
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduction of cryptic metabolites of the endophytic fungus Trichocladium sp. through OSMAC and co-cultivation†
Nam Michael Tran-Conga,
Attila Mándi b,
Tibor Kurtánb,
Werner E. G. Müllerc,
Rainer Kalscheuera,
Wenhan Lind,
Zhen Liu*a and
Peter Proksch*a
b,
Tibor Kurtánb,
Werner E. G. Müllerc,
Rainer Kalscheuera,
Wenhan Lind,
Zhen Liu*a and
Peter Proksch*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich Heine University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: zhenfeizi0@sina.com; proksch@uni-duesseldorf.de
bDepartment of Organic Chemistry, University of Debrecen, P. O. Box 400, H-4002 Debrecen, Hungary
cInstitute of Physiological Chemistry, Universitätsmedizin der Johannes Gutenberg-Universität Mainz, 55128 Mainz, Germany
dState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China
First published on 30th August 2019
Abstract
The endophytic fungus Trichocladium sp. isolated from roots of Houttuynia cordata was cultured on solid rice medium, yielding a new amidepsine derivative (1) and a new reduced spiro azaphilone derivative (3) together with eight known compounds (4–11). Co-cultivation of Trichocladium sp. with Bacillus subtilis resulted in induction of a further new compound (2) and a 10-fold increase of 11 compared to the axenic fungal culture. Moreover, when the fungus was cultivated on peas instead of rice, a new sesquiterpene derivative (13) and two known compounds (12 and 14) were obtained. Addition of 2% tryptophan to rice medium led to the isolation of a new bismacrolactone (15). The structures of the new compounds were elucidated by HRESIMS, 1D and 2D NMR as well as by comparison with the literature. A combination of TDDFT-ECD, TDDFT-SOR, DFT-VCD and DFT-NMR calculations were applied to determine the absolute and relative configurations of 13 and 15. Compounds 7, 11 and 15 exhibited strong cytotoxicity against the L5178Y mouse lymphoma cell line with IC50 values of 0.3, 0.5 and 0.2 μM, respectively.
Introduction
Paclitaxel, a well-known and widely used anti-cancer compound, was first isolated from the bark of Taxus brevifolia by Wani et al. in 1971.1 Later in 1993, Stierle et al. discovered that the endophytic fungus Taxomyces andreanae from the yew tree is able to produce taxol and other taxane derivatives.2 This sparked detailed investigations of natural products from endophytes which confirmed these organisms as a promising source of new bioactive compounds.3 In continuation of our ongoing investigations on bioactive metabolites from fungi,4–6 an endophytic fungus Trichocladium sp. was isolated from roots of the plant Houttuynia cordata known in Vietnam as “fish mint”. Prior to renaming as Trichocladium, Trichocladium species have been listed in the literature under the genus Humicola.7 Fungi of the genus Trichocladium have been reported to produce several bioactive secondary metabolites including inhibitors of serine proteases of the coagulation pathway, asterriquinones CT1, CT3 and CT4,8 a cytotoxic phenolic tetralone, humicolone,9 and inhibitors of human diacylglycerol acyltransferases (DGAT), amidepsines A–K.10In this study, fermentation of Trichocladium sp. on solid rice medium yielded ten natural products (1, 3–11) including two new compounds (1 and 3). Co-cultivation of Trichocladium sp. with Bacillus subtilis led to the production of a new compound (2) and a 10-fold increase of 11. In addition, when the fungus was cultivated on peas instead of rice, a new sesquiterpene derivative (13) and two known compounds (12 and 14) that were not detected when the fungus was grown on rice medium were obtained. Compound 14 is a tryptophan derivative, inspiring us to add 2% tryptophan to rice medium in order to stimulate production of tryptophan derived compounds. Feeding of tryptophan led to the isolation of the new compound 15, which represents a macrolide linked to an anthranilic acid moiety. The latter moiety is likely to be derived from feeding of tryptophan (Fig. 1).
 | ||
| Fig. 1 Structures of isolated compounds from Trichocladium sp. | ||
Results and discussion
Compound 1 was obtained as pale yellow powder. Its molecular formula was determined as C29H27NO13 by HREISMS, indicating seventeen degrees of unsaturation. The NMR data of 1 (Table 1) were similar to those of amidepsine J previously isolated from Humicola sp. FO-2942.10 Six aromatic protons at δH 6.66 (H-12), 6.63 (H-14), 6.57 (H-4), 6.54 (H-6), 6.37 (H-20), and 6.31 (H-22) as well as three aromatic methyl groups at δH 2.37 (Me-16), 2.28 (Me-24), and 2.24 (Me-8) were observed. However, the NMR spectra of 1 showed signals of only one methoxy group at δC 55.8 and δH 3.79, instead of two methoxy groups as found for amidepsine J. The position of this methoxy group at C-19 was evident from the ROESY correlations between the protons of this methoxy group and H-20, and between H-20/21-OH (δH 9.96), 21-OH/H-22, H-22/Me-24. In addition, signals of a valine moiety were replaced by signals of an aspartic acid moiety (from C-25 to C-28) in 1. The COSY correlations between 26-NH/H-26/H-27ab and the HMBC correlations from 26-NH to C-1 together with the ROESY correlation between 26-NH and Me-8 indicated the linkage between the aspartic acid moiety and C-1 through an amide bond. The absolute configuration of the aspartic acid moiety was determined after acid hydrolysis of 1 followed by Marfey's method which indicated that the aspartic acid moiety was present in the L-form. Thus, the structure of compound 1 was elucidated as shown, representing a new amidepsine derivative, for which a trivial name amidepsine L is proposed.| Position | δC, typeb | δH (J in Hz) |
|---|---|---|
| a Measured in DMSO-d6 (1H at 300 MHz and 13C at 75 MHz).b Data were extracted from the HSQC and HMBC spectra. | ||
| 1 | 166.4, C | |
| 2 | 123.2, C | |
| 3 | 155.1, C | |
| 4 | 106.5, CH | 6.57, d (2.1) |
| 5 | 150.6, C | |
| 6 | 113.4, CH | 6.54, d (2.1) |
| 7 | 137.7, C | |
| 8 | 18.9, CH3 | 2.24, s |
| 9 | 165.8, C | |
| 10 | 118.6, C | |
| 11 | 156.1, C | |
| 12 | 107.0, CH | 6.66, d (2.0) |
| 13 | 152.1, C | |
| 14 | 114.1, CH | 6.63, d (2.0) |
| 15 | 137.8, C | |
| 16 | 19.2, CH3 | 2.37, s |
| 17 | 165.7, C | |
| 18 | 112.8, C | |
| 19 | 158.5, C | |
| 20 | 97.0, CH | 6.37, d (2.0) |
| 21 | 160.1, C | |
| 22 | 109.1, CH | 6.31, d (2.0) |
| 23 | 138.0, C | |
| 24 | 19.4, CH3 | 2.28, s |
| 25 | 172.6, C | |
| 26 | 48.8, CH | 4.69, dt (7.6, 6.6) |
| 27 | 36.2, CH2 | 2.78, dd (16.4, 6.6) |
| 2.61, dd (16.4, 6.6) | ||
| 28 | 171.8, C | |
| 11-OH | 10.44, s | |
| 19-OMe | 55.8, CH3 | 3.79, s |
| 21-OH | 9.96, s | |
| 26-NH | 8.47, d (7.6) | |
The molecular formula of 2 was determined as C15H22O5 based on the HRESIMS data. The NMR data of 2 (Table 2) were very similar to those of pestafolide A.11 However, the smaller coupling constants between H-5 and H-6ab (both 3.5 Hz) in 2 compared to those in pestafolide A (10.0 and 5.8 Hz) suggested that the orientation of H-5 was different in 2. This was further supported by the ROESY correlations between H-5 (δH 3.98)/Me-15 (δH 1.30), Me-15/H-6a (δH 2.70), and H-6a/H-5, indicating that these protons were on the same face of the cyclohexenone ring. The remaining substructure and stereochemistry of 2 were identical to those of pestafolide A as concluded after detailed analysis of the 2D NMR spectra of 2. Thus, compound 2 was identified as 5-epi-pestafolide A.
| Position | 2a | 3b | ||
|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| a Measured in CD3OD (1H at 600 MHz and 13C at 150 MHz).b Measured in CDCl3 (1H at 600 MHz and 13C at 150 MHz). | ||||
| 1 | 58.1, CH2 | 4.41, d (15.6) | 57.0, CH2 | 4.55, d (15.8) |
| 4.01, d (15.6) | 4.07, d (15.8) | |||
| 2 | 127.7, C | 126.9, C | ||
| 3 | 200.0, C | 197.4, C | ||
| 4 | 77.2, C | 74.1, C | ||
| 5 | 75.0, CH | 3.98, t (3.5) | 74.7, CH | 5.29, dd (3.6, 2.3) |
| 6 | 37.3, CH2 | 2.70, dd (18.8, 3.5) | 34.3, CH2 | 2.74, dd (19.5, 3.6) |
| 2.44, dd (18.8, 3.5) | 2.47, dd (19.5, 2.3) | |||
| 7 | 149.2, C | 147.2, C | ||
| 8 | 41.8, CH2 | 2.35, d (18.9) | 40.8, CH2 | 2.30, d (18.9) |
| 2.11, d (18.9) | 2.10, d (18.9) | |||
| 9 | 96.4, C | 94.9, C | ||
| 10 | 34.9, CH2 | 1.69, m | 33.8, CH2 | 1.70, m |
| 1.54, m | 1.51, m | |||
| 11 | 20.0, CH2 | 1.92, m | 18.9, CH2 | 1.92, m |
| 1.62, m | 1.63, m | |||
| 12 | 33.4, CH2 | 1.62, m | 32.2, CH2 | 1.61, m |
| 1.20, m | 1.19, m | |||
| 13 | 68.2, CH | 3.80, m | 66.9, CH | 3.77, m |
| 14 | 22.0, CH3 | 1.08, d (6.3) | 21.7, CH3 | 1.09, d (6.4) |
| 15 | 22.7, CH3 | 1.30, s | 23.7, CH3 | 1.40, s |
| 5-OAc | 170.5, C | |||
| 21.1, CH3 | 2.01, s | |||
Comparison of the 1H and 13C NMR data between 2 and 3 (Table 2) revealed the appearance of an additional acetoxy group (δC 170.5 and 21.1, δH 2.01) in 2, which was also evident from the molecular formula of 3 (C17H24O6) being 42 amu larger than that of 2. The downfield-shifted H-5 signal (δH 5.29, dd, J = 3.6, 2.3 Hz) in 3 compared to that in 2 (δH 3.98, t, J = 3.5 Hz) suggested the location of this additional acetoxy group at C-5, which was confirmed by the HMBC correlation from H-5 to the carbonyl carbon of the acetoxy group. The coupling constants of H-5 were similar in 2 and 3, indicating the same stereochemistry at C-5 for 2 and 3. In conclusion, compound 3 was elucidated as 5-O-acetyl-epi-pestafolide A. Storage of 2 in EtOAc over one week did not yield 3 indicating that 3 is a true natural product and not an artifact arising from 2 during workup of the extract.
The remaining known compounds isolated from rice medium were identified as cochliodones A (4), E (5) and H (6),12,13 chateochromin A (7),14 ustilaginoidin D (8),15 5-hydroxyemodin (9),16 methyl 2-O-methyllecanorate (10),17 and colletoketol (11).18–20
In an OSMAC approach, the medium was changed from rice to peas. Investigation of the fungal culture on pea medium led to the isolation of colletodiol (12)20 and asteriquinone CT4 (14)8 as well as of a new compound (13).
Compound 13 was isolated as white amorphous powder. On the basis of the HRESIMS data, the molecular formula of 13 was established as C22H33NO5 with seven degrees of unsaturation. The 1H NMR spectrum (Table 3) of 13 exhibited six methyl groups at δH 1.88 (s, 1-OAc), 1.30 (s, Me-10), 1.03 (s, Me-14), 0.99 (s, Me-15), 0.97 (d, Me-20), and 0.95 (d, Me-19) as well as four oxygenated protons at δH 4.98 (t, H-1), 4.85 (dt, H-13a), 4.50 (ddd, H-13b), and 4.04 (d, H-17). The COSY correlations between H-1/H-2ab/H-3ab, and between H-5/H-6ab/H-7ab together with the HMBC correlations from Me-10 to C-1, C-5, C-8 and C-9, from Me-14 to C-3, C-4, C-5 and C-15, from H-7ab to C-8 and C-11, and from H-13ab to C-8, C-11 and C-12 established a bicyclic sesquiterpene skeleton (from C-1 to C-15) as shown (Fig. 2). In addition, the HMBC correlations from the protons of the methyl at δH 1.88 and H-1 to the carbonyl carbon at δC 173.4 indicated the location of an acetoxy group at C-1. The remaining signals suggested the presence of a valine moiety, which was confirmed by the COSY correlations between H-17/H-18/Me-19(20) and the HMBC correlations from H-17 and H-18 to C-16. The above substructures accounted for six degrees of unsaturation, suggesting the presence of an additional ring. In the HMBC spectrum, H-13ab showed correlations to C-16 while H-17 showed a correlation to C-12, indicating the linkage between the sesquiterpene moiety and the valine moiety through ester and amide bonds to form the third seven-membered ring. The relative configuration of the sesquiterpene part was assigned by coupling constants and NOE relationships. The large value of 2JH-2a/H-3a (13.8 Hz) and the small values of 2JH-1/H-2a and 2JH-1/H-2b (both 2.6 Hz) suggested trans diaxial orientation of H-2a (δH 2.07) and H-3a (δH 1.60) and equatorial orientation of H-1. Assuming that H-1 was on the β-face of the cyclohexane ring, H-2a and H-3b (δH 1.35) are suggested to be on the β-face of the ring whereas H-2b (δH 1.78) and H-3a are orientated on the α-face. The ROESY correlations from Me-10 to H-1, H-2a and Me-15 placed Me-10 on the β-face of the ring whereas the ROESY correlations from H-5 to H-3a and Me-14 placed H-5 on the α-face of the ring. After acid hydrolysis of 13 followed by Marfey's reaction, the valine moiety was determined to have L absolute configuration (17S).
| Position | δC, typeb | δH (J in Hz) |
|---|---|---|
| a Measured in CD3OD (1H at 600 MHz and 13C at 150 MHz).b Data were extracted from the HSQC and HMBC spectra. | ||
| 1 | 75.9, CH | 4.98, t (2.6) |
| 2 | 23.1, CH2 | 2.07, dddd (15.2, 13.8, 4.2, 2.6) |
| 1.78, dddd (15.2, 4.3, 2.6, 2.6) | ||
| 3 | 36.2, CH2 | 1.60, ddd (13.8, 13.8, 4.3) |
| 1.35, ddd (13.8, 4.2, 2.6) | ||
| 4 | 33.7, C | |
| 5 | 46.6, CH | 1.85, dd (12.6, 1.8) |
| 6 | 18.8, CH2 | 1.97, m |
| 1.66, m | ||
| 7 | 22.5, CH2 | 2.35, m |
| 2.11, m | ||
| 8 | 170.3, C | |
| 9 | 42.3, C | |
| 10 | 21.3, CH3 | 1.30, s |
| 11 | 126.3, C | |
| 12 | 172.4, C | |
| 13 | 69.9, CH2 | 4.85, dt (17.2, 2.8) |
| 4.50, ddd (17.2, 3.6, 1.3) | ||
| 14 | 33.6, CH3 | 1.03, s |
| 15 | 21.8, CH3 | 0.99, s |
| 16 | 171.7, C | |
| 17 | 61.1, CH | 4.04, d (7.1) |
| 18 | 31.0, CH | 2.02, m |
| 19 | 19.5, CH3 | 0.95, d (6.8) |
| 20 | 19.1, CH3 | 0.97, d (6.8) |
| 1-OAc | 173.4, C | |
| 22.0, CH3 | 1.88, s | |
 | ||
| Fig. 2 COSY and key HMBC correlations of 13. | ||
In order to elucidate the absolute configuration of the other chirality centers in 13, the solution TDDFT-ECD method was applied on the (1S,5S,9S,17S) and (1R,5R,9R,17S) stereoisomers.21,22 The Merck Molecular Force Field (MMFF) conformational search resulted in 16 and 11 conformers in a 21 kJ mol−1 energy window, respectively, which were reoptimized separately at the B3LYP/6-31+G(d,p), the ωB97X/TZVP23,24 PCM/MeCN, the CAM-B3LYP/TZVP25 PCM/MeCN and the SOGGA11-X/TZVP24,26 SMD/MeCN levels of theory. Interestingly, Boltzmann-averaged ECD spectra of both diastereomers computed for conformers over 1% Boltzmann-population at various levels of theory gave moderate to good mirror-image agreement with the experimental ECD spectrum of 13 indicating that the seven-membered ring with C-17 chirality center has the major contribution to the ECD spectrum22 and suggested (17R) absolute configuration, opposite to that of the Marfey's analysis. However, while almost all low-energy conformers of (1S,5S,9S,17S)-13 gave similarly mirror-image spectra to the experimental one, for (1R,5R,9R,17S)-13 even highly populated conformers (and the lowest-energy ωB97X/TZVP PCM/MeCN one) gave moderate agreement with the measured ECD and the ratio of the Boltzmann-weights of conformers with congruent and mirror-image spectra is comparable at all applied levels of theory. Although some of the best performing DFT functionals were utilized as suggested by Bremond et al. in a recent study,24 no combination of methods could be found, which would suggest the (1R,5R,9R,17S)-13 absolute configuration. With the result of Marfey's analysis in hand, this demonstrated that the small difference in the two ensembles of conformers, having opposite ECD spectra, could not be predicted properly, which is the reason of the failure. Specific optical rotation (SOR) calculations were also performed for the CAM-B3LYP/TZVP PCM/MeOH conformers but resulting in similarly large negative average values with both levo- and dextrorotating low-energy conformers for both stereoisomers, which did not allow a solid assignment of the absolute configuration. The limited amount of 13 did not allow a VCD measurement. Thus, based on the Marfey's reaction and the ECD results, the structure of 13 was tentatively elucidated as shown, for which a trivial name humigriseamide is proposed.
Asteriquinone CT4 (14) is a dimeric tryptophan derivative. In rice approximately 8% of the dry weight consists of proteins whereas the tryptophan content amounts to approximately 1.7% of the total amino acid content.27 Since peas are much richer in proteins compared to rice (20% vs. 8% of the dry weight), we assumed that a higher tryptophan content in peas might be responsible for the formation of 14 during the OSMAC experiment. To test this hypothesis, we added 2% tryptophan to the rice media in order to simulate an excess of tryptophan which resulted in the formation of compound 14 together with another new compound 15 and the known compounds anthranilic acid and N-formyl anthranilic acid.
Compound 15 has the molecular formula C21H25NO8 as deduced from the HRESIMS data, corresponding to ten degrees of unsaturation. The 1H and 13C NMR data of 15 (Table 4) were similar to those of the co-isolated bismacrolactone colletoketol (11)18–20 except for the disappearance of signals of the isolated double bond (C-12/C-13) and the presence of additional signals of one methine group (δC 49.8 and δH 4.57, CH-13), one methylene group (δC 41.8 and δH 3.16, 2.95, CH2-12) and of four aromatic protons at δH 7.81 (H-21), 7.38 (H-19), 6.69 (H-18) and 6.64 (H-20) in 15. An anthranilic acid moiety was established by the COSY correlations between H-18/H-19/H-20/H-21 and the HMBC correlations from H-19 to C-17 (δC 149.2) and from H-21 to C-17 and C-23 (δC 169.7) taking also into account the molecular formula of 15. In addition, the COSY correlations between Hab-12/H-13/13-NH along with the HMBC correlations from H-12ab to C-11 (δC 209.1) and C-14 (δC 172.3), from H-13 to C-11, C-14 and C-17, and from 13-NH to C-14 indicated the attachment of the anthranilic acid moiety at C-13 through an amide bond. Detailed analysis of the 2D NMR spectra of 15 revealed that the remaining substructure of 15 was identical to that of colletoketol (11). Thus, compound 15 was identified as 13-N-(2-carboxyphenyl)colletoketol. Based on the biogenetic relationship and similarity of coupling constants and NOE correlations between 11 and 15, the stereochemistry at C-2, C-8 and C-10 in 15 was suggested to be identical to that in 11, where the absolute configuration had been determined by X-ray analysis.19 In order to confirm this and determine the absolute configuration of the additional C-13 chirality center, the experimental ECD, VCD, 13C NMR chemical shifts and characteristic 3JH,H coupling constants were calculated for the stereoisomers (2R,8R,10R,13R)-15 and (2R,8R,10R,13S)-15.28
| Position | δC, type | δH (J in Hz) |
|---|---|---|
| a Measured in DMSO-d6 (1H at 600 MHz and 13C at 150 MHz). | ||
| 2 | 71.2, CH | 4.68, ddq (11.9, 2.7, 6.1) |
| 3 | 37.1, CH2 | 2.49, ddd (11.9, 8.0, 2.7) |
| 2.33, td (11.9, 8.0) | ||
| 4 | 143.6, CH | 6.67, dt (15.8, 8.0) |
| 5 | 125.1, CH | 5.77, d (15.8) |
| 6 | 164.5, C | |
| 8 | 65.2, CH | 5.08, ddq (11.3, 1.9, 6.3) |
| 9 | 38.4, CH2 | 2.17, dd (14.2, 11.2) |
| 1.96, ddd (14.2, 7.5, 1.9) | ||
| 10 | 73.0, CH | 4.03, d (7.5) |
| 11 | 209.1, C | |
| 12 | 41.8, CH2 | 3.16, dd (18.8, 6.0) |
| 2.95, dd (18.8, 2.0) | ||
| 13 | 49.8, CH | 4.57, ddd (7.1, 6.0, 2.0) |
| 14 | 172.3, C | |
| 15 | 20.0, CH3 | 1.17, d (6.3) |
| 16 | 20.5, CH3 | 1.29, d (6.1) |
| 17 | 149.2, C | |
| 18 | 111.4, CH | 6.69, br d (7.9) |
| 19 | 134.5, CH | 7.38, td (7.9, 1.6) |
| 20 | 115.5, CH | 6.64, br t (7.9) |
| 21 | 131.9, CH | 7.81, dd (7.9, 1.6) |
| 22 | 111.2, C | |
| 23 | 169.7, C | |
| 13-NH | 8.13, d (7.1) | |
The MMFF conformational search of (2R,8R,10R,13R)-15 and (2R,8R,10R,13S)-15 resulted in 197 and 246 conformers, respectively, which were reoptimized at the B3LYP/6-31+G(d,p), the B3LYP/TZVP PCM/CHCl3, the ωB97X/TZVP PCM/MeCN and the mPW1PW91/6-311+(2d,p) SMD/CHCl3 levels of theory, separately. 13C NMR shift calculations performed both at the mPW1PW91/6-311+(2d,p) in vacuo and the mPW1PW91/6-311+(2d,p) SMD/CHCl3 levels for the B3LYP/6-31+G(d,p) conformers gave similar results for the two stereoisomers indicating no preference of any of the two diastereomers.29,30 The ωB97X/TZVP PCM/MeCN conformers (>1%) of (2R,8R,10R,13R)-15 and (2R,8R,10R,13S)-15 showed different conformations for the macrolactone ring, which was also manifested in the dihedral angles ωH-13,C-13,C-12,H-12ab and ωH-13,C-13,NH. Thus the coupling constants 3JH-13,NH, 3JH-12a,H-13 and 3JH-12b,H-13 were selected for calculations, for which 7.1, 6.0 and 2.0 Hz values were measured, respectively. The computed coupling constants of (2R,8R,10R,13R)-15 gave consistently better agreement with the experimental values than those of (2R,8R,10R,13S)-15, which had systematically larger values (Table 5). The largest and decisive difference could be observed for 6.0 Hz 3JH-12a,H-13 experimental value, which was calculated 5.5 Hz for (2R,8R,10R,13R)-15 and 11.2 Hz for (2R,8R,10R,13S)-15. H-13 and H-12a had gauche orientation in (2R,8R,10R,13R)-15 with small ωH-13,C-13,C-12,H-12a dihedral angles and coupling constants, while H-13 and H-12a had near trans-periplanar arrangement in the computed conformers of (2R,8R,10R,13S)-15, which gave rise to large coupling constant (11.2 Hz). The computed 3JH,H coupling constants clearly suggested that 15 has (2R*,8R*,10R*,13R*) relative configuration.
| 3JH-13,NH | 3JH-12a,H-13 | 3JH-12b,H-13 | |
|---|---|---|---|
| Experimental | 7.1 | 6.0 | 2.0 |
| (2R,8R,10R,13R)-15 | 8.6 | 5.5 | 2.9 |
| (2R,8R,10R,13S)-15 | 9.2 | 11.2 | 4.1 |
13C NMR shifts were also computed for the mPW1PW91/6-311+(2d,p) SMD/CHCl3 conformers at the same level and although this level gave worse agreement for both stereoisomers because of the rather limited number of compounds utilized for the NMR parameter generation,31 it gave considerably better agreement for (2R,8R,10R,13R)-15 than for (2R,8R,10R,13S)-15 in line with the result of the coupling constant calculations. ECD calculations performed at various levels for the B3LYP/6-31+G(d,p) and the ωB97X/TZVP PCM/MeCN conformers gave similarly good agreement for both stereoisomers verifying the (2R,8R,10R) absolute configuration of the three stereocenters but giving no information about the C-10 chirality center (Fig. 3 and 4). However, with the combination of the (2R*,8R*,10R*,13R*) relative configuration from the coupling constants and (2R,8R,10R) absolute configuration from the ECD calculations allowed determining the (2R,8R,10R,13R) absolute configuration for 15. VCD measurement and calculations were also carried out to check whether it can differentiate between the (2R,8R,10R,13R) and (2R,8R,10R,13S) stereoisomers independently from other methods (Fig. 5). VCD calculations performed on the B3LYP/TZVP PCM/CHCl3 conformers at the same level gave acceptable agreement for both stereoisomers with a slight preference for the (2R,8R,10R,13R) stereoisomer. Similarity indexes (ESI†)32 computed with the CDSpecTech package33,34 gave considerably higher number (0.335 and a confidence level of 85.4%) for this stereoisomer than for (2R,8R,10R,13S)-15 (0.128). Although this ESI† number is below the limit that Covington and Polavarapu suggested for a safe elucidation of configuration with VCD,34 similarly to the ECD calculations it allowed determining the absolute configuration of 15 as (2R,8R,10R,13R) when combined with the result of the coupling constant calculations.
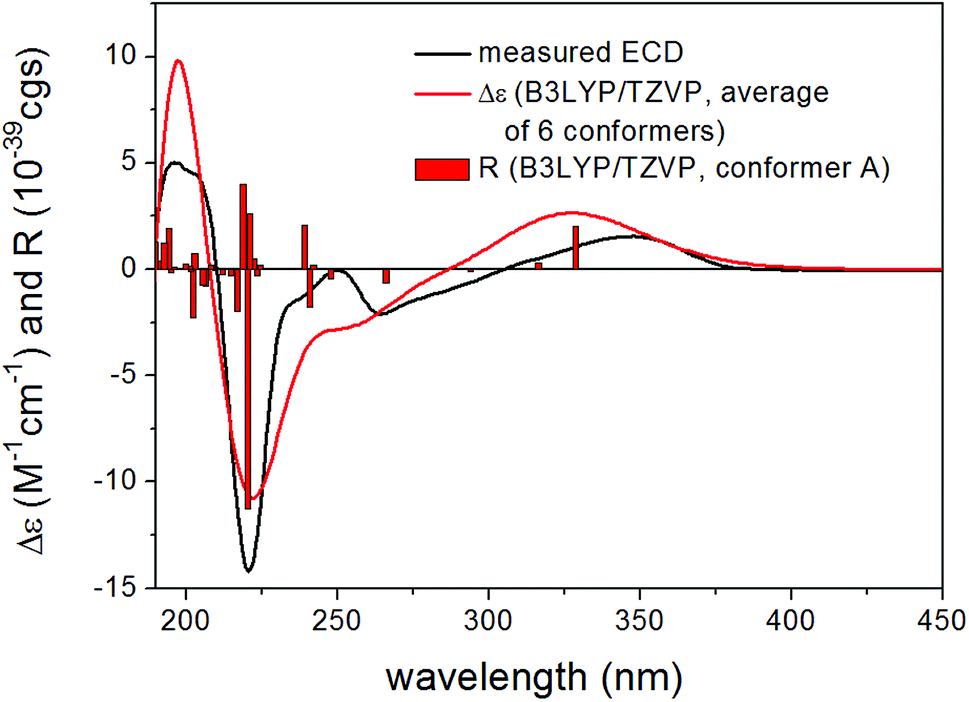 | ||
| Fig. 3 Experimental ECD spectrum of 15 in MeCN compared with the Boltzmann-weighted B3LYP/TZVP PCM/MeCN ECD spectrum of (2R,8R,10R,13R)-15. Level of optimization: ωB97X/TZVP PCM/MeCN. Bars represent the rotatory strength values of the lowest-energy conformer. | ||
 | ||
| Fig. 4 Experimental ECD spectrum of 15 in MeCN compared with the Boltzmann-weighted CAM-B3LYP/TZVP PCM/MeCN ECD spectrum of (2R,8R,10R,13S)-15. Level of optimization: ωB97X/TZVP PCM/MeCN. Bars represent the rotatory strength values of the lowest-energy conformer. | ||
 | ||
| Fig. 5 Experimental VCD spectrum (black line) of 15 in CDCl3 compared with the Boltzmann-weighted B3LYP/TZVP PCM/CHCl3 VCD spectra of (2R,8R,10R,13R)-15 (red line) and (2R,8R,10R,13S)-15 (blue line). | ||
All compounds except for compounds 9 and 14 due to their low amounts were submitted to a cytotoxicity assay using the L5178Y mouse lymphoma cell line and to antimicrobial assay against Mycobacterium tuberculosis, Staphylococcus aureus, Enterococcus faecalis, Acinetobacter baumannii, Pseudomonas aeruginosa and Escherichia coli. Chateochromin A (7), colletoketol (11) and the new compound 15 exhibited strong cytotoxicity with IC50 values of 0.3, 0.5 and 0.2 μM, respectively. Only chateochromin A (7) showed antibacterial activity against E. faecalis with an MIC value of 6.3 μM. The remaining compounds were inactive when tested at a dose of 10 μM.
In conclusion, an axenic fermentation of Trichocladium sp. on rice medium yielded ten natural products including two new compounds (1 and 3). Co-cultivation of the fungus with B. subtilis led to the production of a further new compound (2) and a 10-fold increase of 11. The latter compound is known as a weak broad-spectrum antibiotic,20 implying a possible defense reaction of the fungus against B. subtilis. A switch from solid rice to peas as medium for fungal cultivation resulted in the production of an additional new compound 13, representing a sesquiterpene connected with an amino acid. In addition, a dimeric tryptophan derivative 14 was isolated. These results indicate that a protein rich medium such as peas induces the accumulation of different compounds compared to a carbohydrate rich medium like rice. Addition of tryptophan to the rice medium which was done trying to mimic the higher protein content of peas vs. rice caused the accumulation of the new compound 15 which in addition to colletoketol (11) includes anthranilic acid, the latter probably being a fungal biotransformation product of added tryptophan.35
Experimental section
General experimental procedures
Optical rotations were measured on a PerkinElmer-241 MC polarimeter. ECD spectra were recorded on a J-810 spectropolarimeter. VCD measurements were performed on a BioTools ChiralIR-2X spectrometer at a resolution of 4 cm−1 under ambient temperature for 18 × 3000 scans, respectively. Sample was dissolved in CDCl3 at a concentration of 0.095 M (15), and the solution was placed in a 100 μm BaF2 cell. NMR spectra were recorded on Bruker ARX 300 or AVANCE DMX 600 NMR spectrometers. Mass spectra were obtained from a Finnigan LCQ Deca XP mass spectrometer while high resolution mass spectra were recorded on a FTHRMS-Orbitrap (Thermo-Finnigan) mass spectrometer. A Dionex P580 system was used in combination with a diode array detector (UVD340S) and an Eurosphere 10 C18 column (125 × 4 mm) for HPLC analysis and recording UV spectra. Semi-preparative HPLC was conducted on a Lachrom-Merck Hitachi system (pump L7100, UV detector L7400, Eurospher 100 C18 column, 300 × 8 mm, Knauer Germany). Sephadex-LH20 and Merck MN silica gel 60M (0.04–0.063 mm) were used as stationary phases for column chromatography. TLC plates precoated with silica gel 60 F254 were used for monitoring separation. For spectroscopic measurements spectral grade solvents were used.Fungal material
Houttuynia cordata was grown in a local garden at Willich, Germany. The identification of the plant used in this study was performed by Dr Marc Appelhans (Department of Systematics, Biodiversity and Evolution of Plants with Herbarium, Göttingen University). A voucher specimen of the plant (code GOET038305) has been deposited in Göttingen University Herbarium. Fresh roots were harvested in April 2016 and washed with sterilized water, surface sterilized with 70% ethanol for 1 min, and cut into small pieces (around 1 × 1 × 1 cm) using a flame sterilized blade. These pieces were put on malt agar plates (15 g L−1 malt extract, 15 g L−1 agar, and 0.2 g L−1 chloramphenicol in distilled water, pH 7.4–7.8 with sodium hydroxide or hydrochloric acid), and then incubated at room temperature for several days. A fungus that was growing out from the root segments was identified as Trichocladium sp. using a molecular protocol as described previously.36 Sequence data were submitted to GenBank with the accession number MK241585. A voucher strain (code HCRSW) is kept in the Institute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine University, Düsseldorf, Germany. After surface sterilization we also imprinted the surface sterilized roots on agar plates. Following incubation of these agar plates no fungal growth was detected which proofs that the fungus is an endophyte and not an exophyte.Fermentation and co-cultivation
Fermentation of the fungus was conducted in 11 Erlenmeyer flasks on solid rice medium (100 g rice in 110 mL water followed by autoclaving) at 20 °C at static conditions. After 33 days each flask was extracted with 600 mL EtOAc. The rice medium was cut into small pieces and shaken for 8 hours followed by evaporation of EtOAc.Co-cultivation experiment with Bacillus subtilis (ATCC168) was conducted on solid rice medium. A total of 15 flasks was prepared (5 axenic fungal cultures as controls, 5 co-cultures with bacteria and 5 axenic bacterial cultures as controls). After addition of 10 mL bacteria solution to each flask, the flasks were incubated at 30 °C for three days. Then a constant number of pieces from agar plates containing Trichocladium sp. were transferred to the co-culture flasks. All flasks were inoculated at 20 °C under static conditions. After 33 days 600 mL EtOAc was added to each flask in order to terminate the cultivation.
OSMAC experiments
In addition to rice, peas (Pisum sativum, from Müllers Mühle, Germany) were used as medium (100 g of peas in 110 mL of H2O followed by autoclaving). The cultivation procedure was the same as described for cultivation on rice medium. For the feeding experiment using tryptophan, the rice medium was spiked with 2% tryptophan prior to autoclaving.Extraction and isolation
The crude extract (12 g) obtained from axenic fungal rice cultures was subjected to silica gel vacuum liquid chromatography (VLC) using a step gradient of n-hexane/EtOAc and CH2Cl2/MeOH to give 11 fractions (V1–V11). Fraction V4 (4.6 g) was further fractionated by reversed phase vacuum liquid chromatography using a step gradient of H2O and MeOH to yield 10 subfractions (V4RP1–V4PR10). Subfraction V4RP5 was subjected to a Sephadex LH-20 column using MeOH as eluent followed by purification by semi-preparative HPLC using 55% MeOH–H2O to give 1 (6.4 mg), 9 (1.2 mg) and 11 (4.5 mg). Following a similar procedure as described for V4RP5, 3 (2.4 mg) and 5 (5.2 mg) were isolated from subfraction V4RP6. Compounds 4 (5.4 mg) and 10 (6.4 mg) were isolated from subfraction V4RP7, while 8 (6.8 mg) was obtained from subfraction V4RP9. Compounds 6 (2.8 mg) and 7 (3.2 mg) were obtained from fractions V6 and V9, respectively, after separation by Sephadex LH-20 column chromatography with MeOH as eluent.The chromatographic work up of the OSMAC extracts followed the same procedure as described for the axenic rice cultures. Silica gel vacuum liquid chromatography using a step gradient of n-hexane/EtOAc and CH2Cl2/MeOH was used for separation of the crude extracts obtained from co-cultivation, pea medium and rice medium with addition of tryptophan. A total of ten fractions (CV1–CV10) was obtained from the co-culture extract. Fraction CV4 (2.4 g) was separated by a Sephadex LH-20 column with MeOH as mobile phase followed by purification using semi-preparative HPLC with 45% MeOH–H2O as eluting system to afford 2 (4.3 mg). The pea culture yielded nine subfractions (PV1–PV9) after VLC. Compounds 12 (6.4 mg) and 13 (5.4 mg) were isolated from fraction PV6, while 14 (0.5 mg) was obtained from fraction PV9 by semi-preprative HPLC using a gradient of MeOH–H2O (from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 to 90:10).
:90 to 90:10).
Fraction TV6 (854 mg) was obtained from rice medium after addition of tryptophan. The fraction was submitted to silica gel VLC and purified using reversed phase VLC and a step gradient of H2O and MeOH to give 15 (22.5 mg).
Marfey's analysis
Marfey's analysis was conducted following a previously described protocol.37 Acid hydrolysis of the isolated compounds 1 and 13 was conducted using 0.2 mg of each compound. After addition of 2 mL of 6 N HCl and incubation for 24 h at 110 °C, the acid was removed by drying under a nitrogen stream. 50 μL of each sample was treated with 100 μL of FDNPL (1% N-(5-fluoro-2,4-dinitrophenyl)-L-leucin amide) in acetone together with 20 μL 1 M NaHCO3. Then the mixtures were incubated at 40 °C for 1 h under constant mixing. Next 10 μL of 2 N HCL was added to each vial followed by evaporation to dryness. For injection to HPLC, 1 mL MeOH was added to each vial. L- and D-amino acids which were used as standards were treated in the same manner. Analysis of the derivatized amino acids was performed by comparing their retention times with those of standards.Computational methods
Mixed torsional/low-mode conformational searches were carried out by means of the Macromodel 10.8.011 software using the MMFF with an implicit solvent model for CHCl3 applying a 21 kJ mol−1 energy window.38 Geometry reoptimizations of the resultant conformers [B3LYP/6-31+G(d,p) level in vacuo, B3LYP/TZVP PCM/CHCl3, ωB97X/TZVP and CAM-B3LYP/TZVP with PCM solvent model for MeCN, SOGGA11-X/TZVP SMD/MeCN and mPW1PW91/6-311+G(2d,p) SMD/CHCl3] and DFT VCD, TDDFT ECD and SOR calculations were performed with Gaussian 09 for ECD and SOR using various functionals (B3LYP, BH&HLYP, CAM-B3LYP, PBE0) and the TZVP basis set with the same or no solvent model as in the preceding DFT optimization step.39 ECD spectra were generated as the sum of Gaussians with 3000 cm−1 half-height widths, using dipole-velocity-computed rotational strengths.40 VCD spectra were calculated with 8 cm−1 half-height width and scaled by a factor of 0.98.41 Boltzmann distributions were estimated from the B3LYP, ωB97X, CAM-B3LYP, SOGGA11-X and mPW1PW91 energies. ESI† values were computed with the CDSpecTech 22.0 software package.32–34 The MOLEKEL program was used for visualization of the results.42Cytotoxicity assay
Cytotoxicity against the L51178Y mouse lymphoma cell line was measured using the MTT assay.43 Kahalalide F and 0.1% ethylene glycol monomethyl ether in DMSO were used as positive and negative control, respectively.Antimicrobial assay
Antimicrobial activity of the compounds was conducted using the microdilution method. Compounds were dissolved in DMSO and added to the broth. The resulting final DMSO concentration in the assay was 0.1%. The direct colony suspension method was employed for preparation of the inoculum, and MIC values for each strain were determined according to the recommendations of the Clinical and Laboratory Standards.44 Two known antibiotics, ciprofloxacin and moxifloxacin (analytical standard, Sigma-Aldrich) were used as positive controls.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Deutsche Forschungsgemeinschaft (project number 270650915, GRK 2158). Further support by the Manchot Foundation to P. P. is gratefully acknowledged. The Hungarian authors were supported by the EU and co-financed by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. T. K. and A. M. thank the National Research, Development and Innovation Office (NKFI K120181 and PD121020). The Governmental Information-Technology Development Agency (KIFÜ) is acknowledged for CPU time. We furthermore wish to thank Dr Marc Appelhans (Department of Systematics, Biodiversity and Evolution of Plants with Herbarium, Göttingen University, Germany) for identification of the plant material.References
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- A. Stierle, G. Strobel and D. Stierle, Science, 1993, 260, 214–216 CrossRef CAS.
- A. H. Aly, A. Debbab and P. Proksch, Pharmazie, 2013, 68, 499–505 CAS.
- S. Liu, H. Dai, G. Makhloufi, C. Heering, C. Janiak, R. Hartmann, A. Mándi, T. Kurtán, W. E. Müller, M. U. Kassack, W. Lin, Z. Liu and P. Proksch, J. Nat. Prod., 2016, 79, 2332–2340 CrossRef CAS.
- H. Wang, H. F. Dai, C. Heering, C. Janiak, W. H. Lin, R. S. Orfali, W. E. G. Müller, Z. Liu and P. Proksch, RSC Adv., 2016, 6, 81685–81693 RSC.
- H. Wang, P. M. Eze, S. P. Höfert, C. Janiak, R. Hartmann, F. B. C. Okoye, C. O. Esimone, R. S. Orfali, H. F. Dai, Z. Liu and P. Proksch, RSC Adv., 2018, 8, 7863–7872 RSC.
- X. W. Wang, F. Y. Yang, M. Meijer, B. Kraak, B. D. Sun, Y. L. Jiang, Y. M. Wu, F. Y. Bai, K. A. Seifert, P. W. Crous, R. A. Samson and J. Houbraken, Stud. Mycol., 2018, 93, 65–153 CrossRef PubMed.
- U. Mocek, L. Schultz, T. Buchan, C. Baek, L. Fretto, J. Nzerem, L. Sehl and U. Sinha, J. Antibiot., 1996, 49, 854–859 CrossRef CAS.
- D. Laurent, G. Guella, I. Mancini, M. F. Roquebert, F. Farinole and F. Pietra, Tetrahedron, 2002, 58, 9163–9167 CrossRef CAS.
- J. Inokoshi, Y. Takagi, R. Uchida, R. Masuma, S. Omura and H. Tomoda, J. Antibiot., 2010, 63, 9–16 CrossRef CAS PubMed.
- G. Ding, S. Liu, L. Guo, Y. Zhou and Y. Che, J. Nat. Prod., 2008, 71, 615–618 CrossRef CAS PubMed.
- N. Phonkerd, S. Kanokmedhakul, K. Kanokmedhakul, K. Soytong, S. Prabpai and P. Kongsearee, Tetrahedron, 2008, 64, 9636–9645 CrossRef CAS.
- F. X. Yu, Y. Chen, Y. H. Yang and P. J. Zhao, Phytochem. Lett., 2016, 16, 263–267 CrossRef CAS.
- K. Koyama, S. Natori and Y. Iitaka, Chem. Pharm. Bull., 1987, 35, 4049–4055 CrossRef CAS.
- K. Koyama and S. Natori, Chem. Pharm. Bull., 1988, 36, 146–152 CrossRef CAS.
- N. Morooka, S. Nakano, N. Itoi and Y. Ueno, Agric. Biol. Chem., 1990, 54, 1247–1252 CAS.
- J. Elix and L. Lajide, Aust. J. Chem., 1981, 34, 2005–2011 CrossRef CAS.
- J. MacMillan and T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1973, 1487–1493 RSC.
- U. Höller, A. D. Wright, G. M. König and P. G. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1310–1313 CrossRef.
- T. J. Hunter and G. A. O'Doherty, Org. Lett., 2002, 4, 4447–4450 CrossRef CAS PubMed.
- S. Superchi, P. Scafato, M. Gorecki and G. Pescitelli, Curr. Med. Chem., 2018, 25, 287–320 CrossRef CAS PubMed.
- A. Mándi, I. W. Mudianta, T. Kurtán and M. J. Garson, J. Nat. Prod., 2015, 78, 2051–2056 CrossRef PubMed.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- É. Brémond, M. Savarese, N. Q. Su, Á. J. Pérez-Jiménez, X. Xu, J. C. Sancho-García and C. Adamo, J. Chem. Theory Comput., 2016, 12, 459–465 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. Peverati and D. G. Truhlar, J. Chem. Phys., 2011, 135, 191102 CrossRef PubMed.
- V. W. Padhye and D. K. Salunkhe, Cereal Chem., 1979, 56, 389–393 CAS.
- A. Mándi and T. Kurtán, Nat. Prod. Rep., 2019, 36, 889–918 RSC.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- CHESHIRE CCAT, The Chemical Shift Repository for computed NMR scaling factors, http://cheshirenmr.info/index.htm Search PubMed.
- S. Qiu, E. de Gussem, K. A. Tehrani, S. Sergeyev, P. Bultinck and W. Herrebout, J. Med. Chem., 2013, 56, 8903–8914 CrossRef CAS.
- E. Debie, E. de Gussem, R. K. Dukor, W. Herrebout, L. A. Nafie and P. Bultinck, ChemPhysChem, 2011, 12, 1542–1549 CrossRef CAS PubMed.
- C. L. Covington and P. L. Polavarapu, CDSpecTech: Computer programs for calculating similarity measures of comparison between experimental and calculated dissymmetry factors and circular intensity differentials, version 22.0, 2017, https://sites.google.com/site/cdspectech1/ Search PubMed.
- C. L. Covington and P. L. Polavarapu, Chirality, 2017, 29, 178–192 CrossRef CAS.
- C. K. Wat and G. H. N. Towers, in Biochemistry of plant phenolics. Recent advances in phytochemistry, ed. T. Swain, J. B. Harbone and C. F. Van Sumere, Springer, Boston, MA, 1979, vol 12, Metabolism of the aromatic amino acids by fungi, pp. 371–432 Search PubMed.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591 CrossRef CAS.
- MacroModel, Schrödinger, LLC, 2015, https://www.schrodinger.com/MacroModel Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- A. Mándi, M. M. Swamy, T. Taniguchi, M. Anetai and K. Monde, Chirality, 2016, 28, 453–459 CrossRef PubMed.
- U. Varetto, MOLEKEL 5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
- M. Ashour, R. Edrada, R. Ebel, V. Wray, W. Watjen, K. Padmakumar, W. E. Muller, W. H. Lin and P. Proksch, J. Nat. Prod., 2006, 69, 1547–1553 CrossRef CAS PubMed.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard Ninth ed. CLSI document M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS, 1D and 2D NMR spectra of compounds 1–3, 13 and 15 as well as the results of Marfey's reaction and computational calculations. See DOI: 10.1039/c9ra05469c |
| This journal is © The Royal Society of Chemistry 2019 |