
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubtle structural variation in azine/imine derivatives controls Zn2+ sensitivity: ESIPT-CHEF combination for nano-molar detection of Zn2+ with DFT support†
Somnath Khanra,
Sabyasachi Ta,
Milan Ghosh,
Sudeshna Chatterjee and
Debasis Das *
*
Department of Chemistry, The University of Burdwan, Golapbag, Burdwan, 713104, West Bengal, India. E-mail: ddas100in@yahoo.com; Fax: +91-342-2530452; Tel: +91-342-2533913 ext. 424
First published on 8th July 2019
Abstract
Excited-state intra-molecular proton transfer (ESIPT)-active imine and azine derivatives, structurally characterised by XRD, and denoted L1, L2, L3 and L4, possess weak fluorescence. The interaction of these probes with Zn2+ turns ON the fluorescence to allow its nano-molar detection. Among the four ESIPT-active molecules, L2, L3 and L4 are bis-imine derivatives while L1 is a mono-imine derivative. Among the three bis-imine derivatives, one is symmetric (L3) while L2 and L4 are unsymmetrical. The lowest detection limits (DL) of L1, L2, L3 and L4 for Zn2+ are 32.66 nM, 36.16 nM, 15.20 nM and 33.50 nM respectively. All the probes bind Zn2+ (105 M−1 order) strongly. Computational studies explore the orbital level interactions responsible for the associated photo-physical processes.
Introduction
Design and development of organic molecules for selective recognition of bio-relevant metal ions is a vibrant field of research for its extensive application in biology, ecology and endocrine systems.1–6 Meanwhile, the concept of excited-state intra-molecular proton transfer (ESIPT) processes has been extensively applied in molecular probes, luminescent materials, and molecular logic gates as it offers large Stokes shifts7 to exclude spectral overlap between absorption and emission, highly desirable for fluorescence sensing.8–10 The basis of ESIPT is an intra-molecular hydrogen bond that allows proton transfer from a relatively acidic region of a molecule (hydrogen bond donor, –OH and NH2) to its basic region (hydrogen bond acceptor,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and CO), leading to formation of a five/six membered ring. In most cases, this happens due to the existence of keto–enol tautomerism in the system. In some cases, this leads to dual fluorescence, one at a shorter wavelength due to the enol form and the other at a longer wavelength due to the proton-transferred form.11 Knocking out this proton by a cation arrests this ESIPT process, leading to fluorescence enhancement.
N– and CO), leading to formation of a five/six membered ring. In most cases, this happens due to the existence of keto–enol tautomerism in the system. In some cases, this leads to dual fluorescence, one at a shorter wavelength due to the enol form and the other at a longer wavelength due to the proton-transferred form.11 Knocking out this proton by a cation arrests this ESIPT process, leading to fluorescence enhancement.
The importance of biological zinc needs no introduction.12,13 In humans, its concentration varies from 12 μM (intracellular serum) to ∼0.1–0.5 mM (brain and nerve tissue).14 As Zn2+ strongly interacts with proteins,15 peptides and enzymes,16,17 it regulates functions like gene expression, cellular metabolism, apoptosis, neurotransmission, mammalian reproduction and so on. Moreover, zinc disorders directly affect the metabolic activities of DNA.18–21 The US Food and Nutritional Board recommends a daily dietary intake of Zn2+ from 2 mg (infants, 0–5 years) to 15 mg for adults.22 Its deficiency reduces the ability of the islet cells to produce insulin.23 Excess Zn2+ is neurotoxic and can cause serious neurological disorders like Alzheimer's and Parkinson's disease.24–30 The WHO recommends up to 76 μM Zn2+ in drinking water.31,32 Among a significant number of pioneering Zn2+ sensors,33–36 a few recent reports deserve to be mentioned here. For example, Yoon et al.37 developed both R and S forms of a bis-binaphthol-pyrene conjugate for selective recognition of Zn2+. Bhalla et al.38 have reported a hexaphenylbenzene derivative to recognise Zn2+ in aqueous media. Out of several notable efforts at Zn2+ sensing,39,40 however, few are based on the ESIPT approach.41,42
Herein, an imine (L1) and three azine (L2, L3 and L4) derivatives have been synthesised and structurally characterised by single crystal X-ray diffraction analysis. Out of the three azine derivatives, one is symmetrical (L3) while other two (L2 and L4) are unsymmetrical. Photo-physical interactions of all the molecules with Zn2+ allow selective determination of nano-molar Zn2+.
Experimental
Materials and equipment
High purity bis–tris buffer, 4-diethylamino-2-hydroxy-benzaldehyde, N1-phenyl-ethane-1,2-diamine, 2-hydroxy benzaldehyde, 4-methoxy benzaldehyde, 4-hydroxy benzaldehyde, Zn(OAc)2, ZnCl2, ZnSO4, Zn(PO4)3, Zn(OCl)2 and other chemicals have been purchased from Sigma-Aldrich (India) and used as received. The spectroscopic and analytical grade solvents were used without further purification. Milli-Q Millipore 18.2 MΩ cm−1 water was used as and when required.Absorption spectra were recorded with a Shimadzu Multi Spec 2450 spectrophotometer. A Hitachi F-4500 spectro-fluorometer was used to record steady state emission spectra. The elemental analyses (C, H and N) have been performed on a PerkinElmer 2400 CHN elemental analyser. FTIR spectra were collected using a Shimadzu FTIR (model IR Prestige 21 CE) spectrometer. Electrospray ionisation mass spectra (ESI mass) were recorded using a Thermo Fisher Scientific Exactive Mass Spectrometer using HR (+/−) mode. The pH was measured using a Systronics digital pH meter (model 335). The 1H NMR spectra were collected using a Bruker 400 MHz spectrometer using CDCl3 and DMSO-d6 as solvent. The chemical shift is expressed in ppm using the residual solvent peak as an internal reference. Multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Coupling constants (J) are reported in Hertz (Hz).
Single crystal X-ray diffraction data were collected on a Bruker X8 APEX-II CCD diffractometer at 100(2) K using graphite-monochromated Mo-Kα radiation (0.71073 Å) at 150 K. Data were processed and corrected for Lorentz and polarisation absorption effects. Crystal structures were solved by standard direct methods using SHELXS43 and refined by full-matrix least-squares with SHELXL44 and OLEX2 software.45 Significant crystal parameters and refinement data are presented in Table S1 (ESI†). All non-hydrogen atoms are refined with anisotropic thermal displacements. Hydrogen atoms are included in the structure factor calculation in geometrically idealised positions, with thermal parameters depending on the parent atom, using a riding model. Images were generated by Mercury software.46
Synthesis
Fig. 1 shows the molecules (L1, L2, L3 and L4) that have been synthesised simply by refluxing mixtures of aldehyde and amine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2, mole ratio) as per requirement.
:1 or 1:2, mole ratio) as per requirement.
 | ||
| Fig. 1 ChemDraw structures of new molecules. | ||
N); 1512, ν(CC, stretch); 1331, ν(C–N, stretch) 1250 and 1144, ν(C–O, stretch); [Fig. S1b, ESI†]. 1H NMR (CDCl3, 400 MHz, J, Hz, δ ppm, reference peak 7.26 ppm) [Fig. S1c, ESI†]: 10.730 (1H, s) 9.499 (1H, s), 8.224 (1H, s) 7.970 (1H, s), 7.276 (1H, s), 7.205–7.160 (2H, m), 6.987–6.965 (1H, d, J = 8.8 Hz) 6.736–6.700 (1H, q), 6.643–6.614 (1H, m), 6.181–6.134 (1H, m), 3.715–3.687 (1H, t, J = 5.6 Hz), 3.571–3.541 (1H, t, J = 6.0 Hz), 3.490–3.317 (3H, m), 1.288–1.213 (1H, q), 1.204–1.158 (2H, m). The structure of L1 was confirmed by SC-XRD analysis.
 | ||
| Scheme 1 Synthetic protocol of L1, L2, L3 and L4. | ||
:1 mole ratio and refluxed for 4 h (Scheme 1). The final solution was kept for slow evaporation and yellow crystals of L2 were collected after one week in 91% yield. Molecular formula: C19H16N2O2 (MW = 304.12). Anal. found (%), C, 74.78; H, 5.11 and N, 9.39; calcd (%), C, 74.98; H, 5.30 and N, 9.20. ESI-MS (m/z): [M + H]+, 305.29; [M + Na]+, 327.28 [Fig. S2a, ESI†]. FTIR (KBr, cm−1): 3214.83, ν(O–H); 3012.81 and 2923.69, ν(C–H, aromatic); 1612.19, ν(CN); 1507.98, ν(CC, stretch); 1246.49 and 1172.28, ν(C–O, stretch); 1098.13, ν(N–N, stretch). [Fig. S2b, ESI†]. 1H NMR (CDCl3, 400 MHz, J, Hz, δ ppm, reference peak, 7.26 ppm) [Fig. S2c, ESI†]: 13.406 (1H, s), 9.707 (1H, s), 9.646 (1H, s), 8.740–8.634 (1H, d, J = 1.6 Hz), 8.574 (1H, s), 8.202–8.181 (2H, d, J = 8.4 Hz), 7.879–7.808 (5H, m), 7.585–7.567 (1H, d, J = 7.2), 7.430–7.396 (1H, d, J = 6.4 Hz), 7.283–7.261 (1H, d, J = 9.2), 7.032–7.003 (3H, t, J = 5.8 Hz), 3.902–3.842 (5H, t, J = 12 Hz). The structure of L2 was confirmed by SC-XRD analysis.N); 1474.46, ν(CC, stretch); 1251.60, ν(C–O, stretch); 1122.08, ν(N–N, stretch). [Fig. S3b, ESI†]. 1H NMR (DMSO-d6, 400 MHz, J, Hz, δ ppm, reference peak, 2.5 ppm) [Fig. S3c, ESI†]: 10.450 (1H, s), 7.930 (1H, s), 7.203–7.179 (1H, q), 7.130–7.091 (1H, q), 6.825–6.789 (3H, t, J = 7.2 Hz). The structure of L3 was further confirmed by SC-XRD analysis.N); 1508.14, ν(CC, stretch); 1253.26 and 1158.12, ν(C–O, stretch); 1100.24, ν(N–N, stretch). [Fig. S4b, ESI†]. 1H NMR (DMSO-d6, 400 MHz, J, Hz, δ ppm, reference peak, 2.5 ppm) [Fig. S4c, ESI†]: 10.726 (1H, s) 9.500 (2H, s), 8.547 (1H, s), 7.693–7.671 (2H, d, J = 8.8 Hz), 7.615 (1H, s), 7.305–7.278 (2H, q) 6.863–6.842 (2H, d, J = 8.4 Hz), 6.725–6.704 (2H, q), 6.369 (2H, s).Then L4 was synthesised by refluxing a mixture of L4a (68 mg, 0.5 mmol) and 2-hydroxy-benzaldehyde (61 mg, 0.5 mmol) in methanol for 3 h (Scheme 1). The final solution was kept for slow evaporation and yellow crystals of L4 were observed after one week in 92.7% yield. Molecular formula: C14H12N2O2 (MW, 240.09). Anal. found (%), C, 70.12; H, 4.83 and N, 11.02; calcd (%), C, 69.99; H, 5.03 and N, 11.66. ESI-MS (m/z): [M + H]+, 241.79 [Fig. S4d, ESI†]. FTIR (KBr, cm−1): 3319.09, ν(O–H); 3027.96, ν(C–H, aromatic); 1616.41, ν(CN); 1478.60, ν(CC, stretch); 1216.17 and 1154.94, ν(C–O, stretch); 1100.71, ν(N–N, stretch). [Fig. S4e, ESI†]. 1H NMR (DMSO-d6, 400 MHz, J, Hz, δ ppm, reference peak, 2.5 ppm) [Fig. S4f, ESI†]: 11.427 (1H, s), 11.051 (1H, s) 8.993 (1H, s), 7.940 (1H, s), 7.696–7.677 (1H, d, J = 7.6 Hz), 7.418–7.379 (1H, t, J = 7.8 Hz) 7.222–7.203 (1H, t, J = 3.8), 7.155–7.009 (1H, q), 6.997–6.907 (2H, m), 6.873–6.804 (4H, m).
Ad1: molecular formula, C21H27N3O3Zn; anal. found (%), C, 57.41; H, 6.17 and N, 8.30; calcd (%), C, 58.00; H, 6.26 and N, 9.66. ESI-MS (m/z): [Ad1 + MeOH + Na]+, 486.79 [Fig. S5a, ESI†]. FTIR (KBr, cm−1): 3107.81, ν(C–H, aromatic); 1560.60, ν(CN); 1437.17, ν(CC, stretch); 1247.28 and 1148.75, ν(C–O, stretch) [Fig. S5b, ESI†].
Ad2: molecular formula, C21H19N2O4Zn: anal. found (%), C, 58.41; H, 5.07 and N, 6.01; calcd, C, 58.82; H, 4.47 and N, 6.53. ESI-MS (m/z): [M + Na]+, 451.50 [Fig. S6a, ESI†]. FTIR (KBr, cm−1): 3102.54, ν(C–H, aromatic); 1557.86, ν(CN); 1451.65, ν(CC, stretch); 1249.97 and 1169.97, ν(C–O, stretch); 1066.20, ν(N–N, stretch) [Fig. S6b, ESI†].
Ad3: molecular formula, C18H18N2O6Zn2: anal. found (%), C, 43.68; H, 4.23 and N, 4.97; calcd (%), C, 44.20; H, 3.71 and N, 5.73. ESI-MS (m/z): [M + H]+, 486.18 [Fig. S7a, ESI†]. FTIR (KBr, cm−1): 3098.60, ν(C–H, aromatic); 1601.91 and 1574.94, ν(CN); 1438.32, ν(CC, stretch); 1312.55 and 1171.95, ν(C–O, stretch); 1135.28, ν(N–N, stretch) [Fig. S7b, ESI†].
Ad4: molecular formula, C16H15N2O4Zn: anal. found (%), C, 52.85; H, 3.92 and N, 6.76; calcd (%), C, 52.69; H, 4.15 and N, 7.68. ESI-MS (m/z): [M + H]+, 363.07 [Fig. S8a, ESI†]. FTIR (KBr, cm−1): 3102.54, ν(C–H, aromatic); 1614.49 and 1559.52, ν(CN); 1450.67, ν(CC, stretch); 1286.86 and 1197.53, ν(C–O, stretch); 1111.63, ν(N–N, stretch). [Fig. S8b, ESI†].
Results and discussion
Single crystal X-ray diffraction analysis of L1, L2 and L3
L1, L2 and L3 were well characterised by spectroscopic techniques and their stereo-chemical structures were confirmed by single crystal X-ray diffraction analysis (Fig. 2). Their molecular views along with atom labelling schemes are shown in Fig. S9a–c (ESI†) while their packing patterns are shown in Fig. S10a–c (ESI†). The crystallographic data and refinement parameters are presented in Table S1 (ESI†) while selected bond lengths and angles are listed in Table S2a–c (ESI†). | ||
| Fig. 2 ORTEP views of L1, L2 and L3 and DFT optimised structure of L4. | ||
L1 is triclinic having space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (CCDC 1484206): a, 10.8682(5) (Å); b, 11.1818(6) (Å); c, 16.0637(8) (Å); α (°), 88.542(3); β (°), 73.254(2); γ (°), 70.084(3); volume (Å3), 1754.84(15); Z = 4 (Table S1 (ESI†)). It exists as the enol form in the solid state. The bond lengths N003–C00J, 1.322(2) Å and N005–C00H, 1.295(2) Å indicate the double bond character of C–N whereas N004–C00Z, 1.447(3) Å, N004–C00W, 1.462(2) Å, N3–C00Q, 1.448(3) Å, and N3–C016, 1.536(4) Å indicate the single bond character of C–N bonds in the N-ethyl amino group. The bond lengths O001–C00C, 1.305(2) Å and O002–C00M, 1.291(2) Å indicate the single bond character of C–O in the phenol moiety. The structure is stabilised by intra-molecular H-bonding. The bond angles C00J–N003–C00V, 121.68(18)° and C00H–N005–C00T, 124.16(16)° indicate the sp2 nature of the N003 and N005 centres (Fig. S9a and Table S2a, ESI†).
(CCDC 1484206): a, 10.8682(5) (Å); b, 11.1818(6) (Å); c, 16.0637(8) (Å); α (°), 88.542(3); β (°), 73.254(2); γ (°), 70.084(3); volume (Å3), 1754.84(15); Z = 4 (Table S1 (ESI†)). It exists as the enol form in the solid state. The bond lengths N003–C00J, 1.322(2) Å and N005–C00H, 1.295(2) Å indicate the double bond character of C–N whereas N004–C00Z, 1.447(3) Å, N004–C00W, 1.462(2) Å, N3–C00Q, 1.448(3) Å, and N3–C016, 1.536(4) Å indicate the single bond character of C–N bonds in the N-ethyl amino group. The bond lengths O001–C00C, 1.305(2) Å and O002–C00M, 1.291(2) Å indicate the single bond character of C–O in the phenol moiety. The structure is stabilised by intra-molecular H-bonding. The bond angles C00J–N003–C00V, 121.68(18)° and C00H–N005–C00T, 124.16(16)° indicate the sp2 nature of the N003 and N005 centres (Fig. S9a and Table S2a, ESI†).
L2 is monoclinic having space group P (CCDC 1062760): a, 7.1822(10) (Å); b, 14.0258(18) (Å); c, 15.7770(19) (Å); α (°), 89.987(9); β (°), 90.007(10); γ (°), 77.128(10); volume (Å3), 1549.4(4); Z = 4 (Table S1, ESI†). It exists as the enol form in the solid state. The bond lengths N007–C00I, 1.284(6) Å and N003–C00R, 1.288(6) Å indicate the double bond character of C–N of the aldimine group whereas N006–C00D, 1.280(6) Å and N008–C00T, 1.279(6) Å indicate the C–N bond on the p-methoxy benzaldehyde side. The bond lengths N003–N006, 1.405(6) Å and N007–N008, 1.403(6) Å indicate the single bond character of N–N from the hydrazine moiety. The bond lengths O1–C00J, 1.347(6) Å and O2–C00K, 1.348(6) Å indicate the single bond character of C–O in the phenol moiety. The structure is stabilised by intra-molecular H bonding. The bond angles C00R–N003–N006, 114.2(5)°, C00D–N006–N003, 113.0(5)°, C00I–N007–N008, 113.6(5)°, and C00T–N008–N007, 112.7(5)° indicate the sp2 nature of the N003, N006, N007 and N008 centres (Fig. S9b and Table S2b, ESI†).
L3 is monoclinic having space group P21/c (CCDC 1501269): a, 16.524(3) (Å); b, 5.9394(12) (Å); c, 13.232(2) (Å); α (°), 90; β (°), 113.560(11); γ (°), 90; volume (Å3), 1190.4(4); Z = 4 (Table S1, ESI†). It exists as the enol form in the solid state. The bond lengths N2–C14, 1.277(7) Å and N1–C7, 1.286(7) Å indicate the double bond character in C–N of the aldimine group whereas N004–C00Z, 1.447(3) Å, N1–N1, 1.395(9) Å and N2–N2, 1.409(9) Å indicate the single bond character of N–N in the hydrazine moiety. The bond lengths O1–C1, 1.356(7) Å and O2–C8, 1.350(7) Å indicate the single bond character of C–O in the phenol moiety. The structure is stabilised by intra-molecular H bonding. The bond angles C14–N2–N2, 113.3(6)°, N2–C14–C13, 121.7(6)°, C7–N1–N1, 113.1(6)°, and N1–C7–C6, 121.3(6)° indicate the sp2 nature of the N003, N006, N007 and N008 centres (Fig. S9c and Table S2c, ESI†).
Spectroscopic studies
NR) moieties makes L1, L2, L3 and L4 pH sensitive. Consequently, their emission properties upon interaction with cations, anions and biomolecules have been investigated at different pH. Interestingly, their weak emission (L1: λem, 414 nm, λex, 287 nm; L2: λem, 491 nm, λex, 436 nm; L3: λem, 431 nm, λex, 353 nm and L4: λem, 495 nm, λex, 400 nm) is enhanced significantly in the presence of Zn2+: specifically, the enhancement is maximum for Zn(OAc)2. Other common metal ions, viz. Na+, K+, Ca2+, Al3+, Hg2+, Pb2+, Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Cd2+, Sn2+ and Ag+, do not significantly affect their emission intensities (Fig. 3a–d). Moreover, Zn salts other than Zn(OAc)2, viz. ZnCl2, ZnSO4, Zn(PO4)3, and Zn(OCl)2, show similar fluorescence enhancements in response to Zn2+. In addition, the emission intensity of L1-Zn(OAc)2 (Ad1) is maximum over the pH range 7 to 10 (Fig. S11a–d, ESI†). Hence, the near-biological pH 7.4 was chosen for all experiments. As L1 emits at two different wavelengths, a ratiometric change is observed upon gradual addition of Zn(OAc)2 to L1.
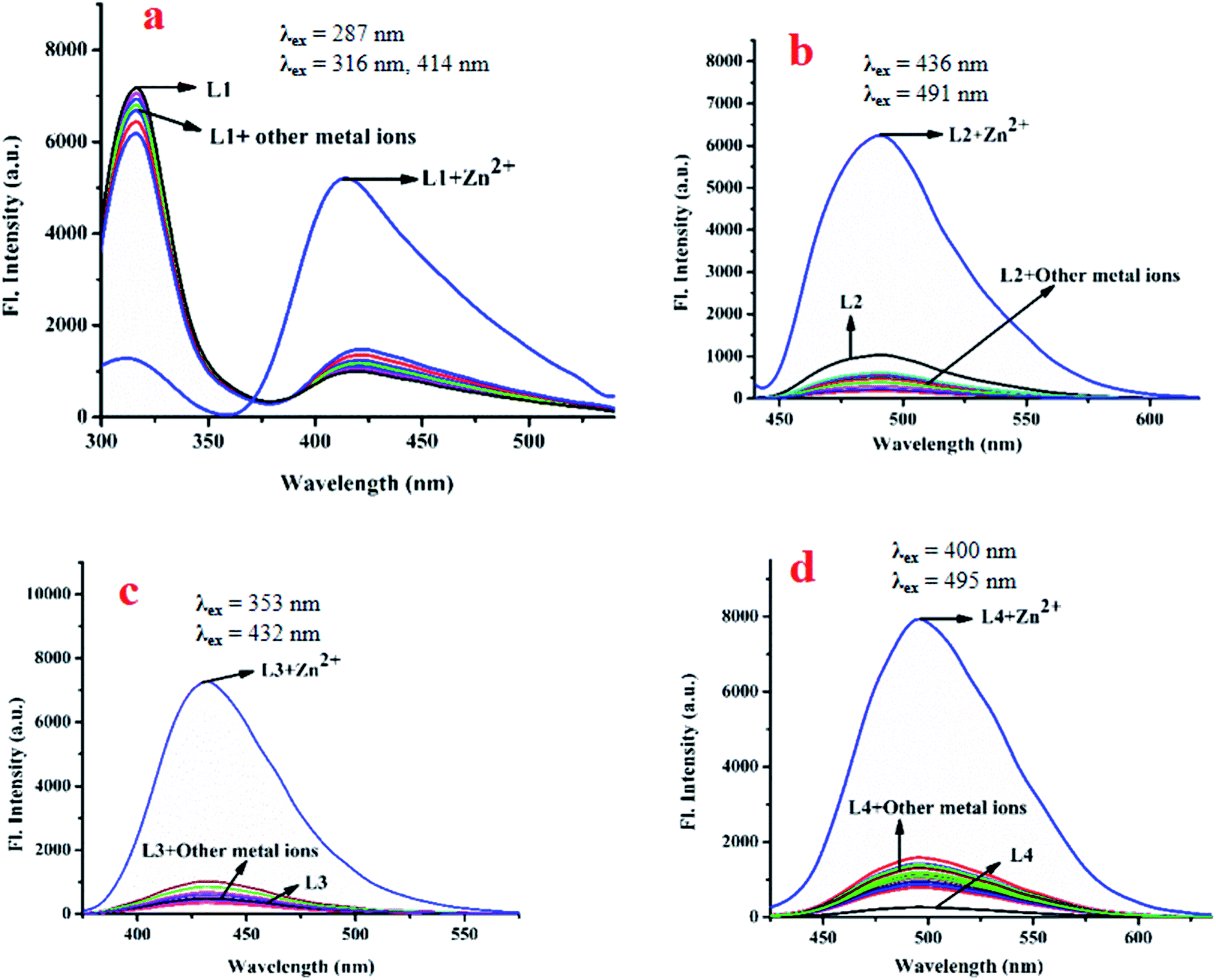 | ||
| Fig. 3 Changes in the emission spectra of L1 (a), L2 (b), L3 (c) and L4 (d) in presence of different metal ions in bis–tris buffered DMSO/H2O (1:1, v/v) media, pH 7.4. | ||
The emission intensity decreases at 316 nm while it increases at 414 nm through an iso-emissive point at 371 nm (Fig. 4a). In all other cases (excluding L1), the emission intensity at the respective wavelength increases upon gradual addition of Zn(OAc)2 (for L2, 491 nm, for L3, 431 nm and for L4, 495 nm) (Fig. 4b–d). Competitive experiments indicate no significant interference from common metal ions during sensing of Zn2+ in the form of Zn(OAc)2 by L1–L4 (Fig. S12a–d, ESI†). The change in emission intensities of L1 at two different wavelengths (414 nm and 316 nm) upon addition of Zn2+ is shown in Fig. S13a (ESI†). Similar plots for L2, L3 and L4 are shown in Fig. S13b–d (ESI†). The linear regions of the plots allow determination of unknown concentrations of Zn2+. The lowest detection limits (DL) of Zn2+ by L1, L2, L3 and L4 are 32.66 nM, 36.16 nM, 15.20 nM and 33.50 nM respectively (Fig. S14a–d, ESI†). These values lie well below the WHO recommended acceptable level of Zn2+ in drinking water (76 μM). The DL of L3 for Zn2+ is the lowest in the series, indicating stronger interaction of L3 with Zn2+, probably due to the presence of two –OH groups. Fluorescence titration enables determination of the association constants (K) of the probes for Zn2+ using the Benesi–Hildebrand equation (Fig. S15a–d, ESI†).
 | ||
| Fig. 4 Changes in emission intensity of L1 (a), L2 (b), L3 (c) and L4 (d) upon gradual addition of Zn(OAc)2; inset: plot of emission intensities vs. added Zn(OAc)2 at respective emission wavelengths. Media and pH are same as in Fig. 3. | ||
The respective association constants are 2.146 × 105 M−1, 2.407 × 105 M−1, 3.28 × 105 M−2 and 1.692 × 105 M−1 for L1, L2, L3 and L4 respectively. The fact that L3 has the highest K is due to the same reason as its having the lowest DL. From the Job plots (Fig. S16a–d, ESI†), ESI mass spectra (Fig. S5a, S6a, S7a and S8a, ESI†), FTIR spectra (Fig. S5b, S6b, S7b and S8b, ESI†), and 1H NMR titration (Fig. 7), plausible binding modes of L1–L4 with Zn2+ are proposed in Scheme 2 and the corresponding adducts (Ad1–Ad4) are presented in Fig. 5. Scheme 2 shows the probable sensing mechanism, assigned to the combined effect of ESIPT and chelation enhanced fluorescence (CHEF).
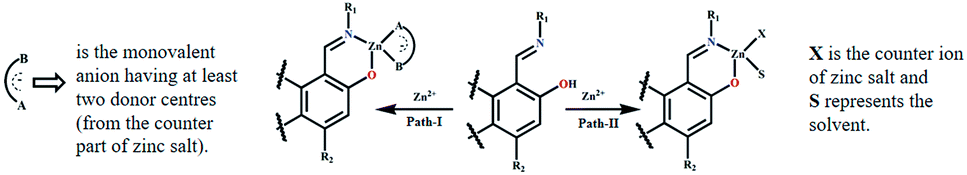 | ||
| Scheme 2 Binding mode of L1, L2, L3 and L4 towards Zn2+ salt. | ||
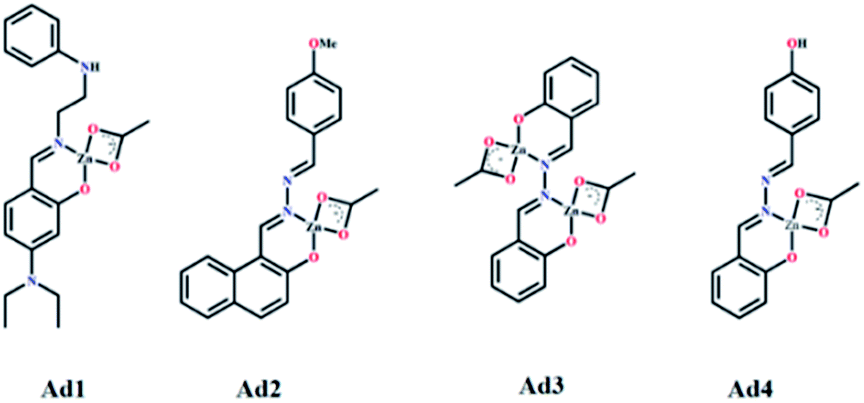 | ||
| Fig. 5 Binding mode of L1, L2, L3 and L4 with Zn(OAc)2 forming adduct Ad1, Ad2, Ad3 and Ad4 respectively. | ||
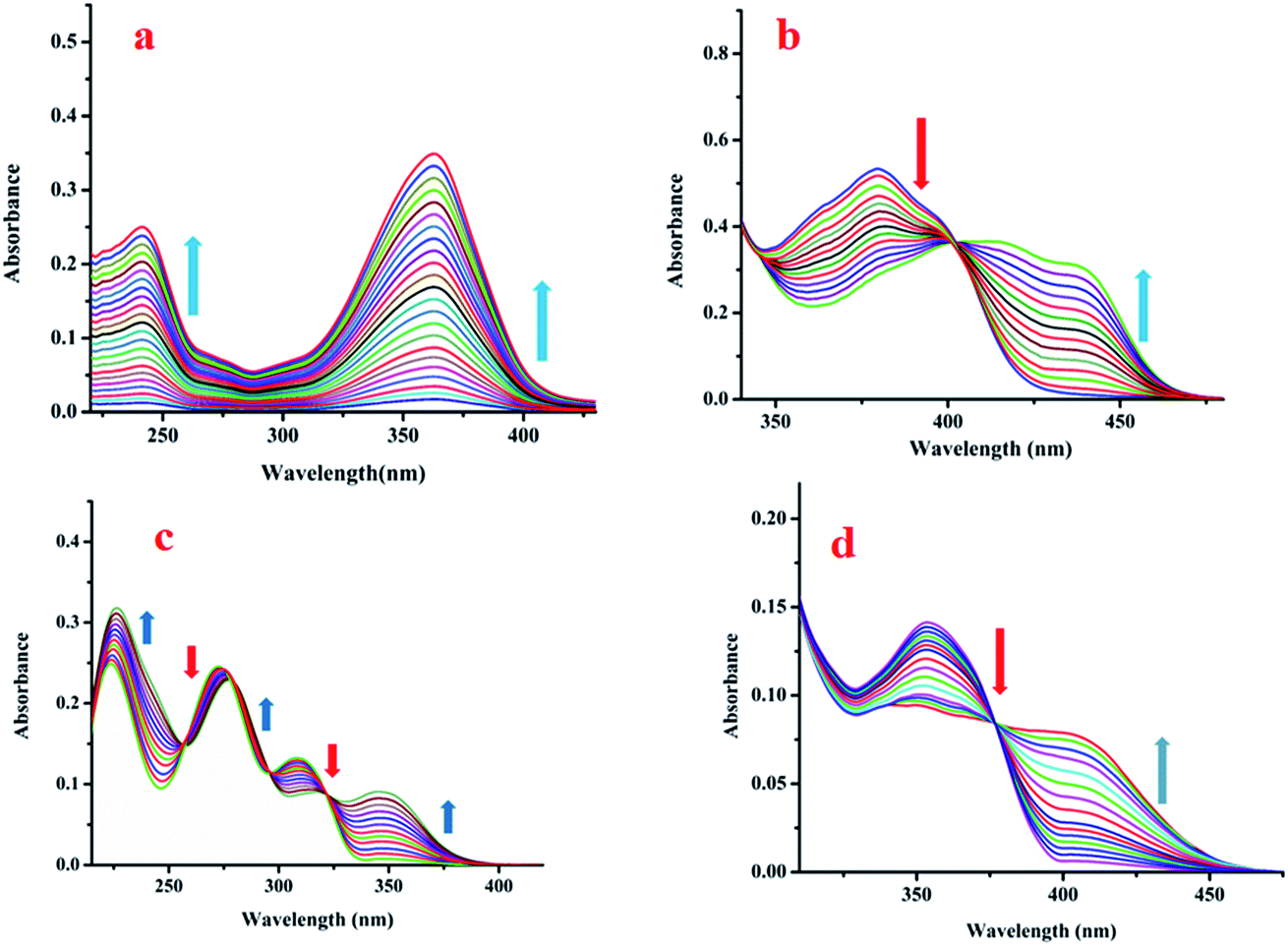 | ||
| Fig. 6 Changes in the absorption spectra of (a) L1, (b) L2, (c) L3 and (d) L4 upon gradual addition of Zn(OAc)2. | ||
Sensing mechanism
The ESI mass spectra of Ad1, Ad2 and Ad4 along with the Job plots (Fig. S16a, b and d, ESI†) support 1:1 (mole ratio) binding stoichiometry between the probe and Zn(OAc)2 whereas the corresponding data for Ad3 indicate 1:2 (mole ratio) stoichiometry (Fig. S16c, ESI†). Thus chelation of L1–L4 to zinc ion leads to CHEF through inhibition of ESIPT and –CHN– isomerisation (Scheme 2 and Fig. 5). 1H NMR titration of L1–L4 with Zn(OAc)2 further supports the proposed mechanism. The FTIR data of the adducts also corroborate the binding mode (Fig. S5b, S6b, S7b and S8b, ESI†). It was observed that the ν(CN) band of free L1, L2, L3 and L4 at 1608, 1612, 1597 and 1616 cm−1 shifted to 1560, 1557, 1574, 1559 cm−1 in the respective adducts Ad1, Ad2, Ad3 and Ad4, in addition to their original peaks in some cases. This indicates involvement of imine-N in chelation. Moreover, the respective ν(O–H) bands at 3370, 3214, 3373 and 3319 cm−1 almost disappear in the case of Ad1, Ad2 and Ad4 while it partially disappears for Ad3. Thus ESIPT protons disappear upon binding of phenol-O to Zn(OAc)2.
1H NMR titration
The 1H NMR titration results unveil the binding mode of L1 with Zn(OAc)2 (Fig. 7a). Upon addition of 0.5 equiv. of Zn2+, the highly de-shielded proton (–OH) at 10.73 ppm almost disappears, indicating interaction of phenol-O with Zn2+, while the imine proton is slightly up-field shifted from 9.49 ppm to 9.41 ppm, suggesting involvement of imine-N in chelation.57 At 1.0 equiv. of Zn(OAc)2 the phenol proton completely disappears, while the imine proton is further shifted from 9.41 ppm to 9.40 ppm. Similar facts are observed for L2 and L4. In the case of L2, the –OH proton at 13.41 ppm gradually disappears upon gradual addition of Zn(OAc)2 while the imine proton experiences a slight up-field shift (Fig. 7b). However, upon addition of 1.0 equiv. Zn(OAc)2 to L3, the –OH peak at 10.45 ppm does not disappear fully, probably due to the presence of two symmetric –OH units that can equally interact with two molecules of Zn(OAc)2. Interestingly, upon addition of two equiv. of Zn(OAc)2, the –OH peak almost disappears and the imine proton is shifted up-field (Fig. 7c). In L4, two –OH protons at 11.427 and 11.051 ppm have been observed (Fig. S4f, ESI†). Upon addition of 0.5 equiv. Zn(OAc)2, the –OH peak ortho to the imine group tends to disappear while the other –OH at the para position of the other ring is slightly up-field shifted from 11.427 to 11.344 ppm. At saturation point both –OH peaks almost disappear (Fig. 7d). | ||
| Fig. 7 1H NMR titration of L1 (a), L2 (b), L3 (c) and L4 (d) with Zn(OAc)2 in DMSO-d6. | ||
Density functional theoretical (DFT) studies
DFT studies have been performed using the TD-SCF/DFT/B3LYP/6-311G level of theory. Only the HOMO and LUMOs are provided to demonstrate the stability of the adducts compared to the free probes. The lowering of the HOMO–LUMO energy gaps in Ad1–Ad4 compared to free L1–L4 indicates stabilisation of the former over the latter. In each case, the oscillator strength (fb) for the S0 → S1 transition of the adduct increases compared to the free probes. This indicates increased probability of absorption/emission as observed in the experimental findings. The electron transition energies for the predominant absorption of the free probes and corresponding Zn(OAc)2 adducts are depicted in Table S3a–d (ESI†).For L1, the HOMO–LUMO energy gap was reduced by 0.04855 eV from 0.16836 eV (free L1) to 0.11987 eV (Ad1) (Fig. 8a). The S0 → S1 transition (HOMO → LUMO) at 363 nm has a low fb, 0.006, which indicates intra-molecular charge transfer (ICT) from phenol moieties to the amide. A hypsochromic shift to 477 nm is observed with a significant increase of fb (0.0237) for Ad1 (S0 → S1 transition, HOMO → LUMO, HOMO → LUMO + 1), suggesting formation of rigid Ad1.49 Another HOMO → LUMO transition along with HOMO → LUMO + 1 have been observed for S0 → S2 with fb = 0.0095. The HOMO–LUMO energy gap in L2 is 0.11953 eV while it is 0.110807 eV in Ad2 (Fig. 8b). Here again the main transition S0 → S1 (HOMO → LUMO, HOMO → LUMO + 1) occurs with a very low value of fb (0.0011), while Ad2 has a higher value of fb (0.2214) for the same electronic transition. Again one interesting observation is that the transition S0 → S2 (HOMO − 1 → LUMO, HOMO → LUMO + 1, HOMO − 1 → LUMO + 2) at 371.56 nm for L2 is well matched with the experimental value of the absorption peak at 380 nm, and S0 → S2 (HOMO → LUMO, HOMO → LUMO + 1) for Ad2 at 430.78 nm is also well matched with the experimental value at 439 nm. We have performed a DFT study of L3 and Ad3, and again observed that energy relaxation is more pronounced in the case of Ad3 than L3. The respective values of the HOMO–LUMO energy gap of L3 and Ad3 are 0.14254 eV and 0.10751 eV (Fig. 8c). Here also the main transition S0 → S1 (HOMO − 1 → LUMO, HOMO → LUMO) occurs with a very low value of fb (0.0032) for L3, as seen from the MO profile. Meanwhile, for Ad3 the transition S0 → S1 (HOMO − 1 → LUMO, HOMO → LUMO) occurs with fb = 0.1602. In the case of L4, the HOMO–LUMO energy gap decreases from 0.13004 eV to 0.113695 eV for the Ad4 adduct (Fig. 8d). The respective fb values for L4 and Ad4 are 0.0010 and 0.0978.
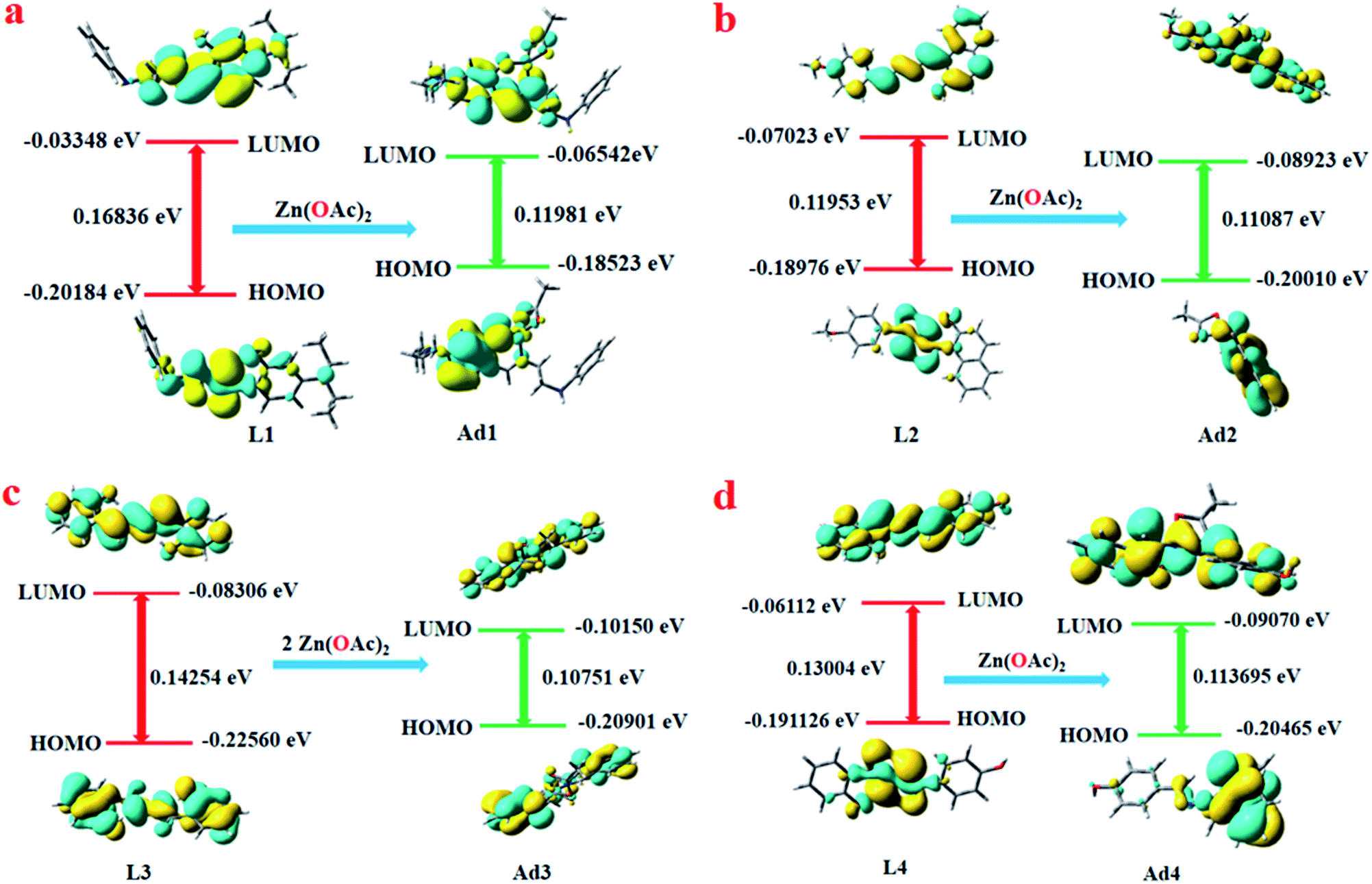 | ||
| Fig. 8 Frontier molecular orbitals of L1 and its adduct Ad1 (a), L2 and its adduct Ad2 (b), L3 and its adduct Ad3 (c), and L4 and its adduct Ad4 (d). | ||
Conclusion
Four simple organic molecules (Schiff base and azine derivatives) have been synthesised and structurally characterised by SC-XRD analysis. Due to the existence of an ESIPT process, a weak fluorescence is observed in the molecules. It is found that the molecules selectively interact with the Zn2+ ion as monitored by fluorescence and absorption spectroscopic studies. The association constants of L1, L2, L3 and L4 with Zn(OAc)2 are 2.146 × 105 M−1, 2.15 × 105 M−1, 3.28 × 105 M−2, and 1.692 × 105 M−1 respectively. The enhancement of fluorescence is due to the arrested ESIPT with the formation of a rigid adduct, leading to a CHEF process. The lowest detection limit is found to be in the nano-molar range. A DFT study elucidates the experimental results. Thus, a new class of ESIPT-CHEF probes for recognition of Zn(II) is established for its ultra-trace level determination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK and SC acknowledge CSIR, New Delhi (India) and DST-INSPIRE (India) for fellowship.Notes and references
- T. Hirano, K. Kikuchi, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2002, 124, 6555–6562 CrossRef CAS PubMed.
- M. W. Hentze, M. U. Muckenthaler and N. C. Andrews, Cell, 2002, 117, 285–297 CrossRef.
- D. Chen, R. J. Letcher, L. T. Gauthier, S. G. Chu, R. McCrindle and D. Potter, Environ. Sci. Technol., 2011, 45, 9523–9530 CrossRef CAS.
- M. Vendrell, D. T. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- Y. M. Yang, Q. Zhao, W. Feng and S. Y. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS.
- Y. Liua, J. Zhaob, Y. Wanga, J. Tian, X. Feic and H. Wanga, J. Mol. Liq., 2017, 233, 303–309 CrossRef.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- A. Weller, Naturwissenschaften, 1955, 42, 175–176 CrossRef CAS.
- J. Z. Zhao, S. M. Ji, Y. H. Chen, H. M. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- C. Azarias, S. Budzak, A. D. Laurent, G. Ulrich and D. Jacquemin, Chem. Sci., 2016, 7, 3763–3774 RSC.
- J. M. Berg and Y. Shi, Science, 1996, 271, 1081–1085 CrossRef CAS.
- F. J. J. R. Da Silva and R. J. P. Williams, The Biological Chemistry of the Elements, Oxford University Press, 2nd edn, New York, 2001 Search PubMed.
- Y. Chen, K. Y. Han and Y. Liu, Bioorg. Med. Chem., 2007, 15, 4537–4542 CrossRef CAS PubMed.
- K. Komatsu, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2005, 127(29), 10197–10204 CrossRef CAS.
- B. L. Vallee and K. H. Falchuk, Physiol. Rev., 1993, 73, 79–118 CrossRef CAS.
- C. B. Freddari, P. Fattoretti, T. Casoli, G. D. Stefano, B. Giorgetti and M. Baliett, Exp. Gerontol., 2008, 43, 389–393 CrossRef.
- N. P. Pavletich and C. O. Pabo, Science, 1991, 252, 809–817 CrossRef CAS.
- C. O. Pabo and R. T. Sauer, Annu. Rev. Biochem., 1992, 61, 1053–1095 CrossRef CAS PubMed.
- K. A. McCall, C. C. Huang and C. A. Fierke, J. Nutr., 2000, 130, 1437S–1446S CrossRef CAS.
- K. Jobe, C. H. Brennan, M. Motevalli, S. M. Goldup and M. Watkinson, Chem. Commun., 2011, 37, 6036–6038 RSC.
- M. Ghosh, A. Ghosh, S. Ta, J. Matalobos and D. Das, ChemistrySelect, 2017, 2, 7426–7431 CrossRef CAS.
- J. Quarterman, C. F. Mills and W. R. Humphries, Biochem. Biophys. Res. Commun., 1966, 25, 354–358 CrossRef CAS.
- E. U. Akkaya, M. E. Huston and A. W. Czarnik, J. Am. Chem. Soc., 1990, 112, 3590–3593 CrossRef CAS.
- D. A. Pearce, N. Jotterand, I. S. Carrico and B. Imperiali, J. Am. Chem. Soc., 2001, 123, 5160–5161 CrossRef CAS.
- Z. Xu, X. Liu, J. Pan and D. R. Spring, Chem. Commun., 2012, 48, 4764–4766 RSC.
- J. Jiang, H. Jiang, X. Tang, L. Yang, W. Dou, W. Liu, R. Fang and W. Liu, Dalton Trans., 2011, 40, 6367–6370 RSC.
- Y. Xu, J. Meng, L. Meng, Y. Dong, Y. Cheng and C. Zhu, Chem.–Eur. J., 2010, 16, 12898–12903 CrossRef CAS.
- L. Salmon, P. Thuery, E. Riviere and M. Ephritikhine, Inorg. Chem., 2006, 45, 83–93 CrossRef CAS.
- D. M. Epstein, S. Choudhary, M. R. Churchill, K. M. Keil, A. V. Eliseev and J. R. Morrow, Inorg. Chem., 2001, 40, 1591–1596 CrossRef CAS.
- B. Kumari, S. Lohar, M. Ghosh, S. Ta, A. Sengupta, P. P. Banerjee, A. Chattopadhyay and D. Das, J. Fluoresc., 2016, 26(1), 87–103 CrossRef CAS.
- R. E. Sturgeon, S. S. Berman, A. Desaulniers and D. S. Russell, Anal. Chem., 1979, 14, 2364–2369 CrossRef.
- K. Aich, S. Goswami, S. Das and C. D. Mukhopadhyay, RSC Adv., 2015, 5, 31189–31194 RSC.
- J. J. Lee, S. A. Lee, H. Kim, L. Nguyen, I. Noh and C. Kim, RSC Adv., 2015, 5, 41905–41913 RSC.
- A. M. Hessels and M. Merkx, Metallomics, 2015, 7, 258–266 RSC.
- P. K. Mehta, E. T. Oh, H. J. Park and K. H. Lee, Sens. Actuators, B, 2017, 245, 996–1003 CrossRef CAS.
- M. E. Shirbhate, Y. Jeong, G. Ko, G. Baek, G. Kim, Y. U. Kwon, K. M. Kim and J. Yoon, Dyes Pigm., 2019, 167, 29–35 CrossRef CAS.
- S. Pramanik, V. Bhalla and M. Kumar, New J. Chem., 2017, 41, 4806–4813 RSC.
- M. Mariyappan, N. Malini, J. Sivamani, G. Sivaraman, M. Harikrishnan, S. Murugesan and A. Siva, J. Fluoresc., 2019, 29, 737–749 CrossRef CAS.
- S. Sinha, B. Chowdhury, N. N. Adarsh and P. Ghosh, Dalton Trans., 2018, 47, 6819–6830 RSC.
- Y. Xu and Y. Pang, Dalton Trans., 2011, 40, 1503–1509 RSC.
- S. Goswami, A. Manna, S. Paul, A. K. Maity, P. Saha, C. K. Quah and H. K. Fun, RSC Adv., 2014, 4, 34572–34576 RSC.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, E64, 112–122 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Ghosh, S. Ta, M. Ghosh, S. Karmakar, A. Banikc, T. K. Dangar, S. K. Mukherjee and D. Das, Dalton Trans., 2016, 45, 599–606 RSC.
- M. Ghosh, S. Ta, M. Banerjee, M. Mahiuddin, S. Khanra, S. K. Hira, P. P. Manna and D. Das, J. Photochem. Photobiol., A, 2018, 367, 32–38 CrossRef CAS.
- T. G. Jo, J. J. Lee, E. Nam, K. H. Bok, M. H. Lim and C. Kim, New J. Chem., 2016, 40, 8918–8927 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FTIR, UV-Vis, fluorescence spectra, X-ray single crystal data and DFT calculation data. CCDC 1484206, 1062760 and 1501269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03652k |
| This journal is © The Royal Society of Chemistry 2019 |