH– | 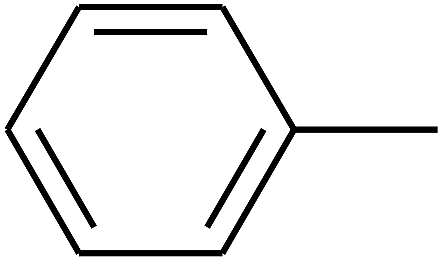 |
N |
| 18 | B | –H | 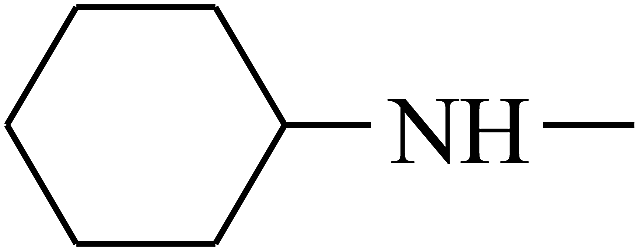 |
 |
C |
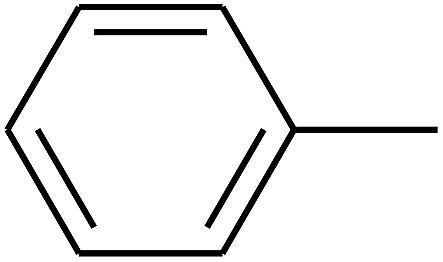
| 19 | B | CH3 | 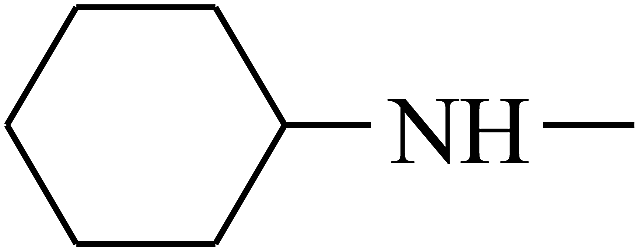 |
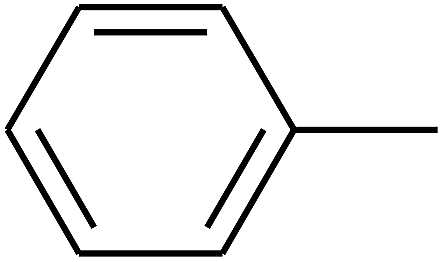 |
C |
| 20 | B | — | 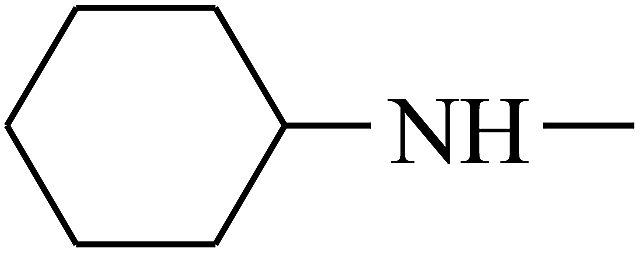 |
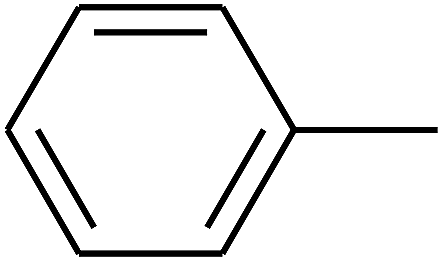 |
N |
| 21 | B | –H | (CH3)3CNH– | 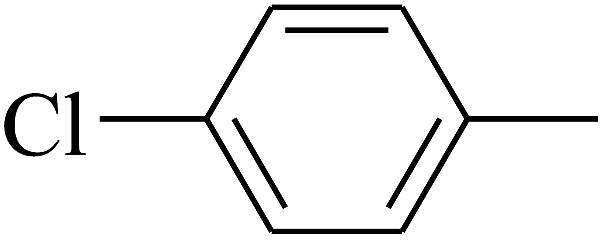 |
C |
| 22 | B | CH3 | (CH3)3CNH– |  |
C |
| 23 | B | — | (CH3)3CNH– |  |
N |
| 24 | B | –H |  |
 |
C |
| 25 | B | CH3 | 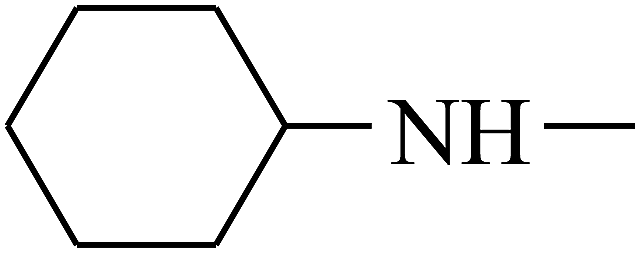 |
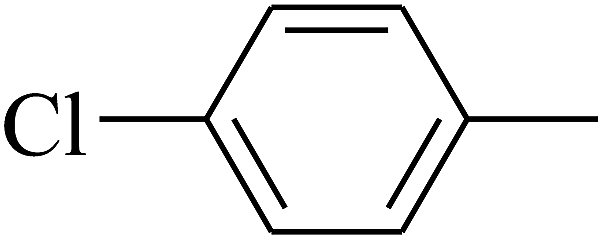 |
C |
| 26 | B | — | 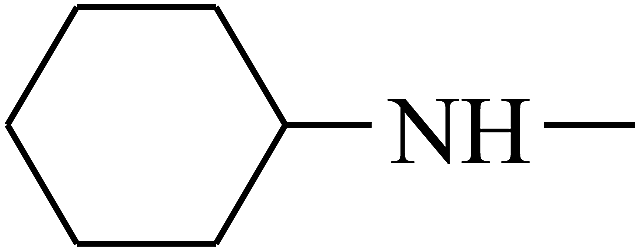 |
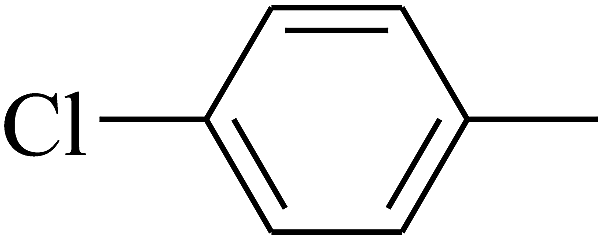 |
N |
| 27 | B | –H | (CH3)3CNH– | 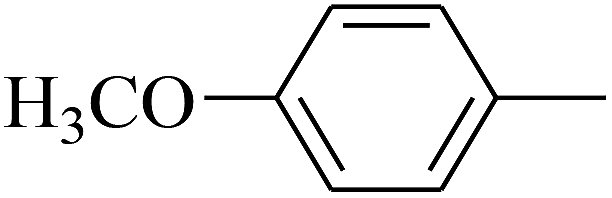 |
C |
| 28 | B | CH3 | (CH3)3CNH– | 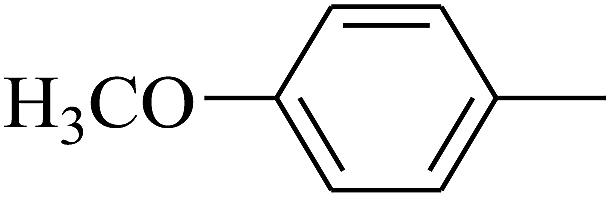 |
C |
| 29 | B | — | (CH3)3CNH– | 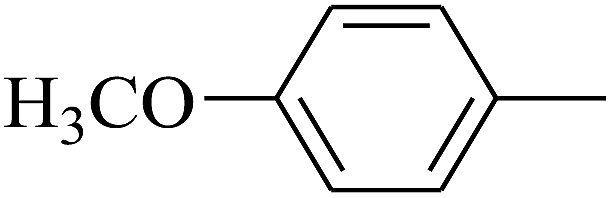 |
N |
| 30 | B | –H | 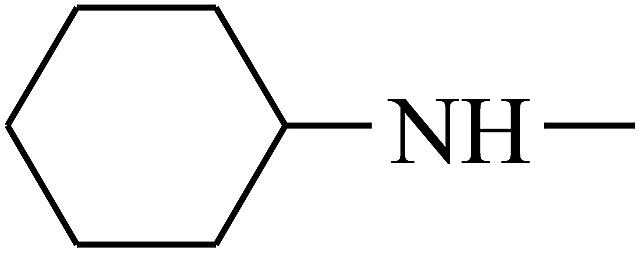 |
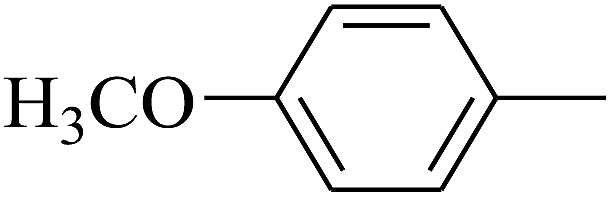 |
C |
| 31 | B | CH3 | 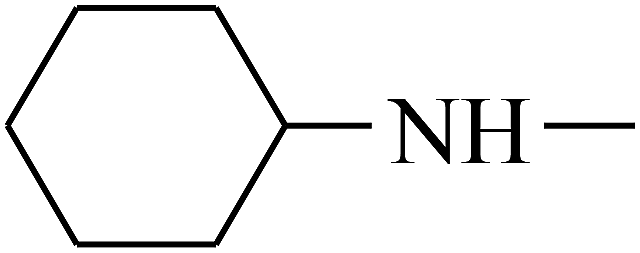 |
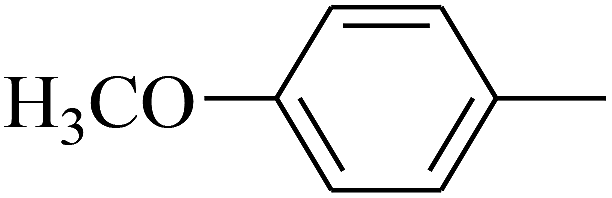 |
C |
| 32 | B | — | 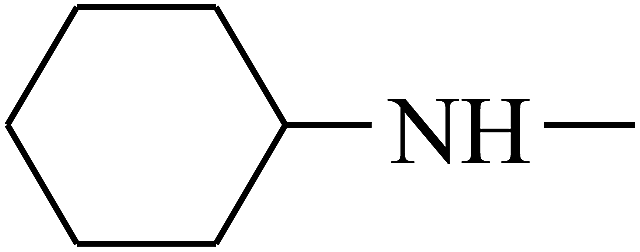 |
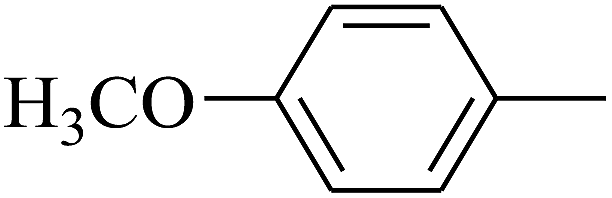 |
N |
| 33 | B | –H | (CH3)3CNH– | H | C |
| 34 | B | CH3 | (CH3)3CNH– | H | C |
| 35 | B | — | (CH3)3CNH– | H | N |
| 36 | B | –H | 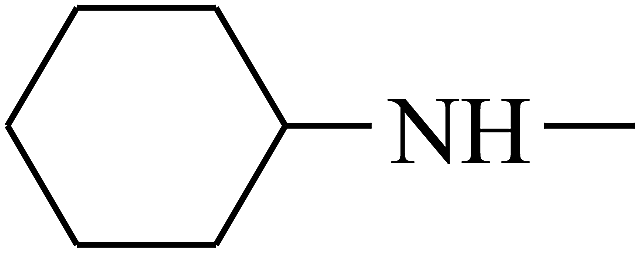 |
H | C |
| 37 | B | CH3 | 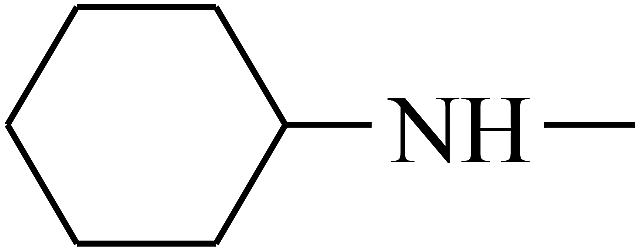 |
H | C |
| 38 | B | — | 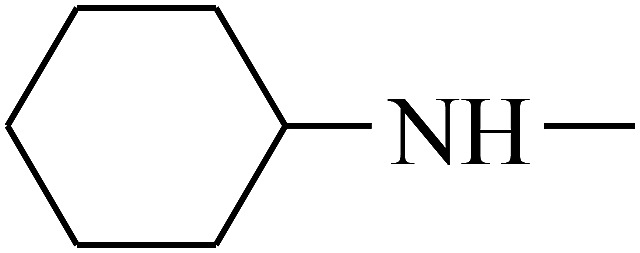 |
H | N |
| 39 | B | –H | (CH3)3CNH– | 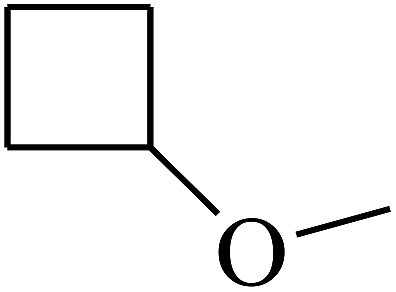 |
C |
| 40 | B | –H | (CH3)3CNH– | 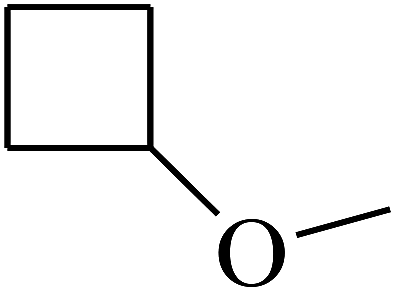 |
C |
| 41 | B | — | (CH3)3CNH– | 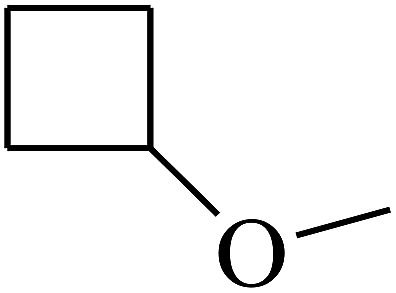 |
N |
| 42 | B | –H | 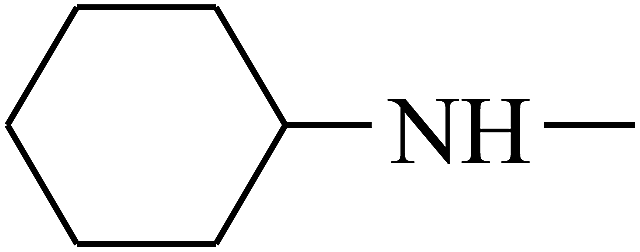 |
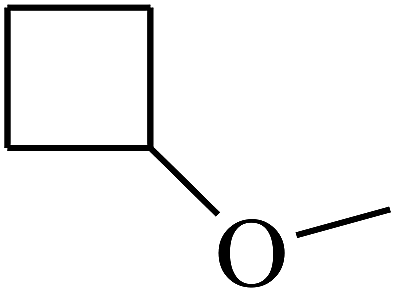
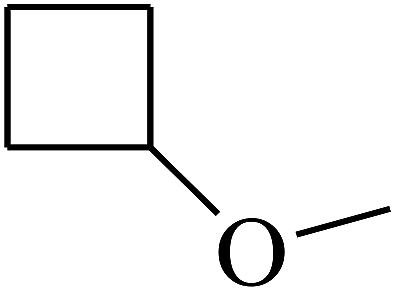 |
C |
| 43 | B | CH3 | 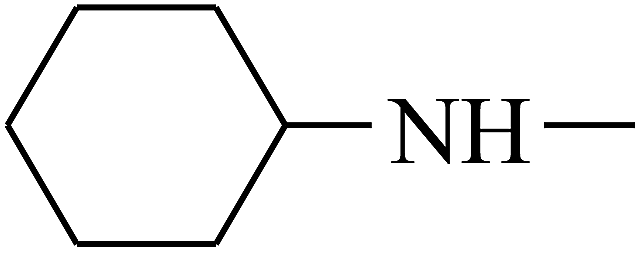 |
 |
C |
| 44 | B | — | 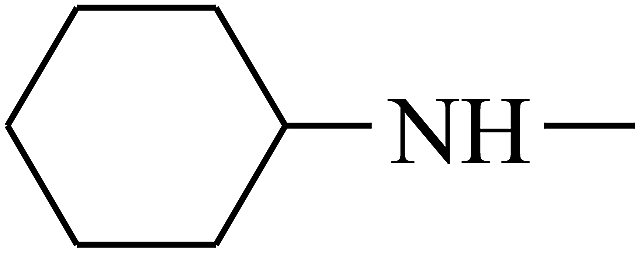 |
 |
N |
| 45 | C | –H |  |
— | — |
| 46 | C | –H |  |
— | — |
| 47 | C | –H |  |
— | — |
| 48 | C | –H | 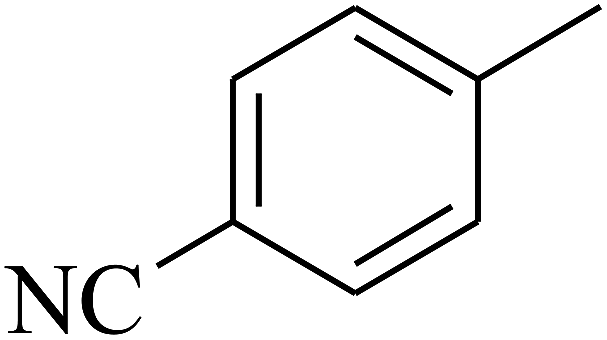 |
— | — |
| 49 | C | –H | 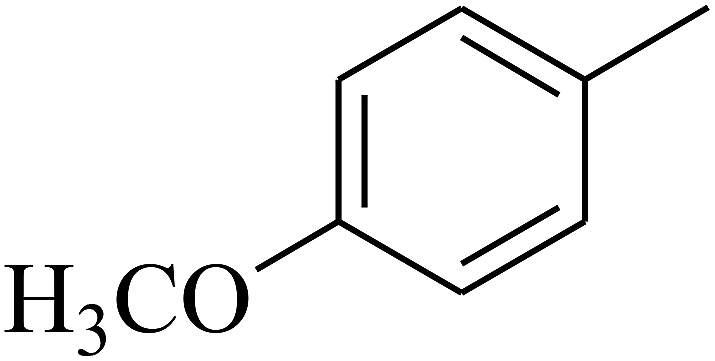 |
— | — |
| 50 | C | –H | 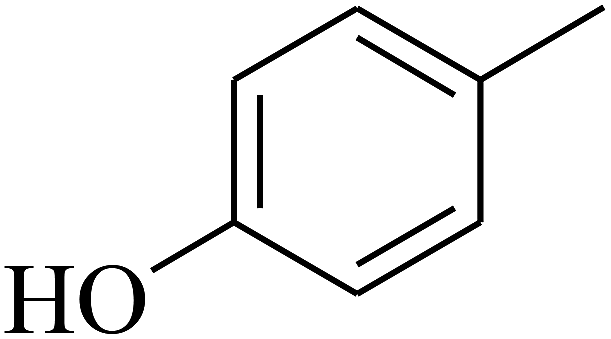 |
— | — |
| 51 | C | –H | 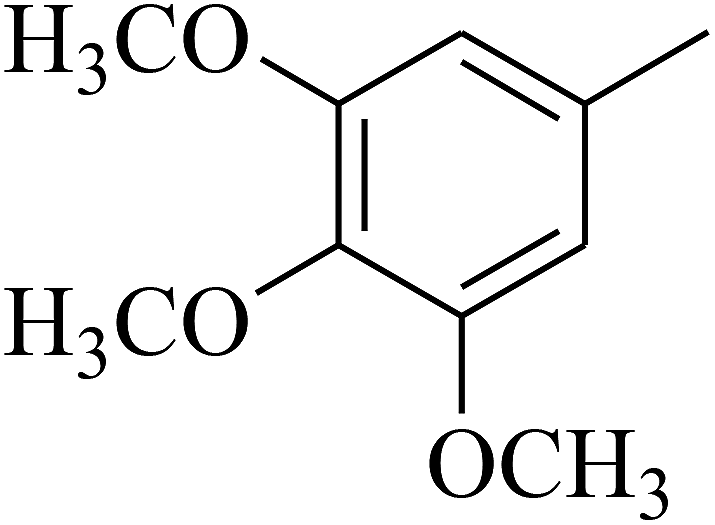 |
— | — |
| 52 | C | –H | 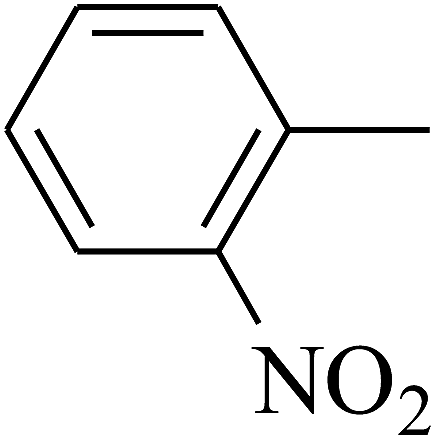 |
— | — |
| 53 | C | –H | 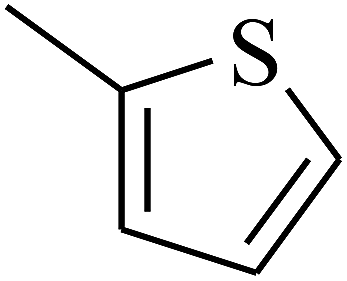 |
— | — |
| 54 | C | –H | 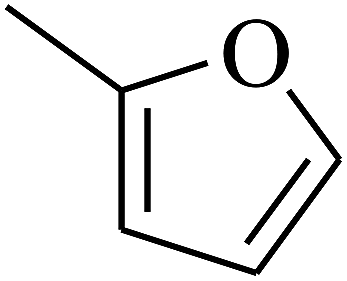 |
— | — |
| 55 | C | –H | 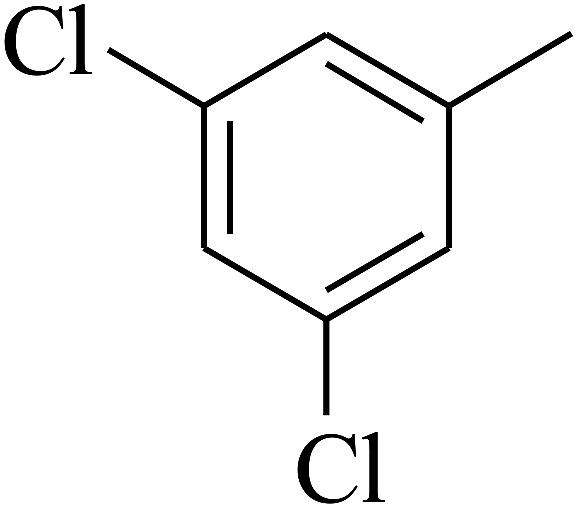 |
— | — |
| 56 | C | –CH3 | 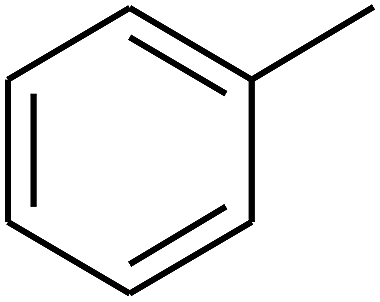 |
— | — |
| 57 | C | –CH3 | 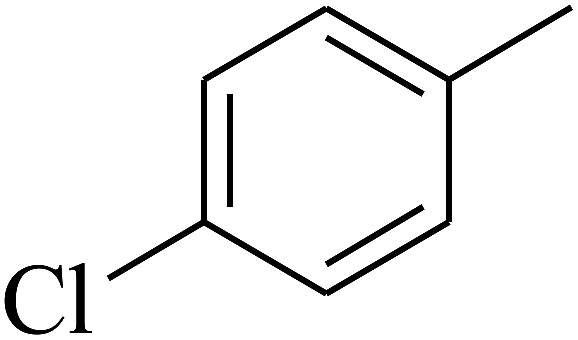 |
— | — |
| 58 | C | –CH3 | 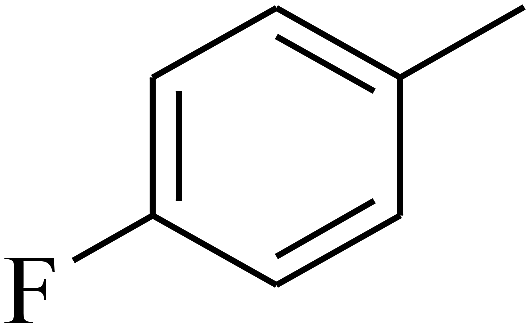 |
— | — |
| 59 | C | –CH3 | 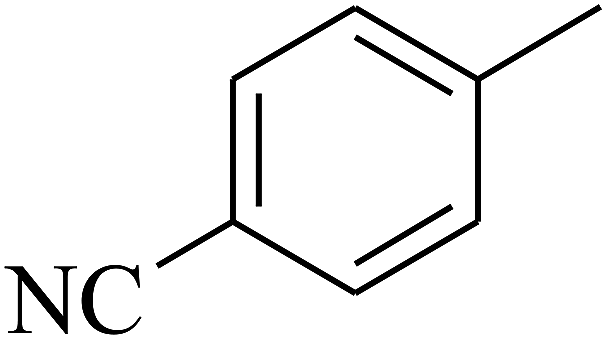 |
— | — |
| 60 | C | –CH3 | 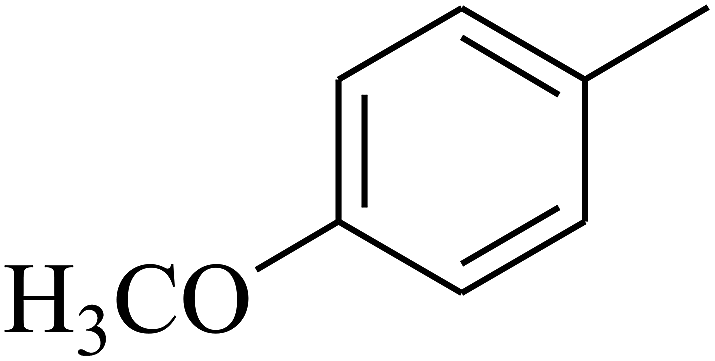 |
— | — |
| 61 | C | –CH3 | 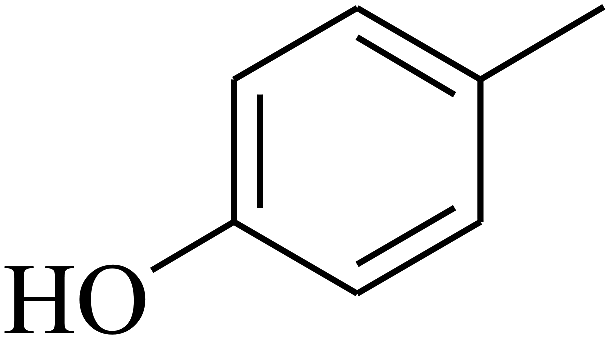 |
— | — |
| 62 | C | –CH3 | 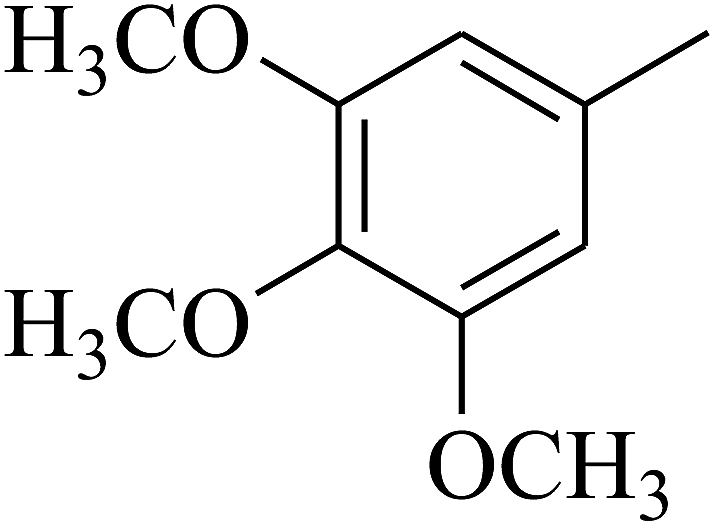 |
— | — |
| 63 | C | –CH3 | 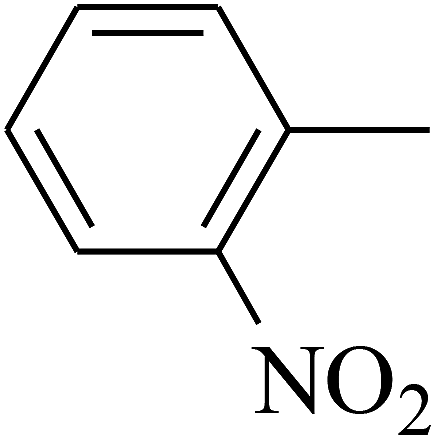 |
— | — |
| 64 | C | –CH3 | 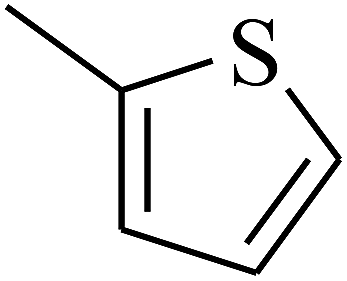 |
— | — |
| 65 | C | –CH3 | 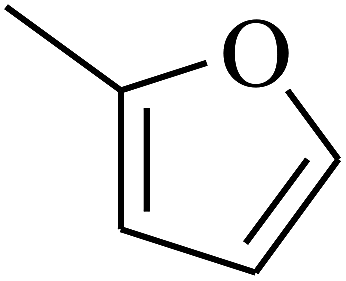 |
— | — |
| 66 | C | –CH3 | 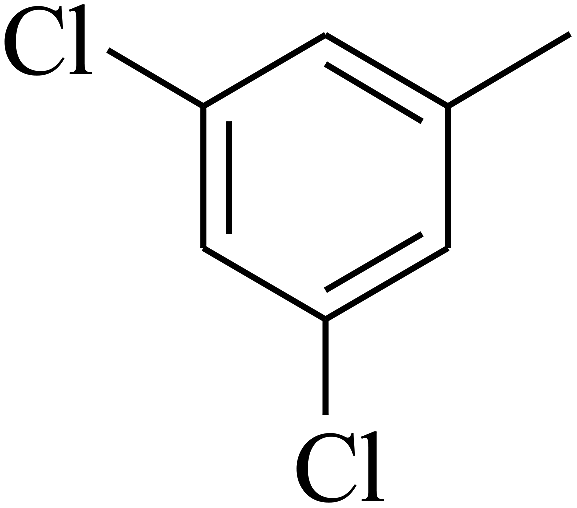 |
— | — |
| 67 | D | –H | 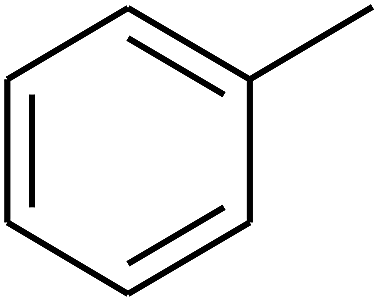 |
— | — |
| 68 | D | –H |  |
— | — |
| 69 | D | –H |  |
— | — |
| 70 | D | –H |  |
— | — |
| 71 | D | –H |  |
— | — |
| 72 | D | –CH3 | 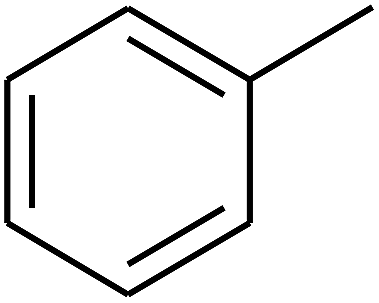 |
— | — |
| 73 | D | –CH3 | 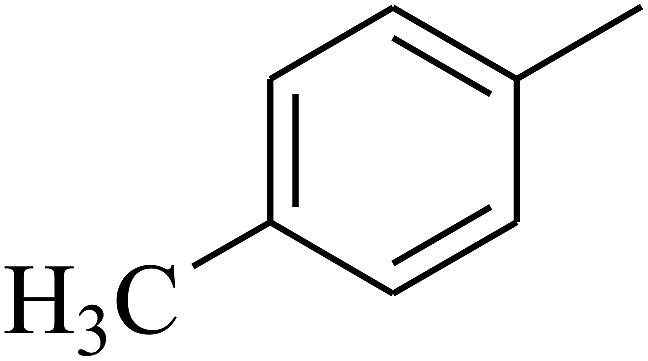 |
— | — |
| 74 | D | –CH3 | 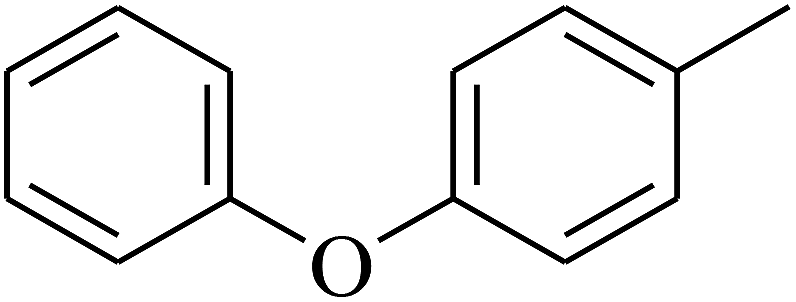 |
— | — |
| 75 | D | –CH3 | 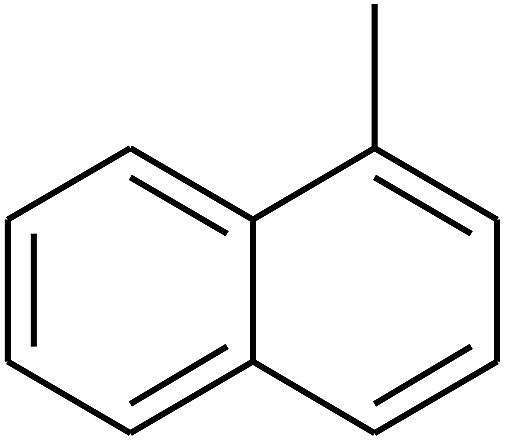 |
— | — |
| 76 | D | –CH3 | 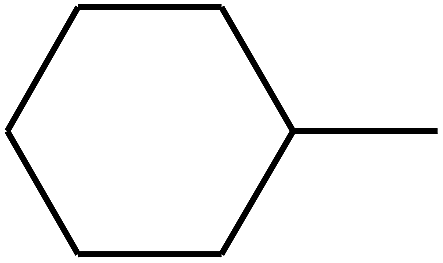 |
— | — |
| 77 | D | –Cl | 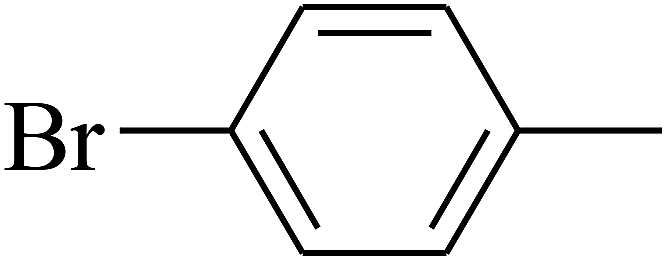 |
— | — |
| 78 | D | –Cl | 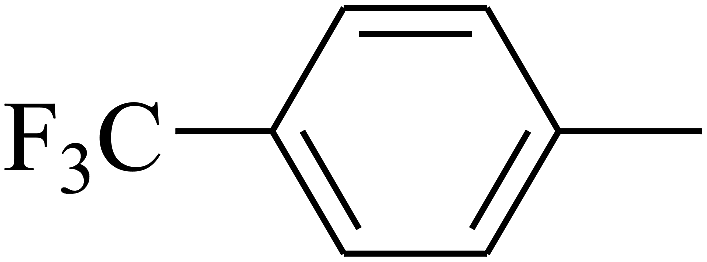 |
— | — |
| 79 | D | –Cl | 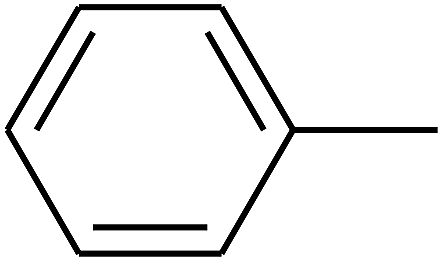 |
— | — |
| 80 | D | –Cl | 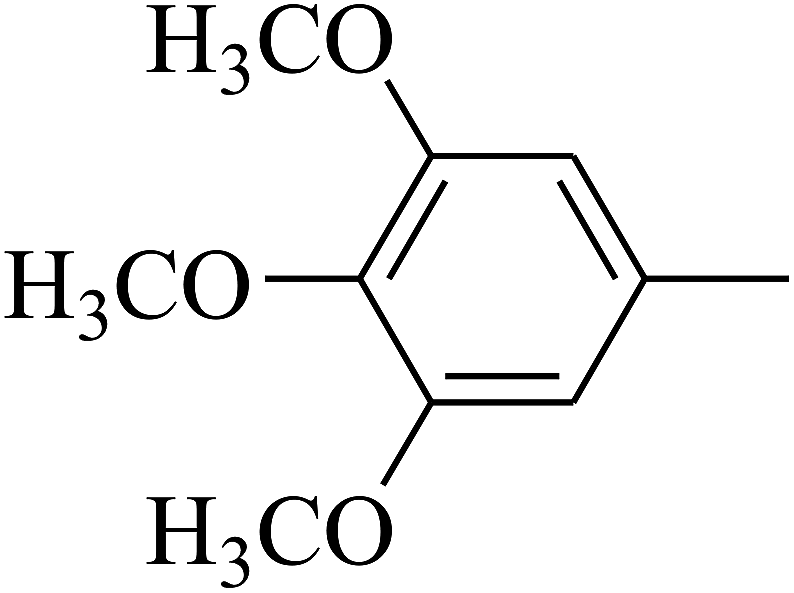 |
— | — |
| 81 | D | –Cl | 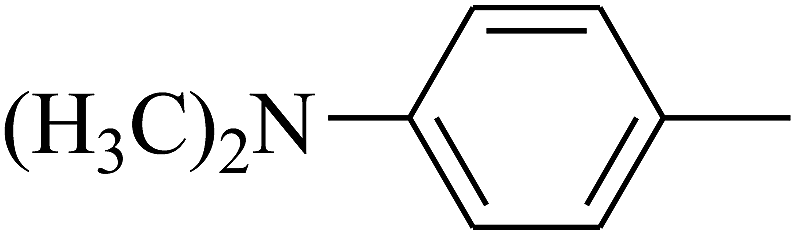 |
— | — |
| 82 | D | –Br | 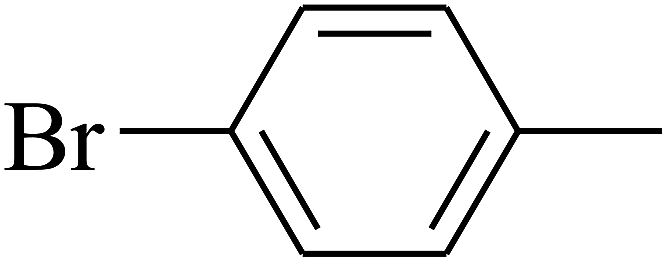 |
— | — |
| 83 | D | –Br | 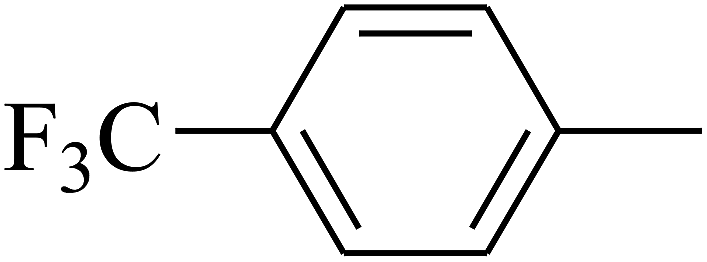 |
— | — |
| 84 | D | –Br | 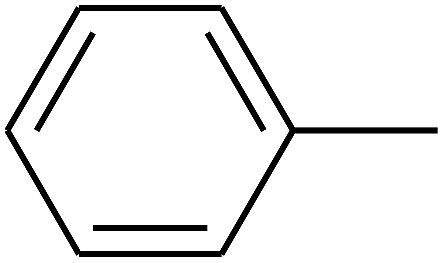 |
— | — |
| 85 | D | –Br | 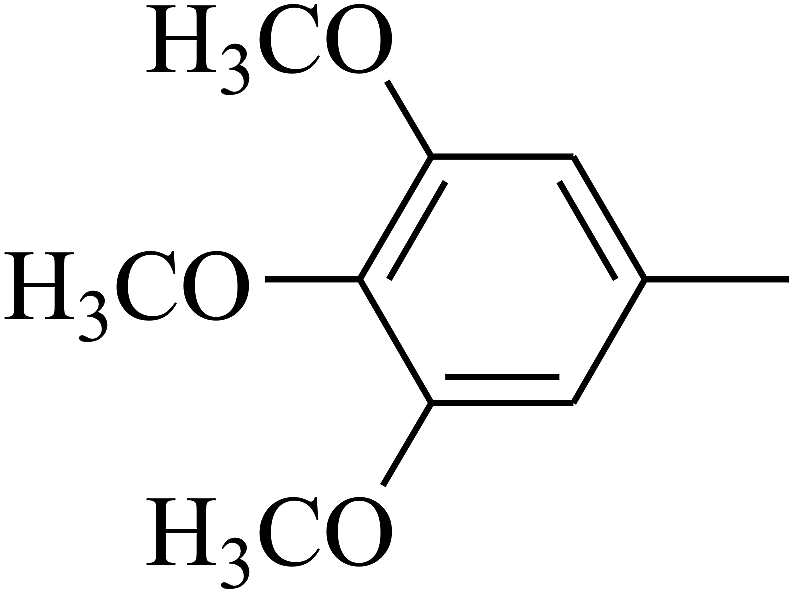 |
— | — |
| 86 | D | –Br | 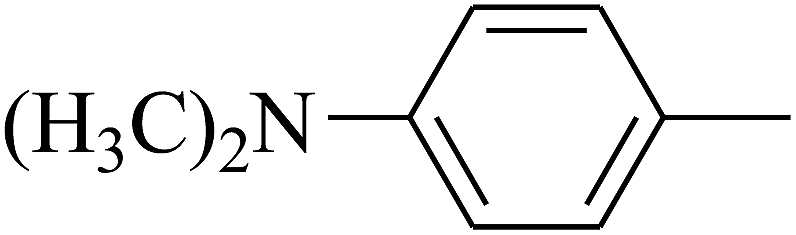 |
— | — |
| 87 | D | –C2H5 | 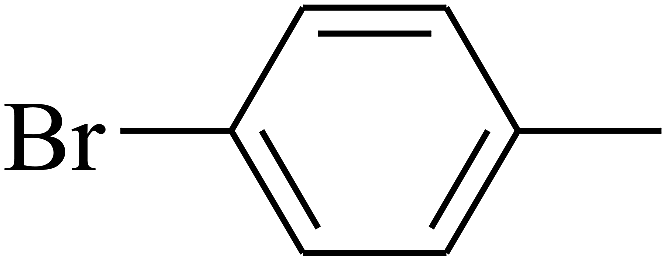 |
— | — |
| 88 | D | –C2H5 | 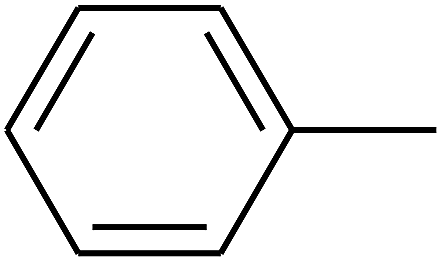 |
— | — |
| 89 | D | –C2H5 | 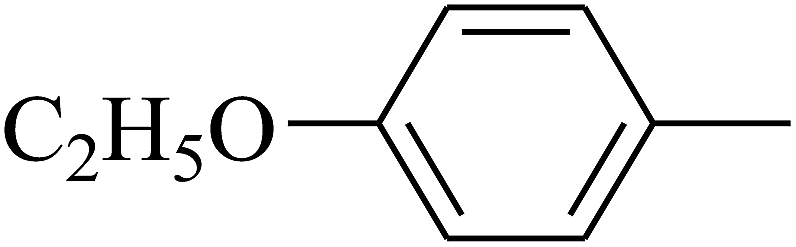 |
— | — |
| 90 | D | –C2H5 | 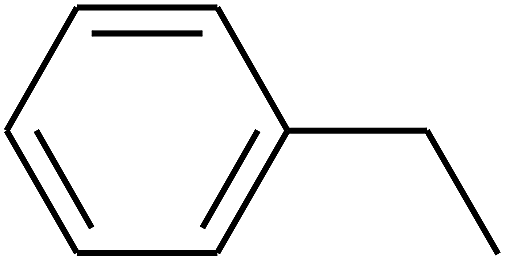 |
— | — |
| 91 | D | –C2H5 | 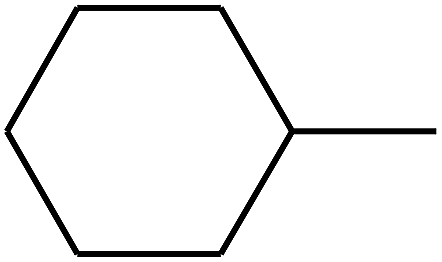 |
— | — |
| 92 | D | –C3H7 | 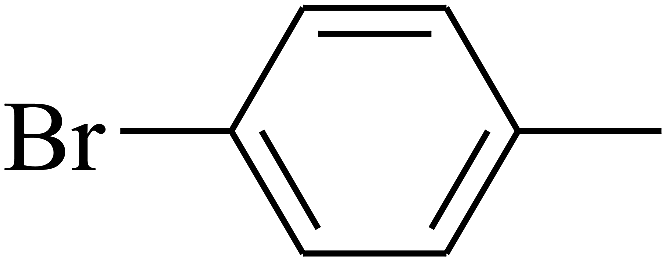 |
— | — |
| 93 | D | –C3H7 | 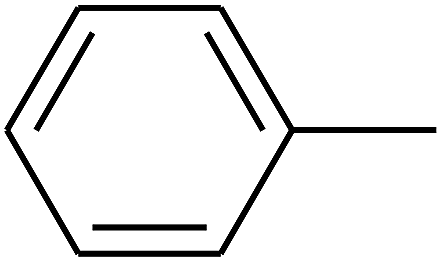 |
— | — |
| 94 | D | –C3H7 | 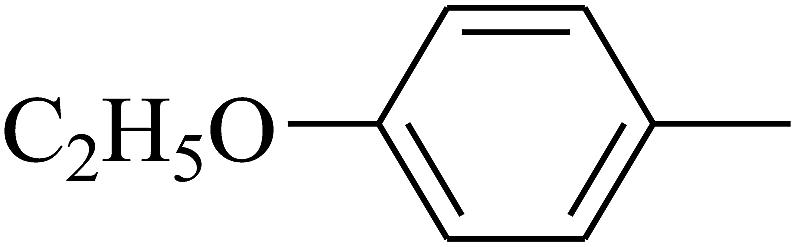 |
— | — |
| 95 | D | –C3H7 | 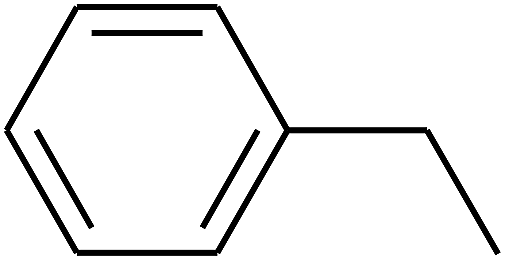 |
— | — |
| 96 | D | –C3H7 | 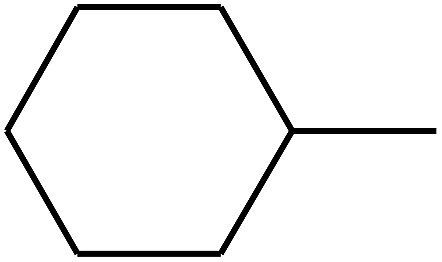 |
— | — |
| 97 | E | 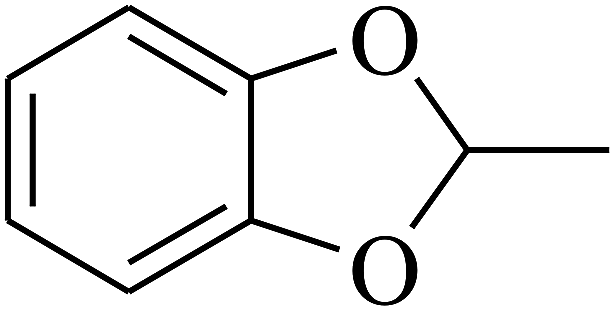 |
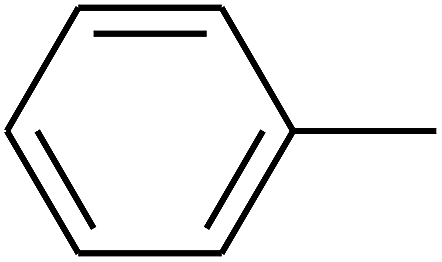 |
— | — |
| 98 | E | 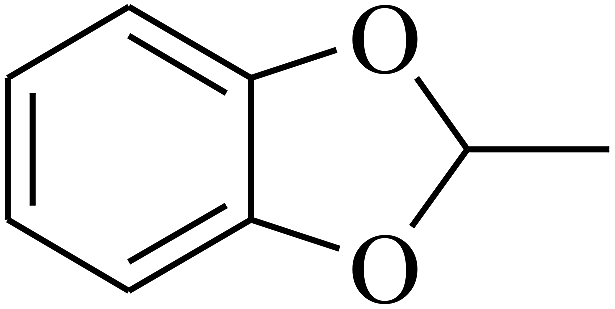 |
 |
— | — |
| 99 | E |  |
 |
— | — |
| 100 | E |  |
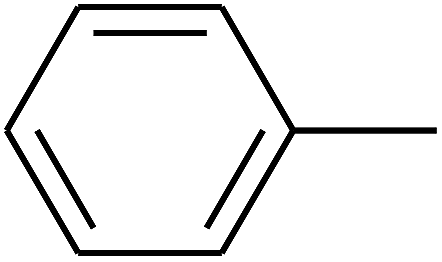 |
— | — |
| 101 | E | 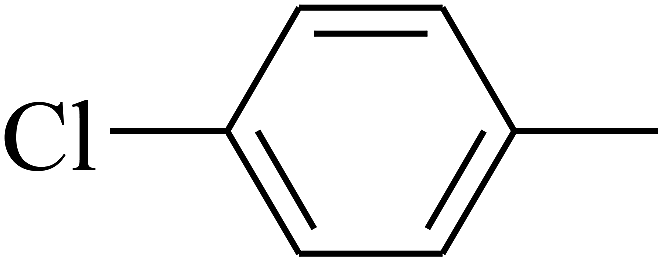 |
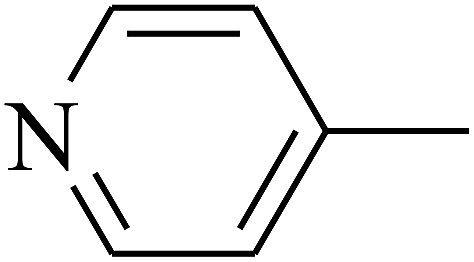 |
— | — |
| 102 | E | 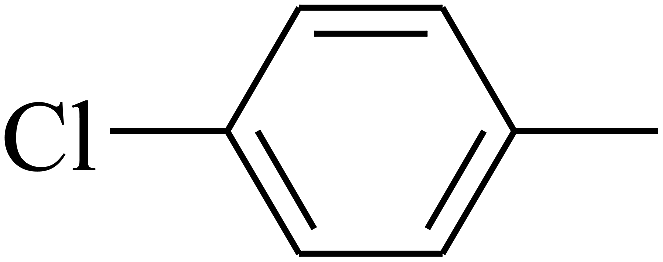 |
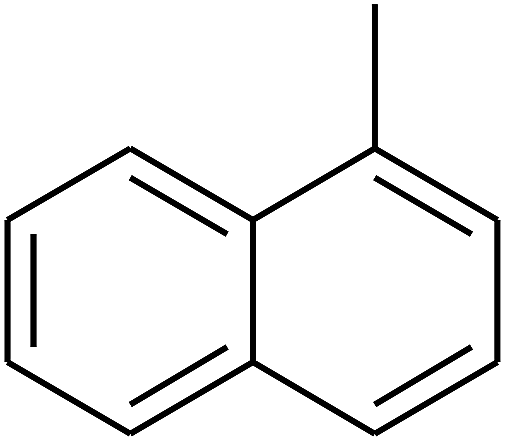 |
— | — |
| 103 | E | 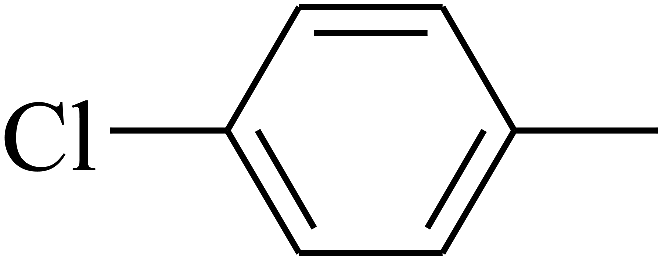 |
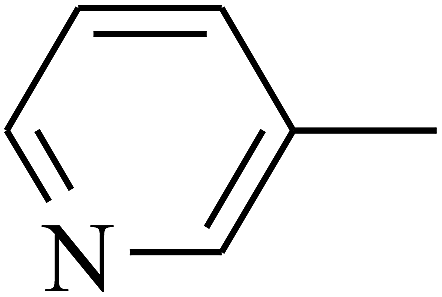 |
— | — |
| 104 | E | 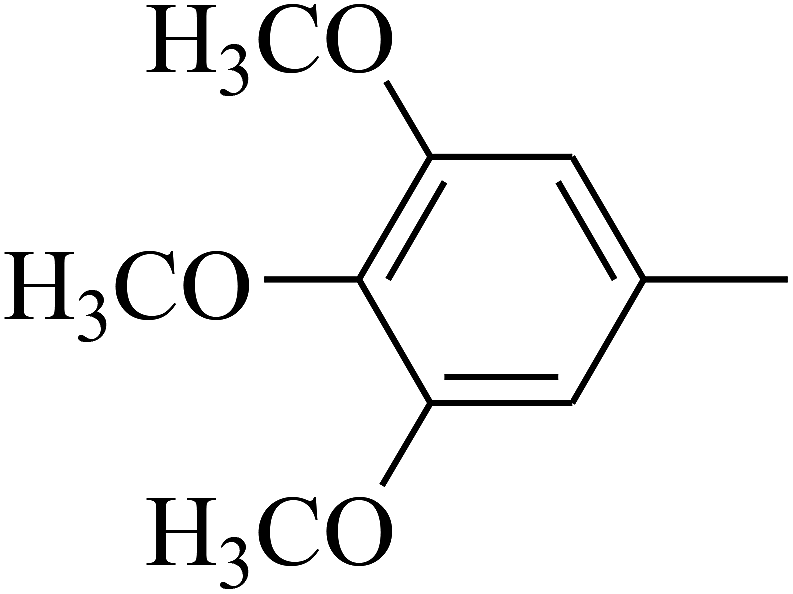 |
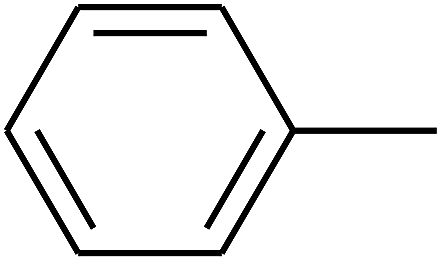 |
— | — |
| 105 | E | 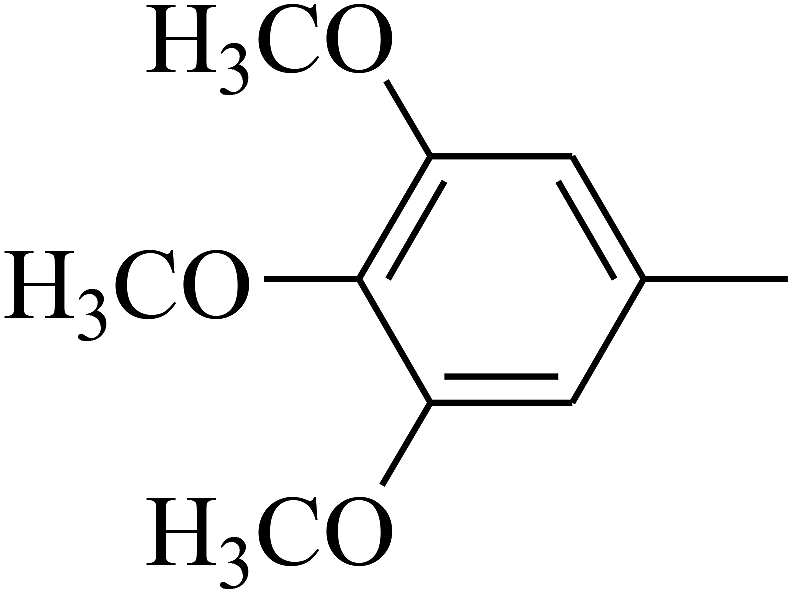 |
 |
— | — |
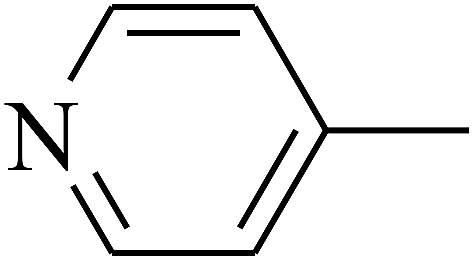
| 106 | E | 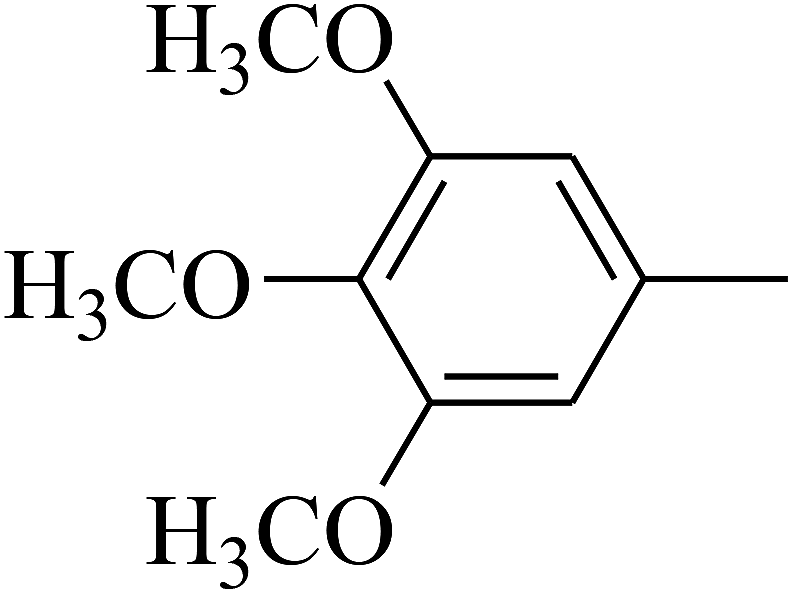 |
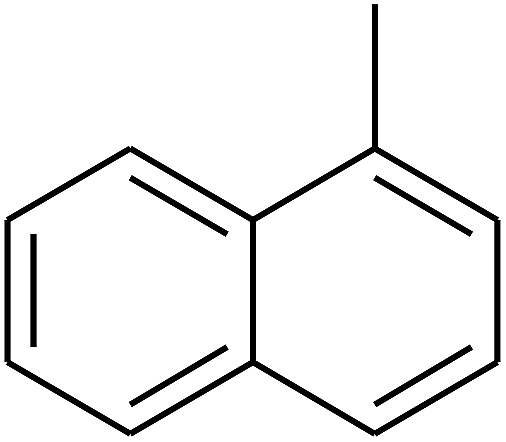 |
— | — |
| 107 | E | 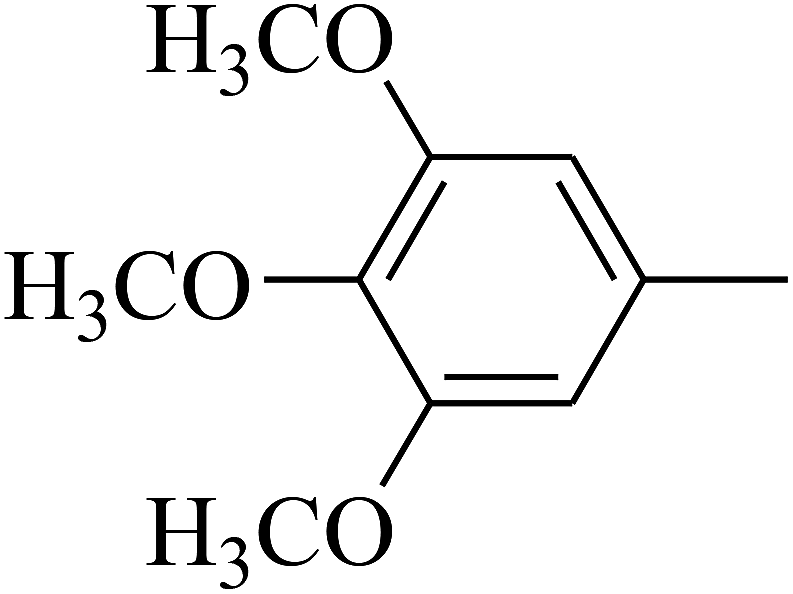 |
 |
— | — |
| 108 | E |  |
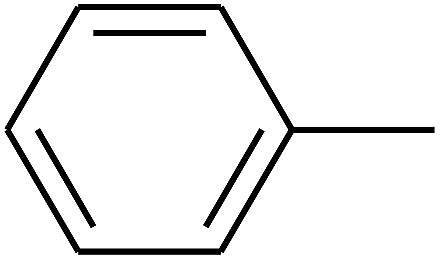 |
— | — |
| 109 | E | 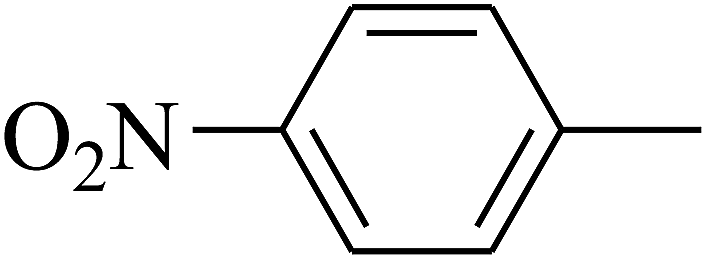 |
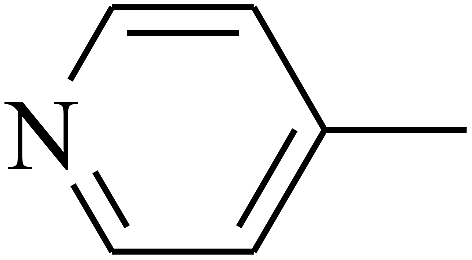 |
— | — |
| 110 | E | 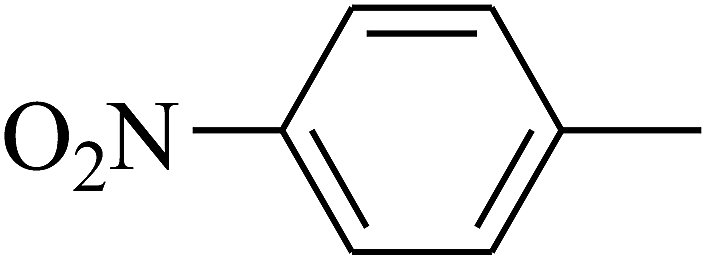 |
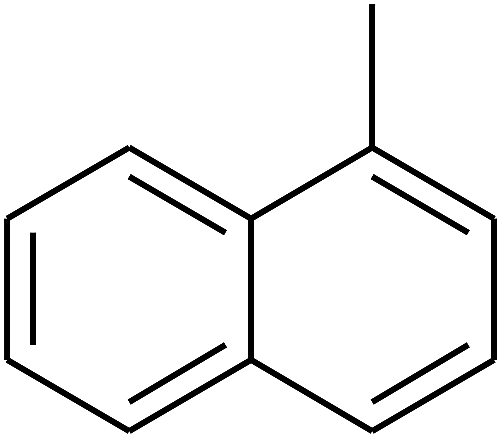 |
— | — |
| 111 | E | 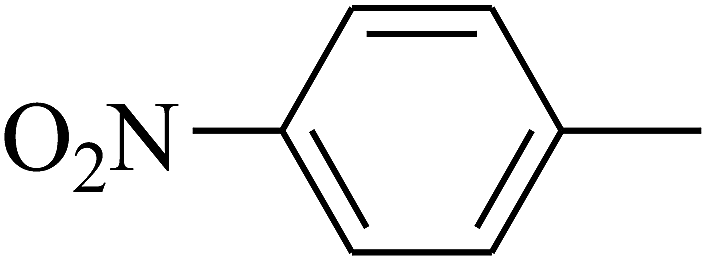 |
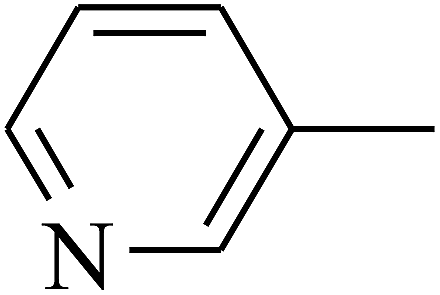 |
— | — |
| 112 | E | 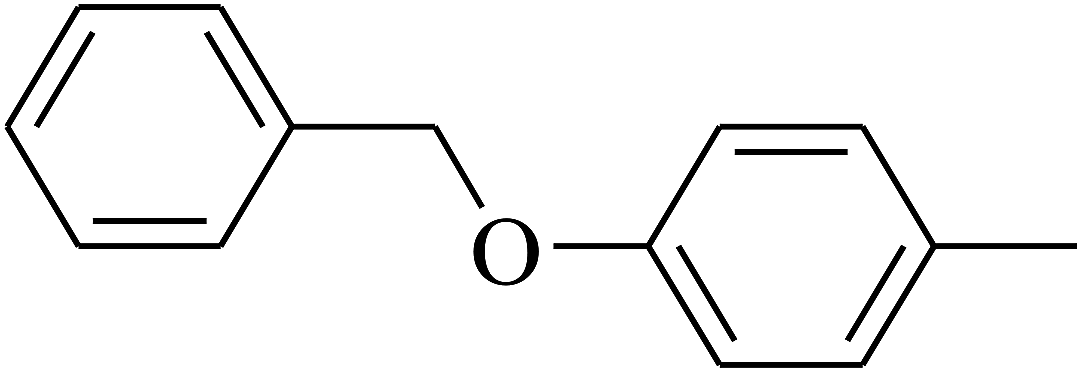 |
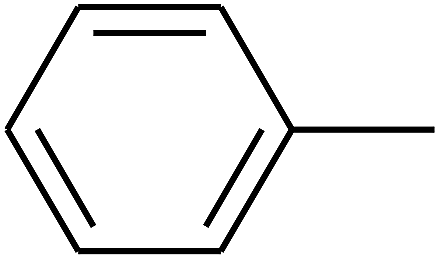 |
— | — |
| 113 | E | 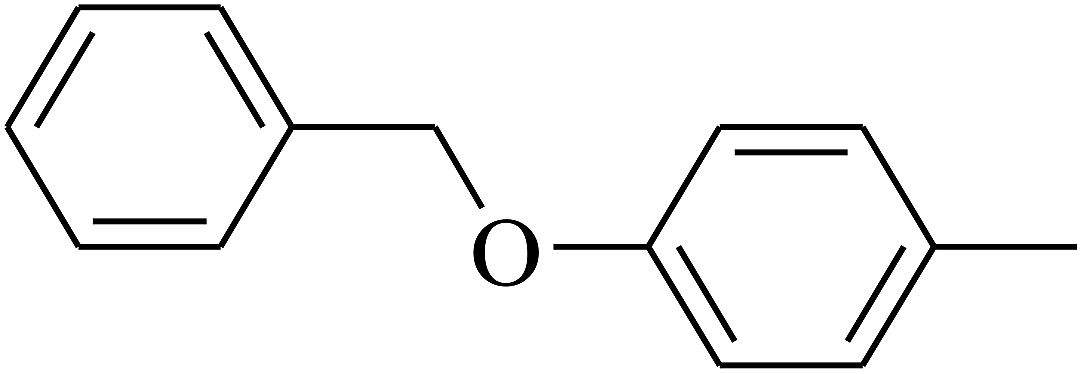 |
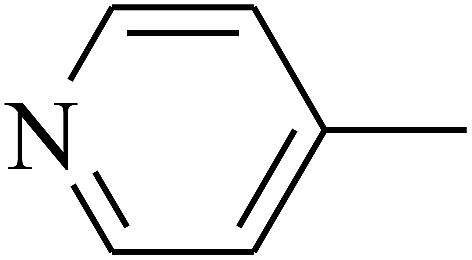 |
— | — |
| 114 | E | 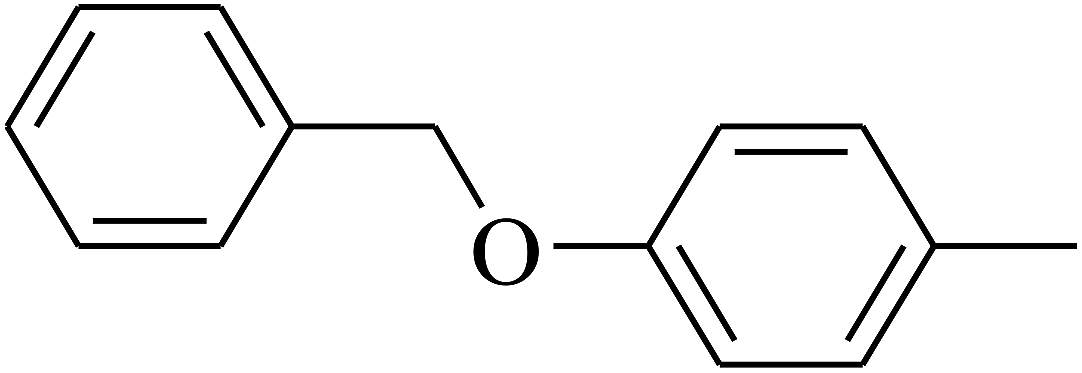 |
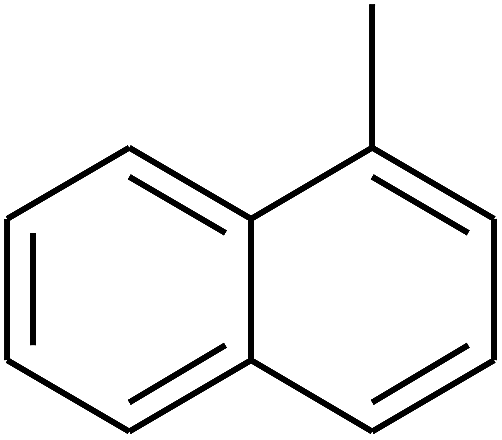 |
— | — |
| 115 | E | 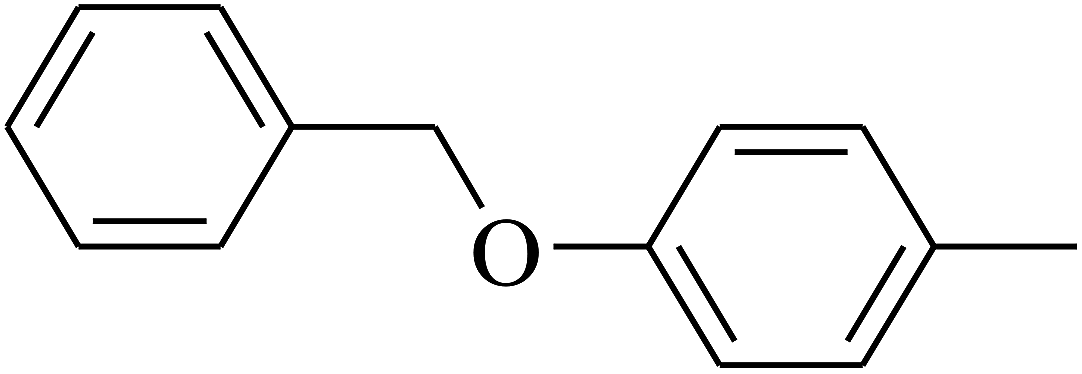 |
 |
— | — |
The 2D structures of these inhibitors were drawn in the LigX module and converted into the 3D conformers and subsequently subjected to the energy minimization step using MMFF force field. The docked structures were ranked as per their binding affinity score called as the S-score. Re-docking experiment to judge and ensure the docking accuracy was also carried out using already optimized inhibitor structures and panC structure. The root mean square deviations and deviations in docking score were accounted in deciding the accuracy of docking protocol.
2.3 Pharmacophore modeling and virtual screening
The top ranked docked conformers of the known PanC inhibitors were used for the construction of the pharmacophore model.43 The phase module of MOE was used to derive the pharmacophoric features on the basis of the structural and energy features of the protein and the inhibitors. The 3D pharmacophoric model thus generated having various pharmacophoric features was used for the virtual screening through virtual screening module of the MOE. The InterBioScreen natural database (https://www.ibscreen.com/natural-compounds)44,45 consisting of 60000 natural compounds was screened against this pharmacophore.
2.4 ADMET analysis
The potential hits obtained in the virtual screening were subjected to the ADMET screening for the predication of drug-like properties with the FAF-Drug2 tool for the ADMET prediction.46 Further, the data of the ADMET studies were filtered and validated for the Lipinski's rule of 5 and the Veber rule.47,48 In the ADMET studies the values of TPSA (<100), logP (<5) and molecular weight (<500) indicates good oral bioavailability, whereas number of rotatable (<25), rigid bonds (<10) and number hydrogen bond acceptors (<10)/donors (<5) indicates good intestinal availability.
2.5 Molecular dynamics (MD) simulation studies
The top-ranked hits with desired ADMET features were subjected to the MD simulations using Gromacs 5.1.2.49,50 The remote server of the Bioinformatics Resources and Applications Facility (BRAF), C-DAC, Pune was used to perform the production phase MD simulations. The topology of the protein was generated using the CHARMM36 all-atom force field51 while the ligand topology was generated with the CHARMM General Force Field (CGenFF) from the CGenFF server available at https://cgenff.paramchem.org. After solvating the system with the simple point charge solvent-216 model in a dodecahedral unit cell the system was neutralized with the addition of appropriate counter ions such as sodium and chloride. The system was subsequently energy minimized and equilibrated with respect to the constant volume and the pressure position restraint dynamics at a constant temperature of 300 K for 100 picoseconds. The system was subsequently subjected to the energy minimization in order to remove the steric clashes. Subsequently, the position restraint dynamics under NVT and NPT (constant volume and pressure) conditions at 300 K for 100 ps was carried out. During the 10 nanosecond production phase MD, the covalent bonds were restrained with the LINCS algorithm52 and the long range electrostatics such as coulombic and Lennard Jones interaction energies were controlled with the cutoff value of 12 A° with the Particle Mesh Ewald method (PME).53 The results of the MD were analyzed with the help of different modules in the GROMACS package.3 Results and discussion
3.1 Homology modeling
The results of Metacyc analysis showed that total 144 unique metabolic pathways are present in the H. pylori with 570 known enzymes. Amongst these enzymes, 172 enzymes are unique to the H. pylori. Further, 24 enzymes amongst these unique enzymes serve as essential enzymes executing the respective metabolic pathway and can serve as promising targets in the drug design. The analysis of these enzymes suggested that the pantothenate synthetase (PanC, Pantoate-β-alanine ligase) is unique to H. pylori, but not expressed in Homo sapiens. The identification of key genes responsible for the survival of the H. pylori through the database of essential genes also revealed that PanC is one of the essential gene. The primary sequence of the panC with 276 amino acids was retrieved from the UniPort database. The results of the preliminary BLAST analysis suggest that best model has the sequence identity of 43.87% with the template of the X-ray crystallographic structure of the panC from Thermotoga maritima (PDB ID: 2EJC). The atomic resolution being the key parameter for the selection of the template structure, this template structure with atomic resolution 2.4 Å was found appropriate. Further, the full genomes of H. pylori and T. maritima were retrieved (https://www.ncbi.nlm.nih.gov/genome) and subjected to species variation studies. It was found that there is 25.4% species similarity suggesting the suitability of species for homology modeling. The differences in the residue composition of the binding site of panC from Thermotoga maritima and homology model of H. pylori were also investigated. The binding site of panC from T. maritime constitutes the residues Pro28, Thr29, Met30, Gly31, Tyr32, Leu33, His34, His37, Leu40, Gln61, Glu66, Tyr71, Leu116, Arg121, His124, Phe125, Tyr145, Phe146, Gly147, Lys149, Asp150, Ala151, Gln152, Gln153, Phe154, Leu157, Ser185, Ser186, Arg187, Tyr241, Arg272 and Ile274. All these residues are also part of the binding site of homology model of H. pylori except few differences such as absence of Tyr32, Phe154 and Ile274 and presence of Ala29, Leu260 and Leu271. Sequence alignment of template and homology model has performed in MOE Sequence Editor in order to check the similarity and identity as shown in Fig. 1. | ||
| Fig. 1 Sequence alignment of model and template. | ||
The homology models generated were subjected to the structure validation tools like Procheck, ProSA and SPDBV.54–56 In order to validate the homology model the Cα deviation and all atom fit were calculated in SPDBV tool. The Cα deviation and all atom fit values 0.42 and 0.1 Å respectively suggest that the model structure is acceptable for further studies. The structural alignment of the homology model and template structure is shown in Fig. 2.
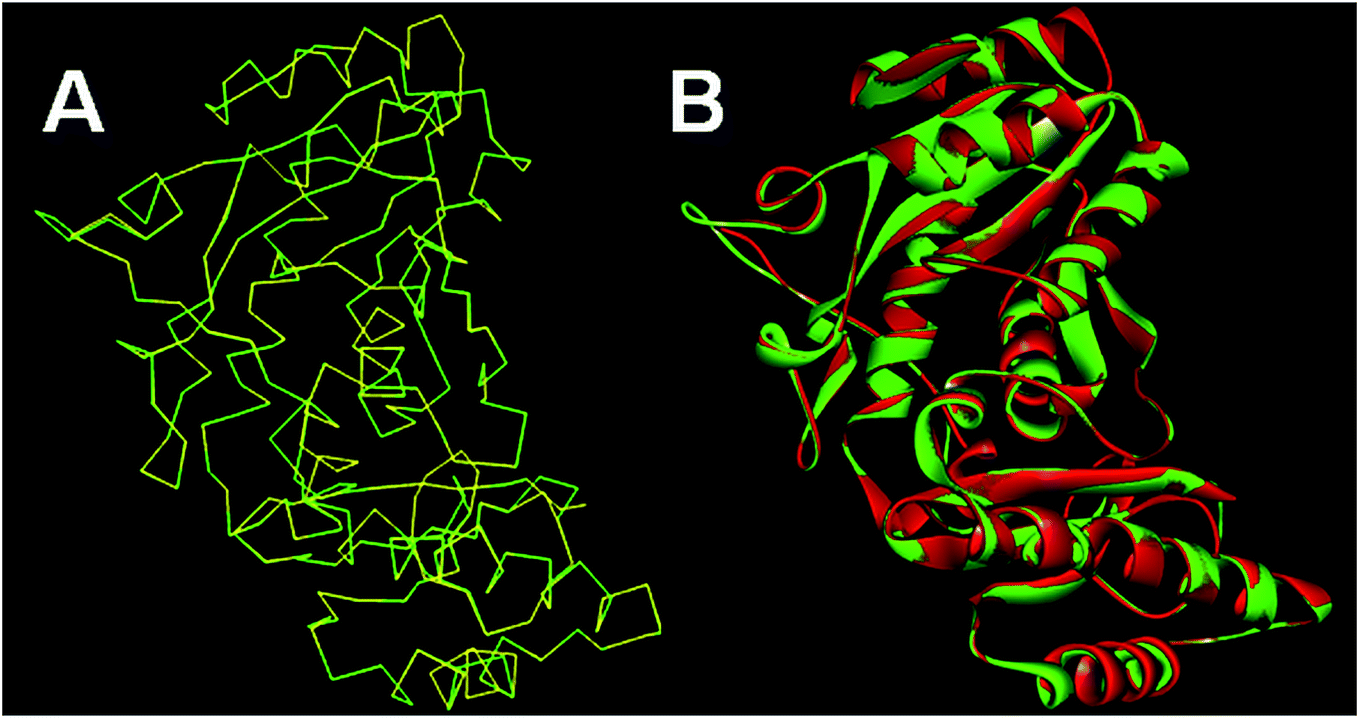 | ||
| Fig. 2 Structural alignment of homology model and template structure (A) Cα backbone alignment; (B) all atom alignment. | ||
The final model was chosen in which the 99.2% of the residues were in the allowed region as shown in the Ramachandran plot (Fig. 3).
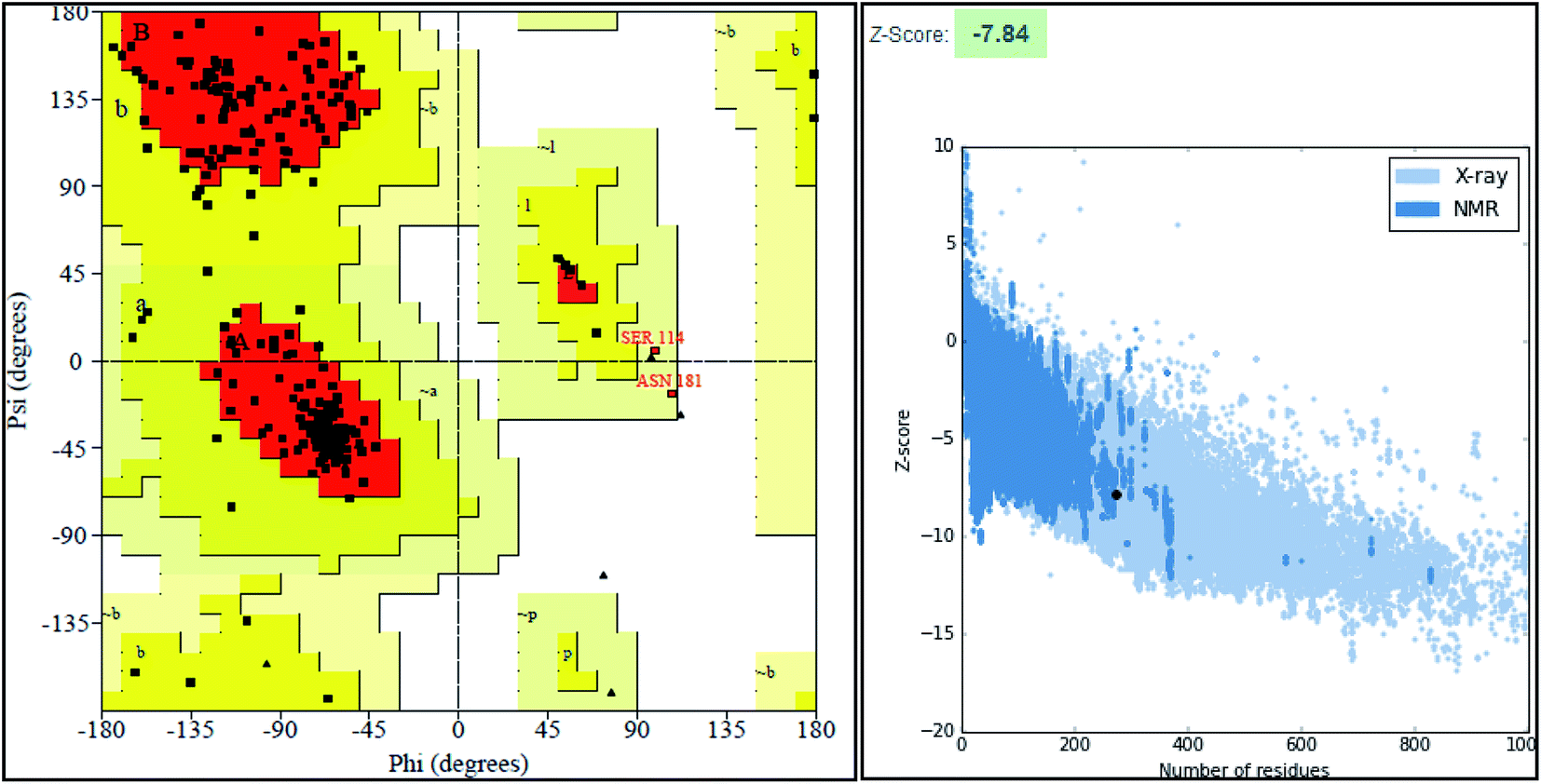 | ||
| Fig. 3 Structure validation parameter Ramachandran plot and ProSa Z score of model structure. | ||
ProSA-web server provides the estimate of errors in experimental and theoretical models of the proteins. It uses the atomic co-ordinates of each residue of the protein and provides the Z score and residue score. The Z score is measure of overall quality of model protein and determined on the basis of Z scores of all experimentally determined protein chains in model protein. The Z score of final model was −7.84 suggesting the overall good model quality. The results of binding site analysis through MOE's site finder module showed that the residues Pro25, Glu63, Gln58, Tyr68, Leu270, Leu271, Phe59, Gln153, His124, Phe125, Asp150, Leu271, Leu270, Ser185 and Lys149 are the key residues in the binding pocket which contribute to the various types of interactions with ligand (Fig. 4). The binding site of the final model correlated well with the binding site of the template structure and most of these residues are present at the binding site of both the structures.
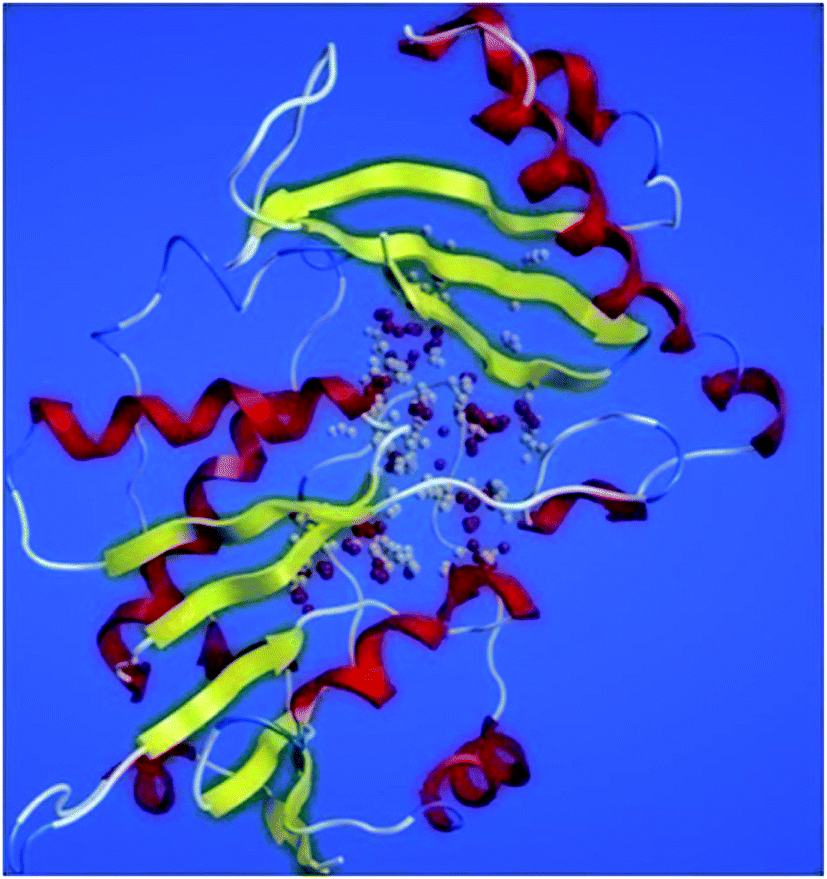 | ||
| Fig. 4 PanC binding site residues predicted by site finder module of MOE. | ||
3.2 Molecular docking
The identification of the key contributing residues in the binding pocket of the panC model was carried out by the measurement of the contribution of interaction energy of each residue with the ligand through the Promodel module of MOE-2013. During the docking studies all the known inhibitors (total 115 such inhibitors) of M. tuberculosis were docked into the binding site of the modeled H. pylori panC. The M. tuberculosis specific panC inhibitors were chosen because of diversity in the scaffolds of these inhibitors. Further, these inhibitors were reported to bind at the catalytic site of M. tuberculosis panC where the pantoate and β-alanine substrates are processed to the patothenate. A similar biochemical mechanism is expected at the binding site of H. pylori panC and for computational design such inhibitors can be a good starting point. The docking results showed that the residues Pro25, Glu63, Gln58, Tyr68, Leu270, Leu271, Phe59, Gln153, His124, Phe125, Asp150, Leu271, Leu270, Ser185 and Lys349 are the key residues in the binding pocket which contribute to the various types of interactions with ligand. The analysis of docking interactions showed that the core scaffold of all the known inhibitors plays an important role in the key interactions. The details of the structures of all the known inhibitors with the docking scores, the interacting residues and the type of key interactions are provided in the Table 2.| Comp. no. | Docking score (kcal mol−1) | Redocking score (kcal mol−1) | Interacting residues | Kinds of interactions | ||
|---|---|---|---|---|---|---|
| H-Bond | vdW | Pi | ||||
| 1 | −4.23 | −4.6 | GLN58, GLN153, ARG187 | 3 | 0 | 1 |
| 2 | −4.07 | −4.34 | MET27, GLU63, TYR68, ASN62, GLU117 | 3 | 1 | 2 |
| 3 | −4.1 | −4.29 | HIS34, LEU116, ASP150, SER186, LEU271 | 2 | 1 | 4 |
| 4 | −4.81 | −4.9 | MET27, TYR68, ASP150, LEU271, ARG121, LEU117, TYR238 | 3 | 1 | 2 |
| 5 | −4.25 | −4.33 | MET27, GLN58, GLN153, ARG121 | 3 | 1 | 2 |
| 6 | −5.1 | −5.19 | MET27, HIS24, GLU64, ASN62, ARG121, GLN58, GLN153, GLU117 | 4 | 1 | 3 |
| 7 | −5.12 | −5.39 | HIS34, HIS124, ARG121, ASP150, LYS149 | 2 | 1 | 5 |
| 8 | −4.57 | −4.63 | MET27, GLU63, TYR68, ASN62, ARG121 | 3 | 1 | 3 |
| 9 | −5.03 | −5.22 | HIS31, HIS34, LYS149, SER186, ARG269, ARG187, SER185 | 6 | 3 | 3 |
| 10 | −6.08 | −6.24 | PRO25, MET27, HIS34, LEU116, ARG269, ARG121, ASP150, GLN153 | 6 | 2 | 1 |
| 11 | −4.71 | −4.87 | AP150, LEU271, LEU260, ARG269 | 3 | 1 | 2 |
| 12 | −4.7 | −4.71 | MET27, LEU116, ARG121, ARG269, GLN153, HIS124 | 5 | 1 | 1 |
| 13 | −4.91 | −5.01 | MET27, HIS34, SER185, SER186, ARG187, ARG269 | 4 | 1 | 2 |
| 14 | −4.54 | −4.57 | GLY28, HIS34, LYS149, LEU271, ARG269 | 4 | 1 | 1 |
| 15 | −4.8995 | −3.6702 | HIS124, ASP150, ARG269, LYS149, LEU271 | 2 | 3 | 2 |
| 16 | −4.2209 | −4.9713 | HIS124, ASP150, LEU271 | 2 | 1 | 2 |
| 17 | −5.8087 | −5.756 | MET27, GLU63, HIS124, ARG187 | 1 | 1 | 3 |
| 18 | −5.2863 | −5.39 | HIS124, ASP150, ARG187, ARG269, LEU271 | 2 | 1 | 4 |
| 19 | −5.1268 | −5.6967 | MET27, PHE59, LYS149, ARG187, HIS124, ARG187 | 1 | 1 | 4 |
| 20 | −4.0056 | −4.1323 | MET27, PHE59, TYR68, GLU63, ARG269, GLN159, GLN58 | 3 | 1 | 4 |
| 21 | −4.2869 | −4.3971 | HIS124, ASP150, LEU271 | 2 | 1 | 1 |
| 22 | −4.4866 | −4.7215 | MET27, PHE59, TYR68, HIS124, LEU271, ASP150 | 2 | 1 | 4 |
| 23 | −4.6603 | −4.6689 | PHE59, HIS124, ASP150, LEU271 | 2 | 1 | 2 |
| 24 | −5.8087 | −5.9781 | PRO25, MET27, LEU37, HIS34, PHE146, GLY147, VAL132 | 2 | 1 | 6 |
| 25 | −4.8805 | −5.0012 | MET27, HIS34, PHE125, HIS124, ARG187 | 2 | 2 | 3 |
| 26 | −4.27 | −4.6732 | GLU63, ARG269, LEU271, LEU260 | 2 | 1 | 2 |
| 27 | −3.9108 | −4.2996 | TYR68, ARG187 | 2 | 1 | 0 |
| 28 | −4.3609 | −4.9383 | GLU63, ARG121, HIS124 | 1 | 1 | 4 |
| 29 | −4.4464 | −4.4861 | GLN58, HIS124, LEU271, ASP150 | 2 | 1 | 2 |
| 30 | −4.4241 | 4.6267 | GLN58, ASP150, LYS149, ARG269, LEU271 | 3 | 1 | 2 |
| 31 | −5.2496 | −5.342 | MET27, TYR68, ARG187, HIS124, ARG121, GLN152, LEU116 | 3 | 2 | 2 |
| 32 | −5.11 | −5.1287 | MET27, GLY28, HIS31, ALA29, HIS34, ARG187, SER186, TYR68 | 4 | 1 | 5 |
| 33 | −3.6937 | −3.8271 | ASP150, LEU271 | 1 | 1 | 1 |
| 34 | −3.935 | −4.135 | HIS124, LEU271, ASP150 | 2 | 1 | 2 |
| 35 | −3.747 | −3.8005 | MET27, HIS34, ASP150, GLN153 | 3 | 1 | 1 |
| 36 | −4.954 | −4.991 | ARG121, ASP150, LEU271 | 2 | 1 | 3 |
| 37 | −4.6733 | −4.7029 | ASP150, ARG269, LEU271 | 1 | 1 | 3 |
| 38 | −4.5544 | −4.6281 | ARG121, ASP150, ALA151, LEU160, LEU271 | 4 | 1 | 3 |
| 39 | −4.2869 | −4.3187 | GLU63, ARG121, HIS124 | 3 | 1 | 2 |
| 40 | −3.9108 | −4.0013 | GLU63, ARG269, LEU260 | 1 | 1 | 2 |
| 41 | −4.4464 | −4.4556 | GLN58, HIS124, LEU271, ASP150 | 2 | 1 | 3 |
| 42 | −3.935 | −4.1032 | HIS124, LEU271, ASP150 | 1 | 1 | 1 |
| 43 | −4.954 | −5.0031 | MET27, HIS34, PHE125, HIS124, ARG187 | 2 | 1 | 2 |
| 44 | −4.6733 | −4.7631 | MET27, HIS34, HIS124, ARG187 | 2 | 1 | 1 |
| 45 | −3.57 | −3.66 | TYR68, ASP150, LEU271, SER186, ARG187 | 3 | 1 | 1 |
| 46 | −4.78 | −4.81 | HIS34, HIS124, PHE125, ALA151, LEU271, LEU260, ASP150, ARG269, LEU271 | 4 | 1 | 5 |
| 47 | −4.08 | −4.12 | GLU63, ALA151, GLN152, GLN153, ARG121, LYS149, HIS124 | 4 | 1 | 2 |
| 48 | −4.84 | −4.93 | LEU116, ARG121, ALA151, LYS149, ALA183, LEU270, ARG269 | 5 | 1 | 5 |
| 49 | −4.16 | −4.14 | PRO69, ALA29, TYR68, ASP150, SER186, TYR190, LEU271, ARG187 | 2 | 1 | 3 |
| 50 | −4.38 | −4.14 | ALA29, MET27, GLN58, HIS124, ASP150, ARG187, LYS149, SER185, SER186 | 4 | 2 | 4 |
| 51 | −4.39 | −4.41 | TYR68, ASP150, LEU271, SER186, ARG187, HIS124, GLN153, LYS149 | 5 | 1 | 5 |
| 52 | −4.25 | −4.31 | MET27, TYR68, LYS149, VAL176, SER185, SER186 | 4 | 1 | 4 |
| 53 | −5.84 | −5.91 | PRO25, TYR26, ALA29, HIS34, GLN153, GLY147, ASP150, SER186 | 3 | 1 | 3 |
| 54 | −5.79 | −5.789 | ARG121, LYS149, GLN152, ASN183, LEU270, LEU271, ARG269 | 3 | 1 | 4 |
| 55 | −5.93 | −6.01 | HIS34, LYS149, SER185, ARG187, ARG269, LEU271 | 3 | 0 | 3 |
| 56 | −5.68 | −5.79 | MET27, PHE59, TYR68, ARG187, ASP150, LEU271 | 3 | 1 | 4 |
| 57 | −5.77 | −5.87 | PRO25, MET27, HIS31, HIS34, LEU37, SER187, VAL128, VAL132, SER186, GLY147 | 3 | 2 | 4 |
| 58 | −4.63 | −4.76 | MET27, GLU63, TYR68, GLN153, ARG121 | 3 | 1 | 3 |
| 59 | −5.1 | −5.12 | PRO25, MET27, HIS34, MET135, ILE131, ARG187, ARG269 | 2 | 2 | 4 |
| 60 | −4.05 | −4.21 | HIS34, TYR68, PHE271 | 1 | 1 | 2 |
| 61 | −5.33 | −5.39 | MET27, GLN58, HIS124, VAL128, TYR145, GLY147, ASP150, GLN153, LEU271 | 4 | 1 | 3 |
| 62 | −5.15 | −5.21 | PRO25, MET27, THR26, ILE131, MET135, LYS149, GLN153 | 4 | 2 | 2 |
| 63 | −7.7 | −7.79 | GLN58, GLU63, GLN153, ARG121, TYR68 | 5 | 1 | 1 |
| 64 | −5.02 | −5.19 | TYR68, ASP150, GLN152, ARG269, SER186 | 3 | 2 | 2 |
| 65 | −3.96 | −4.18 | HIS34, ARG187, ASP150, GLN152, LEU271 | 1 | 1 | 4 |
| 66 | −4.13 | −4.19 | PRO25, MET27, HIS31, SER187, ILE131, ARG187, MET135 | 1 | 2 | 3 |
| 67 | −2.8687 | −2.8729 | PHE59, TYR58, HIS124, ASP150, ARG187, LEU271 | 4 | 1 | 3 |
| 68 | −3.3455 | −3.4411 | LEU30, HIS31, HIS34, LYS149, SER185 | 3 | 1 | 3 |
| 69 | −4.7611 | −4.812 | LEU30, HIS31, HIS34, LYS149, SER185 | 3 | 1 | 3 |
| 70 | −4.1494 | −4.4271 | LEU30, HIS34, LYS149, SER185, SER186, ARG187 | 4 | 1 | 4 |
| 71 | −3.6942 | −3.7316 | GLU63, PHE125, ARG121, HIS124, ARG269 | 2 | 1 | 3 |
| 72 | −4.2448 | −4.3993 | MET27, HIS31, HIS34, LYS149, SER185, ARG187 | 3 | 1 | 4 |
| 73 | −3.5961 | −3.6821 | ASN62, GLU63, HIS124, PHE125, ARG187, ARG269 | 3 | 1 | 3 |
| 74 | −5.7105 | −5.7389 | GLY60, ALA61, ASN62, GLU63, LYS149, ARG187, ARG269, ASN271, ARG121 | 6 | 1 | 3 |
| 75 | −4.1277 | −4.1791 | MET27, HIS31, HIS34, LYS149, SER185, ARG187, LEU271 | 2 | 1 | 4 |
| 76 | −3.9968 | −4.156 | MET27, HIS34, ASP150, LEU271 | 1 | 1 | 3 |
| 77 | −5.0105 | −5.1995 | ASN62, GLU63, ARG121, ARG187, ARG269 | 3 | 1 | 3 |
| 78 | −6.1995 | −6.2677 | ASN62, GLU63, ARG121, LYS149, SER185, ARG187, ARG269 | 8 | 1 | 3 |
| 79 | −3.3602 | −3.3871 | LYS149, ALA183, SER185, ARG187, ARG269, LEU271, ILE202, LEU182 | 4 | 2 | 3 |
| 80 | −4.2577 | −4.3289 | PRO25, MET27, GLY28, HIS31, GLY33, HIS34, LYS149, LEU184 | 4 | 1 | 2 |
| 81 | −4.3562 | −4.3891 | PRO25, HIS31, GLY33, HIS34, LYS149, LEU184 | 2 | 2 | 6 |
| 82 | −4.8416 | −4.9562 | GLN58, GLU63, TYR68, HIS124, ARG269, ARG187 | 3 | 1 | 2 |
| 83 | −4.8644 | −4.9493 | GLU63, GLN58, ASP64, TYR68, HIS124, ARG269, ARG187 | 3 | 1 | 4 |
| 84 | −4.0638 | −4.1452 | MET27, PHE59, TYR68, ARG187 | 2 | 1 | 2 |
| 85 | −5.323 | −5.3671 | GLY28, ALA29, HIS34, SER185, LYS149, ARG269, ARG187, SER186, LEU184 | 5 | 1 | 3 |
| 86 | −5.1447 | −5.1591 | HIS34, PHE59, TYR68, ARG187, LEU184 | 4 | 2 | 3 |
| 87 | −3.2977 | −3.3592 | LYS149, SER185, ARG187, ARG269, LEU270, LEU271 | 3 | 1 | 3 |
| 88 | −2.9499 | −2.8429 | MET27, GLU63, HIS124, VAL128, ARG187, ARG169 | 2 | 1 | 3 |
| 89 | −4.4058 | −4.4851 | PHE59, PRO25, HIS31, GLY33, HIS34, LYS149, LEU184 | 2 | 2 | 6 |
| 90 | −4.4837 | −4.5201 | MET34, LYS149, ALA183, SER185, ARG187, ARG269, LEU271, ILE202, LEU182 | 4 | 2 | 3 |
| 91 | −2.011 | −2.293 | PHE59, TYR58, HIS124, ASP150, ARG187 | 1 | 1 | 2 |
| 92 | −3.4441 | −3.4896 | LYS149, ALA183, SER185, ARG187, ARG269, LEU271, ILE202, LEU182 | 4 | 2 | 4 |
| 93 | −2.9499 | −2.9618 | MET27, GLU63, HIS124, VAL128, ARG187, ARG169 | 2 | 1 | 3 |
| 94 | −3.1448 | −3.1844 | MET27, HIS34, GLU63, HIS124, VAL128, ARG187, ARG169 | 2 | 1 | 4 |
| 95 | −4.4864 | −4.5891 | PRO25, HIS31, HIS34, GLY33, HIS34, LYS149, LEU184, SER185 | 3 | 2 | 6 |
| 96 | −2.9824 | −3.3168 | MET27, GLU63, HIS124, VAL128, ARG187, ARG169 | 2 | 1 | 4 |
| 97 | −3.8659 | −3.962 | GLU63, ARG121, SER185, LEU271, HIS124 | 4 | 1 | 2 |
| 98 | −4.5719 | −4.782 | PRO25, HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 4 | 1 | 3 |
| 99 | −4.5506 | −4.62 | HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 3 | 1 | 3 |
| 100 | −4.0345 | −4.491 | GLU63, ARG121, SER185, LEU271, HIS124 | 3 | 1 | 3 |
| 101 | −4.8854 | −4.887 | PRO25, HIS31, HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 4 | 1 | 3 |
| 102 | −3.7926 | −3.8416 | HIS34, ARG121, SER185, LEU271, HIS124 | 4 | 1 | 1 |
| 103 | −5.305 | −5.4591 | PRO25, HIS31, HIS34, LYS149, SER185, ARG187, ARG269, TYR238, LEU271 | 5 | 1 | 4 |
| 104 | −5.1502 | −5.3819 | PRO25, HIS34, LYS149, SER185, ARG187, ARG269, TYR238, LEU271 | 4 | 1 | 4 |
| 105 | −5.831 | −5.8702 | PRO25, HIS31, HIS34, LYS149, SER185, SER186, ARG187, ARG269, TYR238 | 5 | 1 | 4 |
| 106 | −6.0256 | −6.0482 | PRO25, HIS31, HIS34, LYS149, SER185, SER186, ARG187, ARG269, TYR238, LEU27 | 5 | 1 | 4 |
| 107 | −4.8235 | −4.8491 | MET27, HIS31, HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 5 | 1 | 4 |
| 108 | −6.0962 | −6.173 | PRO25, MET27, HIS31, HIS34, LYS149, SER185, SER186, ARG187, ARG269, TYR238 | 6 | 1 | 4 |
| 109 | −4.0069 | −4.1639 | HIS34, ARG121, SER185, LEU271, HIS124 | 3 | 1 | 3 |
| 110 | −4.9985 | −5.0173 | MET27, HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 4 | 1 | 4 |
| 111 | −4.9124 | −5.0792 | GLU63, GLN58, ASP64, TYR68, HIS124, ARG269, ARG187 | 3 | 1 | 4 |
| 112 | −6.2852 | −6.4021 | PRO25, MET27, HIS31, HIS34, LYS149, SER185, SER186, ARG187, ARG269, TYR238 | 6 | 2 | 4 |
| 113 | −5.1362 | −5.1562 | PRO25, HIS34, LYS149, SER185, ARG269, TYR238, LEU271 | 4 | 2 | 1 |
| 114 | −5.7806 | −5.8492 | PRO25, HIS34, GLY60, LYS149, SER185, ARG187, ARG269, TYR238, LEU271 | 5 | 1 | 4 |
| 115 | −4.7319 | −4.872 | PRO25, HIS34, LYS149, SER185, ARG187, ARG269, TYR238 | 4 | 1 | 3 |
The tetrahydropyrido thieno pyrimidin-4-one derivative 63 having the lowest binding free energy of −7.70 kcal mol−1 was found forming two hydrogen bond interactions with the nitrogen atoms of core scaffold and the residues Glu63 and Tyr68 and two hydrogen bond interactions with the oxygen atom of o-nitro substituent with the Gln58 and Gln153. This inhibitor also forms the hydrophobic π–π stacking interaction between the phenyl ring and Arg121 (Fig. 5). The lowest binding free energy for this ligand may be due to such hydrogen bond formation and hydrophobic interaction. The re-docking of the ligands produced similar interactions with slight variations in the docking scores suggesting the accuracy of docking protocol followed.
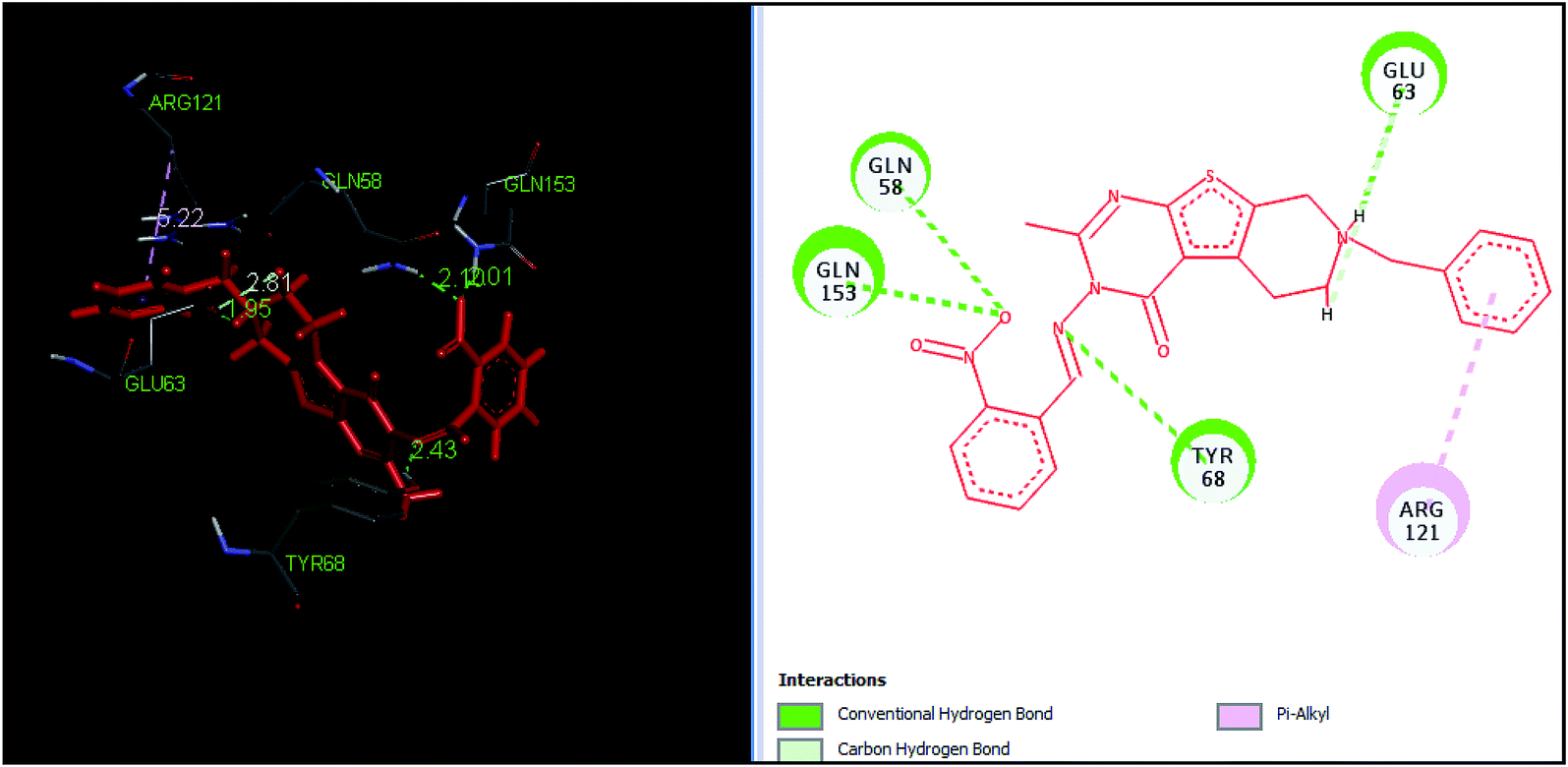 | ||
| Fig. 5 The binding pose and molecular interactions of inhibitor 63 into the active site of the model structure of the PanC. | ||
3.3 Pharmacophore-model construction and virtual screening
These pharmacophoric features provide the important insights of functionalities contributing in the molecular activity of the inhibitors. The key pharmacophoric features such as the hydrogen bond acceptor, the metal ligator, the aromatic ring, the hydrogen bond donor responsible for the inhibitory activity were identified. The pharmacophoric model was constructed on the basis of the key interactions of top ligands with the residues at the binding site of the PanC. For this purpose the set of predefined features such as hydrogen bond acceptor (HBA), aromatic ring (Aro) from such interactions were exploited. The mapping of developed 3D-pharmacophore model with the top docked hit 63 into the active site of the homology model of the PanC is shown in Fig. 6.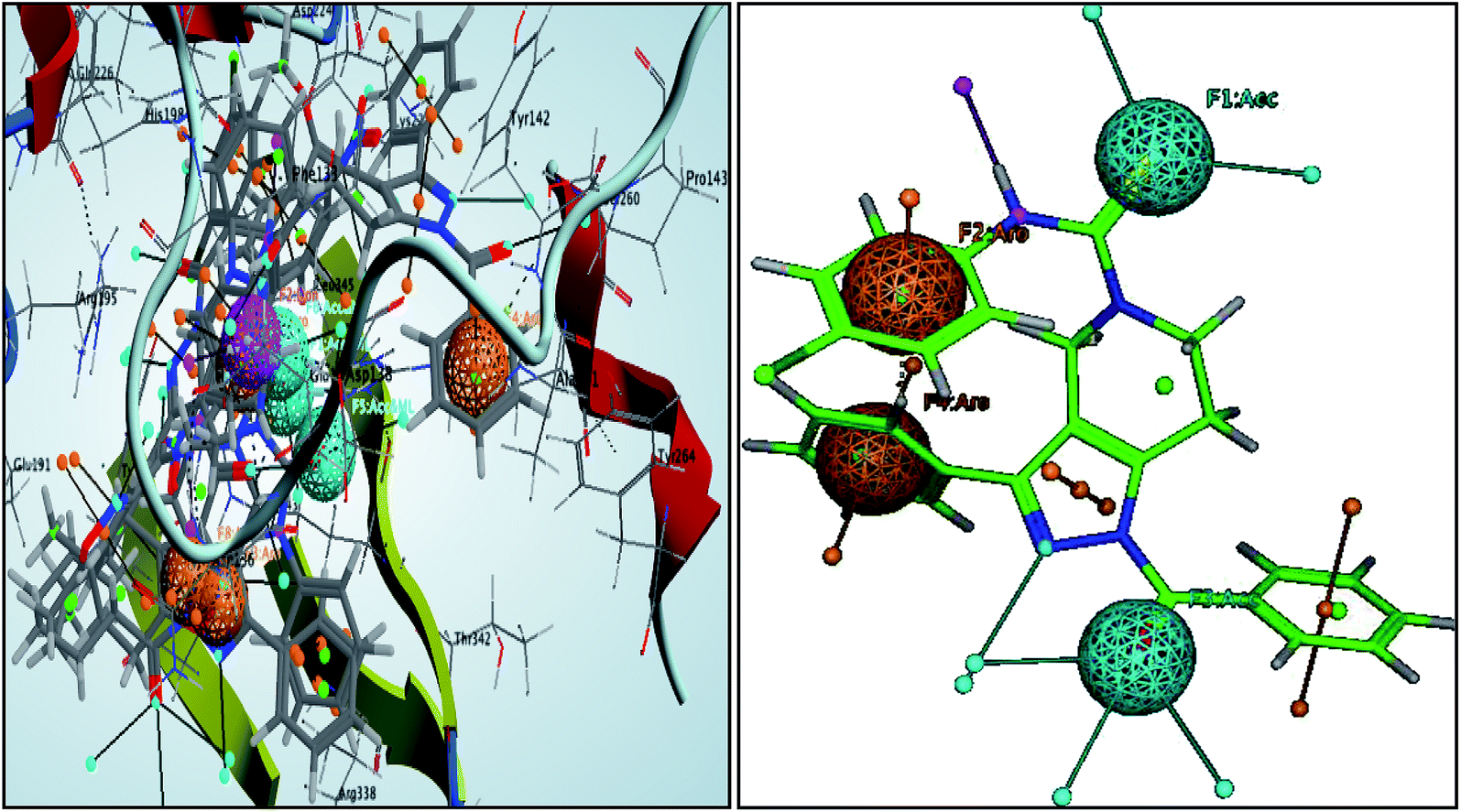 | ||
| Fig. 6 3D-pharmacophore models of the top docked hit 63 into the active site of the homology model of the PanC. | ||
The results of the pharmacophore design suggest that the ligand 63 has four key pharmacophoric features namely two hydrogen bond acceptor sites (F1 & F3, designated as Acc), two aromatic rings (F2 & F4 designated as Aro). The best pharmacophore model was selected on the basis of the highest correlation coefficient, root mean square deviation (rmsd) values and E-value. As per the protocol shown in Fig. 7, virtual screening of InterBioScreen natural compounds database was carried out.
 | ||
| Fig. 7 Schematic representation of the virtual screening protocol. | ||
The pharmacophoric features F1–F4 were chosen as the filtering criteria during virtual screening. The virtual screening experiment gave 20 virtual hits as given in the Table 3.
| Sr. no. | Hits_ID | Docking score (kcal mol−1) | Interacting residues | Kinds of interactions | ||
|---|---|---|---|---|---|---|
| H-Bond | vdW | Pi | ||||
| 1 | STOCK1N-76518 | −6.38 | HIS34, GLU63, ARG121, HIS124, ASP150, ARG269, SER185, SER186, GLU271 | 4 | 3 | 3 |
| 2 | STOCK1N-74127 | −5.45 | THR26, MET27, TYR68, HIS34, GLN58, ASP150, LYS149, SER187 | 3 | 3 | 2 |
| 3 | STOCK1N-73916 | −5.7 | THR26, MET27, HIS34, GLN58, GLU63, LYS149, RG187 | 6 | 2 | 3 |
| 4 | STOCK1N-71293 | −5.81 | THR26, MET27, GLY28, PRO25, ARG121, LYS149, ARG269, LEU271 | 4 | 2 | 2 |
| 5 | STOCK1N-68553 | −6.69 | ASN62, GLU63, ARG121, ARG187, LEU271, ARG269 | 5 | 1 | 1 |
| 6 | STOCK1N-67377 | −7.87 | HIS34, TYR68, ARG121, PHE125, HIS124, ASP150, SER185, TYR238, ARG269 | 5 | 2 | 5 |
| 7 | STOCK1N-64449 | −6.71 | ARG121, HIS124, LYS149, ASP150, SER185, ARG187, ARG269 | 3 | 3 | 3 |
| 8 | STOCK1N-64228 | −6.07 | PRO25, GLN58, ASP150, ARG269, GLN153 | 3 | 2 | 1 |
| 9 | STOCK1N-63827 | −8.27 | PRO25, THR26, MET27, HIS34, GLN58, GLU63, LYS149, ARG187, LEU271 | 5 | 4 | 4 |
| 10 | STOCK1N-63231 | −8.67 | TYR68, MET27, HIS124, ALA151, GLN152, ASP150, LYS149 | 6 | 1 | 4 |
| 11 | STOCK1N-63040 | −9.51 | ASN62, GLU63, ARG121, GLN152, GLN153, ARG187, LEU271, ARG269 | 5 | 3 | 3 |
| 12 | STOCK1N-60270 | −10.7 | LEU30, MET27, HIS34, TYR68, ARG121, ARG268, GLN153, ASP150, PHE146, GLN152, SER186, ARG269 | 6 | 1 | 4 |
| 13 | STOCK1N-59730 | −8.33 | HIS34, TYR68, PHE125, GLU63, LYS149, ARG187, ARG269 | 3 | 3 | 3 |
| 14 | STOCK1N-26126 | −7.87 | ARG121, LYS149, ASP150, SER185, SER186, ARG269, ARG187 | 6 | 1 | 1 |
| 15 | STOCK1N-28765 | −4.65 | ARG121, ASP150, LYS149, SER185, SER186, ARG269 | 6 | 1 | 1 |
| 16 | STOCK1N-32864 | −7.46 | PRO25, GLN58, ASP150, GLN153, ARG269 | 4 | 2 | 1 |
| 17 | STOCK1N-36335 | −4.49 | PHE59, GLU63, ARG187, HIS124, ARG121, ARG269, LEU271 | 5 | 1 | 1 |
| 18 | STOCK1N-44424 | −8.91 | TYR68, ARG121, PHE125, ASP150, ARG187, TYR238, ARG269, LEU271 | 6 | 2 | 3 |
| 19 | STOCK1N-45307 | −7.14 | TYR68, MET27, HIS124, ALA151, GLN152, ASP150, LYS149 | 6 | 1 | 4 |
| 20 | STOCK1N-45539 | −6.39 | ARG121, HIS124, LYS149, ASP150, SER185, ARG187, ARG269, LEU271 | 4 | 1 | 1 |
The virtual hits thus obtained were subjected to docking studies to investigate the interactions at the binding site. The virtual hits STOCK1N-60270, STOCK1N-63040, STOCK1N-44424 and STOCK1N-63231 showed the lowest binding free energy of −10.1, −9.51, −8.91 and −8.67 kcal mol−1 respectively in the docking studies. These virtual hits were found making key hydrogen bond and hydrophobic π–π stacking interactions with residues such as Gln153, Tyr68, Glu63 and Arg121. The analysis of the docking score and the binding poses of the virtual hits it was observed that STOCK1N-60270, STOCK1N-63040, STOCK1N-44424 and STOCK1N-63231 are the best possible hits. The binding poses of these hit molecules at the binding site is shown in Fig. 8.
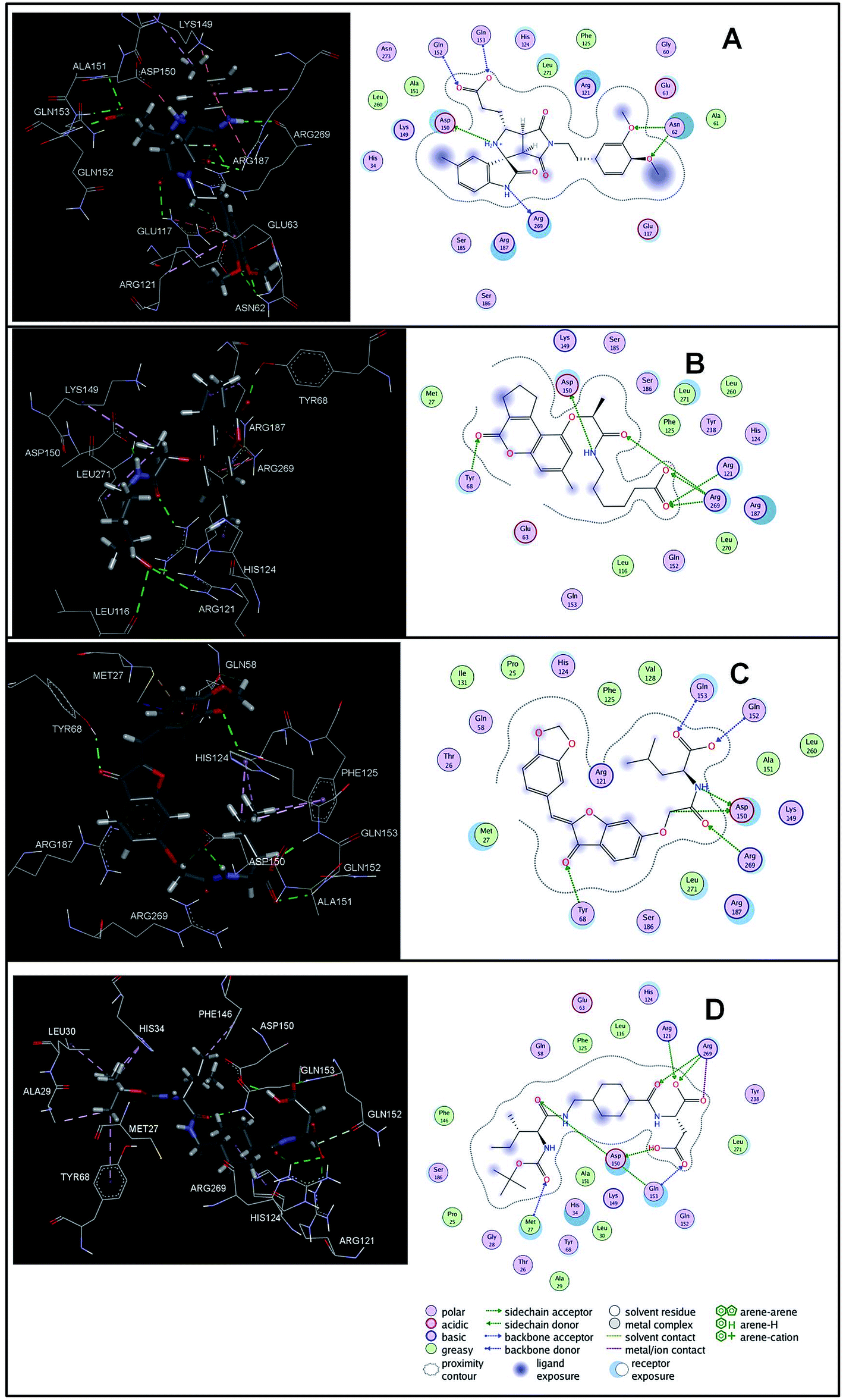 | ||
| Fig. 8 The binding pose of top hits at binding site, (A) STOCK1IN 63040; (B) STOCK1IN 44424; (C) STOCK1IN 63231 and; (D) STOCK1IN 60270. | ||
The hits with matching features of the best docked ligand may have the highest potential to inhibit the pantothenate synthetase of the H. pylori. The docked conformer of the tetrahydropyrido thieno pyrimidin-4-one derivative 63 having lowest binding free energy and the docked conformer of the potential virtual hit STOCK1N-60270 overlaid at the binding site is shown in Fig. 9.
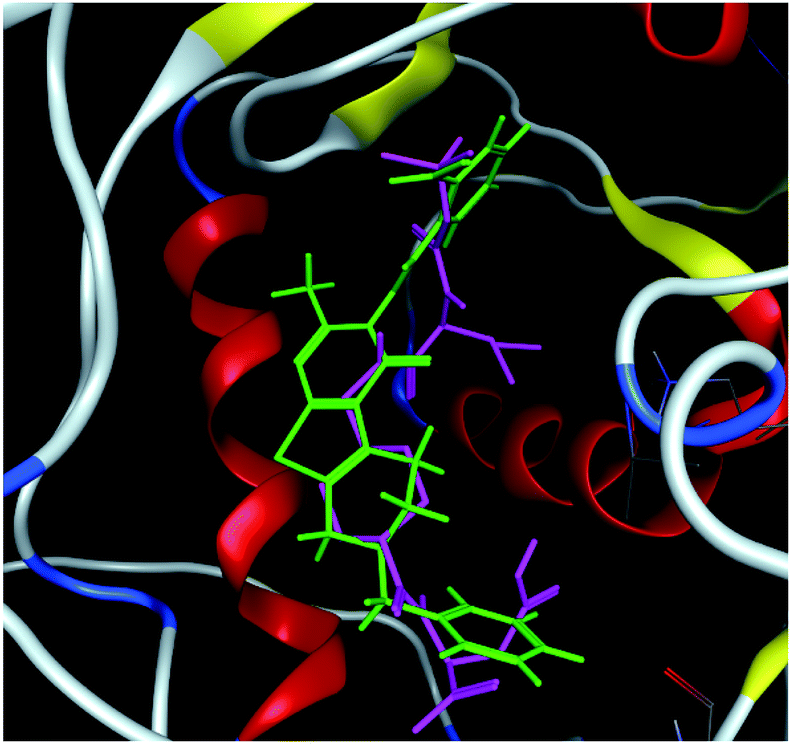 | ||
| Fig. 9 The docked conformers of ligand 63 (magenta) and top virtual hit STOCK1N-60270 (green) at the binding site. | ||
3.4 In silico ADME predictions
The drug-like properties has been predicted by analyzing absorption, distribution, metabolism, elimination and toxicity risk characteristics. These characteristics are calculated and analyzed in terms of various physicochemical parameters and pharmaceutical properties using the FAFDrug2 ADMET prediction tool and the data is summarized in Table 4. The data obtained for the virtual hits with STOCK1N ID numbers 73916, 64228, 63827, 63231, 63040, 60270, 44424 and 45307 were found within the acceptable range of the ADMET criteria.| Sr. no. | Ligand_ID | % ABS | MW | logP |
TPSA | RotatableB | RigidB | HBD | HBA | Rings | Ratio H/C | Toxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a ABS – absorption; TPSA – topological polar surface area; n-ROTB – number of rotatable bonds; MV – molecular volume; MW – molecular weight; logP logarithm of partition coefficient; nON – number of hydrogen bond acceptors; n-OHNH – number of hydrogen bonds donors. |
||||||||||||
| 1 | STOCK1N-76518 | 77.15 | 467.51 | 4.39 | 92.32 | 9 | 24 | 1 | 7 | 2 | 0.31 | Non toxic |
| 2 | STOCK1N-74127 | 81.52 | 354.36 | 2.27 | 79.65 | 7 | 19 | 0 | 6 | 2 | 0.37 | Non toxic |
| 3 | STOCK1N-73916 | 86.73 | 425.48 | 4.45 | 64.55 | 4 | 30 | 0 | 5 | 3 | 0.23 | Non toxic |
| 4 | STOCK1N-71293 | 76.16 | 356.33 | 2.94 | 95.20 | 6 | 19 | 1 | 7 | 2 | 0.37 | Non toxic |
| 5 | STOCK1N-68553 | 80.64 | 260.29 | 2.08 | 82.19 | 6 | 13 | 3 | 3 | 1 | 0.36 | Non toxic |
| 6 | STOCK1N-67377 | 59.45 | 519.59 | 4.48 | 143.63 | 8 | 28 | 5 | 5 | 2 | 0.36 | Non toxic |
| 7 | STOCK1N-64449 | 82.55 | 266.29 | 1.84 | 76.66 | 6 | 10 | 2 | 4 | 1 | 0.46 | Non toxic |
| 8 | STOCK1N-64228 | 78.99 | 431.44 | 4.81 | 87.00 | 6 | 26 | 1 | 6 | 3 | 0.28 | Non toxic |
| 9 | STOCK1N-63827 | 61.59 | 543.57 | 2.73 | 137.43 | 7 | 34 | 4 | 7 | 4 | 0.33 | Non toxic |
| 10 | STOCK1N-63231 | 67.47 | 453.44 | 3.42 | 120.39 | 8 | 25 | 2 | 8 | 2 | 0.38 | Non toxic |
| 11 | STOCK1N-63040 | 62.68 | 507.54 | 2.24 | 134.27 | 8 | 29 | 3 | 7 | 3 | 0.37 | Non toxic |
| 12 | STOCK1N-60270 | 49.96 | 485.57 | 3.07 | 171.13 | 13 | 14 | 5 | 8 | 1 | 0.48 | Non toxic |
| 13 | STOCK1N-59730 | 67.47 | 455.46 | 3.32 | 120.39 | 9 | 21 | 2 | 8 | 2 | 0.38 | Non toxic |
| 14 | STOCK1N-26126 | 85.43 | 514.70 | 3.57 | 68.31 | 2 | 33 | 0 | 5 | 3 | 0.23 | Non toxic |
| 15 | STOCK1N-28765 | 76.94 | 269.26 | 1.88 | 92.93 | 3 | 17 | 2 | 5 | 2 | 0.54 | Non toxic |
| 16 | STOCK1N-32864 | 67.47 | 389.36 | 2.66 | 120.39 | 8 | 16 | 2 | 8 | 2 | 0.47 | Non toxic |
| 17 | STOCK1N-36335 | 61.49 | 587.74 | 2.33 | 137.71 | 3 | 37 | 5 | 8 | 3 | 0.27 | Non toxic |
| 18 | STOCK1N-44424 | 72.49 | 401.45 | 3.51 | 105.84 | 9 | 19 | 2 | 6 | 1 | 0.32 | Non toxic |
| 19 | STOCK1N-45307 | 73.31 | 417.42 | 2.30 | 103.46 | 4 | 30 | 2 | 6 | 2 | 0.41 | Non toxic |
| 20 | STOCK1N-45539 | 71.14 | 473.52 | 5.46 | 109.75 | 6 | 31 | 2 | 6 | 3 | 0.25 | Non toxic |
The value of the topological polar surface area (TPSA) and the logP of the hits indicate that they have very good oral bioavailability. The parameters like the number of rotatable bonds and the number of rigid bonds are linked with the intestinal absorption and showed that all the hits may have good intestinal absorption. In silico assessment of the hits also showed that they have very good pharmacokinetic properties based on their physicochemical values. The structures of the best virtual hits with their docking scores are given in the Fig. 10. The structures of other virtual hits are provided in ESI S1.†
 | ||
| Fig. 10 Structures of the top virtual hits. | ||
3.5 Molecular dynamics simulation
The molecular dynamics simulations offer a more precise estimate of the interactions, energetic and the conformational changes of the ligands at the binding site of the protein and such studies actually simulate the biological environment which is more advantageous than the in vacuo conditions. In order to gain deeper insights into the exact mode of the binding of the best virtual hits and to evaluate the energetic of the ligand enzyme interactions more accurately the MD simulation studies were undertaken. The docked complexes of the panC with the potential hits (STOCK1N-60270, STOCK1N-63040, STOCK1N-44424 and STOCK1N-63231) with the top ranked conformers were used in the 10 ns MD simulations. The results of the MD simulation are evaluated through the measurement of root mean square deviation (RMSD), root mean square fluctuation (RMSF), number of H-bonds formed between the ligand and the key residues and the energetic of each complex. In case of the RMSD measurement the deviations from the original starting positions of the backbone atoms or the ligand atoms are measured during entire MD simulation. Lower deviations are indicative of the better stability. The average RMSD values for the complexes of STOCK1N IDs 60270, 63040, 44424 and 63231 were 0.265 ± 0.002, 0.289 ± 0.002, 0.228 ± 0.003 and 0.1236 ± 0.006 respectively. This suggests better stability of the STOCK1IN-63231 than the other hits. The average RMSD values for the ligands in these complexes 60270, 63040, 44424 and 63231 were 0.165 ± 0.002, 0.189 ± 0.003, 0.178 ± 0.001 and 0.108 ± 0.004 respectively, which suggests the stability of the hit 63231 better than the other hits. The results of the MD studies with RMSD are provided in Fig. 11.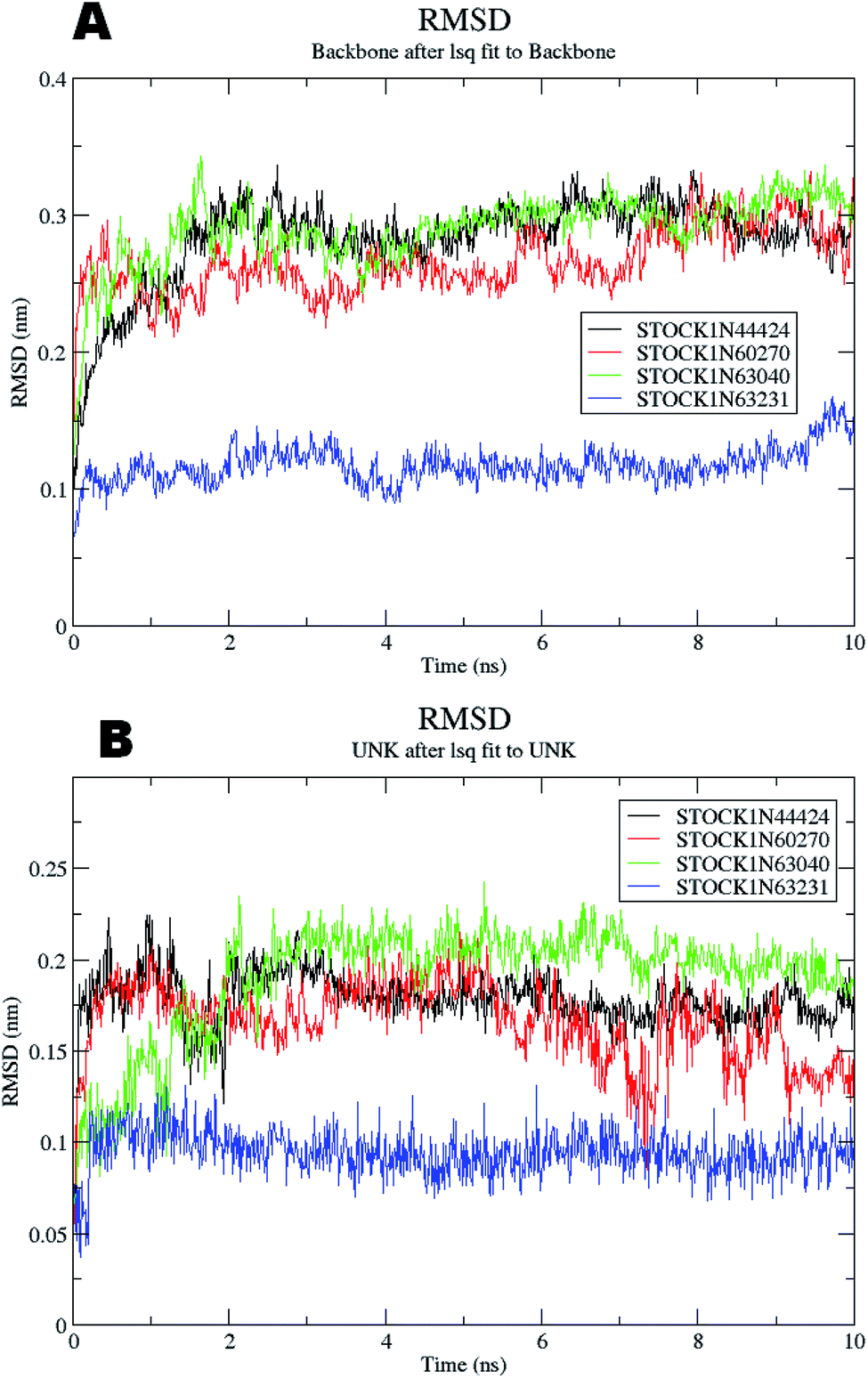 | ||
| Fig. 11 RMSD for panC (A) the backbone atoms and (B) the atoms of virtual hits. | ||
The RMSF is a measure of elasticity of the protein under the investigation in terms of the fluctuations in the protein backbone during the MD simulation. The results of the RMSF evaluations are provided in Fig. 12.
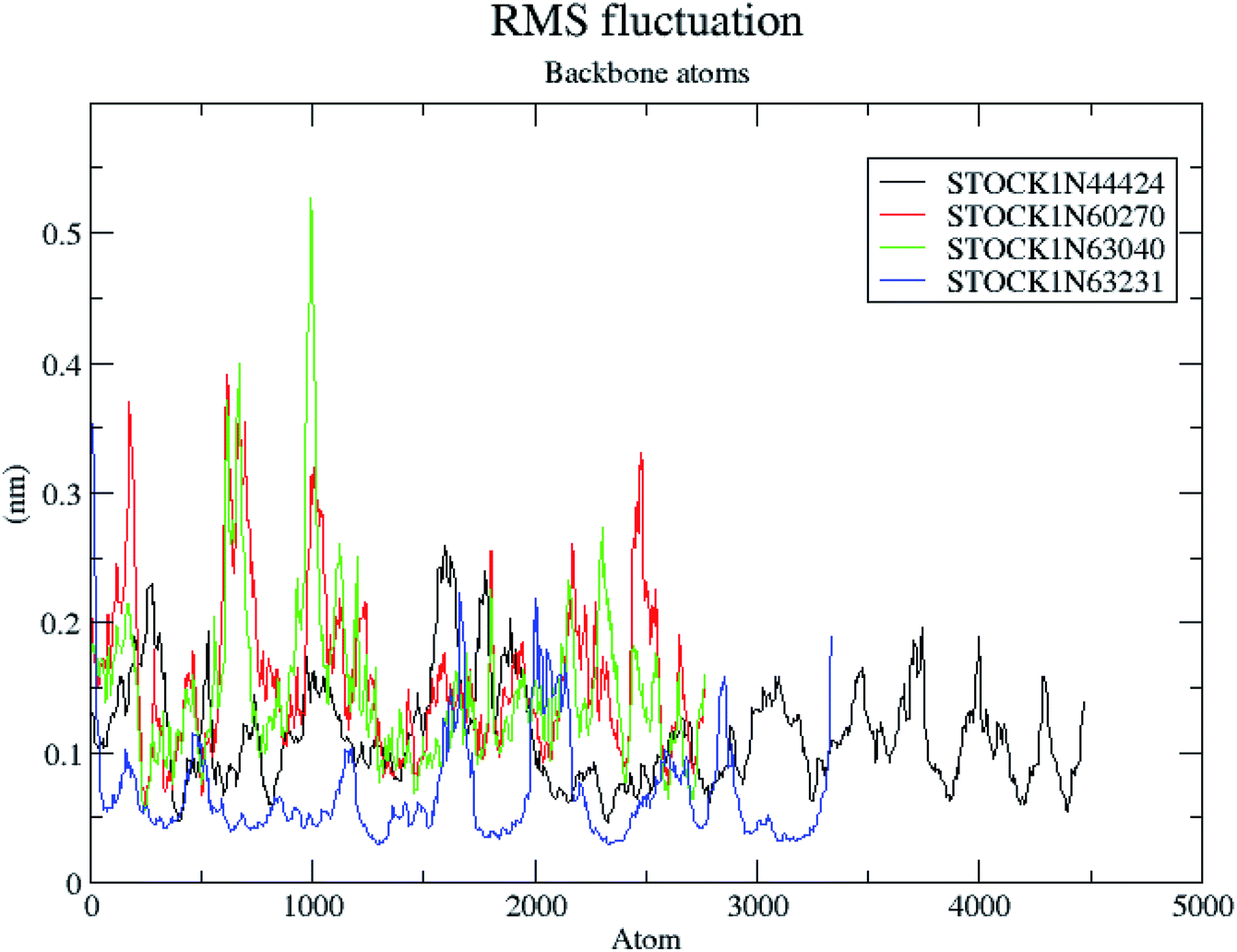 | ||
| Fig. 12 RMS fluctuations in the backbone atoms during the MD simulations. | ||
The RMSF evaluations suggest that the backbone fluctuations are minimal in case of the complex of 63231 suggesting its better stability than the other complexes. The minimal fluctuations here may be attributed to the favorable interactions at the active site. The non bonded interactions such as hydrogen bonds between the ligand and the key residues at the binding site of the protein contribute to the binding affinity and activity of the ligand. The more number of hydrogen bonds between hydrogen bond donor–acceptor atoms of the ligand with such atoms of residues in binding site suggests the more binding affinity. In MD simulation studies the maximum number of hydrogen bonds formed between the ligands 60270, 63040, 44424 and 63231 and residues at binding site are 11, 15, 10 and 3 respectively. The average number of hydrogen bonds formed during entire simulation for the ligands 60270, 63040, 44424 and 63231 are 5.1, 2.1, 1.6 and 1.8 respectively suggesting the better binding affinity of the complex 60270. The results of hydrogen bond analysis are provided in Fig. 13.
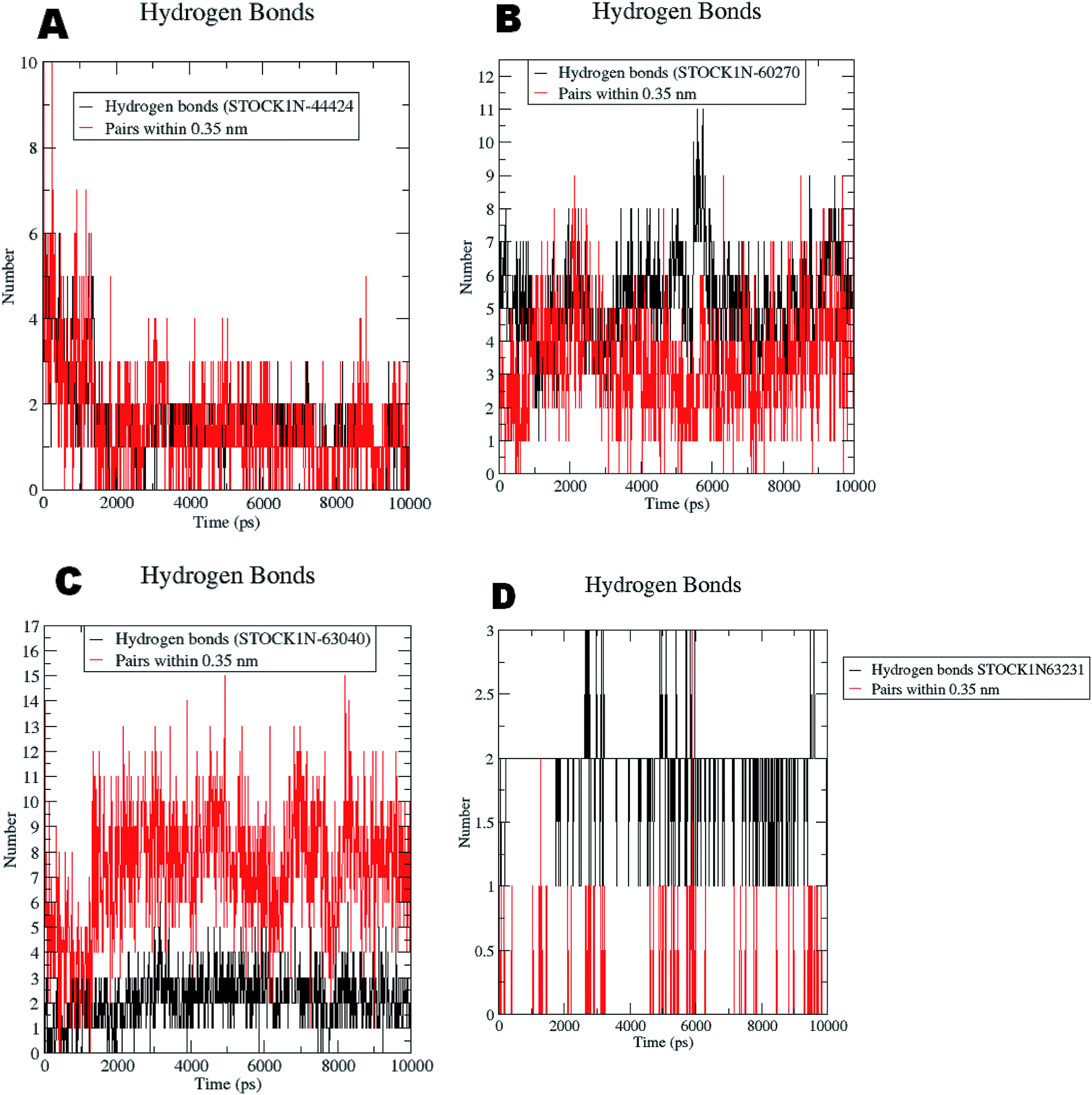 | ||
| Fig. 13 Hydrogen bonds formed with the chosen virtual hits during entire MD simulation (A) 44424; (B) 60270; (C) 63040; and (D) 63231. | ||
The docking studies also support the evaluation of hydrogen bonds. Other non bonded interaction energies such as the short-range coulombic and the Lennard-Jones and the total interaction energies were also computed to understand the strength of the interaction between compounds under study and the panC. The average short-range coulombic and Lennard-Jones and total interaction energies are given in Table 5. The results suggest that only in compound 44424 the short range coulombic interactions occur.
| Compound STOCK1IN ID | Short-range coulombic interaction energy (kJ mol−1) | Short-range Lennard-Jones interaction energy (kJ mol−1) | Total short-range interaction energy (kJ mol−1) |
|---|---|---|---|
| 60270 | 0 | −149.88 ± 4 | −4.794482 × 105 |
| 63040 | 0 | −229.108 ± 6.7 | −4.798895 × 105 |
| 44424 | −87.6557 ± 6.7 | −174.416 ± 5.6 | −4.719041 × 105 |
| 63231 | −80.0583 ± 1.9 | −177.261 ± 4 | −3.89751 × 105 |
This interaction may be due to the presence of the ionizable carboxylate groups extended through the pentyl chain. The Lennard-Jones (LJ) interaction energy estimate suggest that the compound 60270 has the highest LJ interaction energy which may be contributing in the highest activity of this compound. The results of energy evaluations are also provided in Fig. 14.
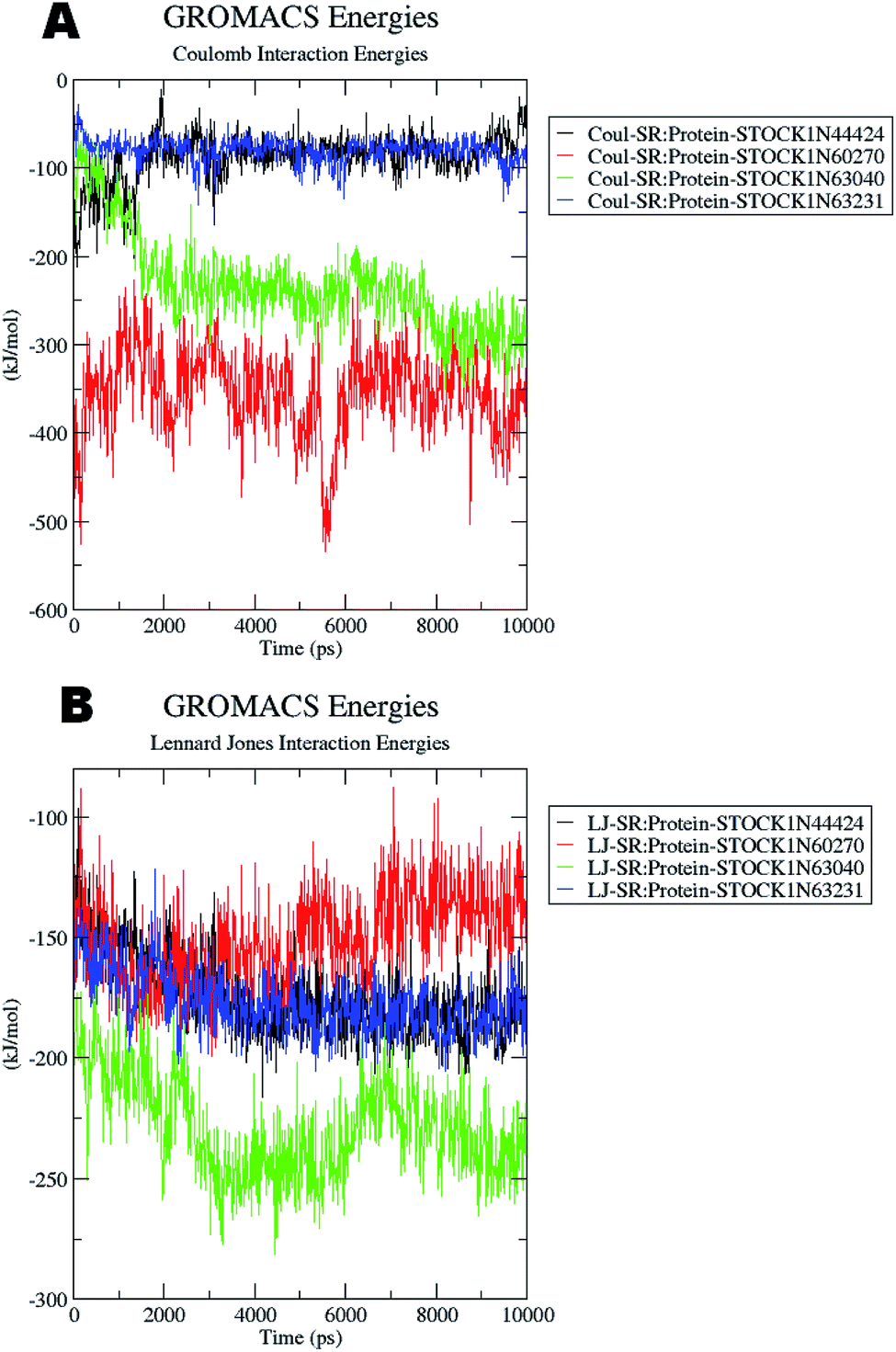 | ||
| Fig. 14 Energy evaluation between the protein and the hit molecules. (A) The short range coulombic interaction energy; (B) the short range Lennard-Jones interaction energy. | ||
On the basis of these MD results such as the number of hydrogen bonds formed during the simulation, RMSD, RMSF and the corresponding energetics of the simulation, the virtual hit STOCK1IN 60270 could be the best ligand as a potential inhibitor of modeled PanC.
4 Conclusion
In the present study the novel comparative pathway analysis approach was employed to find the unique metabolic pathways and to identify the key enzymes in such pathways in the H. pylori. The study has shown that the pantothenate synthetase of the H. pylori can serve as the potential drug target as it is essential for its survival. The combined structure-based drug design approaches namely homology modeling, molecular docking, pharmacophore modeling, virtual screening and molecular dynamics simulations provided the virtual hits with better structural, binding, pharmacokinetic and toxicity features. The virtual hits STOCK1N-60270, 63040, 44424 and 63231 has been identified as potential H. pylori specific panC inhibitors. Based on the docking and MD studies the virtual hit STOCK1IN 60270 which is a succinic acid derivative could serve as a lead for further development of potential inhibitors of the H. pylori specific panC. These virtual hit could be potentially beneficial in gastric carcinomas due to underlying H. pylori infections.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors SAA, HMA and AAA would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for funding through the Research Group Project No. RG-1439-010. Authors are thankful to Dr Zahid Zaheer, Principal, Y. B. Chavan College of Pharmacy, Dr Rafiq Zakaria Campus, Aurangabad 431 001 (M.S.), India for providing the laboratory facility.References
- W.-L. Chang, Y.-C. Yeh and B.-S. Sheu, J. Biomed. Sci., 2018, 25, 68 CrossRef PubMed.
- Cancer (Fact sheet N 297), World Health Organization, February 2014, retrieved 2009-05-11, http://www.who.int/news-room/fact-sheets/detail/cancer Search PubMed.
- J.-Y. Wu, C.-C. Cheng, J.-Y. Wang, D.-C. Wu, J.-S. Hsieh, S.-C. Lee and W.-M. Wang, PLoS One, 2014, 9, e84158 CrossRef PubMed.
- Y. Yamaoka, Helicobacter pylori: molecular genetics and cellular biology, Horizon Scientific Press, 2008 Search PubMed.
- T. T. Binh, V. P. Tuan, H. D. Q. Dung, P. H. Tung, T. D. Tri, N. P. M. Thuan, V. Van Khien, P. Q. Hoan, R. Suzuki and T. Uchida, Gut Pathog., 2017, 9, 46 CrossRef PubMed.
- A. Savoldi, E. Carrara, D. Y. Graham, M. Conti and E. Tacconelli, Gastroenterology, 2018, 155, 1372–1382 CrossRef PubMed.
- F. Mégraud, Ther. Adv. Gastroenterol., 2012, 5, 103–109 CrossRef PubMed.
- K. E. L. McColl and E. El-Omar, Keio J. Med., 2002, 51, 53–56 CrossRef CAS PubMed.
- R. Leonardi and S. Jackowski, EcoSal Plus, 2007, 1–18 Search PubMed.
- L. M. Brown, Epidemiol. Rev., 2000, 22, 283–297 CrossRef CAS PubMed.
- J.-F. Tomb, O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill and B. A. Dougherty, Nature, 1997, 389, 412 CrossRef CAS.
- J.-C. Rain, L. Selig, H. De Reuse, V. Battaglia, C. Reverdy, S. Simon, G. Lenzen, F. Petel, J. Wojcik and V. Schächter, Nature, 2001, 409, 211 CrossRef CAS PubMed.
- R. Häuser, A. Ceol, S. V. Rajagopala, R. Mosca, G. Siszler, N. Wermke, P. Sikorski, F. Schwarz, M. Schick and S. Wuchty, Mol. Cell. Proteomics, 2014, 13, 1318–1329 CrossRef PubMed.
- https://www.ebi.ac.uk/, accessed on 31 October 2018.
- https://www.uniprot.org/uniprot/P56061, accessed on 31 October 2018.
- V. K. Vyas, R. D. Ukawala, M. Ghate and C. Chintha, Indian J. Pharm. Sci., 2012, 74, 1 CrossRef CAS PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157 CrossRef CAS.
- D. K. Yadav, R. Rai, N. Kumar, S. Singh, S. Misra, P. Sharma, P. Shaw, H. Pérez-Sánchez, R. L. Mancera and E. H. Choi, Sci. Rep., 2016, 6, 38128 CrossRef CAS PubMed.
- R. Gaur, D. K. Yadav, S. Kumar, M. P Darokar, F. Khan and R. Singh Bhakuni, Curr. Top. Med. Chem., 2015, 15, 1003–1012 CrossRef CAS PubMed.
- B. S. Kumar, A. Kumar, J. Singh, M. Hasanain, A. Singh, K. Fatima, D. K. Yadav, V. Shukla, S. Luqman and F. Khan, Eur. J. Med. Chem., 2014, 86, 740–751 CrossRef PubMed.
- D. Kumar Yadav, K. Kalani, A. K. Singh, F. Khan, S. K. Srivastava and A. B. Pant, Curr. Med. Chem., 2014, 21, 1160–1170 CrossRef.
- D. K. Yadav, P. Sharma, S. Misra, H. Singh, R. L. Mancera, K. Kim, C. Jang, M. Kim, H. Pérez-Sánchez and E. H. Choi, Arch. Pharmacal Res., 2018, 41, 1178–1189 CrossRef CAS PubMed.
- H. Sun, J. Zhu, F. Chen, S. Zhang, Y. Zhang and Q. You, Eur. J. Med. Chem., 2011, 46, 3942–3952 CrossRef CAS PubMed.
- A. Hussain and C. Verma, J. Cancer Res. Ther., 2019 DOI:10.4103/jcrt.JCRT_47_18.
- S. Pal, V. Kumar, B. Kundu, D. Bhattacharya, N. Preethy, M. Prashanth Reddy and A. Talukdar, Comput. Struct. Biotechnol. J., 2019, 17, 291–310 CrossRef CAS PubMed.
- G. Eren, A. Bruno, S. Guntekin-Ergun, R. Cetin-Atalay, F. Ozgencil, Y. Ozkan, M. Gozelle, S. Gozde Kaya and G. Costantino, J. Mol. Graphics Modell., 2019, 89, 60–73 CrossRef CAS PubMed.
- X. Feng, W. Jia, X. Liu, Z. Jing, Y. Liu, W. Xu and X. Cheng, Comput. Biol. Chem., 2019, 78, 178–189 CrossRef CAS PubMed.
- F. Cheng, W. Li, G. Liu and Y. Tang, Curr. Top. Med. Chem., 2013, 13, 1273–1289 CrossRef CAS PubMed.
- D. K. Yadav, S. Kumar, H. S. Saloni, M. Kim, P. Sharma, S. Misra and F. Khan, Drug Des., Dev. Ther., 2017, 11, 1859 CrossRef CAS PubMed.
- R. Zheng and J. S. Blanchard, Biochemistry, 2001, 40, 12904–12912 CrossRef CAS PubMed.
- R. Caspi, H. Foerster, C. A. Fulcher, P. Kaipa, M. Krummenacker, M. Latendresse, S. Paley, S. Y. Rhee, A. G. Shearer, C. Tissier, T. C. Walk, P. Zhang and P. D. Karp, Nucleic Acids Res., 2008, 36, D623–D631 CrossRef CAS PubMed.
- P. D. Karp, S. M. Paley, M. Krummenacker, M. Latendresse, J. M. Dale, T. J. Lee, P. Kaipa, F. Gilham, A. Spaulding, L. Popescu, T. Altman, I. Paulsen, I. M. Keseler and R. Caspi, Briefings Bioinf., 2010, 11, 40–79 CrossRef CAS PubMed.
- R. Zhang, H. Ou and C. Zhang, Nucleic Acids Res., 2004, 32, D271–D272 CrossRef CAS.
- S. Pundir, M. J. Martin and C. O'Donovan, in Protein Bioinformatics, Springer, 2017, pp. 41–55 Search PubMed.
- G. Hu and L. Kurgan, Sequence Similarity Searching, Curr. Protoc. Protein Sci., 2018, 13, e71, DOI:10.1002/cpps.71.
- Molecular Operating Environment (MOE), 2014.09, Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2014 Search PubMed.
- P. Labute, J. Comput. Chem., 2008, 29, 1693–1698 CrossRef CAS PubMed.
- M. R. Anguru, A. K. Taduri, R. D. Bhoomireddy, M. Jojula and S. K. Gunda, Chem. Cent. J., 2017, 11, 68 CrossRef PubMed.
- M. Kumar, B. Makhal, V. K. Gupta and A. Sharma, J. Mol. Graphics Modell., 2014, 50, 1–9 CrossRef CAS PubMed.
- M. Narender, K. Umasankar, J. Malathi, A. R. Reddy, K. R. Umadevi, A. V. N. Dusthackeer and K. V. Rao, Bioorg. Med. Chem. Lett., 2016, 26, 836–840 CrossRef CAS PubMed.
- G. Samala, P. B. Devi, S. Saxena, N. Meda, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 1298–1307 CrossRef CAS.
- P. B. Devi, G. Samala, J. P. Sridevi, S. Saxena, M. Alvala, E. G. Salina, D. Sriram and P. Yogeeswari, ChemMedChem, 2014, 9, 2538–2547 CrossRef CAS.
- S. Rampogu, M. Son, A. Baek, C. Park, R. M. Rana, A. Zeb, S. Parameswaran and K. W. Lee, Comput. Biol. Chem., 2018, 74, 327–338 CrossRef CAS.
- F. Cheng, W. Li, G. Liu and Y. Tang, Curr. Top. Med. Chem., 2013, 13, 1273–1289 CrossRef CAS PubMed.
- A. Kumar, S. Roy, S. Tripathi and A. Sharma, J. Biomol. Struct. Dyn., 2016, 34, 239–249 CrossRef CAS PubMed.
- D. Lagorce, O. Sperandio, H. Galons, M. A. Miteva and B. O. Villoutreix, BMC Bioinf., 2008, 9, 396 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- A. Hospital, J. R. Goñi, M. Orozco and J. L. Gelpí, Adv. Appl. Bioinf. Chem., 2015, 8, 37 Search PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell Jr, Nat. Methods, 2017, 14, 71 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. G. Petersen, J. Chem. Phys., 1995, 103, 3668–3679 CrossRef CAS.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, W407–W410 CrossRef.
- M. U. Johansson, V. Zoete, O. Michielin and N. Guex, BMC Bioinf., 2012, 13, 173 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03281a |
| This journal is © The Royal Society of Chemistry 2019 |