
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModel Affitin and PEG modifications onto siRNA lipid nanocapsules: cell uptake and in vivo biodistribution improvements
Pauline Resniera,
Elise Lepeltier a,
Anthea Lucrezia Eminaa,
Natacha Galopinb,
Jérôme Bejauda,
Stephanie Davidc,
Caroline Balletd,
Thierry Benvegnud,
Frédéric Pecorarie,
Igor Chourpac,
Jean-Pierre Benoita and
Catherine Passirani*a
a,
Anthea Lucrezia Eminaa,
Natacha Galopinb,
Jérôme Bejauda,
Stephanie Davidc,
Caroline Balletd,
Thierry Benvegnud,
Frédéric Pecorarie,
Igor Chourpac,
Jean-Pierre Benoita and
Catherine Passirani*a
aMINT, UNIV Angers, INSERM 1066, CNRS 6021, Université Bretagne Loire, Angers, France. E-mail: catherine.passirani@univ-angers.fr
bCRCINA, INSERM, Université d'Angers, Université de Nantes, Nantes, France
cEA6295 – Nanomédicaments et Nanosondes, Université François-Rabelais de Tours, UFR de Pharmacie, 31 avenue Monge, F-37200 Tours, France
dUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France
eUMR CNRS 6299 – INSERM U892, CRCNA, F-44007 Nantes, France
First published on 30th August 2019
Abstract
Malignant melanoma is an aggressive tumor, associated with the presence of local and/or distant metastases. The development of gene therapy by the use of small interfering RNA (siRNA) represents a promising new treatment. However, the protection of this biomolecule is necessary in order for it to be intravenously administrated, for example via its incorporation into nanomedicines. In parallel to the passive targeting usually obtained by pegylation, various studies have aimed at developing “smart” nanomedicines to efficiently deliver the drug to tumor sites. In this work, siRNA loaded lipid nanocapsules (LNCs) were modified with DSPE-polyethylene glycol (DSPE-PEG), tetraether-PEG (TE-PEG) and/or with an Affitin model, to assay multiple targeting strategies. The uptake of fluorescently labelled LNCs, nanocarrier integrity and siRNA release into human SK-Mel28 melanoma cells were studied by flow cytometry, conventional confocal microscopy and by confocal spectral imaging in a Förster Resonance Energy Transfer (FRET) mode. Surface modified siRNA LNCs were followed after human plasma incubation and after intravenous injection, in order to compare the stealth properties. Finally, the biodistribution of the different siRNA LNCs in healthy and melanoma tumor bearing mice models was assessed by in vivo biofluorescence imaging (BFI), to evaluate the potential tumor targeting ability. The post-insertion of DSPE-PEG induced a strong decrease of the internalization into melanoma cells compared to TE-PEG modification. Both PEG polymer decorations induced a great plasma protection of siRNA but only DSPE-PEG led to stealth properties, even at low concentration (5 mM). The Affitin grafting by thiolation of DSPE-PEG was validated on siRNA LNCs. DSPE-PEG-Affitin LNCs were not detected in this melanoma tumor model but did not show unspecific accumulation in organs. DSPE-PEG and TE-PEG LNCs induced a significant intratumoral accumulation of modified LNCs.
Introduction
Malignant melanoma is the most aggressive form of melanoma: chemotherapy or immunotherapy still fail to cure this disease with local or distant metastases. In fact, the maximal response rate is obtained with dacarbazine, the reference treatment, but it is limited to 16% of patients. The use of small interfering RNA (siRNA) could offer an opportunity to play on the expression of proteins involved in melanoma resistance. As described in the literature, siRNA can specifically interact with targeted mRNA by RISC (RNA-induced silencing complex) pathways leading to transitory inhibition of protein production in the cytoplasm.1 However, this hydrophilic entity is not able to cross the cellular membrane alone and it is fragile.For a few decades now, the encapsulation or adsorption of siRNA have been mainly based on electrostatic interactions of anionic nucleic acids with cationic lipids or polymers such as DOTAP (1,2-dioleoyl-3-trimethylammoniumpropane), cholesterol, or polyethylenimine (PEI), respectively forming lipoplexes and polyplexes.2,3 Neutral or zwitterionic lipids, such as DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), are usually added for their fusiogenic property, promoting the endosomal escape into the intracellular compartment.4,5 Various nanomedicines, combining these different lipids and/or polymers, have been developed and they have demonstrated protection effects against enzymatic degradation, siRNA crossing through cell membrane and its endosomal escape.6–8 Among them, lipid nanocapsules (LNCs), based on emulsion phase inversion process, were adapted for siRNA encapsulation thanks to the use of DOTAP/DOPE lipids.9,10 This LNC formulation is solvent-free, low-energy consuming, easily realizable and reproducible inducing stable siRNA complexation and monodispersed formulation. Moreover, LNCs showed an efficient transfection efficacy of siRNA on U87MG glioma and SK-Mel28 melanoma cells with a significant inhibition of two different targeted proteins.9,11 With this proof of in vitro transfection, the challenge will be to preferentially accumulate the LNCs into the targeted tumor site, for an efficient in vivo siRNA delivery and a potential therapeutic application.
In the context of melanoma metastases, the intravenous injection route appears as an evidence. In this case siRNA encapsulation into LNCs should provide: (i) protection against blood nucleases and immune cells, (ii) accumulation into tumor site and (iii) penetration into the intracellular compartment.
The formulation of nanoparticles usually includes pharmacokinetic modulating moieties such as polyethylene glycol (PEG). In this study, the surface of the LNCs was modified by different molecules. The pegylation, i.e. the addition of long chains of polyethylene glycol (PEG), is known to play an important role in the efficacy of passive targeting strategy in solid tumor models.12,13 This hydrophilic polymer allows a long blood circulation time in limiting the LNC recognition and the massive destruction by the innate immune system and then the elimination via the liver. LNCs already showed their capacity to accumulate into tumor microenvironment, characterized by leaky junctions of the neovessels and a poor lymphatic drainage, a phenomenon known as the EPR effect (enhanced permeability and retention effect).14 DSPE-PEG (or 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]) has been usually used in LNCs to improve this passive targeting.
In parallel, emerging archaeal tetraether PEG (TE-PEG) molecules are able to form stable and stealth liposomes.15,16 The TE natural lipid has shown a strong stability in several conditions such as high temperature, acidic pH and serum media.17 This lipid found in archaea has been studied for a potential application in drug/gene delivery system. It could be a promising candidate for a new LNC surface modification.
In order to overcome the limits of passive targeting, the active targeting strategies by post-insertion of innovative moieties into nanoparticles to improve their half-life time, their targeting efficiency or stimuli-sensitivity, and their intracellular delivery is a hot topic.18 For instance, ligands as sugars, peptides or antibodies grafted on nanomedicine surface are used to specifically recognize tumor antigens. Archaeal extremophilic proteins from the Sul7d family,19 such as Sac7d, were recently developed as scaffolding to generate small artificial affinity proteins (Affitins).20–28 Affitins appear as an alternative to the use of antibodies for an active targeting approach.
In this study, surface modifications of siRNA LNCs were performed with an innovative TE-PEG polymer and an Affitin model, compared with a classic DSPE-PEG LNCs, in order to evaluate the potential tumor targeting of siRNA LNCs, after intravenous administration: Fig. 1. Flow cytometry and confocal fluorescence microscopy were used to determine the LNC internalization capacity in SK-Mel28 human melanoma cells. The LNC and siRNA integrities after SK-Mel28 incubation was evaluated by fluorescence confocal spectral imaging (FCSI) using the co-loading of two fluorophores: a FRET couple (DiI/DiD) and a fluorescently labelled siRNA (siRNA Alexa488) were used. Moreover, the protection against nucleases and blood behavior were determined in vitro and in vivo on mice. Finally, biodistribution studies of modified siRNA LNCs on healthy and melanoma tumor grafted nude mice were performed to evaluate the elimination and the potential tumor targeting of these surface modified LNCs.
 | ||
| Fig. 1 The different performed and studied surface functionalizations. | ||
Results
Formulation and stability of siRNA LNCs
The encapsulation of siRNA into LNCs was performed with or without lecithin (Lipoid®). The size and zeta potential stability were followed over time after formulation and storage at 4 °C (Fig. 2). The absence of lecithin into the siRNA loaded LNCs induced a long term stability compared to the initial formulation containing Lipoid®. Indeed, the formulation with lipoid®, adapted from the initial LNC process, led to an important size decrease and a zeta potential increase: at day 0, the diameter was around 100 nm and reached 60 nm after 15 days, with a zeta potential near neutrality at day 0 and a zeta potential around +40 mV after 2 weeks. The optimization of the process, by Lipoid® elimination, led to a long-term stability of size and zeta potential, for at least one month with a mean diameter of 80 nm and a zeta potential of around +23 mV. That is why, in this study, siRNA LNCs were formulated without Lipoid® for all the following experiments.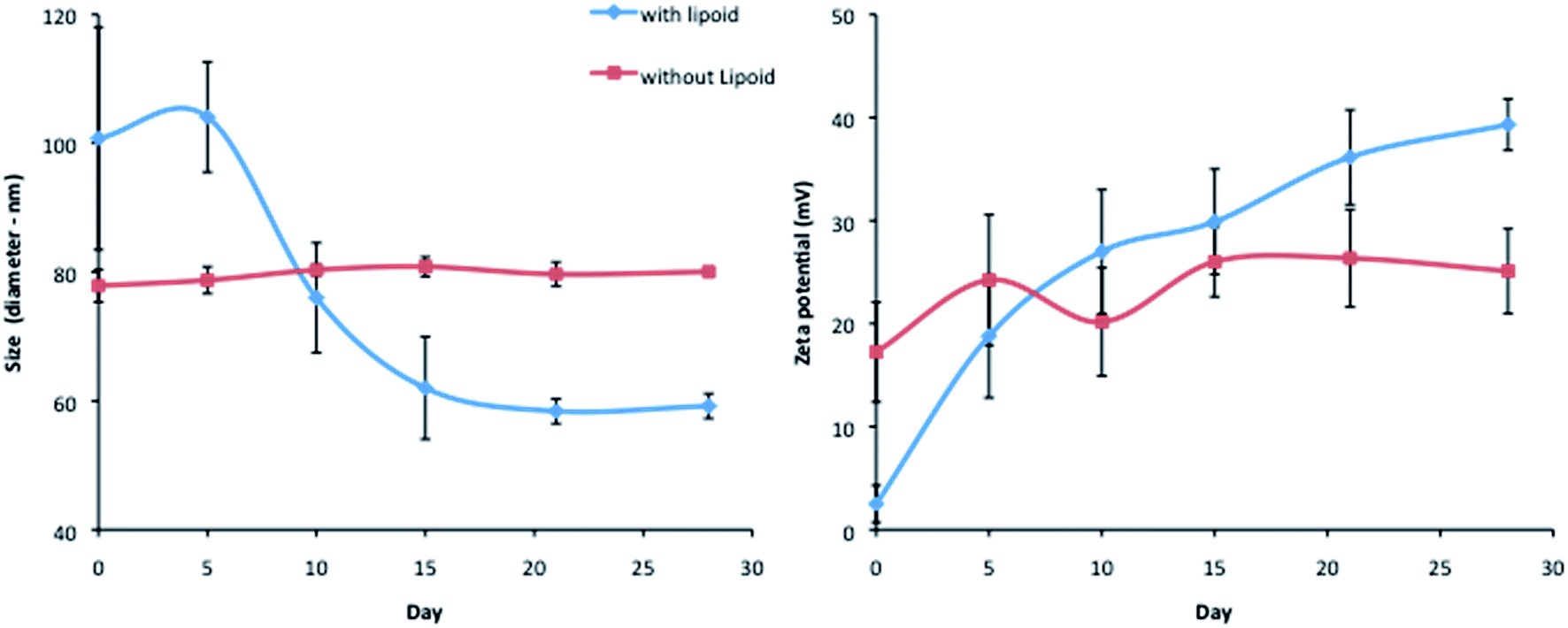 | ||
| Fig. 2 Stability evaluation of siRNA LNCs with or without Lipoid®. Size (left) and zeta potential (right) were determined using DLS (Zetasizer, Malvern). siRNA LNCs with Lipoid® showed a time influence in terms of size and in zeta potential. At the opposite, long-term stability was observed for siRNA LNCs without Lipoid®, characterized by a diameter of 80 nm and a zeta potential of +23 mV. | ||
Behavior of FRET siRNA LNCs in melanoma cells
siRNA delivery by LNCs was studied on living melanoma cells by fluorescence confocal multispectral imaging: FCSI (Fig. 3). In order to follow both LNC integrity and siRNA distribution, triple-labelled LNCs were formulated by loading them with the DiI/DiD FRET couple and with the siRNA-Alexa488 (see experimental section). With a 488 nm excitation, both Alexa488 fluorescence and FRET of DiI/DiD couple were simultaneously recorded (black spectrum in Fig. 3A: I669 nm/I569 nm ratio 1.28). Once LNCs were completely disintegrated, for instance by dissolving them in organic solvents, the FRET was lost and the DiD emission band disappeared (I669 nm/I569 nm ratio decreased to 0.19, data not shown).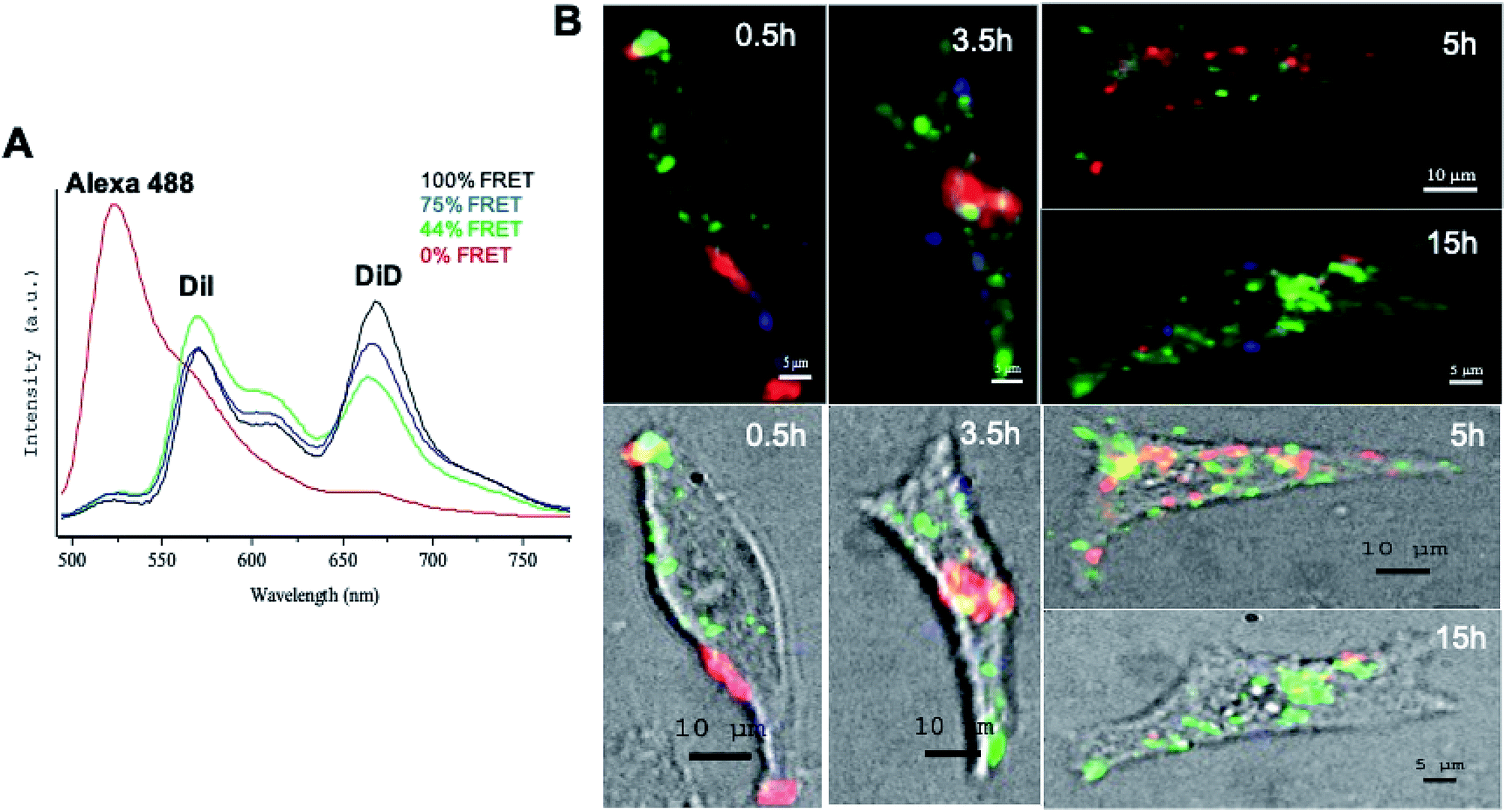 | ||
| Fig. 3 Behavior of siRNA LNCs in SK-Mel28 cells over time, followed by fluorescence confocal spectral imaging, FCSI. (A) Model spectra of triple-labelled control LNCs (100% of the FRET signal in black), partially degraded LNCs (75% of the FRET signal in blue and 44% of the FRET signal in green) and Alexa 488-labeled siRNAs (0% of the FRET signal in red). (B) Spectral maps (upper panel) and spectral maps merged with white light images of SK-Mel28 cells (lower panel) over time. The red color represents free siRNA (corresponding to the red spectrum) and the green and blue colors represent the degraded LNCs (corresponding to the green and blue spectra respectively). | ||
Using the above considerations, spectral analysis allowed us to distinguish the intracellular situations corresponding to the three model spectra (Fig. 3A) and to generate the corresponding merged maps shown in pseudocolors (Fig. 3B): (i) blue corresponded to 75% of the initial FRET signal (high DiD signal); (ii) green corresponded to 44% of the FRET signal (high DiI signal), and (iii) red corresponded to free Alexa488-siRNAs, not co-localized with DiI/DiD (0% of FRET).
The results showed that after 0.5 h of incubation, LNCs were mostly observed in the cell membrane and only few of them were found in the cytosol. Compared to the initial LNCs, a partial FRET decrease was observed, with a FRET signal of 75% for the LNCs in the cell membranes and of 44% in the cytosol (respectively blue and green spectra in Fig. 3A and blue and green zones in Fig. 3B). Red zones were observed in membranes, explained by free siRNA released from LNCs.
At 3.5 h, the cell cytosol was significantly enriched with degraded LNCs (green zones). As a result of this degradation, a massive accumulation of free siRNA released from LNCs (large red zones) was observed in the cytosol (Fig. 3B). A fraction of LNCs (blue zones) was still observed mainly at the periphery of the cell membrane. At 5 h, the siRNA was observed in numerous small zones in the cytosol. The dissociation of LNCs in cytosol led to rare blue zones, even on membranes. At 15 h, only a few free siRNA (red spots) were observed in the cell, close to its periphery, while the DiI fluorescence (green) released from the degraded LNCs was still readily detected in the large zones of cytosol.
Surface modified LNCs
The formulation of siRNA loaded LNCs led to nanoparticles of 75 nm in diameter and characterized by a positive zeta potential (+13 mV) (Table 1). The post-insertion of TE-PEG (Table 2) did not induce any modification. With DSPE-PEG, described in Table 2, at 5 and 10 mM concentrations, the diameter increased gradually and the zeta potential became negative (−10 mV; −16 mV respectively).| Formulations | Diameter (nm) | PDI | Zeta potential (mV) | EE (%) |
|---|---|---|---|---|
| a Results (n = 3) are expressed as mean measure ± standard deviation.29 No difference was demonstrated on encapsulation efficiency30 using ANOVA 1W, post hoc Tukey. | ||||
| LNCs | 74 ± 4 | 0.05 ± 0.01 | +13 ± 6 | 35 ± 5 |
| TE-PEG LNCs | 74 ± 3 | 0.074 ± 0.04 | +15 ± 1 | 30 ± 5 |
| 5 mM DSPE-PEG LNCs | 85 ± 2 | 0.13 ± 0.05 | −10 ± 4 | 33 ± 4 |
| 10 mM DSPE-PEG LNCs | 94 ± 14 | 0.16 ± 0.08 | −16 ± 4 | 36 ± 7 |
| 10 mM DSPE-PEG-Affitins LNCs | 103 ± 13 | 0.16 ± 0.02 | −16 ± 4 | 36 ± 3 |
| Polymer | Structure | PEG length (kDa) |
|---|---|---|
| TE-PEG | 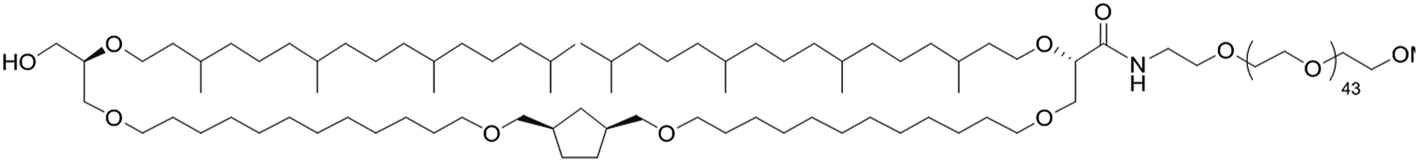 |
2000 |
| DSPE-PEG | 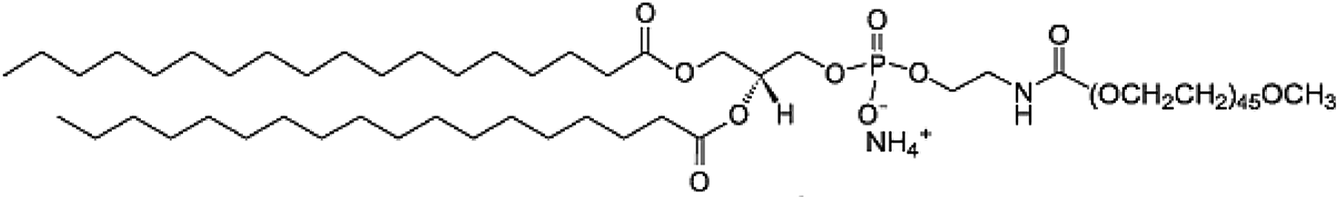 |
2000 |
| malDSPE-PEG | 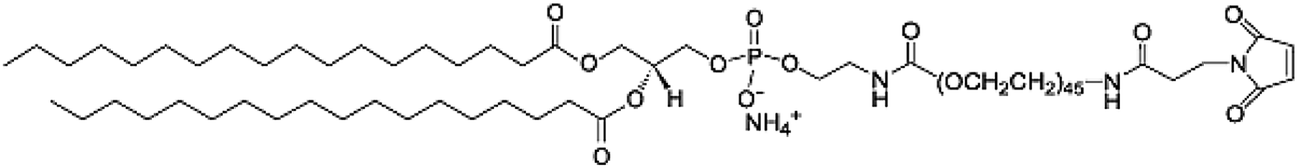 |
2000 |
The presence of a maleimide group on DSPE-PEG led to the same nanoparticle diameter, but with a lower negative zeta potential (−9 mV versus −16 mV) (data not shown). The incorporation of fluorescent probes DiD, DiI or Alexa488 siRNA did not modify the physico-chemical properties of siRNA LNCs (data not shown).
The Affitin was grafted on 10 mM malDSPE-PEG siRNA LNCs. Turbidity and protein detection confirmed the efficient grafting of the Affitins on DSPE-PEG LNC surface, as previously obtained with the grafting of an antibody by the same chemical reaction in Bourseau et al.31 The resulting formulation had a larger diameter than 10 mM DSPE-PEG LNCs (103 and 94 nm, respectively), leaving the other parameters unchanged (Table 1). All the different surface modified siRNA LNCs were monodispersed (PDI < 0.2) and the encapsulation efficiency of siRNA was evaluated around 35%.
Cellular uptake into melanoma cells
The internalization of LNCs and siRNA was evaluated by fluorescence microscopy and flow cytometry, after surface modification by different PEG (5 mM DSPE-PEG, 10 mM DSPE-PEG, 10 mM TE-PEG, 10 mM DSPE-PEG-Affitins) (Fig. 4 and 5). After 48 h of incubation, TE-PEG LNCs were observed in SK-Mel28 cells via the Alexa488 and DiD fluorescences. On the opposite, no signal was detected after incubation with the other formulations (5, 10 mM and DSPE-PEG-Affitins) (Fig. 4). To quantify the proportion of positive cells, flow cytometry was performed after quenching of Alexa488 with trypan blue to discriminate the adsorbed from internalized siRNA (Fig. 5). The analysis confirmed the large proportion of positive cells after incubation with non-modified and TE-PEG post-inserted LNCs (Fig. 5B). After 4 h, 90% of cells were Alexa488 positive with non-modified and TE-PEG LNCs and the percentage was constant until 48 h. In this case, the proportion of absorbed siRNA was limited to 20% at 4 h and decreased over time to a few percent (Fig. 5B). For DSPE-PEG LNCs, flow cytometry proved the significant lower percentage of internalized cells by siRNA (lower than 10%). Moreover, compared to 10 mM DSPE-PEG LNCs, the presence of Affitin did not modify the behavior of siRNA LNCs on melanoma cells. | ||
| Fig. 4 Cell uptake of siRNA LNCs in melanoma cells according to the surface modification. After two days of incubation, fluorescence microscopy was performed with siRNA LNCs on SK-Mel28 human melanoma cells. Cells are fixed on Labtek slide and nucleus staining was performed with DAPI (blue). Double fluorescent probes are used to follow siRNA LNCs: lipophilic DiD (red) and Alexa488 siRNA (green). Analysis confirmed the internalization of siRNA LNCs without surface modification or after post-insertion of TE-PEG. On the opposite, DSPE-PEG LNCs showed no fluorescent signal. White scale bar = 100 μm. | ||
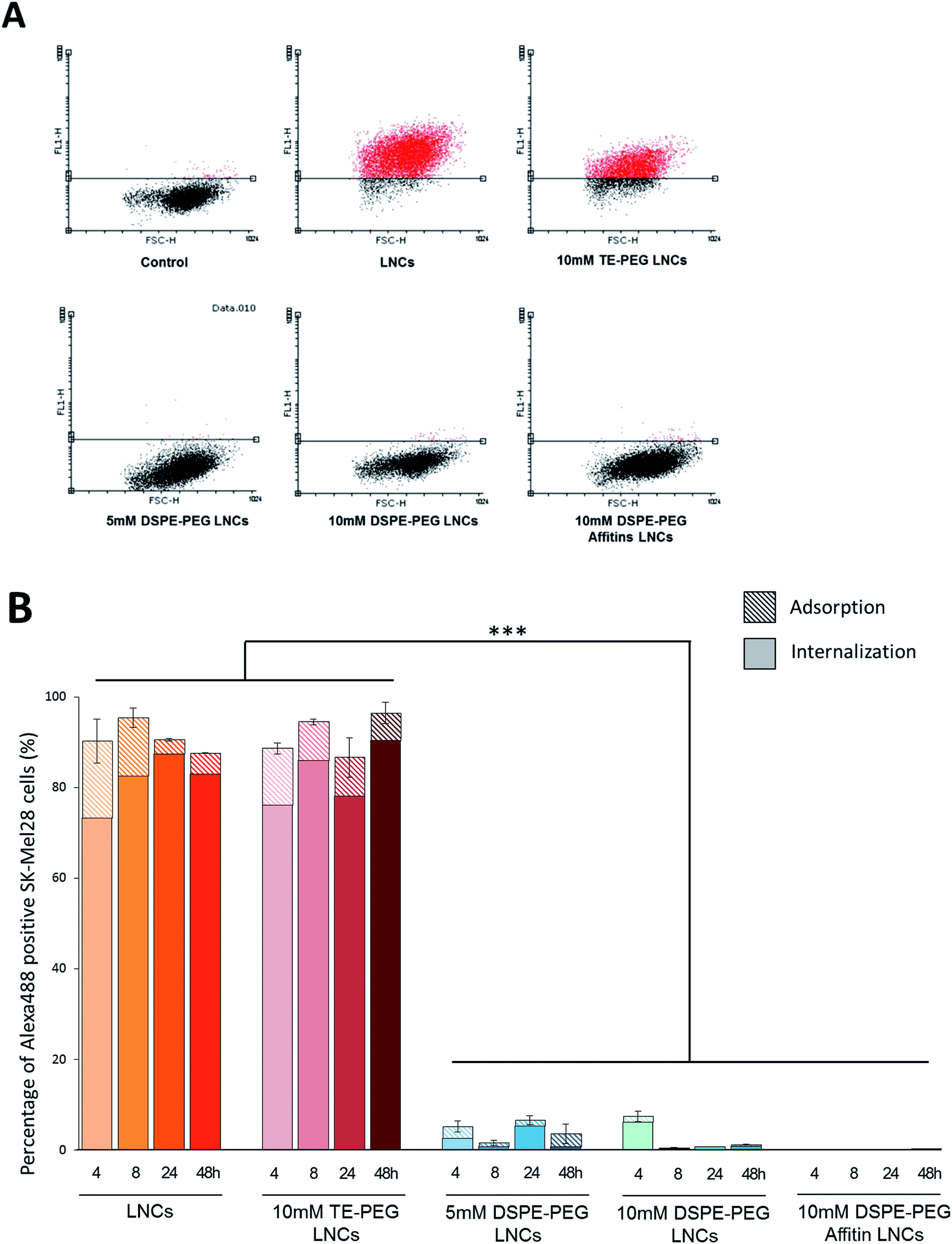 | ||
| Fig. 5 Evaluation of siRNA positive cells by flow cytometry analysis over time. (A) Dot plot analysis evidenced the large uptake of Alexa488 siRNA into SK-Mel28 cells for LNCs and TE-PEG LNCs. (B) Histograms showed the difference between internalized and adsorbed (hatched part of bar) siRNA, after trypan blue quenching. Data confirmed the large proportion of positive cells in LNCs and TE-PEG LNCs, with a low adsorption phenomenon (<10%), contrary to 5 and 10 mM DSPE-PEG LNCs and 10 mM DSPE-PEG-Affitins LNCs at 48 h. The mean percentage of fluorescent cells was compared to control cells without any fluorescence ± standard error of mean SEM (n = 3). Statistical analysis was performed with ANOVA 1W, post hoc Tukey, ***p = 0.001. | ||
Plasma protection
The siRNA protection was evaluated after incubation with human plasma, at physiological temperature (Fig. 6). In control conditions with siRNA alone, a fast degradation by plasmatic nucleases was observed after 30 min. LNCs demonstrated their efficacy to protect the siRNA against nucleases for at less 8 h, and longer protection times up to 24 h or 48 h were observed for all PEGylated LNCs (TE-PEG, 5 and 10 mM DSPE-PEG). The binding of Affitin to DSPE-PEG LNCs did not modify the protection effect against nucleases up to 24 h.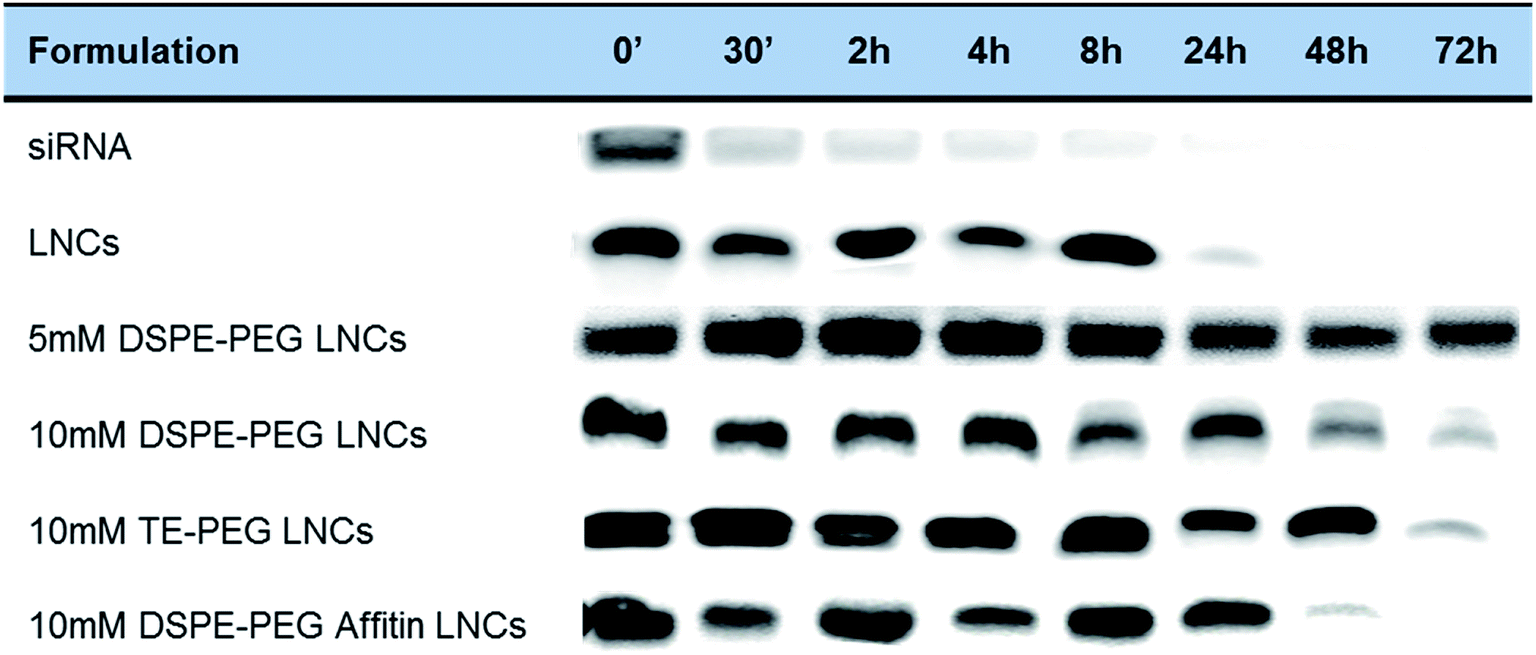 | ||
| Fig. 6 Evaluation of plasmatic protection of siRNA LNCs with different surface modifications. UV light detection of siRNA (thanks to ethidium bromide electrophoresis) after incubation with human plasma at 37 °C showed the efficient nuclease protection of siRNA by pegylated LNCs. | ||
Biodistribution on healthy mice
Biofluorescence imaging on animals was performed to evaluate the biodistribution of DiD siRNA LNCs with or without surface modification, after i.v. injection in healthy mice (Fig. 7). For non-modified siRNA LNC, the maximal signal was obtained after 1 h injection and induced a strong hepatic accumulation after 3 h and until 8 h (Fig. 7A). The 5 mM DSPE-PEG post-inserted LNCs showed an important liver localization between 5 h and 24 h with a maximum at 8 h. The highest concentration of DSPE-PEG (10 mM) on LNCs seemed to limit liver accumulation: the fluorescence signal was diffused (Fig. 7A). However, after 48 h, organ fluorescence evidenced the high amount of 5 and 10 mM DSPE-PEG LNCs, especially in liver, spleen, kidneys and bladder (Fig. 7B). Surprisingly, a very high fluorescence signal was observed in ovary with 5 and 10 mM DSPE-PEG LNCs. The Affitin grafting modified slightly the biodistribution, with a liver accumulation between 1 h and 24 h (Fig. 7A). However, the global fluorescence signal of DSPE-PEG-Affitin LNCs showed no important retention in undesirable organs (Fig. 7B). Concerning the TE-PEG siRNA LNCs, biofluorescence imaging and organ dissection demonstrated the accumulation into liver over time with a higher signal at 48 h (Fig. 7). To conclude, the analysis of the organs confirmed the high level of fluorescence into different organs such as spleen, kidney, lung, ovary, heart or bladder for all pegylated LNCs (5 mM, 10 mM DSPE-PEG and TE-PEG LNCs) (Fig. 7B). | ||
| Fig. 7 Evaluation of siRNA LNC biodistribution after intravenous injection in healthy NRMI nude female mice. (A) LNCs (yellow), 5 mM DSPE-PEG LNCs (blue), 10 mM DSPE-PEG LNCs (green), 10 mM DSPE-PEG-Affitin LNCs (purple) and 10 mM TE-PEG LNCs (red), loaded by a fluorescent tracer, DiD, were injected intravenously in healthy mice (n = 4). In vivo biofluorescence imaging (BFI) was evaluated at different times (15 min, 30 min, 1, 3, 5, 8, 24 and 48 hours after injection) from decubitus dorsal view, to follow biodistribution. (B) Two days after injection, biodistribution in organs was evaluated, proving the high liver accumulation for pegylated LNCs compared to LNCs and 10 mM DSPE-PEG-Affitins LNCs that were fully eliminated. Mean fluorescence ± standard error of the mean (SEM). | ||
Biodistribution on subcutaneous melanoma mice model
Biofluorescence imaging was performed on subcutaneous melanoma mice model (Fig. 8). In order to better visualize the tumor, animals were put on the left flank. A diffuse distribution of fluorescence with a liver accumulation was observed for unmodified LNCs. No specific fluorescence signal was co-localized with the tumor site, confirming the important hepatic accumulation (Fig. 8B). Biofluorescence imaging of DSPE-PEG LNCs showed a fast tumor accumulation (after 1 h and 5 h, for 10 mM and 5 mM DSPE-PEG LNCs respectively). This passive accumulation of siRNA LNCs modified by DSPE-PEG was confirmed by tumor fluorescence at 48 h (Fig. 8). Concerning DSPE-PEG-Affitin LNCs, no signal was observed in the tumor site, demonstrating a large elimination of this formulation after 48 h compared to the other formulations (Fig. 8B). Finally, LNCs modified with TE-PEG showed a persistent tumor localization 24 h after intravenous injection (Fig. 8A). Interestingly, at 48 h, TE-PEG LNCs showed the highest fluorescence signal in tumor compared to unmodified and DSPE-PEG-Affitin LNCs (Fig. 8B).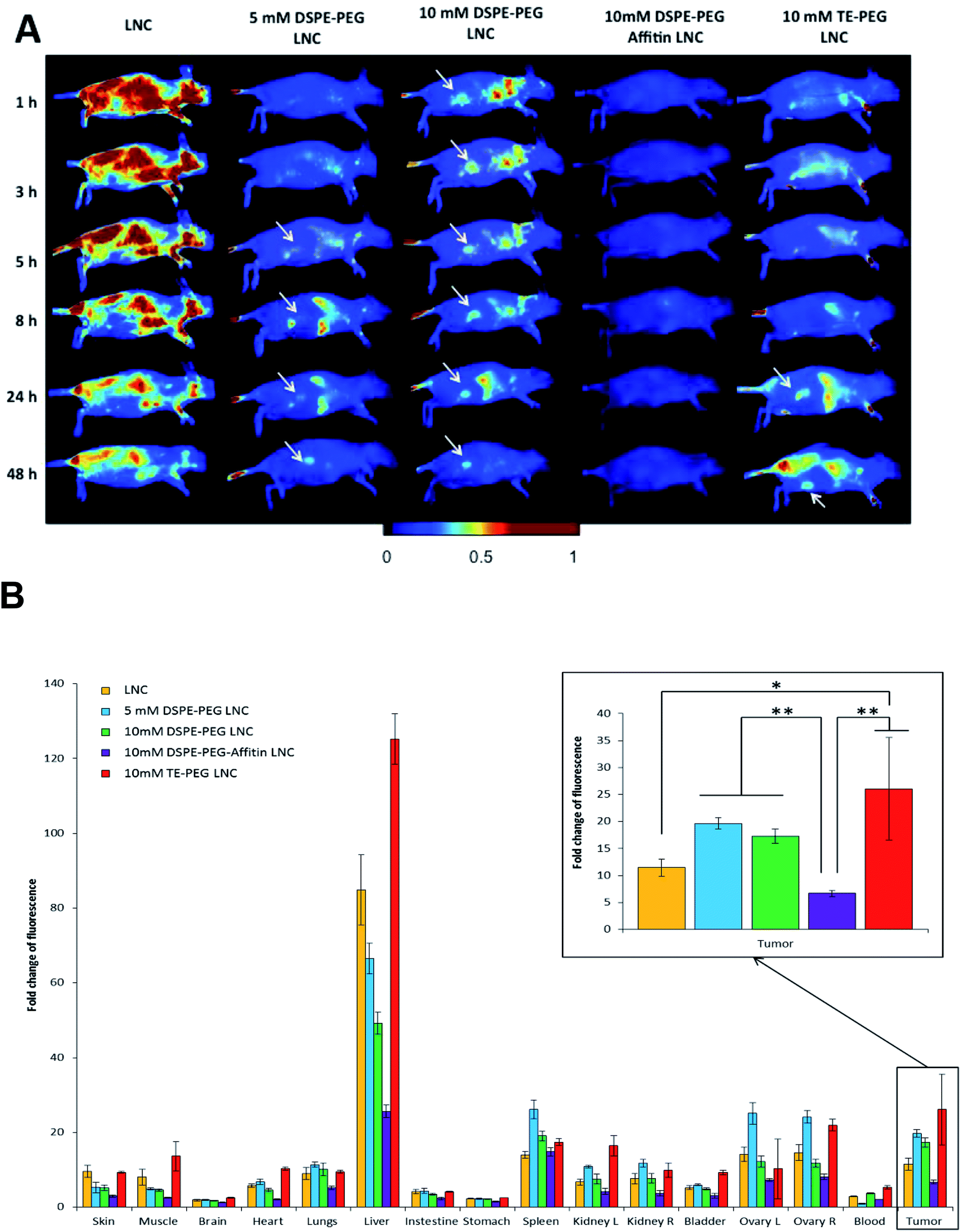 | ||
| Fig. 8 Evaluation of siRNA LNC biodistribution after intravenous injection in subcutaneous tumor bearing NRMI nude female mice. (A) LNCs (yellow), 5 mM DSPE-PEG LNCs (blue), 10 mM DSPE-PEG LNCs (green), 10 mM DSPE-PEG-Affitin LNCs (purple) and 10 mM TE-PEG LNCs (red), loaded by a fluorescent tracer, DiD, were injected intravenously in mice (n = 4) and vivo biofluorescence imaging (BFI) were taken at different times (1, 8, 24 and 48 hours after injection) from lateral views to follow their biodistribution and the potential tumor targeting. BFI revealed the specific tumor targeting in subcutaneous melanoma model for pegylated LNCs over time. (B) Two days after injection; biodistribution in organs proved the high liver accumulation for LNCs and TE-PEG LNCs compared to DSPE-PEG LNCs and 10 mM DSPE-PEG-Affitins LNCs. Fluorescence analysis in tumor demonstrated the best result for TE-PEG and DSPE-PEG LNCs. Mean fluorescence ± standard error of the mean (SEM), ANOVA 1W, post hoc Tukey, *p < 0.05 and **p < 0.01. | ||
Discussion
LNCs have been developed 20 years ago: the process is based on inversion phase of emulsion by mixing phospholipids, triglycerides, tensio-active polymers, salt and water.32 Firstly used for the encapsulation of lipophilic compounds, such as paclitaxel,33,34 LNCs were adapted more recently for the encapsulation of hydrophilic moieties via lipoplexes (DNA, siRNA).35 However, stability over time was not satisfying, especially in terms of size, such as described in our previous works.9 Some experiments led to the hypothesis of a saturated LNC surface, induced by the large amount of phospholipids, explaining the low stability of the system. This optimization of the process was assessed by the elimination of the lecithin (Fig. 1) (patent: FR 4185991, 24 Sept 2014).The siRNA loaded LNC cell uptake was studied overtime on living melanoma cells, by fluorescence confocal multispectral imaging (FCSI). The loss of FRET signal could correlate with a nanocapsule degradation. After a short incubation time (0.5 h) at 37 °C, a part of the siRNA LNCs seemed to be degraded before or by the contact with the cell membrane, explained by a decrease of the DiD component from the FRET signal. This element may be due to a fusion of LNCs with the cell membrane, supported by DOTAP/DOPE lipids, and leading to the efficient cytoplasm delivery of siRNAs. Indeed, localization of fluorescence was diffused into the cytoplasm: concentrated spots could be observed after endocytosis phenomenon such as with calcium phosphate particles developed notably by Xie et al.36 siRNA transfection seemed to reach a maximum at 3.5 h, where the siRNAs were stained over large cytosolic regions. At 5 h, the siRNAs seemed to be dispatched in the whole cell and the fluorescence signal decreased clearly at longer incubation time (15 h), indicating a siRNA release. The same intracellular trafficking and kinetic were observed for calcium phosphate particles with a maximal cytoplasmic loading after 4 h of incubation.36 Furthermore, the cellular trafficking of blank LNCs, without any drug, was investigated by Paillard et al.37 Interestingly, results clearly showed blank LNCs internalized through an energy dependent mechanism such as clathrin- or caveolin-mediated endocytosis pathways. The presence of new excipients in siRNA LNC formulation, cationic DOTAP and zwitterionic DOPE lipids6 would allow additional fusogenic properties to the nanosystem: indeed DOPE is well known to undergo transition from bilayer to inverted hexagonal structures at low pH known to catalyze the fusion process with the endosomal membrane.38,39
The surface modification of nanomedicines with innovative polymers represents an important challenge to improve tumor targeting after intravenous injection. In this work, various surface coatings (DSPE-PEG, TE-PEG and Affitin protein grafting) were applied to siRNA LNCs and validated without siRNA loading modification. In order to control the fluorescence and avoid variation between the different formulations, all biofluorescent imaging experiments were performed using one batch of fluorescent LNCs. This precaution taken, the altered biodistribution of PEG LNCs, compared to unmodified LNCs, could not be justified by a different fluorescence intensity between formulations. The biodistribution pattern obtained for PEG LNCs showed weak variations, excepted for the liver, as already observed in a previous study.40 Supplementary blood analysis confirmed the presence of PEG LNCs, 1 h and 3 h after intravenous injection, such as for DNA LNCs.12 This result could be explained by a difference of diffusion into the blood circulation, with a lower peripherical distribution.40 However, the addition of DSPE-PEG induced considerable limitation of in vitro cell uptake, avoiding intracellular siRNA delivery. This result could be explained by a steric hindrance between grafted chains and cell membrane according to PEG concentration, length and conformation.41–43 The comparison of two PEG concentrations, in biodistribution and tumor targeting studies, evidenced the advantage of choosing the lowest tested concentration of PEG. A theoretical calculation estimated that 5 mM led to a surface coating of 10 molecules per nm2 or approximatively 3000 PEG molecules per LNC.12
The grafting of Affitins on the LNC surface, a small artificial model binding protein, modified the behavior of LNCs as already observed for nanomedicines modified with peptides.41,44 As a first step toward active targeting with LNCs, we wanted to study if an Affitin model, i.e. without any affinity for a melanoma biomarker, had intrinsic properties compatible with our goals. In this work, the Affitin did not alter the plasma protection of siRNA in 10 mM DSPE-PEG LNCs. Moreover, Affitin grafted on LNCs was associated with the lowest fluorescence signal 48 h after injection, confirming the rapid elimination and the absence of accumulation in undesirable organs in healthy and melanoma bearing mice. The 10 mM DSPE-PEG-Affitin LNCs induced, at early times, a higher hepatic accumulation than for the 10 mM DSPE-PEG LNCs, probably due to the reduced stealth effect of the PEG with the covalent peptide grafting or/and due to a potential recognition of the peptide by the kupffer cells.
The 10 mM DSPE-PEG-Affitin LNC showed the lowest fluorescence signal in tumor site: even if the Affitin model used had no specificity for a melanoma biomarker, a passive targeting was at least expected. A study of Rangger et al., showed the necessity to study the balance between pegylation and ligand grafting to achieve an efficient and optimized targeting.41 Indeed, in our study, the Affitin conjugation to the PEG moiety significantly reduced the in vivo tumor uptake compared with pegylated LNC: the tumor accumulation was in fact comparable to LNC without any surface modification. The Affitin model grafting probably prevented the stealth effect of PEG, reducing thus the passive accumulation. In conclusion, these results highlight one of the most challenging problems in the delivery of nanomedicines: developing high-quality formulations that are fully characterized and with a specific accumulation of nanoparticles in tumor.45
Finally, the tetraether-PEG (TE-PEG) appears as an innovative solution to replace DSPE-PEG, with a low influence on cell uptake and a good plasma protection compared to non-modified LNCs. This archaeal lipid is known to improve the stability of liposomes.17 Interestingly, TE-PEG LNCs had a short half-life time in blood, but the biodistribution profile evidenced a higher tumor accumulation overtime. This efficacy could be explained by a different PEG conformation on the LNC surface. Beyond classic surface modifications of nanomedicines providing longevity in blood stream, some efforts have been performed to develop “smart” nanocarriers,18,46 via a stimuli-sensitive drug delivery system in response to various internal stimuli such as pH, temperature, or external signals, as magnetism or ultrasounds.18 This TE-PEG appears as a good candidate to create a “smart” targeting with a pH-sensitive link, adapted to the melanoma acidic microenvironment.16,47 Moreover, Affitins display a high stability for acidic pH,20 and future works should test the efficiency of TE-PEG-Affitin formulations using a specific melanoma biomarker Affitin.
The challenges and barriers to the systemic delivery of siRNA are numerous such as specificity, degradation, penetration of the cell membrane and endosomal escaping. Various natures of nanoparticles have been studied for siRNA delivery such mesoporous silica nanoparticles,48 gold nanoparticles,49 carbon nanotubes,50 polymeric nanoparticles51 or lipid-based nanoparticles52 with respective disadvantages: in vivo toxicity, poor stability, limited biocompatibility, high cost of large scale production. In this study, lipid nanocapsules have been used, demonstrating a good stability over time, a plasma protection, an impressive cell internalization, with a release of siRNA into the cytosol. Moreover, in this work, the versatility of the surface functionalization has been demonstrated with a significant tumor accumulation for the TE-PEG LNCs.
Experimental
siRNA LNC formulation
The complexes were characterized by the charge ratio (CR), i.e., the ratio between the positive charges of the lipids and negative charges of the nucleic acids (±ratio), fixed to 5 considering our previous results.10 These pre-formulated lipoplexes were added during the last phase inversion temperature (PIT) of LNC formulation leading to the formation of siRNA LNCs.
For in vitro experiments, the DiD concentration was fixed at 200 μg mL−1 of LNC suspension or corresponding to 1.36 mg of DiD per grams of Labrafac®. Regarding the FRET experiments, the DiD concentration was unchanged and the ratio DiD/DiI was fixed at 1/2 (mol/mol), corresponding to a DiD concentration at 400 μg mL−1 of LNC suspension. For biodistribution on animals, the DiD concentration was fixed at 33 μg mL−1 of LNC suspension or 0.23 μg mL−1 of Labrafac.
The adequate volume of DiD or DiD/DiI solubilized in acetone was incorporated in Labrafac® and acetone was evaporated at room temperature. The formulation process was unchanged, and formulation was stored at 4 °C, protected from light. For siRNA fluorescent LNCs, a fluorescent Alexa488 siRNA (λex = 488 nm; λem = 524 nm, Eurogentec) was used.
Purification and post-insertion of PEG polymers
Purifications were performed in water (obtained from a Milli-Q system, Millipore, Paris, France) with PD10 Sephadex column (Amersham Biosciences Europe, Orsay, France) to remove the non-encapsulated siRNA and lipoplexes.9 At the end, the NaCl concentration was adjusted to physiological concentration (150 mM).The polymers used for post-insertion were 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG) (mean molecular weight MMw = 2805 g mol−1), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethyleneglycol)-2000] (malDSPE-PEG) (mean molecular weight MMw = 2942 g mol−1) (Avanti Polar Lipids, Inc, Alabaster, USA) and tetraether-[methoxy(polyethyleneglycol)-2000] (TE-PEG) (MMw = 3217 g mol−1) synthesized according to Barbeau et al.17 Polymers and siRNA LNCs were co-incubated 4 h at 37 °C with a final polymer concentration adjusted to 5 or 10 mM.
Affitin coupling on DSPE-PEG LNCs
The Affitin H4 used in this study is specific to hen egg white lysozyme, and this “model” Affitin has been described previously.21 To allow the covalent coupling via thiol/maleimide chemistry, a cysteine residue was added to C-terminus of this Affitin by directed mutagenesis. The resulting Affitin (H4-Ct) was coupled to LNCs according to Bourseau et al.31 Briefly, a solution at 1 mg mL−1 (corresponding to 110 μM) of Affitin was prepared in water and mixed with DTT (dithiothreitol, Sigma-Aldrich), a strong reducing agent, at 0.012 mg mL−1 (corresponding to 80 μM) under stirring at room temperature during 1 h. Affitins with DTT were purified using PD10 Sephadex column (Amersham Biosciences Europe, Orsay, France). PEGylated siRNA LNCs with 10 mM malDSPE-PEG were mixed with purified Affitins under stirring at room temperature overnight. DSPE-PEG-Affitin LNCs were purified by CL4B sepharose column (Sigma Aldrich). The presence of LNCs was evidenced with turbidity lecture at 580 nm and Affitins by microBCA test (Thermo Fisher, Waltham, MA, USA) according to the manufacturer protocol.Characterization of siRNA LNCs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 v/v dilution ratio with deionized water.53
:200 v/v dilution ratio with deionized water.53Cell culture
The SK-Mel28 human melanoma cell lines were grown in Roswell Park Memorial Institute (RPMI) 1640 medium (Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum (Lonza, Verviers, Belgium), 1% antibiotics (10 units of penicillin, 10 mg of streptomycin, 25 μg of amphotericin B per mL; Sigma-Aldrich, Saint louis, USA) and 1% non-essential amino acids (Lonza). Cell lines were cultured according to ATCC protocol and maintained at 37 °C in a humidified atmosphere with 5% CO2.Cellular uptake
Melanoma cells were seeded onto 6-well plates at the density of 50 × 104 cells per well and precultured overnight. Before transfection, the medium was changed to a fresh medium, without serum. The cells were treated with double fluorescent siRNA LNCs (DiD and Alexa488 siRNA) at siRNA concentration of 0.25 μg mL−1 for 4 h to 48 h. Immunostaining was performed to evaluate the internalization of LNCs and siRNA into SK-Mel28 cells. After kinetic point (4, 8, 24 and 48 h), cells were washed twice with PBS (Lonza) and incubation with trypan blue (Lonza) was performed during 1 min following by three PBS washing. Cell suspensions were fixed with PBS/azide 0.02%/formaldehyde 1%, protected from light and stored at 4 °C. Analyses were performed with a FACScalibur flow cytometer (BD Bioscience, San José, USA) in collaboration with PACeM platform (Angers).
Fluorescence confocal spectral imaging (FCSI)
To follow the nanocarrier behavior in the presence of melanoma cells, siRNA-loaded LNCs were formulated with three different fluorochromes. Two fluorescent probes, DiI and DiD were used respectively as donor and acceptor partners to obtain a FRET (Fluorescence/Förster Resonance Energy Transfer) signal. DiI (λex = 549 nm, λem = 565 nm) was excited with a 488 nm laser. With this excitation wavelength, the DiD fluorescence (λex = 644 nm, λem = 665 nm) can mainly result from FRET. To observe FRET, the LNCs were co-loaded with DiI:DiD mixture at 2:1 molar ratio. In addition, the LNCs were loaded with siRNA conjugated to Alexa488 fluorophore (λex = 488 nm, λem = 525 nm). Therefore, with the triple-labelled LNCs, we simultaneously excited the DiI/DiD FRET couple and the green fluorescence of Alexa488 in order to follow both LNC integrity and siRNA distribution.
SK-Mel28 cells were seeded onto CC2 Labtek 4 chambers (Dominique Dutcher, France) at the density of 30 × 103 cells per chamber and precultured for 96 h. The medium was then replaced by a suspension of triple-labelled siRNA LNCs in the culture medium without serum at the concentration of 0.50 μg mL−1 of siRNA and the cells incubated for 0.5, 3.5, 5, or 15 h at 37 °C, 5% CO2. After treatment, SK-Mel28 cells were washed twice in HBSS supplemented with Ca2+ and Mg2+. Then the media chambers were removed, some HBSS supplemented with Ca2+ and Mg2+ was added to keep the cells alive during the fluorescence confocal spectral imaging (FCSI), and finally the cells were recovered by a lamella.
The FCSI measurements were carried out using a LabRam confocal microspectrometer (Horiba Jobin Yvon, Villeneuve d'Ascq, France) equipped with an automated X–Y–Z scanning stage, a low dispersion grating (300 grooves per mm) and an air-cooled CCD detector. The excitation wavelength used was a 488 nm line of an Ar+ laser (Melles Griot, France) and collected in a confocal mode, through the 50× LWD objective (NA 0.5). For each cell analysis, an equatorial optical section (x–y plane situated at half-thickness of the cell) was scanned with a step of 0.7 μm that provided maps containing typically ≈900 spectra. The cellular auto-fluorescence was completely neglected, because of the absence of any significant fluorescence of the untreated cells under the conditions used (laser power on the sample ca. 5 μW, 0.03 s per spectrum). No sample photodegradation was observed.
Both acquisition (n = 3 to 5) and treatment of multispectral maps were performed with LabSpec software. Subcellular LNC and siRNA distribution maps were established via analysis of both the intensity and shape of intracellular fluorescence spectra. Briefly, each experimental spectrum was fitted using the least-squares method to a sum of three reference spectra described below. The fitting errors were below 5% (typically 2–4%).
Plasma protection
siRNA, siRNA LNCs and surface modified formulations containing 30 μg siRNA were incubated for different time intervals in 50% (v/v) Normal Human Serum (NHS) at 37 °C.30 Following incubation, samples were stored at −20 °C. Electrophoresis into 1% (m/v) agarose gels in TBE buffer containing ethidium bromide (EtB) was performed at 125 V for 20 min and the resulting gels were photographed under UV light.In vivo experiments
Statistical analysis
Comparisons between all groups, supposed with normal distribution, were performed using a classical analysis of variance (one-way ANOVA) followed by a Tukey's post hoc analysis. The encapsulation efficiencies for modified LNCs and non-modified LNCs were compared using a t-test. Statistical significance was ascribed to a threshold p-value of 0.05 (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).Conclusions
Lipid nanocapsules (LNCs) previously demonstrated their capacity to efficiently encapsulate siRNAs. In this study, FRET experiments proved the efficient time-dependency delivery of siRNA into the cytosol of human melanoma cells, thanks to a strong interaction/fusion of LNCs with the cell membranes. In addition, different surface modifications were performed. While the tested DSPE-PEG LNCs were not able to penetrate into melanoma cells, results showed the efficient internalization of siRNA loaded LNCs modified by TE-PEG. The biodistribution on melanoma mice model showed the best tumor targeting for pegylated LNCs at 48 h, notably for TE-PEG LNCs. Moreover, this TE-PEG, recently modified thanks to the addition of pH sensitive link between the PEG and TE molecule, could represent an innovative moiety to develop smart nanomedicines. Finally, the results obtained with an Affitin are encouraging, as its presence on LNCs did not induce in vivo undesirable side effect. The use of a melanoma specific Affitin combined with an optimized balance of DSPE or TE-PEG/Affitin would allow an efficient smart targeting.Conflicts of interest
There are no conflicts to declare.Abbreviations
| BFI | Biofluorescence imaging |
| DiD | 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate |
| DiI | 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate |
| DOPE | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine |
| DOTAP | 1,2-Dioleoyl-3-trimethylammoniumpropane |
| DSPE-PEG | 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] |
| DTT | Dithiothreitol |
| FRET | Fluorescence/Förster resonance energy transfer |
| i.v. | Intravenous |
| LNCs | Lipid nanocapsules |
| malDSPE-PEG | 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethyleneglycol)-2000] |
| siRNA | Small interfering RNA |
| TE-PEG | Tetraether-polyethylene glycol |
Acknowledgements
The authors would like to thank the SFR ICAT and the PACEM platform for flow cytometry and especially Catherine Guillet for her expertise in this field. We are also grateful to Dr Joel Eyer and Dr Franck Letournel for their collaboration in using the Maestro II, Dr Anne Clavreul for her expertise in fluorescence microscopy, and Dr Guillaume Bastiat for his help on statistical analysis. This work was supported by special grants from the “Association de la Recherche contre le Cancer” (Reference no.: SFI20121205972) and by “La Ligue contre le cancer 49 et 35” (Reference no.: R12164NN). P. Resnier and C. Passirani are the inventors of a French patent (FR 4185991, 24 Sept 2014) about the formulation of stable siRNA LNCs, owned by University of Angers. F. Pecorari is inventor of a patent application (PCT/IB2007/004388), owned by Institut Pasteur and Centre National de Recherche Scientifique (CNRS), related to generation of Affitins.References
- A. Fire, et al., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- Q. Lin, J. Chen, Z. Zhang and G. Zheng, Lipid-based nanoparticles in the systemic delivery of siRNA, Nanomedicine, 2014, 9, 105–120 CrossRef CAS.
- G. Tiram, A. Scomparin, P. Ofek and R. Satchi-Fainaro, Interfering cancer with polymeric siRNA nanomedicines, J. Biomed. Nanotechnol., 2014, 10, 50–66 CrossRef CAS.
- Z. Du, M. M. Munye, A. D. Tagalakis, M. D. I. Manunta and S. L. Hart, The Role of the Helper Lipid on the DNA Transfection Efficiency of Lipopolyplex Formulations, Sci. Rep., 2014, 4, 7107 CrossRef PubMed.
- D. C. Litzinger and L. Huang, Phosphatidylethanolamine liposomes: drug delivery, gene transfer and immunodiagnostic applications, Biochim. Biophys. Acta, 1992, 1113, 201–227 CrossRef CAS.
- P. Resnier, T. Montier, V. Mathieu, J.-P. Benoit and C. Passirani, A review of the current status of siRNA nanomedicines in the treatment of cancer, Biomaterials, 2013, 34, 6429–6443 CrossRef CAS PubMed.
- S. David, B. Pitard, J.-P. Benoît and C. Passirani, Non-viral nanosystems for systemic siRNA delivery, Pharmacol. Res., 2010, 62, 100–114 CrossRef CAS PubMed.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J.-P. Benoit, Progress in developing cationic vectors for non-viral systemic gene therapy against cancer, Biomaterials, 2008, 29, 3477–3496 CrossRef CAS PubMed.
- P. Resnier, et al., Efficient in vitro gene therapy with PEG siRNA lipid nanocapsules for passive targeting strategy in melanoma, Biotechnol. J., 2014, 9, 1389–1401 CrossRef CAS PubMed.
- S. David, et al., siRNA LNCs–a novel platform of lipid nanocapsules for systemic siRNA administration, Eur. J. Pharm. Biopharm., 2012, 81, 448–452 CrossRef CAS.
- P. Resnier, et al., EGFR siRNA lipid nanocapsules efficiently transfect glioma cells in vitro, Int. J. Pharm., 2013, 454, 748–755 CrossRef CAS PubMed.
- M. Morille, et al., Long-circulating DNA lipid nanocapsules as new vector for passive tumor targeting, Biomaterials, 2010, 31, 321–329 CrossRef CAS PubMed.
- S. David, et al., In vivo imaging of DNA lipid nanocapsules after systemic administration in a melanoma mouse model, Int. J. Pharm., 2012, 423, 108–115 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J. Control. Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- C. Laine, et al., Folate-equipped pegylated archaeal lipid derivatives: synthesis and transfection properties, Chemistry, 2008, 14, 8330–8340 CrossRef PubMed.
- T. Benvegnu, L. Lemiegre and S. Cammas-Marion, New generation of liposomes called archaeosomes based on natural or synthetic archaeal lipids as innovative formulations for drug delivery, Recent Pat. Drug Deliv. Formulation, 2009, 3, 206–220 CrossRef CAS.
- J. Barbeau, S. Cammas-Marion, P. Auvray and T. Benvegnu, Preparation and Characterization of Stealth Archaeosomes Based on a Synthetic PEGylated Archaeal Tetraether Lipid, J. Drug Delivery, 2011, 2011, 396068 Search PubMed.
- A. Jhaveri, P. Deshpande and V. Torchilin, Stimuli-sensitive nanopreparations for combination cancer therapy, J. Control. Release, 2014, 190, 352–370 CrossRef CAS PubMed.
- V. Kalichuk, et al., The archaeal “7 kDa DNA-binding” proteins: extended characterization of an old gifted family, Sci. Rep., 2016, 6, 37274 CrossRef CAS PubMed.
- G. Behar, et al., Tolerance of the archaeal Sac7d scaffold protein to alternative library designs: characterization of anti-immunoglobulin G Affitins, Protein Eng. Des. Sel., 2013, 26, 267–275 CrossRef CAS PubMed.
- A. Correa, et al., Potent and specific inhibition of glycosidases by small artificial binding proteins (affitins), PLoS One, 2014, 9, e97438 CrossRef.
- B. Mouratou, et al., Remodeling a DNA-binding protein as a specific in vivo inhibitor of bacterial secretin PulD, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17983–17988 CrossRef CAS PubMed.
- G. Behar, S. Pacheco, M. Maillasson, B. Mouratou and F. Pecorari, Switching an anti-IgG binding site between archaeal extremophilic proteins results in Affitins with enhanced pH stability, J. Biotechnol., 2014, 192(Pt A), 123–129 CrossRef CAS PubMed.
- G. Behar, A. Renodon-Corniere, B. Mouratou and F. Pecorari, Affitins as robust tailored reagents for affinity chromatography purification of antibodies and non-immunoglobulin proteins, J. Chromatogr. A, 2016, 1441, 44–51 CrossRef CAS PubMed.
- C. S. M. Fernandes, et al., Affitins for protein purification by affinity magnetic fishing, J. Chromatogr. A, 2016, 1457, 50–58 CrossRef CAS PubMed.
- V. Kalichuk, et al., A novel, smaller scaffold for Affitins: Showcase with binders specific for EpCAM, Biotechnol. Bioeng., 2018, 115, 290–299 CrossRef CAS PubMed.
- S. Pacheco, G. Behar, M. Maillasson, B. Mouratou and F. Pecorari, Affinity transfer to the archaeal extremophilic Sac7d protein by insertion of a CDR, Protein Eng. Des. Sel., 2014, 27, 431–438 CrossRef CAS.
- G. Behar, et al., Whole-bacterium ribosome display selection for isolation of highly specific anti-Staphyloccocus aureus Affitins for detection- and capture-based biomedical applications, Biotechnol. Bioeng., 2019, 116, 1844–1855 CrossRef CAS PubMed.
- G. Grazia, I. Penna, V. Perotti, A. Anichini and E. Tassi, Towards combinatorial targeted therapy in melanoma: from pre-clinical evidence to clinical application (review), Int. J. Oncol., 2014, 45, 929–949 CrossRef CAS PubMed.
- W. J. Kim, et al., Anti-angiogenic inhibition of tumor growth by systemic delivery of, J. Control. Release, 2006, 114, 381–388 CrossRef CAS PubMed.
- E. Bourseau-Guilmain, et al., Development and characterization of immuno-nanocarriers targeting the cancer stem cell marker AC133, Int. J. Pharm., 2012, 423, 93–101 CrossRef CAS PubMed.
- B. Heurtault, P. Saulnier, B. Pech, J.-E. Proust and J.-P. Benoit, A novel phase inversion-based process for the preparation of lipid nanocarriers, Pharm. Res., 2002, 19, 875–880 CrossRef CAS.
- J. Hureaux, et al., Lipid nanocapsules: ready-to-use nanovectors for the aerosol delivery of paclitaxel, Eur. J. Pharm. Biopharm., 2009, 73, 239–246 CrossRef CAS.
- E. Roger, F. Lagarce, E. Garcion and J.-P. Benoit, Reciprocal competition between lipid nanocapsules and P-gp for paclitaxel transport across Caco-2 cells, Eur. J. Pharm. Sci., 2010, 40, 422–429 CrossRef CAS PubMed.
- A. Vonarbourg, et al., The encapsulation of DNA molecules within biomimetic lipid nanocapsules, Biomaterials, 2009, 30, 3197–3204 CrossRef CAS PubMed.
- Y. Xie, et al., PEGylated carboxymethyl chitosan/calcium phosphate hybrid anionic nanoparticles mediated hTERT siRNA delivery for anticancer therapy, Biomaterials, 2014, 35, 7978–7991 CrossRef CAS.
- A. Paillard, F. Hindre, C. Vignes-Colombeix, J.-P. Benoit and E. Garcion, The importance of endo-lysosomal escape with lipid nanocapsules for drug subcellular bioavailability, Biomaterials, 2010, 31, 7542–7554 CrossRef CAS.
- A. Elouahabi and J.-M. Ruysschaert, Formation and intracellular trafficking of lipoplexes and polyplexes, Mol. Ther., 2005, 11, 336–347 CrossRef CAS PubMed.
- Y. Ma, et al., Enhanced bactericidal potency of nanoliposomes by modification of the fusion activity between liposomes and bacterium, Int. J. Nanomed., 2013, 8, 2351–2360 CrossRef PubMed.
- A.-L. Laine, et al., Conventional versus stealth lipid nanoparticles: formulation and in vivo fate prediction through FRET monitoring, J. Control. Release, 2014, 188, 1–8 CrossRef CAS PubMed.
- C. Rangger, et al., Influence of PEGylation and RGD loading on the targeting properties of radiolabeled liposomal nanoparticles, Int. J. Nanomed., 2012, 7, 5889–5900 CrossRef CAS PubMed.
- Y. Li, M. Kröger and W. K. Liu, Endocytosis of PEGylated nanoparticles accompanied by structural and free energy changes of the grafted polyethylene glycol, Biomaterials, 2014, 35, 8467–8478 CrossRef CAS PubMed.
- E. C. Cho, Q. Zhang and Y. Xia, The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles, Nat. Nanotechnol., 2011, 6, 385–391 CrossRef CAS PubMed.
- W. Dai, et al., A comprehensive study of iRGD-modified liposomes with improved chemotherapeutic efficacy on B16 melanoma, Drug Deliv., 2015, 22, 10–20 CrossRef CAS PubMed.
- A. D. Friedman, S. E. Claypool and R. Liu, The smart targeting of nanoparticles, Curr. Pharm. Des., 2013, 19, 6315–6329 CrossRef CAS PubMed.
- N. T. Huynh, E. Roger, N. Lautram, J.-P. Benoit and C. Passirani, The rise and rise of stealth nanocarriers for cancer therapy: passive versus active targeting, Nanomedicine, 2010, 5, 1415–1433 CrossRef CAS PubMed.
- Y. Kato, et al., Acidic extracellular microenvironment and cancer, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- K. Möller, et al., Highly efficient siRNA delivery from core–shell mesoporous silica nanoparticles with multifunctional polymer caps, Nanoscale, 2016, 7, 4007 RSC.
- S. Guo, et al., Enhanced Gene Delivery and siRNA Silencing by Gold Nanoparticles Coated with Charge-Reversal Polyelectrolyte, ACS Nano, 2010, 4, 5505–5511 CrossRef CAS PubMed.
- L. Kirkpatrick, et al., Carbon Nanotubes: Solution for the Therapeutic Delivery of siRNA?, Materials, 2012, 5, 278–301 CrossRef PubMed.
- G. Cavallaro, C. Sardo, E. F. Craparo, B. Porsio and G. Giammona, Polymeric nanoparticles for siRNA delivery: production and applications, Int. J. Pharm., 2017, 525, 313–333 CrossRef CAS.
- T. S. Zatsepin, Y. V. Kotelevtsev and V. Koteliansky, Lipid nanoparticles for targeted siRNA delivery – going from bench to bedside, Int. J. Pharm., 2016, 11, 3077–3086 CAS.
- G. Bastiat, et al., A new tool to ensure the fluorescent dye labeling stability of nanocarriers: a real challenge for fluorescence imaging, J. Control. Release, 2013, 170, 334–342 CrossRef CAS PubMed.
- E. van Bracht, et al., Enhanced cellular uptake of albumin-based lyophilisomes when functionalized with cell-penetrating peptide TAT in HeLa cells, PloS one, 2014, 9, e110813 CrossRef PubMed.
- G. K. Srivastava, et al., Trypan Blue staining method for quenching the autofluorescence of RPE cells for improving protein expression analysis, Exp. Eye Res., 2011, 93, 956–962 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |