
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt, nickel and copper complexes with glycinamide: structural insights and magnetic properties†
Darko Vušak a,
Neven Smrečkia,
Biserka Prugovečki*a,
Ivica Đilovića,
Inka Kirasića,
Dijana Žilićb,
Senada Muratovićb and
Dubravka Matković-Čalogovića
a,
Neven Smrečkia,
Biserka Prugovečki*a,
Ivica Đilovića,
Inka Kirasića,
Dijana Žilićb,
Senada Muratovićb and
Dubravka Matković-Čalogovića
aDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia. E-mail: biserka@chem.pmf.hr
bLaboratory for Magnetic Resonances, Division of Physical Chemistry, Ruđer Bošković Institute, Bijenička 54, HR-10000 Zagreb, Croatia
First published on 12th July 2019
Abstract
Ten new compounds of Co, Ni and Cu with glycinamide (HL = glycinamide): [Co(H2O)2(HL)2]Cl2 (1a), [Co(H2O)2(HL)2]Br1.06Cl0.94 (1b), [Co(H2O)2(HL)2]I2 (1c), [Ni(H2O)2(HL)2]Cl2 (2a), [Ni(H2O)2(HL)2]Br0.94Cl1.06 (2b), [Ni(H2O)2(HL)2]I2 (low and room temperature polymorph, 2cLT and 2cRT), [CuCl2(HL)2] (3a), [CuBr1.3Cl0.7(HL)2] (3b) and {[Cu(HL)2]2[Cu2I6]}n (3c), as well as glycinamide hydroiodide (H2LI) and a new polymorph of glycinamide hydrochloride (β-H2LCl) were prepared and characterized by single-crystal X-ray diffraction, infrared spectroscopy, thermal analysis (TG/DTA) and ESR spectroscopy. 1a, 1b, 2a and 2b are isostructural, as well as 1c and 2cRT, while the Cu compounds (3a–c) have entirely different molecular structures. All investigated compounds are mononuclear with exception of the 1D coordination polymer 3c. Compound 3c contains copper ions in the mixed oxidation state Cu(I) and Cu(II) with interesting magnetic properties. Paramagnetic behaviour was found in 1a, 1b, 3a and 3b. Temperature induced polymorphic transformation was observed in 2c. Compounds 1a and 3a showed moderate antiproliferative activity and selectivity toward the human breast tumor cell line MCF-7.
Introduction
The orchestrated transport, exchange and incorporation of various metal ions in different metalloproteins is vital for their function. Metal ions are constituents of many proteins and have either catalytic or structural functions. They are usually coordinated by the side chain functionalities of peptides (histidyl, carboxylate, hydroxyl or amide groups), solvent molecules and ions.1,2 The search for small molecules with the desired structural and functional plasticity to perform, or even enhance, bioinspired processes – from mimicking and electron transfer to recognition and catalysis – has become an integral part of everyday research.3–9 Derivatization of ubiquitous amino acids seems to be an obvious synthetic choice to provide coordination environments complementary to those found in metalloproteins.10 Accurately determined structures of metal complexes with ligands analogous to those of amino acids side chains are useful in protein crystallography for interpretation and validation of protein structural data.11–15Amino acids/amino acid derivatives and their metal complexes possess various biological activities such as antiretroviral,16 antibacterial and antifungal,17–20 and antiproliferative effects on tumor cells,21 with potential applications in biomedicine. Copper coordination compounds, especially those with mixed oxidation states, are also of special interest because of their magnetic properties.22–24 Cobalt and nickel polynuclear compounds showed interesting ferro- and antiferromagnetic properties, specifically compounds containing the carboxylic group, such as amino acids and their derivatives.24–26 There are fewer published papers on magnetic measurements and structural studies of such cobalt and nickel compounds than for copper compounds.
Glycinamide (HL) is the simplest amino acid amide, being cheap, readily available and easily synthesized. In bio-systems its derivative glycinamide ribonucleotide is known as an intermediate in de novo biosynthesis of purine.27 Moreover, glycil-prolyl-glycinamide and its metabolites (glycine, glycinamide, proline, glycil-proline and prolyl-glicinamide) were tested in vitro as potential HIV-1 replication inhibitors. It was shown that only glycil-prolyl-glycinamide and glycinamide showed a pronounced inhibitory effect.16
Glycinamide is capable of building various hydrogen bonding architectures, having four N–H hydrogen atoms in the neutral form as potential HB-donors and an amide oxygen as the acceptor. In the Cambridge Structural Database (CSD)28 there are only six structures containing the glycinamide fragment: glycinamide hydrochloride,29 two rhodium complexes,30,31 a bimetallic (Mn, Cr) ferrimagnet,32 a ruthenium complex33 and an iridium complex.34 This is surprising because coordination of metal ions by amide groups of simple amides, peptides and proteins is of great interest due to their importance in biological systems.35 Different modes of coordination to the metal ion were found. In the reported rhodium(III) complexes glycinamide acts as a monodentate ligand coordinating rhodium through the amine nitrogen atom. In the manganese complex glycinamide acts as a bidentate N,O-coordinating ligand through the amine nitrogen and amide oxygen atoms, while in the ruthenium and iridium complexes the glycinamidato group acts as a bidentate N,N′-coordinating ligand through nitrogen atoms from amide and amino groups (Scheme 1).
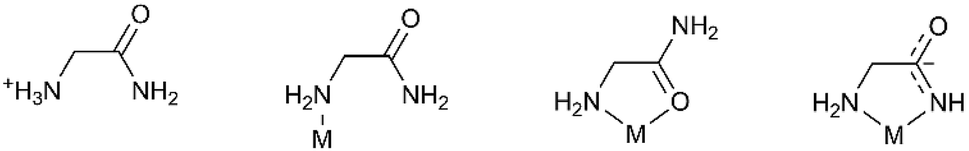 | ||
| Scheme 1 Structural diagrams of H2L+ and HL/L− modes of binding in complexes in the solid state. | ||
As a part of our ongoing research on preparation and structural investigation of metal complexes with amino acids and their derivatives,21,36–39 we have prepared various copper(II) and nickel(II) complexes with N-alkyliminodiacetamide.40 In order to expand our knowledge on the properties of amino acetamide complexes in the solid state, we report synthesis and solid-state characterization (X-ray structural analysis, IR and ESR spectroscopy, TG/DTA analysis) of cobalt, nickel and copper complexes with glycinamide. Structural characterization of glycinamide hydroiodide (H2LI) and a new polymorph of glycinamide hydrochloride (β-H2LCl) is also given. Reactions of H2LCl with cobalt(II), nickel(II) and copper(II) halides, acetate or hydroxides yielded ten novel compounds: nine mononuclear [Co(H2O)2(HL)2]Cl2 (1a), [Co(H2O)2(HL)2]Br1.06Cl0.94 (1b), [Co(H2O)2(HL)2]I2 (1c), [Ni(H2O)2(HL)2]Cl2 (2a), [Ni(H2O)2(HL)2]Br0.94Cl1.06 (2b), [Ni(H2O)2(HL)2]I2 (low and room temperature polymorphs, 2cLT and 2cRT), [Ni(H2O)2(HL)2]I2 (2c); [CuCl2(HL)2] (3a); [CuBr1.3Cl0.7(HL)2] (3b) and a 1D coordination polymer {[Cu(HL)2]2[Cu2I6]}n (3c).
Results and discussion
Synthesis and properties of the complex compounds
Reactions of H2LCl with metal halides and sodium bicarbonate were performed in aqueous solutions, and the reactions of H2LCl with metal hydroxides mechanochemically by neat grinding (NG), Scheme 2. Synthesis of 1c was performed in an aqueous solution by using cobalt(II) acetate and surplus of potassium iodide. When metal bromides were used as reactants mixed halide compounds 1b, 2b and 3b were obtained (bromide ions originated from the metal bromide, while the chloride ions originated from glycinamide hydrochloride). Cobalt(II) and nickel(II) gave water-soluble compounds 1a–c, 2a,b and 2cRT of the general formula [M(H2O)2(HL)2]X2 (M = Co, Ni; X = Cl, Br/Cl, I). On the other hand, copper(II) gave different and less soluble compounds [CuX2(HL)2] (X = Cl, Br/Cl) (3a and 3b). A partial reduction of copper(II) to copper(I) occurred when KI was introduced into the solution of CuCl2, H2LCl and NaHCO3 leading to the formation of a 1D coordination polymer {[Cu(HL)2]2[Cu2I6]}n (3c). Compound 3a can also be prepared by NG mechanochemical synthesis, using Cu(OH)2 and H2LCl, offering a very fast and clean route to the desired product (Scheme 2).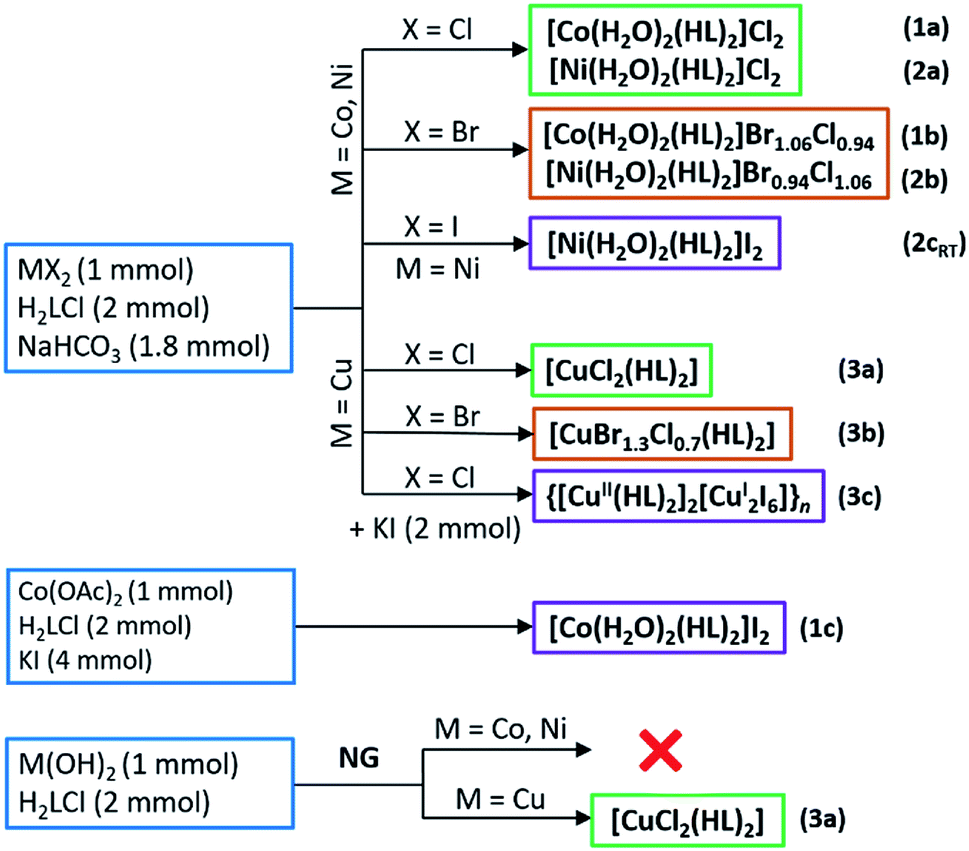 | ||
| Scheme 2 Preparation of the glycinamide complexes with Co, Ni and Cu. | ||
All compounds are air-stable. Thermal stability of the cobalt(II) and nickel(II) compounds (1a–c, 2a,b and 2cRT) is characterized by the initial loss of coordinated water molecules in the range 100–120 °C, followed by further decomposition starting between 220 and 265 °C. Copper(II) compounds (3a–c) are less stable than cobalt(II) and nickel(II) compounds and decompose at significantly lower temperatures in the range 160–195 °C. Full thermal analyses data are given in Table S1 (ESI†).
Infrared spectra of the compounds are characterized by the presence of very strong and sharp bands of the carbonyl group stretching, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)41,42 occurring in the range of 1674–1644 cm−1. Comparing the spectra of cobalt(II), nickel(II) and copper(II) complexes with chlorides and bromides, the ν(CO) bands occur at the highest wavenumbers in the spectra of copper(II) complexes. Carbonyl stretching in protonated glycinamide, β-H2LCl, was observed at higher wavenumber then in any of the complexes, at 1688 cm−1, showing weakening of the CO bond upon coordination to the metal ion. The amide II band,41 which appears at 1594 cm−1 in the spectrum of β-H2LCl, was found in the similar region in the spectra of all compounds (1570–1600 cm−1) and at 1555 cm−1 for compound 3c. The bands of antisymmetric and symmetric stretching of the amide amino groups are observed in the range 3300–3100 cm−1, indicating that these are involved in hydrogen bonding, as evidenced by the crystal structures of all nine complexes. A sharp band of medium intensity, which was assigned as O–H stretching, ν(OH, H2O), was observed at roughly 3430 cm−1 in the spectra of compounds 1a–c, 2a–c. The band is, of course, absent in the spectra of compounds 3a–c. IR spectra of representative compounds are given in Fig. S1 (ESI†).
O)41,42 occurring in the range of 1674–1644 cm−1. Comparing the spectra of cobalt(II), nickel(II) and copper(II) complexes with chlorides and bromides, the ν(CO) bands occur at the highest wavenumbers in the spectra of copper(II) complexes. Carbonyl stretching in protonated glycinamide, β-H2LCl, was observed at higher wavenumber then in any of the complexes, at 1688 cm−1, showing weakening of the CO bond upon coordination to the metal ion. The amide II band,41 which appears at 1594 cm−1 in the spectrum of β-H2LCl, was found in the similar region in the spectra of all compounds (1570–1600 cm−1) and at 1555 cm−1 for compound 3c. The bands of antisymmetric and symmetric stretching of the amide amino groups are observed in the range 3300–3100 cm−1, indicating that these are involved in hydrogen bonding, as evidenced by the crystal structures of all nine complexes. A sharp band of medium intensity, which was assigned as O–H stretching, ν(OH, H2O), was observed at roughly 3430 cm−1 in the spectra of compounds 1a–c, 2a–c. The band is, of course, absent in the spectra of compounds 3a–c. IR spectra of representative compounds are given in Fig. S1 (ESI†).
Molecular and crystal structures of β-H2LCl and H2LI
Both H2LCl polymorphs crystallize in monoclinic space groups, α in P21/c and β in P21/m, while H2LI crystallizes in the orthorhombic crystal system, space group Pca21 (Table S2, ESI†). ORTEP drawings of β-H2LCl and H2LI are given in Fig. S2 (ESI†). Selected bond distances and angles in the crystal structures of α-H2LCl,29 β-H2LCl and H2LI are presented in Table S3 (ESI†). Structures of the [H2L]+ ions are different in the two polymorphs: torsion angle N1–C1–C2–N2 is 149.64(15)° in α-H2LCl, and 180° in β-H2LCl and H2LI. In α-H2LCl, centrosymmetric [H2L]+ dimers [graph-set R22(10)] are bridged by eight chloride ions thus forming sheets parallel to the crystallographic (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) plane (Fig. 1a). In β-H2LCl and H2LI chains of [H2L]+ ions [C(4)] are mutually connected via Cl− or I− ions (Fig. 1b and c). Each [H2L]+ ion is hydrogen bonded to four Cl− or I− ions in a 3D charge-assisted hydrogen bond framework (Fig. 1b, c and Table S4, ESI†). Fingerprint plots and Hirshfeld surface analysis for H2LCl polymorphs are given in Fig. S3 (ESI†). Most of the intermolecular contacts in α-H2LCl and β-H2LCl are similar, however the most notable difference between the two structures is in the surrounding of the oxygen atom.
) plane (Fig. 1a). In β-H2LCl and H2LI chains of [H2L]+ ions [C(4)] are mutually connected via Cl− or I− ions (Fig. 1b and c). Each [H2L]+ ion is hydrogen bonded to four Cl− or I− ions in a 3D charge-assisted hydrogen bond framework (Fig. 1b, c and Table S4, ESI†). Fingerprint plots and Hirshfeld surface analysis for H2LCl polymorphs are given in Fig. S3 (ESI†). Most of the intermolecular contacts in α-H2LCl and β-H2LCl are similar, however the most notable difference between the two structures is in the surrounding of the oxygen atom.
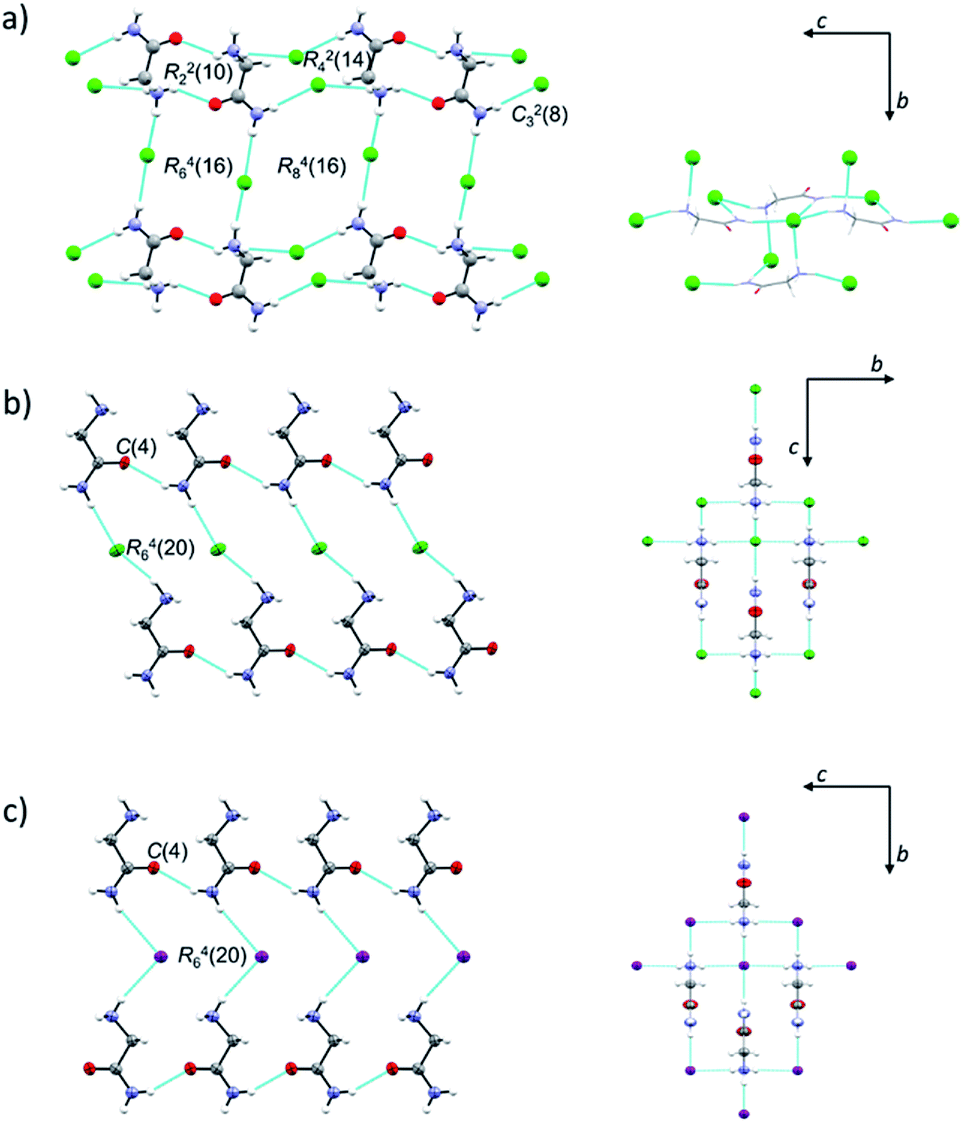 | ||
| Fig. 1 HB-interactions in crystal structures of: (a) α-H2LCl; (b) β-H2LCl and (c) H2LI. In the α-H2LCl polymorph, a layer of [H2L]+ and Cl− ions is parallel to the crystallographic (10) plane, while in β-H2LCl and H2LI an array of [H2L]+ ions is parallel to the crystallographic (010) and (001) plane, respectively (picture on the left). Hydrogen bonding of [H2L]+ with four surrounding halides is presented on the right. Atoms are represented as small spheres of arbitrary radii (α-H2LCl and all hydrogen atoms) or ellipsoids (at 50% probability level; β-H2LCl and H2LI). | ||
In β-H2LCl the oxygen atom is in contact with CH2 and NH2 groups of the neighbouring H2L+ ion, while in α-H2LCl the oxygen atom is surrounded by two –NH3+ groups, having more H⋯H contacts.
Molecular and crystal structures of 1a–c, 2a,b, 2cLT and 2cRT
In cobalt(II) and nickel(II) compounds [M(H2O)2(HL)2]X2 (M = Co, Ni; X = Cl, Br/Cl, I), the metal(II) cation is octahedrally coordinated by two N,O-donating glycinamide ligands and two water molecules (Fig. S4, ESI†). 1a, 1b, 2a and 2b are isostructural and crystallize in the tetragonal crystal system. 1c and 2cRT are also isostructural and crystallize in the orthorhombic crystal system (Tables S2 and S5, ESI†). Two glycinamide molecules are bound to the metal ion via amido O and amino N-atoms in a cis-fashion, and two water molecules occupy the axial coordination sites. Selected bond distances and angles in [M(H2O)2(HL)2]X2 (M = Co, Ni; X = Cl, Br/Cl, I) can be found in Tables S6 and S7 in ESI.†The same building block, a dimer, is found in the isostructural Co(II) and Ni(II) compounds 1a, 1b, 2a and 2b (Fig. 2a). A dimer consists of complex ion pairs mutually connected by four hydrogen bonds of the Ow–H⋯O type [graph-set R22(6)] (Fig. 2a, Tables S8 and S9, ESI†). Halide ions (Cl or Br/Cl) are placed between almost perpendicular dimers forming chains parallel to crystallographic a axis (Fig. S5, ESI†). The remaining hydrogen bond donors N–H and O–H are used for counter ion hydrogen bonding, thus forming a three dimensional framework.
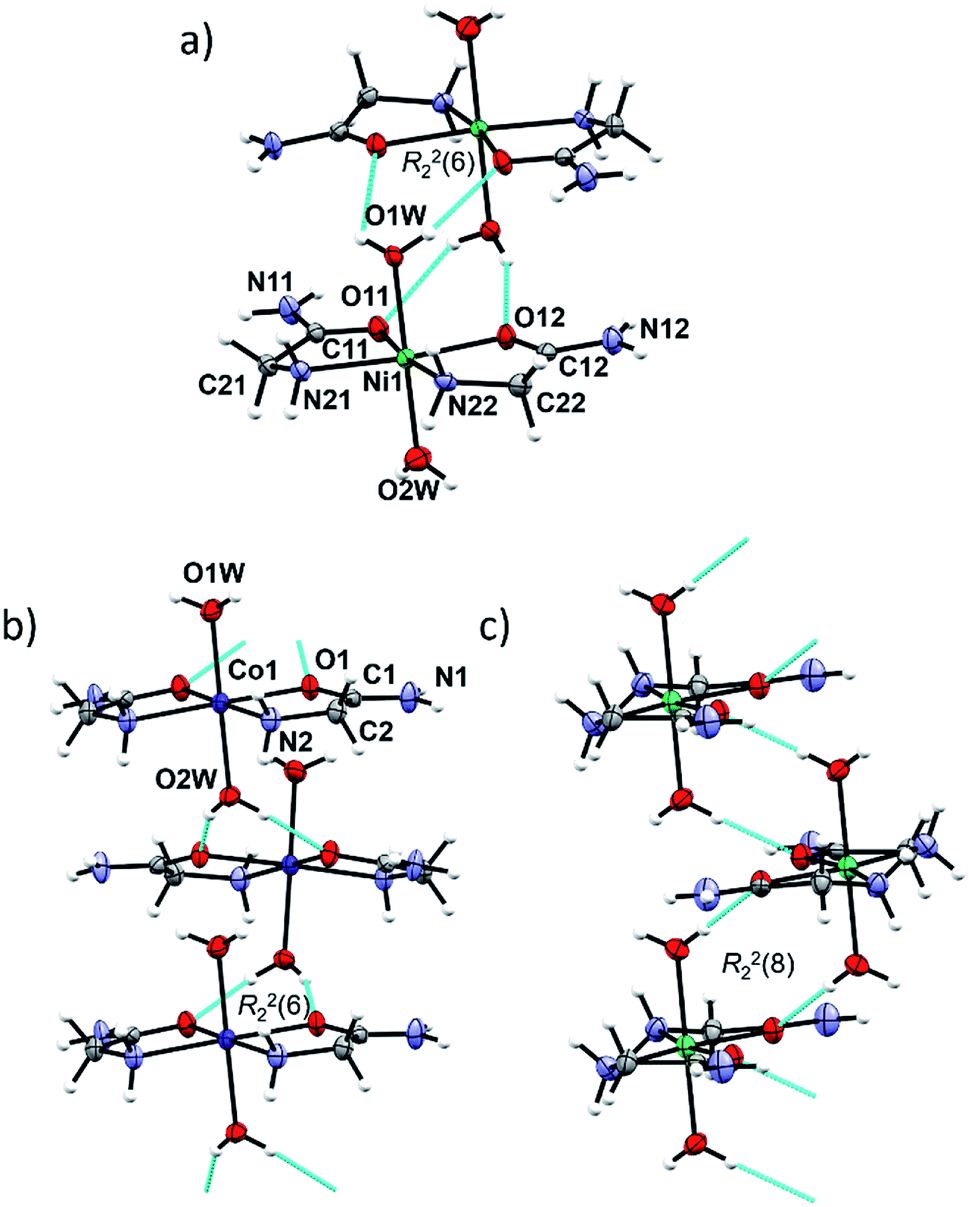 | ||
| Fig. 2 (a) Discrete centrosymmetric dimers in [Ni(HL)2(H2O)2]Br0.94Cl1.06 (2b) formed through intermolecular Ow–H⋯O hydrogen bonds (arrays of cyan cylinders). (b) Zig-zag chain of complex ions of [Co(HL)2(H2O)2]2+ in 1c. (c) Zig-zag chain of complex ions of [Ni(HL)2(H2O)2]2+ in 2cLT. Atom labelling is analogous to 1c, except for water molecules (O1W) which are symmetrically equivalent. | ||
In 1c, 2cRT and 2cLT the complex ions are connected by Ow–H⋯O hydrogen bonds forming zig-zag chains along the a-axis (Fig. 2b and c, Tables S8 and S9, ESI†). In 1c and 2cRT six-membered hydrogen bond rings are formed, R22(6) (Fig. 2b), while in 2cLT the hydrogen bond rings are eight-membered, R22(8) (Fig. 2c). Iodide ions are hydrogen bonded by N–H and O–H groups from three neighbouring cations thus forming a three dimensional framework (Fig. S6–S9, Tables S8 and S9, ESI†).
Two polymorphs 2cRT and 2cLT are both cis-octahedral complexes with axial positions occupied by water molecules. At room temperature the orthorhombic polymorph 2cRT is the stable one, while at low temperature (<220 K) it transforms into the monoclinic polymorph 2cLT. The main difference between the two is the orientation of water molecules (rotation by approx. 90°), which consequently changes the intermolecular interactions, as seen in Hirshfeld surface plots (Fig. S10, ESI†). In 2cRT one water molecule (O1w) forms Ow–H⋯O hydrogen bonds with two carbonyl oxygen atoms of adjacent complexes, while the other water molecule forms two Ow–H⋯I hydrogen bonds. After rotation by 90° at low temperature, both water molecules form both Ow–H⋯O and Ow–H⋯I hydrogen bonds (Table S9, ESI†).
Molecular and crystal structures of 3a–c
The octahedral coordination environment around the Cu(II) ions in the structures of 3a and 3b consists of two N,O-bidentate glycinamide ligands and two halide ions (Cl or Br/Cl) (Fig. 3 and S11, ESI†). Cu(II) complex molecules are trans isomers. In the crystal structures of 3a and 3b all amide H-atoms participate in hydrogen bonds with the neighboring halide ions (in total, every halide ion is hydrogen bonded by two amide N–H and one amino N–H hydrogen bond donor) forming a very dense three dimensional framework (Fig. S12, S13 and Table S11, ESI†).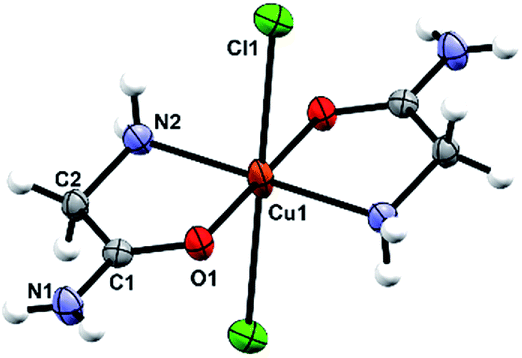 | ||
| Fig. 3 Molecular structure of 3a with the atom labelling scheme. Cu(II) is located on the center of inversion (space group P21/n). | ||
In terms of the crystal structure, the most interesting compound is 3c. It is a 1D coordination polymer built up of dinuclear copper [Cu2I6]4− and [Cu(HL)2]2+ units (Fig. 4a). The connectivity within this polymer is unique among the copper complexes since it is the only copper complex where the [Cu2I6]4− unit links four Cu(II) complex units, in this case [Cu(HL)2]2+ (Fig. 4b). The two bridging atoms within the [Cu2I6]4− unit are I2 and I2ii (ii = x + 1, y − 1, z − 1) with the corresponding Cu2–I bond lengths of 2.6696(12) and 2.7228(9) Å. The [Cu2I6]4− unit (Cu(I) oxidation state) and the four [Cu(HL)2]2+ units are connected through the I1 and I3 bridging atoms (and their centrosymmetrically related atoms). Cu(II)–I bonds are longer and amount to 3.1632(8) Å and 3.2963(8) Å (Table S12, ESI†). These two copper centers have different coordination geometries having the main influence on the bond lengths. Those octahedrally coordinated generally have Cu–I distances greater than 3 Å although the radius of Cu(II) is smaller than of Cu(I). These results are in agreement with similar CuI/CuII mixed oxidation state complexes.23,43–45 Coordination preferences of both Cu centres are also fulfilled: Cu(I) ions are tetrahedrally and Cu(II) ions octahedrally coordinated.
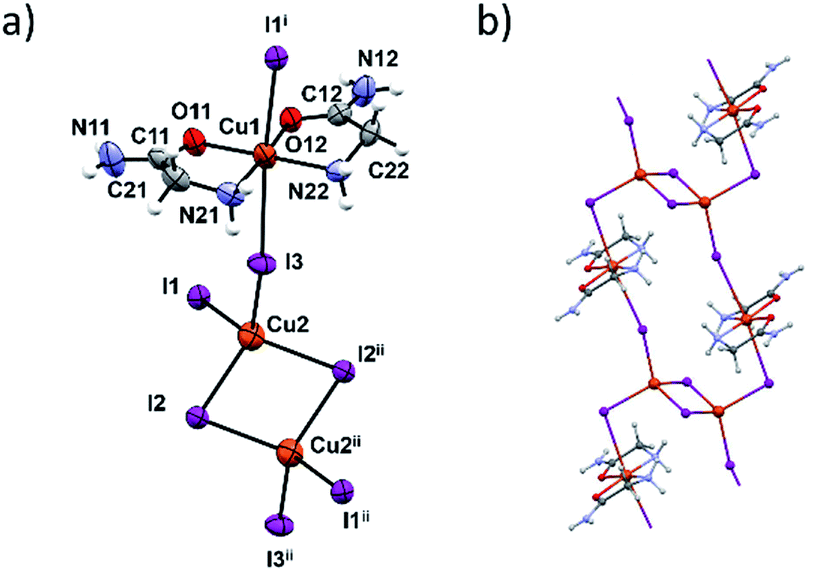 | ||
| Fig. 4 (a) Molecular structure of 3c with atom labelling scheme. Cu(II) are coordinated octahedrally in the cis-HL fashion. Two Cu(I) ions, Cu2 and Cu2ii are both coordinated tetrahedrally with a shared edge. Symmetry operators i = −x, y − 1, −z; ii = x + 1, y − 1, z − 1. (b) double chain of the coordination polymer. | ||
In the [Cu(HL)2]2+ unit two glycinamide molecules bidentately chelate Cu(II) ions in a cis-fashion while iodide ions are found in axial positions. The inner chelate bond lengths indicate partial electron delocalization in the amide group, and the Jahn–Teller effect also influences elongation of the Cu1–I bonds.46
Neighbouring 1D chains are connected by hydrogen bonds between amide N–H bifurcated donors and O- and axial I-atom acceptors. These interactions are almost perpendicular to the chain propagation, Fig. S14 and S15 (ESI†). Inside one chain the glycinamide amino groups serve as N–H donors to iodide ions that are coordinated to the Cu(I) ions (Table S13, ESI†).
All compounds have two chelate rings in the equatorial plane, however the ring conformations are somewhat different. In isostructural 1a, 1b, 2a, 2b, as well as in 3c one 5 membered chelate ring adopts an envelope, and the other a half chair conformation. In 1c and 2cRT both rings are planar, while in 3a, 3b and 2cLT the chelate rings are in a half chair conformation (Table 1). A more detailed conformational analysis is given in Table S14 (ESI†).
| Compound | 5 membered chelate ring conformations |
|---|---|
| 1a, 1b, 2a, 2b and 3c | Envelope and half chair |
| 1c and 2cRT | Planar |
| 3a, 3b and 2cLT | Half chair |
Magnetic properties
1a–b, 2a–c, 3a–c were investigated by X-band electron spin/paramagnetic resonance (ESR/EPR) spectroscopy.Ni(II) complexes (2a, 2b and 2cRT, 2cLT (<220 K)) were ESR silent i.e. from room down to 4 K no ESR signal was detected. This effect could be related to a high value of the zero-field splitting (ZFS) parameter of the Ni(II) ion or with the spin-relaxation phenomena.47 Additionally, it is also possible that, due to Jahn–Teller distortion, Ni(II) ions have a low-spin configuration (S = 0), instead of high-spin (S = 1) expected for octahedral complexes.
Representative spectra of 1a, 3a and 3c, obtained at several selected temperatures, are shown in Fig. 5 while the corresponding spectra of 1b and 3b are shown in Fig. S16, ESI.†
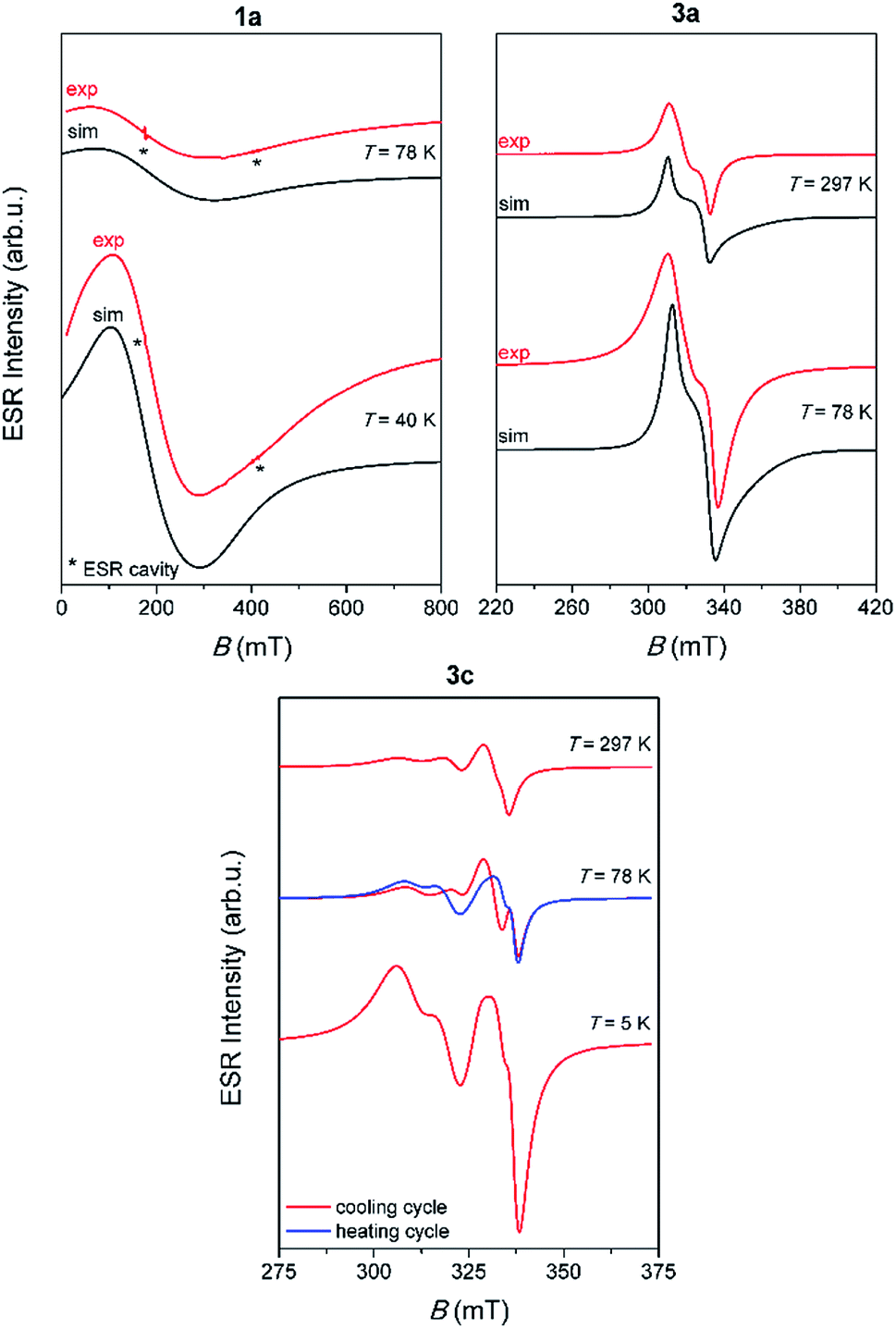 | ||
| Fig. 5 Experimental (red lines) and simulated (black lines) ESR spectra of polycrystalline samples of the investigated complexes. The ESR intensities of the spectra at different temperatures are presented in the real ratios. The narrow lines labeled with asterisks originate from the ESR cavity. | ||
The simulation of the spectra was performed by EasySpin software48 using the following form of the spin-Hamiltonian for Cu(II) and Co(II) ions:49
| H = μBB·g·S | (1) |
In eqn (1), the constant μB is the Bohr magneton, g is the g-tensor, B is the magnetic field vector and S is the spin operator. Hyperfine interaction between electron spin S = 1/2 and nuclear spin I = 3/2 for Cu(II) ion was not detected probably due to weak interactions between Cu(II) ions (the nearest Cu⋯Cu distances are around 6.5 Å). For octahedral Co(II) ions in the high-spin state S = 3/2, it is assumed that magnetic anisotropy is very large and therefore only the lowest states (m = 1/2 and m = −1/2) are thermally occupied.50 As a result only one ESR line with very anisotropic g-values is observed.51 Also, hyperfine interaction for Co(II) ions were not detected. Therefore, the spectra for both Cu(II) and Co(II) ions were simulated using anisotropic g-tensor and allowing only linewidth for assumed Lorentzian lineshape to change with temperature. The obtained g-values, together with the parameters used for the simulations, are given in Table S15 (ESI†). For 3a and 3b complexes, it was necessary to include gstrain values in the simulation. Namely, small variations in the local geometry in Cu(II) octahedra can cause distribution of ESR parameters around some average g-values, described by the gstrain parameter.52 This effect is not observed for 1a and 1b complexes because of their very broad ESR lines. The obtained g-values are the same for 3a and 3b complexes, as expected due to their similar crystal structures. Here obtained results for the g-values are in agreement with the g-values for Cu(II) and Co(II) ions that can be found in the literature.49–51,53
Contrary to the paramagnetic behaviour of 1a, 1b, 3a and 3b samples, 3c shows the most interesting magnetic behaviour, due to its linear 1D structure which contains dinuclear copper units [Cu2I6]4− (Cu⋯Cu distance in the dimer is 3.2057 Å). Beside the unusual ESR spectra, it was noticed that when the compound was heated from 5 K to 78 K, the spectra show different patterns compared to the spectra recorded when the compound was cooled from 78 K to 5 K, Fig. 5. This observation points to possible interesting magnetic behaviour of this compound. Further investigation of 3c should also include magnetic susceptibility measurement.
Biological assays
Antiproliferative activities of 1a and 3a were tested on human lung (H 460), breast (MCF-7) and colon carcinoma (HCT116) cell lines (paragraph biological activity in the ESI†). The tested compounds showed moderate antiproliferative activity towards the MCF-7 cell line, and minor to negligible activity towards HCT116 and H 460 cell lines. However, the effects of the two compounds were almost identical, pointing to negligible structural influence on their biological/antitumor activity (Table S16, ESI†).Experimental
Materials and methods
All chemicals for the syntheses were purchased from commercial sources (Aldrich, Acros or Alfa Aesar) and used as received without further purification. Glycinamide hydrochloride was prepared by aminolysis of chloroacetamide according to the method of E. Fischer.54 CHN analyses were performed on a PerkinElmer 2400 Series II CHNS analyzer in the Analytical Services Laboratories of the Ruđer Bošković Institute, Zagreb, Croatia. The IR spectra were obtained in the range 4000–450 cm−1 on a PerkinElmer Spectrum Two™ FTIR-spectrometer in the ATR mode. TGA measurements were performed at a heating rate of 10 °C min−1 in the temperature range of 25–800 °C, under an oxygen flow of 100 mL min−1 on a Mettler-Toledo TG/SDTA 851e instrument. Approximately 5–10 mg of each sample was placed in a standard alumina crucible (70 μL). The NMR spectra of the ligand were recorded on a Bruker AV 600 spectrometer, operating at 600.130 MHz for the 1H nucleus and at 150.903 MHz for the 13C nucleus. The samples of the ligand were measured in DMSO-d6 solutions at 298 K, using 5 mm NMR tubes. The chemical shifts in ppm were referenced to TMS.The ESR measurements were performed on a Bruker Elexsys 580 FT/CW spectrometer from room down to liquid helium temperature. The microwave frequency was around 9.7 GHz with the magnetic field modulation amplitude of 0.5 mT and modulation frequency of 100 kHz. 3a and 3b complexes, due to observed passage effect55 at low temperatures, were recorded with modulation amplitude of 0.1 mT and modulation frequency of 1 kHz.
Synthetic procedures
β-H2LCl. CAUTION – the experiment should be performed in a fume hood!
Chloroacetamide (18.6 g; 0.2 mol) was mixed with a concentrated ammonia solution (200 mL) and the mixture was heated at 100 °C for 30 min. The reaction mixture was then concentrated at ≈80 °C‡ until the product started to crystallize (the final volume was about 20–30 mL) and immediately mixed with ethanol (200 mL). The reaction mixture was left to stand overnight in a refrigerator and the product was filtered off, washed with ethanol (50 mL) and air-dried. Additional amount of the product can be obtained by evaporation of the filtrate at room temperature.§ When prepared in this manner, the product can be used without any further purification.
White crystals, yield: 16.8 g (76%); mp 210 °C. 1H NMR (DMSO-d6, δ, ppm): 3.50 (s, 2H) CH2, 7.49 (s, br, 1H) NHa, 8.05 (s, br, 1H) NHb, 8.27 (s, br, 3H) NH3+. 13C NMR (DMSO-d6, δ, ppm): 39.85 CH2, 167.66 CONH2. IR (ATR, cm−1): 3364(w), 3263(w), 3184(w), 2997(m), 2893(w), 2773(w), 2566(w), 1688(s), 1594(m), 1578(m), 1464(s), 1417(s), 1314(s), 1151(w), 1093(m), 1038(m), 891(s), 826(m), 528(w), 479(w).
H2LI. Green crystals of H2LI were obtained from a solution containing CoI2 (0.156 g, 0.5 mmol), H2LCl (0.110 g, 1.0 mmol) and NaHCO3 (0.076 g, 0.9 mmol) and 10 mL of water in a very low yield. Crystals decomposed after several weeks.
[Co(H2O)2(HL)2]Cl2 (1a). Cobalt(II) chloride hexahydrate (0.24 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Rose-red crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4Cl2Co: C 15.30, H 5.14, N 17.84%. Found: C 15.42, H 4.66, N 17.83%. IR (ATR, cm−1): 3424(w), 3277(s), 3245(s), 3129(m), 2956(w), 2929(w), 2781(w), 1665(vs), 1595(s), 1461(m), 1422(m), 1343(w), 1313(w), 1196(w), 1134(vs), 1043(s), 944(w), 858(w), 767(w), 656(s), 601(s), 545(w), 488(w).
[Co(H2O)2(HL)2]Br1.06Cl0.94 (1b). Cobalt(II) bromide (0.22 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Rose-red crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4Br1.06Cl0.94Co: C 13.32, H 4.47, N 15.53%. Found: C 13.31, H 3.98, N 15.34%. IR (ATR, cm−1): 3428(w), 3272(s), 3243(s), 3144(m), 2951(w), 2925(w), 2771(w), 1659(vs), 1586(s), 1455(m), 1417(m), 1340(w), 1312(w), 1193(w), 1129(s), 1040(s), 939(w), 856(w), 754(w), 635(s), 587(s), 542(w), 488(w).
[Co(H2O)2(HL)2]I2 (1c). Cobalt(II) acetate dihydrate (0.108 g, 0.5 mmol), glycinamide hydrochloride (0.111 g, 1.0 mmol) and potassium iodide (0.166 g, 1.0 mmol) were mixed in 10 mL of water. Pink crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4I2Co: C 9.67, H 3.25, N 11.27%. Found: C 9.72, H 3.41, N 11.25%. IR (ATR, cm−1): 3335(s), 3317(s), 3273(s), 3242(s), 3145(s), 2929(m), 2784(w), 1662(s), 1596(s), 1461(m), 1420(m), 1312(m), 1193(w), 1131(m), 1042(s), 940(w), 853(w), 766(w), 658(m), 596(m), 544(w), 493(w).
[Ni(H2O)2(HL)2]Cl2 (2a). Nickel(II) chloride hexahydrate (0.24 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Light blue crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4Cl2Ni: C 15.31, H 5.14, N 17.86%. Found: C 15.27, H 4.68, N 17.74%. IR (ATR, cm−1): 3432(w), 3285(s), 3250(s), 3114(m), 2958(w), 2933(w), 2786(w), 1677(vs), 1595(s), 1463(m), 1422(m), 1341(w), 1313(w), 1193(w), 1134(s), 1042(s), 946(w), 860(w), 768(w), 659(s), 605(s), 548(w), 495(w).
[Ni(H2O)2(HL)2]Br0.94Cl1.06 (2b). Nickel(II) bromide (0.22 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Light blue crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4Br0.94Cl1.06Ni: C 13.51, H 4.53, N 15.75%. Found: C 13.66, H 4.06, N 15.81%. IR (ATR, cm−1): 3437(w), 3280(s), 3247(w), 3142(m), 2956(w), 2927(w), 2778(w), 1663(vs), 1592(s), 1458(m), 1418(m), 1338(w), 1312(w), 1191(w), 1131(s), 1040(s), 941(w), 858(w), 755(w), 640(s), 592(s), 544(w), 495(w).
[Ni(H2O)2(HL)2]I2, (2cRT). Nickel(II) iodide (0.31 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol), sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. Blue crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O4I2Ni: C 9.67, H 3.25, N 11.28%. Found: C 9.35, H 3.64, N 11.40%. IR (ATR, cm−1): 3343(s), 3322(s), 3275(s), 3179(s), 2939(m), 2758(w), 1646(s), 1596(s), 1575(s), 1461(m), 1411(m), 1318(m), 1297(m), 1193(w), 1120(m), 1036(s), 935(w), 846(w), 766(w), 682(m), 594(s), 556(m), 505(m).
[CuCl2(HL)2] (3a). Copper(II) chloride dihydrate (0.17 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Dark blue crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O2Cl2Cu: C 17.00, H 4.28, N 19.82%. Found: C 17.18, H 3.81, N 19.82%. IR (ATR, cm−1): 3290(m), 3217(w), 3144(m), 2987(w), 2953(w), 2757(w), 1674(m), 1632(vs), 1579(vs), 1462(m), 1416(m), 1343(m), 1290(w), 1180(w), 1122(vs), 1101(vs), 1056(m), 948(m), 857(w), 777(m), 692(s), 651(s), 563(m), 509(m), 460(m).
[CuBr1.3Cl0.7(HL)2] (3b). Copper(II) bromide (0.22 g, 1.0 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. The mixture was stirred for few minutes, until the effervescence subsided, and was left to stand at room temperature. Dark blue crystals, suitable for X-ray structural analysis, were obtained. Anal. calc. for C4H16N4O2Br1.3Cl0.7Cu: C 14.11, H 3.55, N 16.46%. Found: C 13.97, H 4.18, N 16.18%. IR (ATR, cm−1): 3282(m), 3209(w), 3137(m), 2981(w), 2949(w), 2746(w), 1669(m), 1633(vs), 1572(vs), 1456(m), 1415(m), 1339(m), 1290(w), 1177(w), 1118(vs), 1100(vs), 1055(m), 946(m), 852(w), 755(m), 677(s), 646(s), 560(m), 506(m), 460(m).
{[CuII(HL)2]2[CuI2I6]}n (3c). Copper(II) chloride dihydrate (0.17 g, 1 mmol), glycinamide hydrochloride (0.22 g, 2.0 mmol) and sodium bicarbonate (0.15 g, 1.8 mmol) were mixed in 10 mL of water. After the effervescence subsided, solid potassium iodide (0.35 g, 2 mmol) was added into the solution. The colour changed from dark blue to olive-green, leading to brown crystals, suitable for X-ray structural analysis. Anal. calc. for C4H12N4O2I3Cu2: C 7.32, H 1.84, N 8.54%. Found: C 7.44, H 2.12, N 8.36%. IR (ATR, cm−1): 3380(m), 3310(vs), 3267(vs), 3201(s), 3119(s), 2953(w), 2912(w), 1672(m), 1644(vs), 1555(vs), 1457(m), 1401(m), 1321(w), 1293(w), 1174(w), 1108(s), 1054(m), 1034(m), 929(w), 852(w), 687(w), 620(m), 583(m), 548(m), 480(m).
Crystallization of complex compounds
All compounds crystallized from aqueous solutions by slow evaporation of solvent at room temperature. 1a, 1b, 2a, 2b, 3a, and 3b crystalized after several days. Coordination polymer 3c crystallized within minutes upon addition of potassium iodide due to very low solubility. On the other hand, 1c and 2cRT are highly soluble in water, hence crystallization occurred after several months.X-ray crystallography
The single-crystal X-ray diffraction data of β-H2LCl, H2LI, 1a–c, 2a, 2b, 2cLT, 2cRT, 3a–c were collected by ω-scans on an Oxford Diffraction Xcalibur3 CCD diffractometer with graphite-monochromated MoKα radiation. Data reduction was performed using the CrysAlis software package.56 Solution, refinement and analysis of the structures were done using the programs integrated in the WinGX system.57 All structures were solved by the direct methods using SHELXS and the refinement procedure was performed by the full-matrix least-squares method based on F2 against all reflections using SHELXL.58,59 The non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located in the difference Fourier maps. Because of poor geometry for some of them they were placed in calculated positions and refined using the riding model. In structures 1b, 2b and 3b bromide and chloride ions statistically occupy almost the same site (slightly longer distances are associated with the bromide ion). Displacement parameters of these ions were restrained to the same values. Occupancies were refined to the final ratios Br/Cl: 1.06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.94 in 1b, 0.94:1.06 in 2b, and 1.3:0.7 in 3b. Geometrical calculations were done using PLATON.60 Drawings of the structures were prepared using PLATON and MERCURY program.61 The crystallographic data are summarized in Tables S2 and S5, see ESI.†
763 reflections measured, 2660 unique (Rint = 0.024). Final R(F, I > 2σ(I)) value was 0.0169, wR2(F2, I > 2σ(I)) = 0.0426, S = 1.13. CCDC 1915368.†188 reflections measured, 2501 unique (Rint = 0.036). Final R(F, I > 2σ(I)) value was 0.0238, wR2(F2, I > 2σ(I)) = 0.0538, S = 1.03. CCDC 1915361.†338 reflections measured, 4042 unique (Rint = 0.030). Final R(F, I > 2σ(I)) value was 0.0269, wR2(F2, I > 2σ(I)) = 0.0592, S = 1.08. CCDC 1915364.†070 reflections measured, 2662 unique (Rint = 0.028). Final R(F, I > 2σ(I)) value was 0.0223, wR2(F2, I > 2σ(I)) = 0.0485, S = 1.10. CCDC 1915369.†873 reflections measured, 1159 unique (Rint = 0.016). Final R(F, I > 2σ(I)) value was 0.0151, wR2(F2, I > 2σ(I)) = 0.0479, S = 0.98. CCDC 1915371.† (no. 2), Z = 2, 7861 reflections measured, 3084 unique (Rint = 0.039). Final R(F, I > 2σ(I)) value was 0.0259, wR2(F2, I > 2σ(I)) = 0.0686, S = 0.84. CCDC 1915365.†
:0.94 in 1b, 0.94:1.06 in 2b, and 1.3:0.7 in 3b. Geometrical calculations were done using PLATON.60 Drawings of the structures were prepared using PLATON and MERCURY program.61 The crystallographic data are summarized in Tables S2 and S5, see ESI.†
763 reflections measured, 2660 unique (Rint = 0.024). Final R(F, I > 2σ(I)) value was 0.0169, wR2(F2, I > 2σ(I)) = 0.0426, S = 1.13. CCDC 1915368.†188 reflections measured, 2501 unique (Rint = 0.036). Final R(F, I > 2σ(I)) value was 0.0238, wR2(F2, I > 2σ(I)) = 0.0538, S = 1.03. CCDC 1915361.†338 reflections measured, 4042 unique (Rint = 0.030). Final R(F, I > 2σ(I)) value was 0.0269, wR2(F2, I > 2σ(I)) = 0.0592, S = 1.08. CCDC 1915364.†070 reflections measured, 2662 unique (Rint = 0.028). Final R(F, I > 2σ(I)) value was 0.0223, wR2(F2, I > 2σ(I)) = 0.0485, S = 1.10. CCDC 1915369.†873 reflections measured, 1159 unique (Rint = 0.016). Final R(F, I > 2σ(I)) value was 0.0151, wR2(F2, I > 2σ(I)) = 0.0479, S = 0.98. CCDC 1915371.† (no. 2), Z = 2, 7861 reflections measured, 3084 unique (Rint = 0.039). Final R(F, I > 2σ(I)) value was 0.0259, wR2(F2, I > 2σ(I)) = 0.0686, S = 0.84. CCDC 1915365.†Conclusions
New cobalt(II), nickel(II) and copper(II) compounds with glycinamide were prepared and characterized by X-ray crystallography, IR spectroscopy and thermal analysis.Cobalt(II) and nickel(II) compounds 1a–1c and 2a–2c have an analogous chemical composition, [M(HL)2(H2O)2]X2. In these compounds two glycinamide ligands coordinate Co(II) or Ni(II) ions in the N,O-bidentate chelating mode arranged in a cis-configuration, while water molecules occupy the axial coordination sites. Halide ions are counter-ions in all six mentioned complexes. Copper compounds 3a and 3b are trans isomers with two N,O-bidentate glycinamide ligands in the equatorial plane and two halide ions coordinated at the axial coordination sites.
Interestingly, bromide and chloride ions are almost equally preferred by cobalt(II) and nickel(II) complex ions, hence crystal structures are disordered at the halide ion position. Bromide and chloride ions occupy the same positions in the Br/Cl ratio close to 1:1 (1.06:0.94 in Co(II) complexes; 0.94:1.06 in Ni(II) compounds). Copper(II) ion, on the other hand, has slightly more preference towards the bromide ion, which resulted in Br to Cl ratio 1.3:0.7. Copper showed interesting chemical behavior in reaction of copper(II) ions, glycinamide and iodide ions. Copper(II) was partially reduced to copper(I) and the coordination polymer 3c was formed with mixed oxidation states of copper. 1D polymer 3c contains double chains of copper(II) coordinated by two glycinamide ligands in a cis configuration bridged by [Cu2I6]4− species. 3c also showed possible interesting magnetic properties which will be investigated in more details in further research. 1a and 3a were tested for antiproliferative activity. Both compounds showed no activity towards HCT116 and H 460 cell lines, but moderate activity (GI50 just above 10 μmol dm−3) and selectivity was found towards the MCF-7 cell line.
CCDC 1915361–1915372 contain the supplementary crystallographic data for this paper.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support by the Croatian Science Foundation (grant no. IP-2014-09-4274 and IP-2018-01-3168) is gratefully acknowledged. The authors are grateful to Dr Marijeta Kralj and Dr Lidija Uzelac for testing compounds for antiproliferative activity.Notes and references
- C. Kutzscher, P. Müller, S. Raschke and S. Kaskel, in The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, ed. S. Kaskel, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn, 2016, Chiral Linker Systems, pp. 387–418 Search PubMed
.
- I. Dokmanić, M. Šikić and S. Tomić, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64(3), 257–263 CrossRef PubMed
- P. J. Almhjell and J. H. Mills, Curr. Opin. Struct. Biol., 2018, 51, 170–176 CrossRef CAS PubMed
- K. Užarević, I. Halasz, I. Đilović, N. Bregović, M. Rubčić, D. Matković-Čalogović and V. Tomišić, Angew. Chem., Int. Ed., 2013, 52, 5504–5508 CrossRef PubMed
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2014, 14, 23–26 CrossRef PubMed
- J. Aizenberg and P. Fratzl, Adv. Mater., 2009, 21, 387–388 CrossRef CAS
- J. Aizenberg, Adv. Mater., 2004, 16, 1295–1302 CrossRef CAS
- E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133–165 CrossRef CAS
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109(10), 4921–4960 CrossRef CAS
- D. L. Stone, D. K. Smith and A. C. Whitwood, Polyhedron, 2004, 23, 1709–1717 CrossRef CAS
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1432–1443 CrossRef CAS
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 857–867 CrossRef CAS
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 401–411 CrossRef CAS
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 849–859 CrossRef
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 678–682 CrossRef
- E. Andersson, P. Horal, A. Jejcic, S. Höglud, J. Balzarini, A. Vahle and B. Svennerholm, Antimicrob. Agents Chemother., 2005, 49(1), 40–44 CrossRef CAS
- Z. H. Chohan, M. Arif, M. A. Akhtar and C. T. Supuran, Bioinorg. Chem. Appl., 2006, 1–13 Search PubMed
- D. Kannan and M. N. Arumugham, Int. J. Res. Controlled Release, 2012, 2(4), 10–17 Search PubMed
- X. Li, Z. Zhang, C. Wang, T. Zhang, K. He and F. Deng, J. Inorg. Biochem., 2011, 105, 23–30 CrossRef CAS
- X. Liu, X. Li, Z. Zhang, Y. Dong, P. Liu and C. Zhang, Biol. Trace Elem. Res., 2013, 154, 150–155 CrossRef CAS
- D. Vušak, B. Prugovečki, D. Milić, M. Marković, I. Petković, M. Kralj and D. Matković-Čalogović, Cryst. Growth Des., 2017, 17, 6049–6061 CrossRef
- S. M.-F. Lo, S. S.-Y. Chui, L.-Y. Shek, Z. Lin, X. X. Zhang, G. Wen and I. D. Williams, J. Am. Chem. Soc., 2000, 122, 6293–6294 CrossRef CAS
- D. A. Firmin, E. R. Quilano, R. Cameron, A. K. Pant, E. D. Stevens and C. J. O'Connor, Inorg. Chim. Acta, 1990, 172, 211–220 CrossRef CAS
- A. V. Pestov, P. A. Slepukhin and V. N. Charushin, Russ. Chem. Rev., 2015, 84, 210–333 CrossRef
- T.-F. Liu and Z.-X. Wang, Inorg. Chem. Commun., 2013, 30, 84–87 CrossRef CAS
- W. Wen, X. Jimin and X. Yawen, J. Coord. Chem., 2009, 62, 373–379 CrossRef CAS
- T. Adam, Klin. Biochem. Metab., 2005, 13(34), 177–181 Search PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- B. Ganguly, M. K. Kesharwani, N. Basarić, E. Suresh, A. K. Biswas and K. Mlinarić-Majerski, J. Mol. Graphics Modell., 2013, 46, 52–58 CrossRef CAS
- A. Fehn, S. Mihan, K. Polborn and W. Beck, Z. Anorg. Allg. Chem., 1997, 623, 665–675 CrossRef CAS
- R. Krämer, M. Maurus, R. Bergs, K. Polborn, K. Sünkel, B. Wagner and W. Beck, Chem. Ber., 1993, 126, 1969–1980 CrossRef
- N. Usuki, M. Yamada, M. Ohba and H. Ōkawa, J. Solid State Chem., 2001, 159, 328–335 CrossRef CAS
- Y. Ilan and M. Kapon, Inorg. Chem., 1986, 25, 2350–2354 CrossRef CAS
- M. Graf, K. Karaghiosoff, P. Mayer and W. Beck, Z. Anorg. Allg. Chem., 2013, 639(7), 1117–1121 CrossRef CAS
- W. Kaim, B. Schwederski and A. Klein, Bioinorganic chemistry-Inorganic elements in the chemistry of life, John Wiley & Sons, Chichester, 2nd edn, 2013 Search PubMed
- J. Pejić, D. Vušak, G. Szalontai, B. Prugovečki, D. Mrvoš-Sermek, D. Matković-Čalogović and J. Sabolović, Cryst. Growth Des., 2018, 18(9), 5138–5154 CrossRef
- M. Tašner, B. Prugovečki, Ž. Soldin, S. Prugovečki, L. Rukavina and D. Matković-Čalogović, Polyhedron, 2013, 52, 268–275 CrossRef
- M. Tašner, B. Prugovečki, D. Mrvoš-Sermek, B. Korpar-Čolig, G. Giester and D. Matković-Čalogović, Acta Chim. Slov., 2008, 55(4), 928–934 Search PubMed
- M. Tašner, D. Mrvoš-Sermek, E. Hajdarpašić and D. Matković-Čalogović, Sec. Nat. Math. Biotech. Sci., MASA, 2018, 39(2), 91–101 Search PubMed
- N. Smrečki, O. Jović, B.-M. Kukovec, E. Šimunić, S. Vuk, A. Skuhala, M. Babić, T. Rončević, N. Ilić, I. Kekez, D. Matković-Čalogović and Z. Popović, Inorg. Chim. Acta, 2018, 471, 521–529 CrossRef
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., Hoboken, 7th edn, 2005 Search PubMed
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, Inc., Hoboken, 6th edn, 2009 Search PubMed
- S. Myllyviita and R. Sillanpää, J. Chem. Soc., Dalton Trans., 1994, 2125–2128 RSC
- B. Freckmann and K.-F. Tebbe, Z. Naturforsch., B: J. Chem. Sci., 1980, 35, 1319–1321 Search PubMed
- S. Zhang, Y. Cao, H. Zhang, X. Chai, Y. Chen and R. Sun, J. Solid State Chem., 2008, 181, 3327–3336 CrossRef CAS
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS
- C. Ruiz-Perez, P. A. L. Luis, F. Lloret and M. Julve, Inorg. Chim. Acta, 2002, 336, 131–136 CrossRef CAS
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS
- O. Kahn, Molecular Magnetism, VCH Publishers, Inc., 1993 Search PubMed
- A. Carrington and A. D. McLachlan, Introduction to Magnetic Resonance, Harper and Row, New York, 1967 Search PubMed
- D. Žilić, K. Molčanov, M. Jurić, J. Habjanič, B. Rakvin, Y. Krupskaya, V. Kataev, S. Wurmehl and B. Büchner, Polyhedron, 2017, 126, 120–126 CrossRef
- B. Szymańska, D. Skrzypek, D. Kovala-Demertzi, M. Staninska and M. A. Demertzis, Spectrochim. Acta, Part A, 2006, 63(3), 518–523 CrossRef
- J.-S. Park, T.-J. Park, K.-H. Kim, K. Oh, M.-S. Seo, H.-I. Lee, M.-J. Jun, W. Nam and K.-M. Kim, Bull. Korean Chem. Soc., 2006, 27, 193–194 CrossRef CAS
- E. Fischer, Ber. Dtsch. Chem. Ges., 1903, 36, 2982–2992 CrossRef CAS
- G. R. Eaton, S. S. Eaton, D. P. Barr and R. T. Weber, Quantitative EPR, Springer, Vienna, 2010 Search PubMed
- CrysAlisPro Software System, Version 1.171.38.41, Rigaku Oxford Diffraction, 2015 Search PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, M. Towler, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: crystal and molecular structure data of β-H2LCl, H2LI, 1a–c, 2a,b, 2cRT, 2cLT, 3a–c, TG analysis of complexes, Hirshfeld surface analysis of α and β-H2LCl, crystallographic data, bond lengths and angles, torsion angles, geometric parameters of intermolecular hydrogen bonds, conformational analysis of chelate rings. CCDC 1915361–1915372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03693h |
| ‡ The reaction mixture becomes orange-coloured when overheated, leading to a very impure yellow-orange product which is not easily purified. |
| § Crystals obtained by slow evaporation of the filtrate were suitable for X-ray structural analysis. |
| This journal is © The Royal Society of Chemistry 2019 |