
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAspect ratio-controlled preparation of α-CaSO4·0.5H2O from phosphogypsum in potassium tartrate aqueous solution†
Xiangbin Suna,
Genlei Zhang *ab and
Peng Cui*a
*ab and
Peng Cui*a
aSchool of Chemistry and Chemical Engineering, Anhui Province Key Laboratory of Controllable Chemistry Reaction and Material Chemical Engineering, Hefei University of Technology, Tunxi Road 193, Hefei 230009, PR China. E-mail: genleizhang@hfut.edu.cn; cuipeng@hfut.edu.cn
bSchool of Materials Science and Engineering, Hefei University of Technology, Tunxi Road 193, Hefei 230009, PR China
First published on 11th July 2019
Abstract
In this study, a simple and efficient strategy is developed to synthesize rod-shaped α-CaSO4·0.5H2O crystals with tunable aspect ratio from industrial phosphogypsum only in potassium tartrate aqueous solution at a low temperature. Industrial phosphogypsum can be effectively converted into rod-shaped α-CaSO4·0.5H2O crystals with the assistance of potassium tartrate, and the aspect ratio of α-CaSO4·0.5H2O crystals gradually decreases from 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1 with increasing the concentration of potassium tartrate. The formation process of the rod-shaped α-CaSO4·0.5H2O crystals in this system involves the dissolution of CaSO4·2H2O and nucleation of α-CaSO4·0.5H2O crystals. The tartrate ions from potassium tartrate in this system preferentially bind to (001) and (002) facets of α-CaSO4·0.5H2O crystals, inhibiting the growth of α-CaSO4·0.5H2O crystals along the c-axis and controlling its morphology and aspect ratio.
:1 to 1:1 with increasing the concentration of potassium tartrate. The formation process of the rod-shaped α-CaSO4·0.5H2O crystals in this system involves the dissolution of CaSO4·2H2O and nucleation of α-CaSO4·0.5H2O crystals. The tartrate ions from potassium tartrate in this system preferentially bind to (001) and (002) facets of α-CaSO4·0.5H2O crystals, inhibiting the growth of α-CaSO4·0.5H2O crystals along the c-axis and controlling its morphology and aspect ratio.
1 Introduction
Industrial phosphogypsum (I-PG) is mainly composed of calcium sulfate dihydrate (CaSO4·2H2O) and it is the main byproduct of the production process of phosphoric acid.1–3 Annual emissions of I-PG around the world have exceeded one billion tons over the last three years and the huge amount of I-PG not only occupies farmland but also causes severe pollution of the surrounding environment.3 In recent years, high-value utilization of I-PG has drawn significant attention and so far, the most effective way to achieve high-value utilization of I-PG is to produce α-calcium sulphate hemihydrate (α-CaSO4·0.5H2O).4–6To date, methods for preparing α-CaSO4·0.5H2O crystals from CaSO4·2H2O mainly include autoclave hydrothermal,7–10 atmospheric salt solution,11–14 reverse microemulsion15 and alcohol–water solution.16–18 The autoclave hydrothermal method involves high temperature (usually above 120 °C) and certain pressure,19,20 which are not conducive to industrialization. Whereas the crystallization temperature of α-CaSO4·0.5H2O crystals can be significantly decreased in salt solutions with high concentration by atmospheric salt solution method.21–23 However, the high-concentration salt solutions can lead to serious equipment corrosion.24,25 The reverse microemulsion and alcohol–water strategies that require a large amount of surfactants such as CTAB, SDS and organic alcohols such as ethylene glycol and glycerine.26,27 Although these surfactants and organic alcohols are advantageous for the synthesis of α-CaSO4·0.5H2O crystals with various morphologies and sizes, the close adsorptions to the surface of α-CaSO4·0.5H2O crystals cause great trouble for subsequent separation and cleaning. Therefore, the exploration of simple, low-cost and environmentally friendly crystal transformation method is one of the research hotspots of the high-value utilization of I-PG.
The prepared α-CaSO4·0.5H2O crystals are wildly used in construction industry, special binder, 3D print and medical materials.28–39 Mechanical strength is one of the most important properties for α-CaSO4·0.5H2O crystals that we pay more attention to, which is closely related to the crystal morphology.40,41 Presently, other crystal structures of α-CaSO4·0.5H2O crystals besides 1D structure have rarely been reported.4 And the 1D α-CaSO4·0.5H2O crystals with a lower aspect ratio (length-to-diameter) have better mechanical strength than these with higher one.42 To control the aspect ratio of 1D α-CaSO4·0.5H2O crystals, various crystallization (synthetic) methods have been proposed.43,44 The α-CaSO4·0.5H2O whiskers can be obtained by autoclave hydrothermal method in the presence of Mg2+, but its aspect ratio cannot be efficiently controlled and the reaction temperature up to 150 °C.9,18 Rod-shaped α-CaSO4·0.5H2O crystals were successfully produced from flue gas desulfurization in Ca(NO3)2 solution and its size could be significantly controlled via adjusting the Ca(NO3)2 concentration.42 However, regardless of the size of the rod-shaped α-CaSO4·0.5H2O crystals, its aspect ratio remained unchanged. Although CaSO4·2H2O crystals can be easily converted into α-CaSO4·0.5H2O crystals with 1D structure in many inorganic salt systems, the aspect ratio of the obtained α-CaSO4·0.5H2O crystals cannot be effectively regulated without any other organic modifiers. Using organic acid as the modifiers, rod-shaped α-CaSO4·0.5H2O crystals with different aspect ratios were successfully synthesized in CaCl2 aqueous solution containing a large amount of glycerol.14,45 What is more, α-CaSO4·0.5H2O crystals having a controlled aspect ratio was obtained employing surfactants such as CTAB and SDS as modifiers.15,27 However, the adjustable range of the aspect ratio is small, and the use of a large amount of surfactants brings great difficulty to the subsequent cleaning and purification work.
In this study, a simple, eco-friendly, and efficient route was developed to synthesize rod-shaped α-CaSO4·0.5H2O crystals with different aspect ratios in potassium tartrate (PT) aqueous solution. The conversion process and mechanism of the rod-shaped α-CaSO4·0.5H2O crystals were investigated. The selective use of PT was demonstrated to be vital for the aspect ratio of the obtained rod-shaped α-CaSO4·0.5H2O crystals, and as its concentration increased, the aspect ratio of α-CaSO4·0.5H2O crystals gradually decreased. The preferential adsorption of PT on the (001) and (002) facets of the obtained α-CaSO4·0.5H2O crystals inhibited the growth of the crystal along the c-axis, and thereby realized the control of the aspect ratio.
2 Experimental
2.1 Materials
The I-PG was obtained from Anhui Liuguo Chemical Co. Ltd, Anhui Province, China. Analytical reagent grade of sulfuric acid (H2SO4), potassium tartrate (PT) and ethanol were all purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai, China. PT aqueous solutions with different PT concentrations were prepared using purified water.2.2 Preparation of rod-shaped α-CaSO4·0.5H2O crystals
2.3 Characterizations
The X-ray fluorescence spectroscopy (XRF) used in this study was consisted of a 40 kV X-ray tube with a Rh anode target excitation source (Shimadzu; Japan). The scanning electronic microscopy (SEM) images were taken out by using a SU8020 scanning electron microscope (Hitachi; Japan) with an accelerating voltage of 10 kV. The transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) equipped with selected area electron diffraction (SAED) were performed using JEOL 2100F microscope (JEOL; Japan) operated at 200 kV. The energy dispersive X-ray spectroscopy (EDS) analysis was conducted using the TEM to identify the elemental composition of samples. The X-ray diffraction (XRD) spectra were operated via a Rigaku D/Max-2500 X-ray diffractometer (Rigaku; Japan) with a Cu Kα radiation (λ = 0.15406 nm) operated at 40 kV and 80 mA. The thermogravimetry and differential scanning calorimetry (TG-DSC) were conducted at a heating rate of 10°C min−1 (NET-ZSCH; Germany) to distinguish α-CaSO4·0.5H2O from the hemihydrate phase. The Fourier transform infrared spectroscopy (FT-IR) were recorded on (LX10-8813; USA) FT-IR spectrometer under the transmission scheme by using the KBr pellet technique. The X-ray photoelectron spectroscopy (XPS) data were recorded by ESCALAB250Xi (Thermo Scientific; USA) with using a monochromatized Al Kα (h = 1486.6 eV) X-ray source. All the spectra were corrected using C 1s signal located at 284.70 eV.3 Results and discussion
The I-PG from Liuguo Chemical Co., Ltd. is mainly composed of prismatic-sheet crystals with a width of 40–60 μm and a length of 80–120 μm, and its scanning electronic microscopy (SEM) images are shown in Fig. 1a. In addition, a large amount of fine debris was covered on the prismatic-sheet structure, and many impurity elements in the I-PG were detected by X-ray fluorescence spectroscopy (XRF) and the result was recorded in Table S1.† Fortunately, the fine debris and impurity elements in the I-PG could be effectively removed by H2SO4 aqueous solution while the structure and size of the prismatic-sheet crystals remained almost unchanged, which were demonstrated by SEM, transmission electron microscopic (TEM) and energy dispersive X-ray spectroscopy (EDS), and the results shown in Fig. 1b, c and S1,† respectively. To further characterize the H2SO4-treated I-PG, we used a number of tools to analyze the elemental distribution of the crystal and identify the structure. The EDS elemental mapping images of the obtained H2SO4-treated I-PG (Fig. 1d) confirmed that Ca, S and O were homogeneously distributed throughout the prismatic-sheet crystal. The high-resolution TEM (HRTEM) image of the obtained H2SO4-treated I-PG (Fig. 1e) showed that the observed d-spacing measured from the prismatic-sheet crystal was 0.763 nm, which was close to that for the (020) facet of the CaSO4·2H2O crystals.46 The corresponding selected area electron diffraction (SAED) image (inset in Fig. 1e) implied that the prismatic-sheet crystals possess a special single-crystalline feature. The X-ray diffraction (XRD) pattern of the obtained H2SO4-treated I-PG was shown in Fig. 1f, and the main diffraction peaks at 11.66°, 20.78°, 23.41°, 29.14° and 31.18° were assigned to CaSO4·2H2O (020), (021), (040), (041) and (−221) facets (JCPDS 33-0311),8 respectively. Furthermore, its thermogravimetry and differential scanning calorimetry (TG-DSC) analysis result (Fig. S2†) indicated that the thermal stability of the H2SO4-treated I-PG was the same as that of CaSO4·2H2O crystals.20 The above characterization results indicated that the obtained H2SO4-treated I-PG was CaSO4·2H2O crystals with prismatic-sheet structure.13 | ||
| Fig. 1 (a) SEM image of I-PG; (b) SEM and (c) TEM images of the H2SO4-treated I-PG, and (d) the corresponding elemental mapping images showing the distribution of Ca, S and O; (e) HRTEM and SAED (inset) images and (f) XRD pattern of the H2SO4-treated I-PG. | ||
The morphology of the obtained crystals prepared from the H2SO4-treated I-PG that used PT as the inducer was initially determined by electron microscopy. Typical SEM and TEM images of the crystals prepared in 50 mM PT aqueous solution for 6 h are shown in Fig. 2a and b. The SEM image (Fig. 2a) showed that the product consists of uniform rod-shaped crystals with a yield approaching 100%. The average length of the as-prepared rod-shaped crystals is about 88 μm (Fig. 2a) with a width of about 9 μm (Fig. 2b), that is, its aspect ratio is about 10:1. The HRTEM image (inset in Fig. 2b) of the crystal showed that the observed d-spacing were 0.598 nm and 0.344 nm, closing to that for (002) and (020) facets of α-CaSO4·0.5H2O, respectively.44 The XRD pattern of the as-prepared rod-shaped crystals was shown in Fig. 2c, and the main diffraction peaks at 14.91°, 25.61°, 26.82°, 29.77° and 31.52° were assigned to (200), (020), (−121), (400) and (204) facets of CaSO4·0.5H2O crystals (JCPDS 41-0224), respectively.41 The thermogravimetry and differential scanning calorimetry (TG-DSC) analysis of the obtained rod-shaped crystals was shown in Fig. 2d. The TG curve of the as-prepared rod-shaped crystals showed a 6.27% weight loss between 100 °C and 500 °C, which is consistent with the H2O mass fraction in CaSO4·0.5H2O crystals.5 Moreover, the endothermic peak appearing at 128.5 °C on the DSC curve also confirmed that the as-prepared crystals may be composed of CaSO4·0.5H2O crystals.20 The XRD and TG-DSC results indicate that the as-prepared rod-shaped crystals are CaSO4·0.5H2O crystals. It is well known that there are two types of CaSO4·0.5H2O crystals, namely α-CaSO4·0.5H2O and β-CaSO4·0.5H2O, and an exothermic peak at 154.9 °C on the DSC curve indicates that the as-prepared CaSO4·0.5H2O crystals are α-CaSO4·0.5H2O crystals.4,47
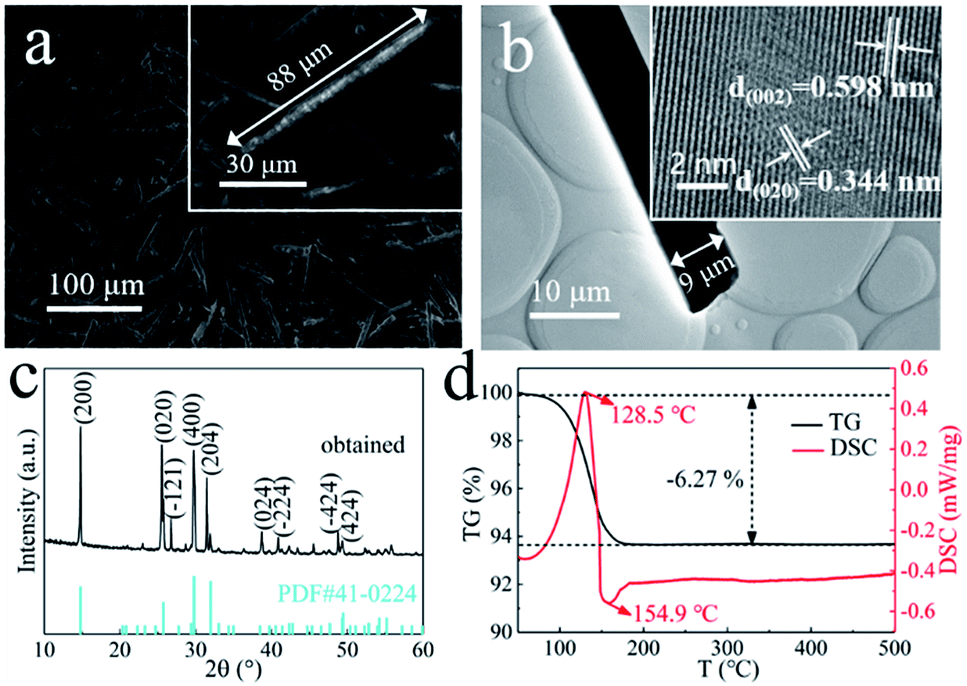 | ||
| Fig. 2 (a) SEM and (b) TEM images of the α-CaSO4·0.5H2O crystals prepared in 50 mM PT aqueous solution for 6 h; (c) XRD pattern and (d) TG-DSC analysis of the obtained α-CaSO4·0.5H2O crystals. The inset images in (b) show the corresponding HRTEM image of obtained crystal. | ||
To gain in-depth understanding of the conversion mechanism of the prismatic-sheet CaSO4·2H2O crystals to rod-shaped α-CaSO4·0.5H2O crystals, the conversion process was carefully investigated by collecting the SEM images (Fig. 3) of the intermediates at different reaction time. As shown in Fig. 3a of the intermediates obtained after the 0.5 h synthesis, a large amount of convex/concave regions (the yellow dotted area in Fig. 3a) and small particles (the yellow circles in Fig. 3a) with the diameter of less than 0.2 μm on the surface of the prismatic-sheet crystals could be observed. The XRD information of the intermediate (the black spectrum in Fig. 3e) indicated that the dissolution and nucleation behaviors of CaSO4·2H2O crystals occurred at this stage. As the reaction prolonged to 1 h, small particles with the diameter of less than 3 μm and a small proportion of short rod crystals appeared at the convex/concave regions (Fig. 3b). And at this point, it was learned from the XRD pattern (the purple spectrum in Fig. 3e) that part of the CaSO4·2H2O crystals has been converted to α-CaSO4·0.5H2O crystals. When the reaction time proceeded to 2 h, the initial prismatic-sheet CaSO4·2H2O crystals were entirely consisted with rod-shaped crystals (Fig. 3c), and this intermediate is mostly consisted of α-CaSO4·0.5H2O crystals (the yellow XRD spectrum in Fig. 3e). Further extension of reaction time to 4 h, the rod crystals in Fig. 3c were detached from its nucleation region and eventually evolved into a structurally intact uniform rod-shaped α-CaSO4·0.5H2O crystals (Fig. 3d and the green XRD spectrum in Fig. 3e). This time-dependent morphological changes suggest an attached process of the formation of rod-shaped α-CaSO4·0.5H2O crystals, where it started from the dissolution of the CaSO4·2H2O crystals and the initial form of small particles, then gradually grew into short rod-shaped crystals and finally lead to the uniform rod-shaped α-CaSO4·0.5H2O crystals (Fig. 2a).
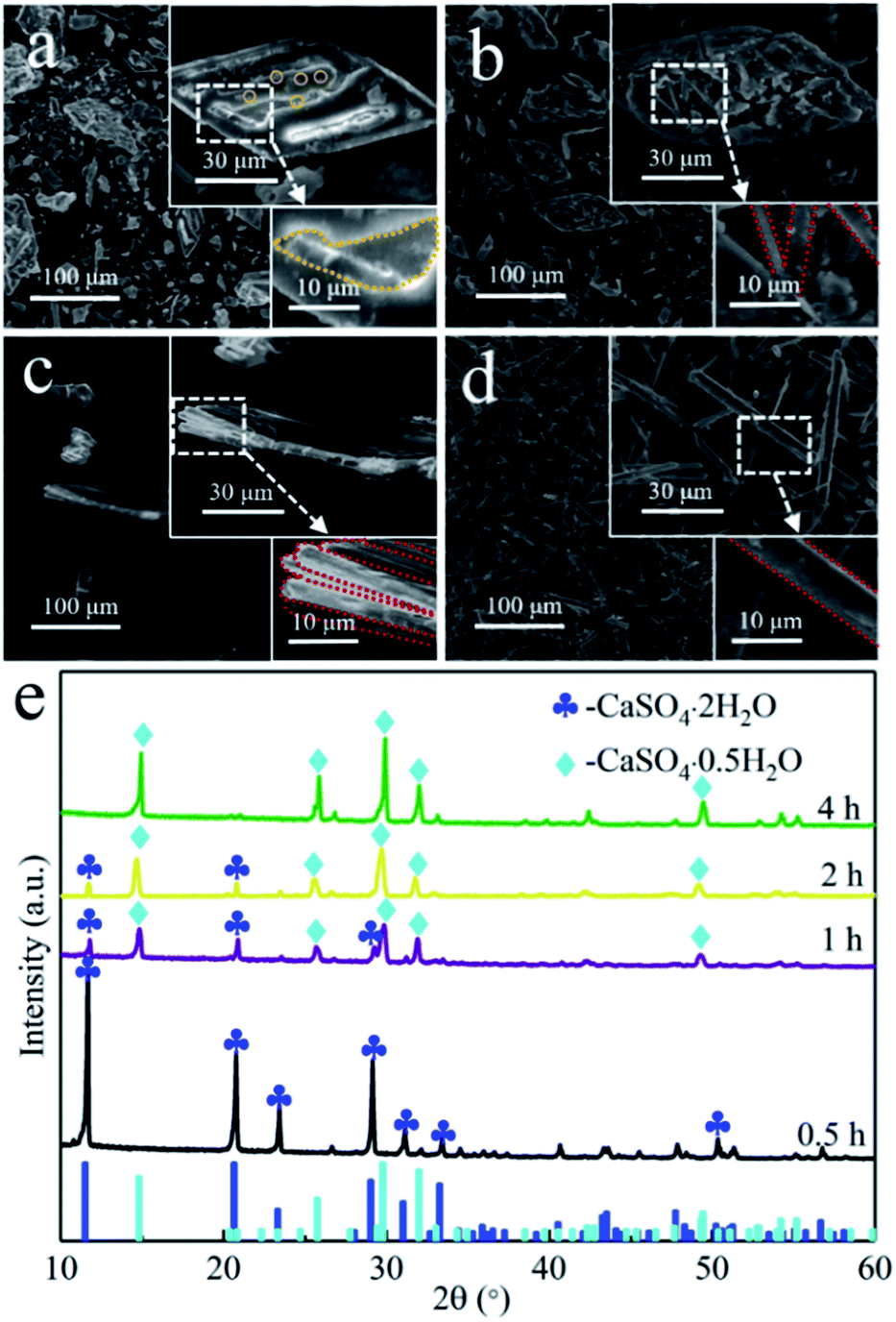 | ||
| Fig. 3 SEM images of the intermediates of rod-shaped α-CaSO4·0.5H2O crystals intermediates obtained after proceeding the reaction for (a) 0.5, (b) 1, (c) 2, and (d) 4 h; (e) XRD patterns of corresponding intermediates. | ||
In this study, the prismatic-sheet CaSO4·2H2O crystals was successfully converted into rod-shaped α-CaSO4·0.5H2O crystals in PT aqueous solution. However, we found that there were no changes on the morphology and physicochemical properties of the prismatic-sheet CaSO4·2H2O crystals when the PT was absent, and as shown in Fig. 4a and the XRD pattern in Fig. 4f. That is, the PT used as crystallize modifier in the formation of rod-shaped α-CaSO4·0.5H2O crystals. As combining indicated by Fig. 4b and f, the yield of the rod-shaped α-CaSO4·0.5H2O crystals increased substantially when the PT was present. Surprisingly, the length and aspect ratio of the rod-shaped α-CaSO4·0.5H2O crystals were depended on the concentration of PT, as shown in Fig. 4b–e, when the concentrations of PT were 20 mM, 50 mM, 100 mM and 200 mM, respectively, the lengths of the obtained rod-shaped α-CaSO4·0.5H2O crystals were 105 μm, 88 μm, 33 μm and 21 μm, and the corresponding widths were 2 μm, 9 μm, 11 μm and 20 μm in turn. That is, the aspect ratios of the rod-shaped ones were about 52:1, 10:1, 3:1 and 1:1, respectively (Fig. S3†). In addition, the rod-shaped α-CaSO4·0.5H2O crystals also could be obtained while used NaCl instead of PT, but its aspect ratio could not be effectively regulated via the concentration of NaCl (Fig. S4 and S5†). The EDS elemental mapping images of the obtained rod-shaped α-CaSO4·0.5H2O crystals (Fig. 5a) confirmed that carbonaceous species were adsorbed onto the surface of the α-CaSO4·0.5H2O crystals, which was also confirmed by the difference between the fourier transform infrared spectroscopy (FT-IR) spectra of the pure α-CaSO4·0.5H2O crystals and the obtained rod-shaped α-CaSO4·0.5H2O crystals in this work (Fig. 5b). As shown in Fig. 5b, three adsorption peaks at 1584 cm−1, 1404 cm−1 and 1385 cm−1 in the FT-IR spectrum of the obtained α-CaSO4·0.5H2O crystals (blue curve in Fig. 5b) should be assigned to the asymmetric and symmetric stretching vibrations of –COO–,44,48 and the three peaks were all shifted to lower wavenumbers as compared with those in the FT-IR spectrum of PT. This result indicated that tartrate ions from PT were absorbed on the surface of the obtained α-CaSO4·0.5H2O crystals. The adsorption behavior of PT was also illustrated by X-ray photoelectron spectroscopy (XPS). Compared to the C 1s XPS spectrum of pure α-CaSO4·0.5H2O in Fig. 5c, two new signal peaks appeared at around 288.41 eV and 286.41 eV in the C 1s XPS spectrum of the obtained α-CaSO4·0.5H2O crystals, which should be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH groups.14 This clearly indicated the adsorption of PT on the obtained α-CaSO4·0.5H2O surfaces considering the similarity of the C 1s XPS spectra of the obtained α-CaSO4·0.5H2O and PT. However, the C 1s peaks of CO and C–OH groups in the obtained α-CaSO4·0.5H2O were positively shifted about 0.25 eV as compared with that in PT, implying that the existence of the electronic effect between tartrate groups and Ca atoms.49–51 Furthermore, this speculate was also determined by the negative shift of the Ca 2p XPS peaks of the obtained α-CaSO4·0.5H2O comparing to that of the pure α-CaSO4·0.5H2O in Fig. 5c.52–54 One of the reasons for the dependence of the aspect ratio of the rod-shaped α-CaSO4·0.5H2O crystals on PT may be related to the preferential adsorption of PT on the crystal surfaces, which is caused by the difference in the properties of the rod-shaped α-CaSO4·0.5H2O facets.
O and C–OH groups.14 This clearly indicated the adsorption of PT on the obtained α-CaSO4·0.5H2O surfaces considering the similarity of the C 1s XPS spectra of the obtained α-CaSO4·0.5H2O and PT. However, the C 1s peaks of CO and C–OH groups in the obtained α-CaSO4·0.5H2O were positively shifted about 0.25 eV as compared with that in PT, implying that the existence of the electronic effect between tartrate groups and Ca atoms.49–51 Furthermore, this speculate was also determined by the negative shift of the Ca 2p XPS peaks of the obtained α-CaSO4·0.5H2O comparing to that of the pure α-CaSO4·0.5H2O in Fig. 5c.52–54 One of the reasons for the dependence of the aspect ratio of the rod-shaped α-CaSO4·0.5H2O crystals on PT may be related to the preferential adsorption of PT on the crystal surfaces, which is caused by the difference in the properties of the rod-shaped α-CaSO4·0.5H2O facets.
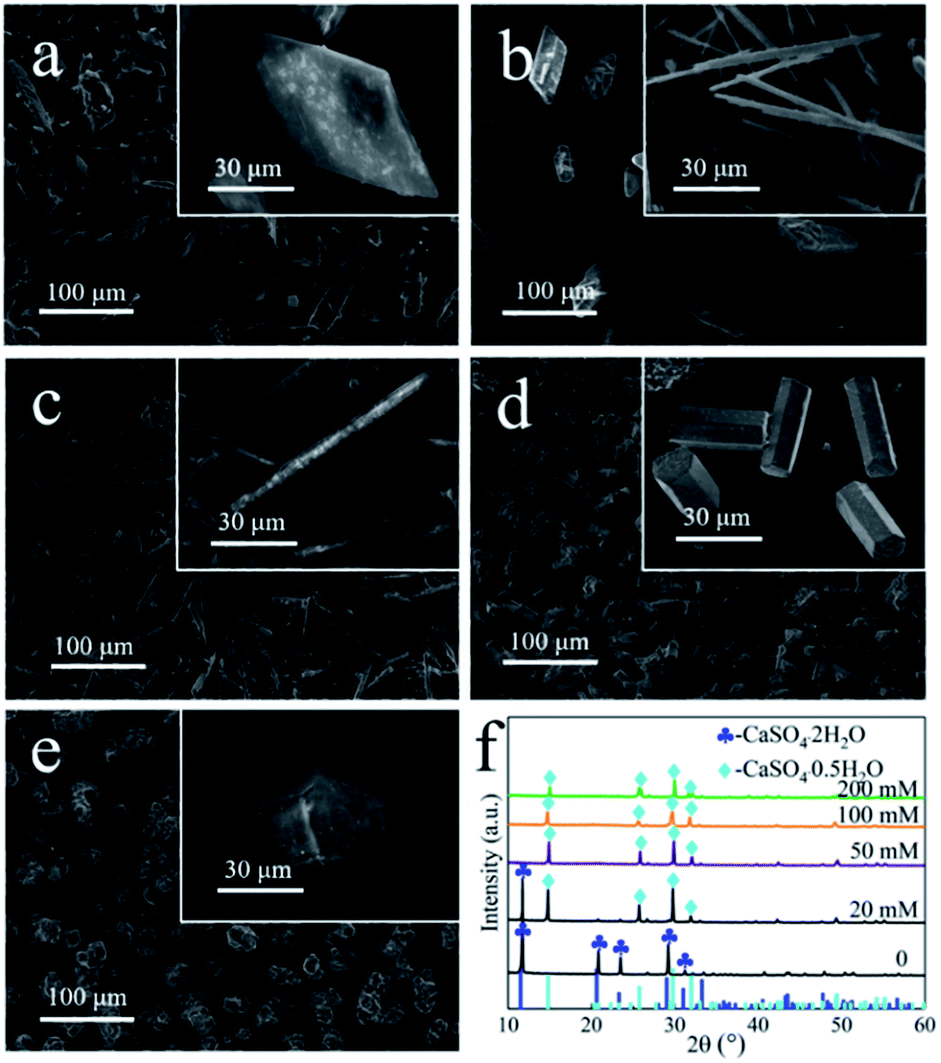 | ||
| Fig. 4 SEM images of samples that prepared in PT aqueous solution with different PT concentrations: (a) 0 mM, (b) 20 mM, (c) 50 mM, (d) 100 mM and (e) 200 mM; (f) XRD patterns of the corresponding samples. | ||
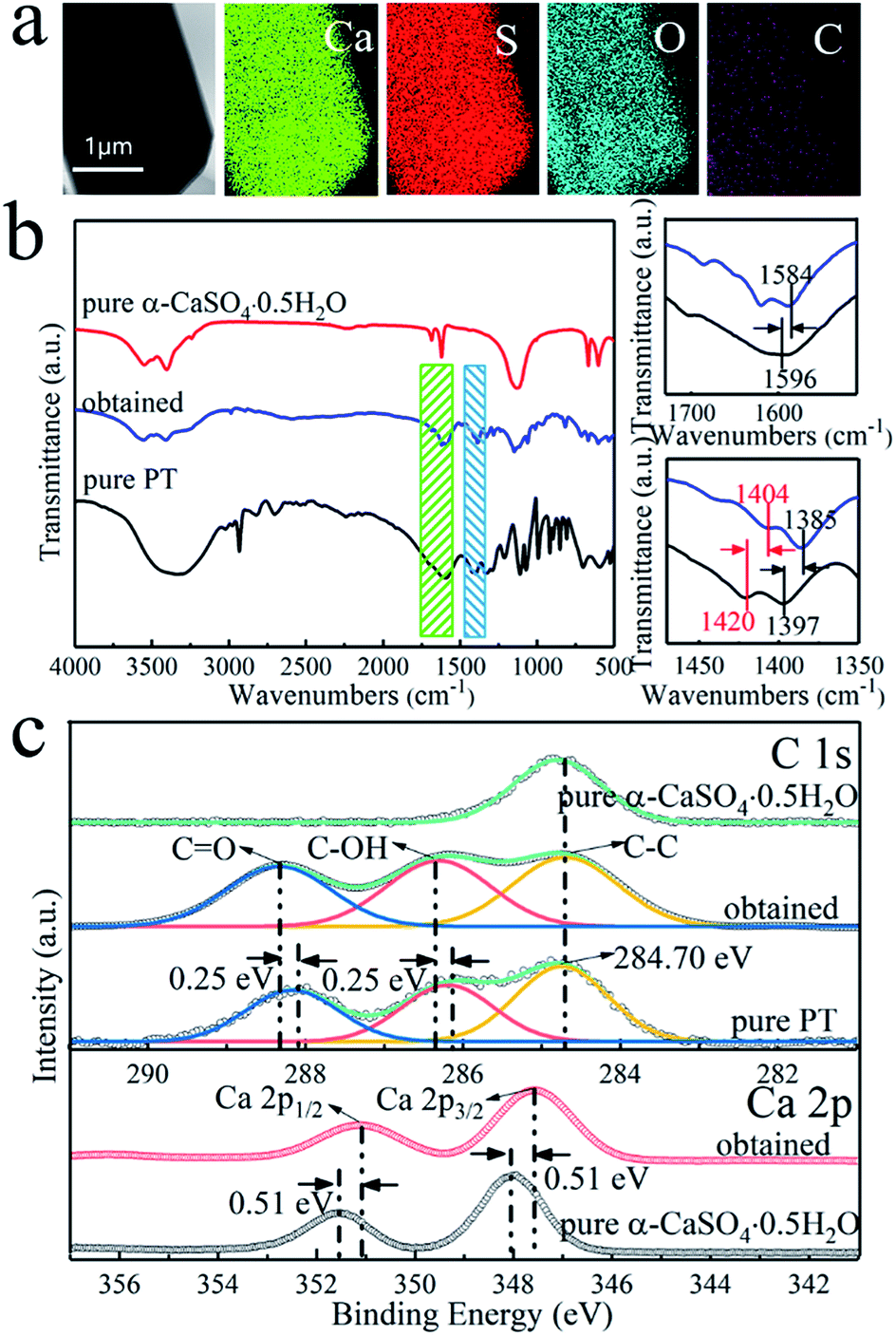 | ||
| Fig. 5 (a) Elemental mapping images of the obtained rod-shaped α-CaSO4·0.5H2O crystals prepared in 50 mM PT aqueous solution; (b) FT-IR spectra of PT, pure α-CaSO4·0.5H2O crystals and the obtained crystals; (c) C 1s and Ca 2p XPS spectra of PT, pure α-CaSO4·0.5H2O and obtained crystals. | ||
The molecular structure of the rod-shaped α-CaSO4·0.5H2O crystals deduced by Material Studio 5.0 software55 was shown in Fig. 6. The 3D model of the α-CaSO4·0.5H2O along the c-axis direction in Fig. 6a was derived a 2 × 2 × 2 supercell molecular structure model (a = 12.0275 Å, b = 6.9312 Å, c = 12.6919 Å), while the facet models in Fig. 6b–d were the top-view charts of (001), (002), (200), (400), (020), and (020) facets cut from the 3D model, ignoring the thickness of the facets. The 3D model in Fig. 6a showed that the obtained rod-shaped α-CaSO4·0.5H2O was consisted of –Ca–SO4–Ca–SO4–Ca– chains (partial structure of –Ca–SO4–Ca– chain in Fig. S6†), where every S atom was covalently bonded to the four O atoms. The distribution of Ca, S, O and H on different facets of the rod-shaped α-CaSO4·0.5H2O crystals were displayed in Fig. 6b–d, and the corresponding statistical results were listed in Table 1. The data in Table 1 showed that the distribution densities of Ca atoms on (001), (002), (200), (400) and (020) facets were 0.0900, 0.0900, 0.0568, 0.0568, and 0.0066, respectively. The denser distribution of Ca atoms on the top facets as (001) and (002) than that on the side facets as (200), (400) and (020) indicated that tartrate ions would preferentially adsorb on top facets of the rod-shaped α-CaSO4·0.5H2O crystals, which is further demonstrated by the C element content of the side and top facets of α-CaSO4·0.5H2O crystals (Fig. S7†). The preferential adsorption of tartrate ions reduced the specific surface energy of (001) and (002) facets, and led to an increased exposure of the two facets in the rod-shaped α-CaSO4·0.5H2O crystals, which hindered the crystal growth along the c-axis and reduced the exposure of (200), (400) and (020) side facets.12 Consequently, the length and width of the α-CaSO4·0.5H2O crystals gradually decreased and increased as the concentration of PT increased, respectively. Hence, using PT as a crystal modifier could effectively tune the aspect ratio of the rod-shaped α-CaSO4·0.5H2O crystals in H2O.
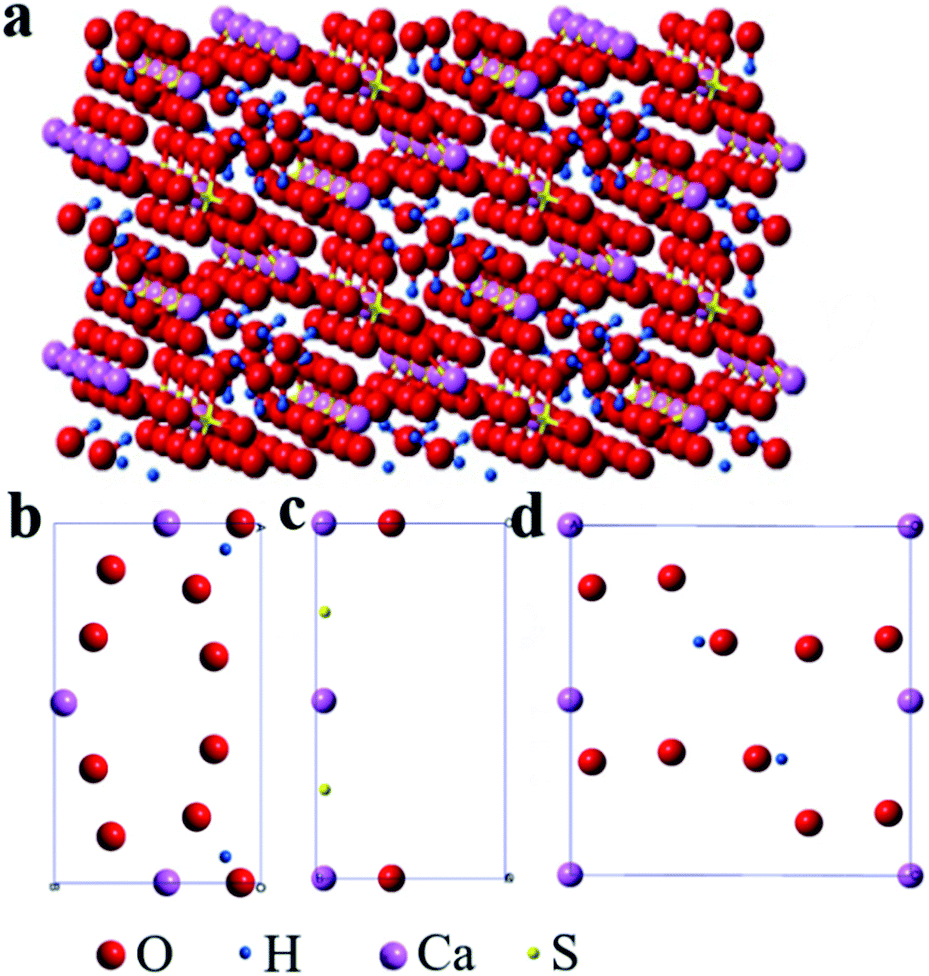 | ||
| Fig. 6 Molecular model of the obtained rod-shaped α-CaSO4·0.5H2O crystals (a) 3D model (2 × 2 × 2 supercell) along the c-axis direction and (b–d) facet slice models: (b) (001) and (002), (c) (200) and (400), (d) (020). | ||
| Facets | Element | Vertex (1/8) | Edge (1/4) | Surface (1/2) | Density/(Å2) |
|---|---|---|---|---|---|
| Ca | 0 | 3 | 0 | 0.0900 | |
| (001) | S | 0 | 0 | 0 | 0 |
| (002) | O | 0 | 2 | 8 | 0.0540 |
| H | 0 | 0 | 2 | 0.0120 | |
| Ca | 2 | 1 | 0 | 0.0568 | |
| (200) | S | 0 | 0 | 2 | 0.0114 |
| (400) | O | 0 | 2 | 0 | 0.0057 |
| H | 0 | 0 | 0 | 0 | |
| (020) | Ca | 4 | 2 | 0 | 0.0066 |
| S | 0 | 0 | 0 | 0 | |
| O | 0 | 0 | 10 | 0.0328 | |
| H | 0 | 0 | 2 | 0.0066 |
4 Conclusions
Summary, we have developed a simple and eco-friendly strategy for the synthesis of rod-shaped α-CaSO4·0.5H2O crystals having a tunable aspect ratio from the industrial phosphogypsum in potassium tartrate aqueous solution. The selective use of potassium tartrate was demonstrated to be vital for the formation of rod-shaped α-CaSO4·0.5H2O crystals. Once the potassium tartrate was present, the industrial phosphogypsum could be successfully converted into rod-shaped α-CaSO4·0.5H2O crystals at a lower temperature. As the potassium tartrate concentration increased from 20 mM to 200 mM, the crystals length decreased from 105 μm to 21 μm, and corresponding aspect ratio decreased sharply from 52:1 to 1:1. The process of the formation of the rod-shaped α-CaSO4·0.5H2O crystals was explored and discussed, where it started from the dissolution of the CaSO4·2H2O crystals and the initial form of small particles, then gradually grew into short rod-shaped crystals and finally lead to the uniform rod-shaped α-CaSO4·0.5H2O crystals. The preferential adsorption of tartrate ions on the top facets of α-CaSO4·0.5H2O crystals as (001) and (002) inhibit the growth of the crystal along the c-axis, and thereby realized the regulation of α-CaSO4·0.5H2O crystals aspect ratio. We expect our study will provide a new idea for the recycling of industrial phosphogypsum.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by Natural Science Foundation of China (21806065) and Doctoral Special Research Foundation of HFUT (JZ2018HGBZ0122 and JZ2018HGBZ0087). We also thank the support from Anhui Province Key Laboratory of Advanced Catalytic Materials and Reaction Engineering (45000-411104/007) and Anhui Liuguo Chemical Co. Ltd (W2018JSKF0563).References
- P. M. Rutherford, M. J. Dudas and R. A. Samek, Sci. Total Environ., 1994, 149, 1–38 CrossRef CAS.
- T. Hanan, C. Mohamed, F. A. López, F. J. Alguacil and L. D. Aurora, J. Environ. Manage., 2009, 90, 2377–2386 CrossRef PubMed.
- C. R. Cánovas, F. Macías, R. Pérez-López, M. D. Basallote and R. Millán-Becerro, J. Cleaner Prod., 2018, 174, 678–690 CrossRef.
- Q. S. Chen, C. Y. Jia, Y. Li, J. Xu, B. H. Guan and M. Z. Yates, Langmuir, 2017, 33, 2362–2369 CrossRef CAS.
- G. M. Jiang, W. Y. Fu, Y. Z. Wang, X. Y. Liu, Y. X. Zhang, F. Dong, Z. Y. Zhang, X. M. Zhang, Y. M. Huang, S. Zhang and X. S. Lv, Environ. Sci. Technol., 2017, 51, 10519–10525 CrossRef CAS PubMed.
- G. M. Jiang, J. X. Li, Y. L. Nie, S. Zhang, F. Dong, B. H. Guan and X. S. Lv, Environ. Sci. Technol., 2016, 50, 7650–7657 CrossRef CAS PubMed.
- H. L. Fu, C. Y. Jia, Q. S. Chen, X. T. Cao and X. M. Zhang, Particuology, 2018, 40, 98–104 CrossRef CAS.
- F. Hao, X. F. Song, T. J. Liu, Y. X. Xu and J. G. Yu, J. Cryst. Growth, 2018, 495, 29–36 CrossRef.
- S. C. Hou, J. Wang, X. X. Wang, H. Y. Chen and L. Xiang, Langmuir, 2014, 30, 9804–9810 CrossRef CAS.
- W. P. Zhao, C. H. Gao, G. Y. Zhang, J. Xu, C. X. Wang and Y. M. Wu, New J. Chem., 2016, 40, 3104–3108 RSC.
- C. Y. Jia, Q. S. Chen, Z. Xu, W. Hao, G. M. Jiang and B. H. Guan, Ind. Eng. Chem. Res., 2016, 55, 9189–9194 CrossRef CAS.
- Q. J. Guan, Y. H. Hu, H. H. Tang, W. Sun and Z. Y. Gao, J. Colloid Interface Sci., 2018, 530, 292–301 CrossRef CAS PubMed.
- H. L. Fu, B. H. Guan and Z. B. Wu, Fuel, 2015, 150, 602–608 CrossRef CAS.
- J. W. Mao, G. M. Jiang, Q. S. Chen and B. H. Guan, Colloids Surf., A, 2014, 443, 265–271 CrossRef CAS.
- K. Bao, B. H. Guan, M. Z. Yates and Z. B. Wu, Langmuir, 2012, 28, 14137–14142 CrossRef.
- Z. Y. Pan, Y. Lou, G. Y. Yang, X. Ni, M. C. Chen, H. Z. Xu, X. G. Miao, J. L. Liu, C. F. Hu and Q. Huang, Ceram. Int., 2013, 39, 5495–5502 CrossRef CAS.
- B. H. Guan, G. M. Jiang, H. L. Fu, Y. Li and Z. B. Wu, Ind. Eng. Chem. Res., 2011, 50, 13561–13567 CrossRef CAS.
- X. L. Mao, X. F. Song, G. M. Lu, Y. Z. Sun, Y. X. Xu and J. G. Yu, Ind. Eng. Chem. Res., 2014, 53, 17625–17635 CrossRef CAS.
- Y. B. Tang and J. M. Gao, Langmuir, 2017, 33, 9637–9644 CrossRef CAS.
- W. L. Teng, J. S. Wang, J. S. Wu, Y. C. Du, X. J. Jia, H. Y. Li and T. N. Wang, J. Cryst. Growth, 2018, 496, 24–30 CrossRef.
- L. C. Yang, Z. B. Wu, B. H. Guan, H. L. Fu and Q. Q. Ye, J. Cryst. Growth, 2009, 311, 4518–4524 CrossRef CAS.
- B. H. Guan, H. L. Fu, Y. Jie, G. M. Jiang, K. Bao and Z. B. Wu, Fuel, 2011, 90, 36–41 CrossRef CAS.
- L. C. Yang, B. H. Guan, Z. B. Wu and X. F. Ma, Ind. Eng. Chem. Res., 2009, 48, 7773–7779 CrossRef CAS.
- Q. J. Guan, H. H. Tang, W. Sun, Y. H. Hu and Z. G. Yin, Ind. Eng. Chem. Res., 2017, 56, 9831–9838 CrossRef CAS.
- T. Kamimura, S. Hara, H. Miyuki, M. Yamashita and H. Uchida, Corros. Sci., 2006, 48, 2799–2812 CrossRef CAS.
- X. L. Mao, X. F. Song, G. M. Lu, Y. Z. Sun, Y. X. Xu and J. G. Yu, Ind. Eng. Chem. Res., 2015, 54, 2781–4787 Search PubMed.
- X. L. Mao, X. F. Song, G. M. Lu, Y. X. Xu, Y. Z. Sun and J. G. Yu, Chem. Eng. J., 2015, 278, 320–327 CrossRef CAS.
- S. Grudén, M. Sandelin, V. Rasanen, P. Micke, M. Hedeland, N. Axén and M. Jeansson, Eur. J. Pharm. Biopharm., 2017, 114, 186–193 CrossRef.
- Z. X. Zhou, F. Buchanan, C. Mitchell and N. Dunne, Mater. Sci. Eng., C, 2014, 38, 1–10 CrossRef CAS.
- Y. Shen, S. Z. Yang, J. L. Liu, H. Z. Xu, Z. L. Shi, Z. Q. Lin, X. Z. Ying, P. Guo, T. Lin, S. G. Yan, Q. Huang and L. Peng, ACS Appl. Mater. Interfaces, 2014, 6, 12177–12188 CrossRef CAS.
- D. Pförringer, N. Harrasser, M. Beirer, M. Crönlein, A. Stemberger, M. V. Griensven, M. Lucke, R. Burgkart and A. Obermeier, Materials, 2018, 11, 935–943 CrossRef.
- C. C. Chen, C. W. Wang, N. S. Hsueh and S. J. Ding, J. Alloys Compd., 2014, 585, 25–31 CrossRef CAS.
- H. L. Tan, S. B. Yang, P. Y. Dai, W. Y. Li and B. Yue, Int. J. Nanomed., 2015, 10, 4341–4350 CAS.
- S. P. Zhang, X. X. Xu, S. A. Memon, Z. J. Dong, D. X. Li and H. Z. Cui, Constr. Build. Mater., 2018, 167, 253–262 CrossRef CAS.
- N. Li, L. L. Xu, R. Wang, L. Li and P. M. Wang, Cem. Concr. Res., 2018, 108, 103–115 CrossRef CAS.
- X. M. Chen, J. M. Gao, C. B. Liu and Y. S. Zhao, Constr. Build. Mater., 2018, 190, 53–64 CrossRef CAS.
- V. Antunes, A. Candeias, M. J. Oliveira, M. Lorena, A. I. Seruya, M. L. Carvalho, M. Gil, J. Mirão, J. Coroado and V. Gomes, Microchem. J., 2016, 125, 290–298 CrossRef CAS.
- K. H. Lee and K. H. Yang, Constr. Build. Mater., 2016, 127, 442–449 CrossRef CAS.
- A. Farzadi, V. Waran, M. Solati-Hashjin, Z. A. A. Rahman, M. Asadi and N. A. A. Osman, Ceram. Int., 2015, 41, 8320–8330 CrossRef CAS.
- C. J. Liu, Q. Zhao, Y. G. Wang, P. Y. Shi and M. F. Jiang, Appl. Surf. Sci., 2016, 360, 263–269 CrossRef CAS.
- B. G. Ma, W. D. Lu, Y. Su, Y. B. Li, C. Gao and X. Y. He, J. Cleaner Prod., 2018, 195, 396–405 CrossRef CAS.
- G. M. Jiang, H. Wang, Q. S. Chen, X. M. Zhang, Z. B. Wu and B. H. Guan, Fuel, 2016, 174, 235–241 CrossRef CAS.
- Y. Q. Zhang, D. Wang, L. L. Zhang, Y. Le, J. X. Wang and J. F. Chen, Ind. Eng. Chem. Res., 2017, 56, 14053–14059 CrossRef CAS.
- G. M. Jiang, Q. S. Chen, C. Y. Jia, S. Zhang, Z. B. Wu and B. H. Guan, Phys. Chem. Chem. Phys., 2015, 17, 11509–11515 RSC.
- F. Li, J. L. Liu, G. Y. Yang, Z. Y. Pan, X. Ni, H. Z. Xu and Q. Huang, J. Cryst. Growth, 2013, 374, 31–36 CrossRef CAS.
- K. Gupta, S. Singh and M. S. R. Rao, Cryst. Growth Des., 2016, 16, 3256–3261 CrossRef CAS.
- K. Park, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2003, 6, 991–995 CrossRef.
- J. M. Ouyang, H. Zheng and S. P. Deng, J. Cryst. Growth, 2006, 293, 118–123 CrossRef CAS.
- Y. T. Song, P. J. Swedlund, C. W. Zou and R. D. Hamid, Chem. Geol., 2013, 347, 114–122 CrossRef CAS.
- G. F. Moreira, E. R. Peçanha, M. B. M. Monte, L. S. L. Filho and F. Stavale, Miner. Eng., 2017, 110, 96–103 CrossRef CAS.
- Y. Song, D. Zemlyanov, X. Chen, H. C. Nie, Z. Y. Su, K. Fang, X. H. Yang, D. Smith, S. Byrn and J. W. Lubach, Mol. Pharmaceutics, 2015, 13, 483–492 CrossRef.
- L. O. Filippov, V. V. Severov and I. V. Filippova, Int. J. Miner. Process., 2013, 123, 120–128 CrossRef CAS.
- W. Kai, Y. N. Qin, L. Fan, M. Q. Li, L. Qiao, L. Fei, J. R. Feng, Y. Chao, G. Peng and S. J. Guo, Small Methods, 2018, 2, 1700331–1700339 CrossRef.
- C. Y. Tang, N. Zhang, Q. Shao, X. Q. Huang and X. H. Xiao, Nanoscale, 2019, 12, 5145–5150 RSC.
- Material Studio 5.0, Molecular Simulations Inc. (MSI), Genetics Computer Group (GCG), Synopsys Scientific and Oxford Molecular Group (OMG), 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03569a |
| This journal is © The Royal Society of Chemistry 2019 |