
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO oxidization catalyzed by B, N, and their co-doped fullerenes: a first-principles investigation†
Boya Gao and
Gang Chen *
*
School of Physics and Technology, University of Jinan, Shandong 250022, China. E-mail: ss_cheng@ujn.edu.cn
First published on 12th July 2019
Abstract
Using the elastic band method based on first-principles calculations, we have carefully studied the catalytic properties of B, N, and their co-doped fullerenes. During oxidization of CO, both C59B and C59N can be oxidized to form durable oxide catalysts for successive CO oxidizations, the rate determining steps of which have 0.59 and 0.80 eV barriers, respectively. In CO-rich conditions, the C59N may remain in the entire reaction cycle with a 0.44 eV rate determining barrier. Both BN-pair doped fullerene and B-rich B3N doped fullerene can also be oxidized during the process of catalyzing CO oxidizations, and the oxides can then be repeatedly used as catalysts in successive CO oxidizations with rate determining barriers of approximately 0.42 eV. The central B in the N-rich C56BN3 is protected by its surrounding N atoms against oxidization to remain as a durable catalyst, the rate determining barrier of which is 0.63 eV for catalyzing CO oxidization. These results for the B and N doped fullerenes, and especially for the B–N co-doped fullerenes, could help in the design of high-performance non-metal catalysts, calling for further detailed experimental investigations.
1. Introduction
The increased energy demand and the corresponding rapid consumption of various fuels have attracted extraordinary research interests that are focused on environmental pollutants. Among these CO has been identified as a poisonous pollutant species and has already aroused intensive research attention during the past few decades. It is usually generated by incomplete combustion of various fossil energy sources, for example emissions from vehicle exhaust fumes, various industrial processes, the consumption of fossil energy materials, and so forth. It can bind to hemoglobin more strongly than O2 to reduce oxygen availability for different organs, resulting in impaired concentration, slow reflexes, and confusion. In addition, CO in fuel cells is a critical and harmful issue, especially with regard to the large-scale application of fuel cells as sustainable energy carriers, which could poison the electrode making it inactive. With regard to cleaning, effective heterogeneous catalysts for CO oxidization play a critical role. Usually, precious metals or their related alloys or oxides are needed for catalysis of the CO oxidization reaction, for example Pt, Au, Ru, Rh, and Pd.1–23 However, these metal catalysts are expensive and usually need to be used at a high temperature to achieve an efficient operation. Finding inexpensive alternatives to replace these precious metal-based catalysts is therefore highly desirable. Therefore, investigation of low-priced metal-free catalysts has gained tremendous research interest in recent decades.Recently, carbon or carbon-based nanomaterials have been found to have superior catalytic properties, and a significant amount of research has been focused on exploring non-metal catalysts. These carbon nanomaterials, which have sp2-like hybridization networks including graphene, nanotubes, fullerene, and so forth, have abundant free-flowing π electrons to provide reactions requiring electrons, and this makes them potential base materials for exploring high-performance metal-free catalysts. However, the π electrons in ideal sp2-like nanostructures, such as C60, are too inert to be directly used to catalyze reactions. In recent years, studies have shown that breaking the integrity of the π electron distribution in heteroatom doped sp2-like nanostructures could activate π electrons, giving them superior catalytic properties.24–27 Owing to the difference in the number of valence electrons, B and N are the most frequently studied heteroatom doping species. The doped C60 structures with a maximum number of doped B and N were previously studied by Manaa et al.28–30 N doping can cause conjugation of the lone-pair electrons with the carbon π electrons to allow the surrounding C atoms to become active for O2 adsorption. In contrast, the doping of the electron deficient B atom in carbon networks can only provide a vacant 2pz orbital for conjugation with carbon π electrons, resulting in B becoming an active site for O2 adsorption. Hu et al.31 and Lin et al.32 studied the CO oxidizations catalyzed by N doped carbon nanotubes using density functional theory (DFT) calculations. Chen and his co-workers reported DFT studies on CO oxidizations catalyzed by N doped C60 (ref. 33) and penta-graphene.34 In addition, B, N, P, and Si-doped graphene also shows superior reactivities for catalyzing CO oxidizations.35–39 Despite the great progress that has been made in the search for heteroatom doped carbon nanostructure catalysts, studies providing detailed reaction paths are still highly desired to give deeper insights into the catalytic origin, aiming to help researchers design high-performance and durable metal free catalysts. To the best of our knowledge, comparative theoretical studies on the catalytic properties of B, N and their co-doped C60 are limited and there are no detailed studies on the catalytic properties of the corresponding oxides of doped C60, which are the by-products of the pioneering CO oxidizations.
In this paper, by using B or N doped C60, and their co-doped fullerenes as prototype nanostructure catalysts, we carefully studied the minimum energy reaction paths of CO oxidizations by using the elastic band method based on first-principles calculations. Our studies suggest that the carbon-based nanostructures would experience oxidization during catalyzing CO oxidizations. The catalytic properties of these oxides are investigated for the first time, as these could then act as durable recycling high-performance catalyst species for successive CO oxidization cycles. Furthermore, our studies on the BN-pair, BN3, and B3N doped C60 could shed light on the development of metal-free catalysts by using small B–N co-doping motifs, either in stoichiometric or B- and N-rich conditions.
2. Computational details
We have carried out detailed minimum energy reaction path studies based on first-principles calculations which were performed within the framework of spin-polarized DFT as implemented in the Vienna ab initio simulation package (VASP).40 The projector augmented-wave method was employed and a cutoff energy of 400 eV was used for the plane wave bases to construct the wave functions.41 The exchange–correlation energy was calculated by using generalized gradient approximation (GGA) with the Perdew, Burke, and Ernzerhof (PBE) parameterized functional.42 In order to mimic the zero-dimensional materials, a supercell with a vacuum space of more than 10 Å was employed to clearly separate the cluster and its periodic images in order to eliminate the interaction between them. For such a large supercell, only the Γ point was used to integrate the electronic properties in the first Brillouin zone. The convergence criterion of the electronic properties was set to 10−5 eV. The positions of the atoms were fully optimized until the maximum force acting on each atom converged to 0.01 eV Å−1. For the minimum energy path, we first carried out a 16-image nudged elastic band method (NEB) calculation. Based on the optimized NEB path, we re-optimized the reaction path by using the elastic band method with a dense image set around the saddle point along the reaction path, with the purpose of obtaining a sense of the accurate energy barrier.43–45 Along the entire reaction cycle, we compared the calculated barriers and chose the largest one as the rate determining barrier. The adsorption energy was calculated by referring to the free-standing gas molecule and nanostructure:| Eads = Efullerene+molecule − Efullerene − Emolecule |
In which Efullerene+molecule, Efullerene, and Emolecule were the total energies of the molecule adsorbed structure, the doped fullerene, and the isolated molecule, respectively. A negative value indicates an exothermic process. In addition, we also calculated the frequencies of the initial, transition, and final states of all of the studied reaction paths. The vibration properties were studied by using the linear response method as implemented in the VASP code.
3. Results and discussion
3.1 Catalytic properties of B and N doped fullerenes
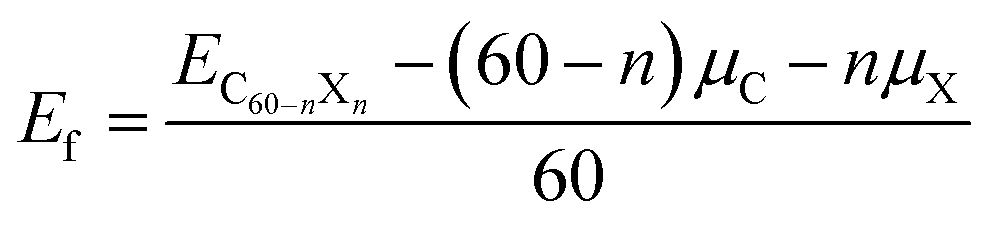
In which EC60−nXn is the total energy of the doped configuration. X represents B or N. μC and μX are the chemical potentials of C in graphene and X in its bulk. Although C60, C59B, and C59N were calculated to have formation energies of 0.373, 0.387, and 0.380 eV, they have previously been fabricated in experiments. In order to estimate their stabilities, we performed first-principles molecular dynamics simulations (FPMD) at 1000 K, which last for 5 ps with a time step of 1 fs. The geometrical structures are found to be retained during our FPMD studies, suggesting their stability. Once they were formed in the experiments, they could be then used as catalysts for CO oxidization. Both CO and O2 could be exothermically adsorbed onto C59B, the lowest energy adsorption configurations for which are shown in Fig. 1a and b, respectively. The corresponding adsorption energies are −0.80 and −1.02 eV. Our elastic band method studies show that CO adsorption is barrierless. The reaction energy, such as the energy barrier, is defined as:
| Ereaction = Einter − Einitial |
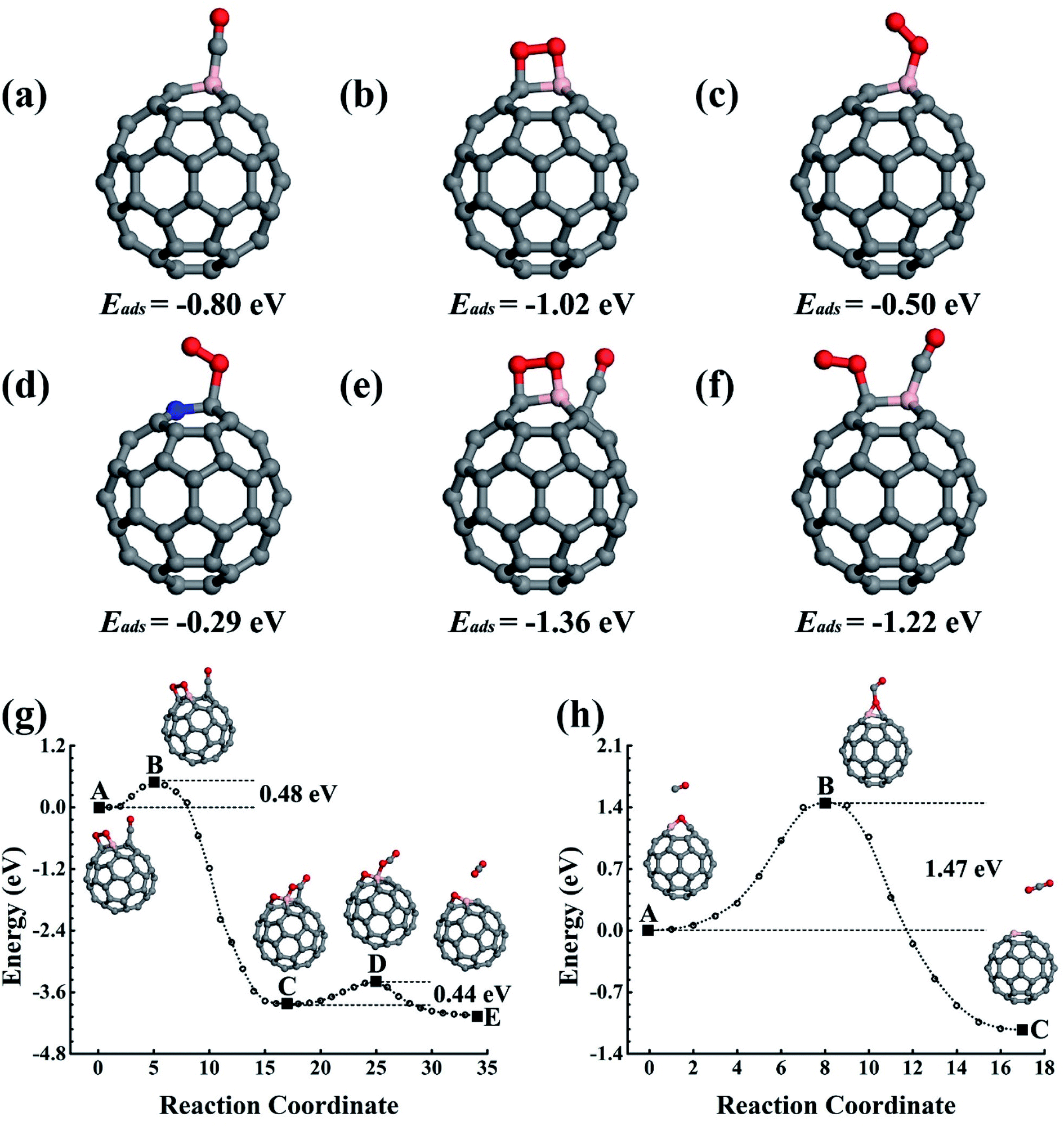 | ||
| Fig. 1 The structures of CO adsorption (a), O2 adsorption (b) and (c), and CO and O2 co-adsorption (e) and (f) on C59B. (d) The lowest energy structure of O2 molecules on C59N. The adsorption energies are also given. (g) and (h) Show the reaction paths for CO oxidization catalyzed by C59B. The initial, final, saddle, and meta-stable intermediate states are illustrated in the inset figures. C, B, N, and O are represented by the grey, pink, blue, and red balls. | ||
In which the Einter and Einitial are the calculated total energies of the intermediate state along the reaction path and the initial reaction state, respectively. A positive value indicates there is an energy cost to activate the reaction. However, the O2 must first overcome the 0.06 eV energy barrier to cap onto the B atom in an end-on adsorption configuration (see Fig. 1c), then reach the final chemisorption state by overcoming a 0.05 eV energy barrier. In the final adsorption state, the O atoms connect with the B and its adjunct C atoms. The O–O bond is roughly parallel to the B–C bond below. The bonding between O and C59B results in a charge transfer of approximately 0.86e (calculated using the Bader analysis technique46) from C59B to O2 to elongate the O–O bond by 0.19 Å to activate the O2, which is close to previously reported results.33,47–49 In our calculation, the O2 is found in the 3Σg− triplet ground state before adsorption onto C59B. When it gets close enough to C59B to adsorb onto it, a charge transfer would occur. In the final adsorption state, the charge gain elongates the O–O bond and transfers it into the 1Δg singlet excited state. In contrast to the gas adsorption on C59B, the C59N can only adsorb O2, for which our elastic band method calculations did not find a barrier. The adsorption energy was calculated to be −0.29 eV. The O2 could only be adsorbed in an end-on adsorption configuration as shown in Fig. 1d with one of its O atoms being connected to the adjacent C atom around the N dopant, gaining approximately a 0.36e charge to elongate the O–O bond by 0.06 Å. The O2 is then transferred from the 3Σg− triplet ground state to the 1Δg singlet excited state.
The reaction product C59BO, as shown in Fig. 2a, could be further used as a catalyst. First, we consider the case of a CO molecule approaching the O atom in the C59BO oxide, which was found to have an energy barrier of approximately 1.47 eV in the converged reaction path shown in Fig. 1h, this limits its occurrence. Interestingly, the B atom in C59BO oxide is still able to bond an O2 molecule. There is only a 0.02 eV energy barrier for O2 adsorption, which forms a 1.54 Å BO bond in the end-on adsorption configuration shown in Fig. 2b, the adsorption energy is −0.76 eV. The O–O bond was found to be elongated by 0.08 Å. A charge gain of approximately 0.64e was confirmed by our Bader charge analysis for O2, which helps to transfer it from a 3Σg− triplet state to a 1Δg singlet state. As shown in Fig. 2g, a CO could approach the adsorbed O2 to form the O–O–C–O complex species through the Langmuir–Hinshelwood (LH) oxidization mechanism by overcoming the 0.63 eV energy barrier, which would evolve to release a CO2 molecule under a 0.21 eV activation energy, leaving a single O atom adsorbed on the B atom. Providing that it is used in the CO-rich surrounding conditions, the produced atomic O could be removed from the C59BO oxide through oxidization of one more of the CO molecules (the barrier is 0.11 eV). Otherwise, it would directly react with the doped fullerene to form C59BO2 oxide (see the structures shown in Fig. 2c), which require a 0.17 eV activation energy. It should be mentioned here that the C59BO2 oxide could also be obtained from the lowest energy chemisorption state of O2 on C59B under a 0.24 eV activation energy for O2 dissociation and then after the oxidization process. After this, one more O2 could rapidly be attached onto the C59BO2 oxide (see Fig. 2d for adsorption configuration), which could then be used to oxidize one more CO under a 0.37 eV activation energy along the converged pathway presented in Fig. 2h to produce a CO2 molecule and an atomic O. For the O2 adsorption on the C59BO2 oxide, a charge of approximately 0.70e is transferred from the oxide to the O2 molecule, making the triplet-singlet state transition which elongates the O–O bond by 0.10 Å. The produced atomic O would either oxidize one more CO or directly react with C59BO2, depending on whether it is protected by CO molecules or not. The oxidization of CO requires an activation of 0.11 eV, while the barrier is 0.09 eV for the oxidization of C59BO2 to form C59BO3 oxide (see Fig. 2e for the geometrical configuration). Again, an O2 molecule could be adsorbed onto the B atom through an exothermal adsorption process, forming a 1.23 eV lower end-on chemisorption state, which is also transferred into the 1Δg singlet excited state by gaining a ∼0.73e charge. Correspondingly, the O–O is elongated by 0.10 Å. As shown in Fig. 2i and j, the C59BO3 oxide could be repeatedly used as a catalyst for CO oxidization. Therefore, in the process of CO oxidization on C59B, the doped fullerene would have the chance to change to the new catalyst species, C59BO3 oxide. The corresponding rate determining barrier is only 0.59 eV for the CO oxidization cycles catalyzed by C59BO3 oxide, showing a good catalytic performance. In addition, we have also calculated the CO adsorption on nicked C59BOx (x = 1–3) oxides, which are only weakly adsorbed and could be easily removed from the C59BOx oxides to leave the B atom for O2 adsorption. The superior catalytic properties of the C59BOx oxides are mainly associated with the activation of the adsorbed O2. In addition, in Fig. 2k and l, we present the optimized reaction pathway starting from another CO–O2 co-adsorption state as shown Fig. 1f. The CO would be oxidized by the LH oxidization mechanism releasing an CO2 molecule and producing the C59BO oxide, for which the higher energy barrier is 0.51 eV. Again, the produced C59BO oxide could be further used to catalyze CO oxidizations through the reactions studied in Fig. 2g–j to finally obtain the C59BO3 oxide. Also, for the C59B oxide materials, we have carried out FPMD simulations. The C59BO, C59BO2, and C59BO3 were found to be well preserved in our 5 ps simulations at 1000 K, suggesting their stabilities.
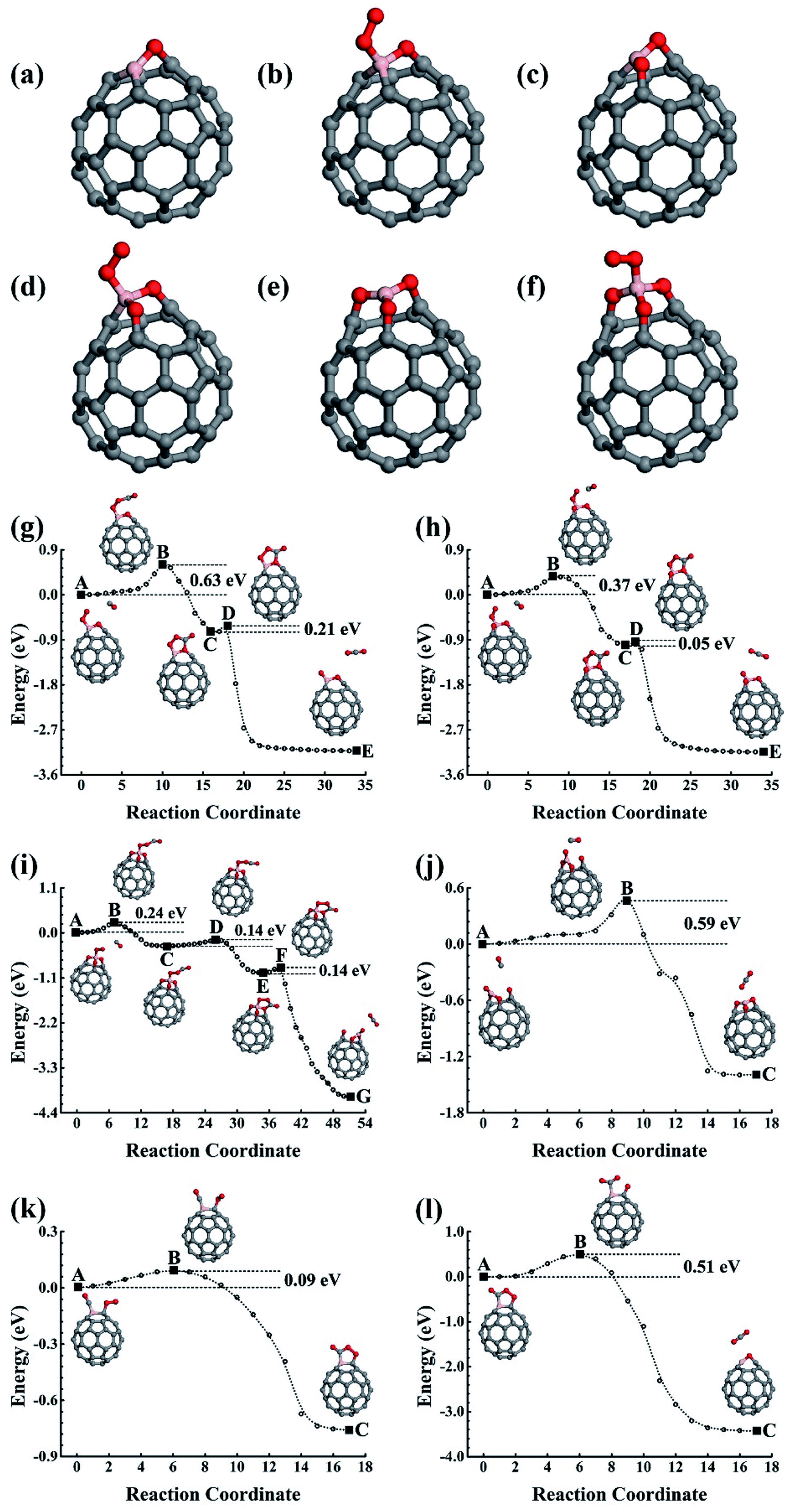 | ||
| Fig. 2 (a)–(f) Show the structural skeleton illustrations for C59BO, C59BO2, C59BO3 oxides, and the O2 molecule adsorption configurations on the different oxide structures of C59B, respectively. (g)–(l) Show the reaction paths for CO oxidization on C59B oxide catalysts. The initial, final, saddle, and meta-stable intermediate states are illustrated by the inset images. C, B, and O are represented by the grey, pink, and red balls. | ||
 | ||
| Fig. 3 (a) Structural skeleton illustrations for the C59NO oxide; and (b) the lowest energy structure of O2 molecules on the C59N oxide. (c) The structure of C59NO2. (d)–(f) The optimized reaction paths for CO oxidization on C59N and its oxides catalysts. The initial, final, saddle, and meta-stable intermediate states are illustrated in the inset images. C, N, and O are represented by the grey, blue, and red balls. | ||
In addition to the singly doped fullerene, a low doping concentration structure will probably also be generated during the fabrication of doped fullerene. Previous studies have shown that the B(N) dopants prefer to separately substitute the carbon atom in fullerene.28–30 For example, the best doping configurations for the two B and N atoms are that the dopants substitutionally occupy the opposite sites of a C6 hexagon. In our studies, we have studied the catalytic properties of the best doping configurations of C58B2 and C58N2. Like the singly doped fullerene, they evolve into the oxide materials after the first few catalytic cycles for catalyzing CO oxidizations. When compared to the oxides of C59B and C59N, our calculations do not show obvious synergetic effects on the catalytic properties of the C58B2 and C58N2 oxides (see Fig. S1 and S2 in the ESI†).
3.2 BN-pair doped fullerene
In the experiment, the B and N can also co-doped in fullerene, and the B–N bond is probably formed in the lowest energy doping configuration. The B–N bonding obviously affects their effects upon the co-doped nanostructures, which could be used to engineer the physical and chemical properties of the carbon nanostructures. With the purpose of helping design high-performance nanostructures in the experiments, we also carried out careful studies on the BN-pair doped fullerene to shed light on the catalytic properties of the low-concentration substitution doping configurations of the B–N co-doped carbon nanostructures. The stability of the BN-pair doped fullerene has also been confirmed using FPMD simulations performed for 5 ps at 1000 K.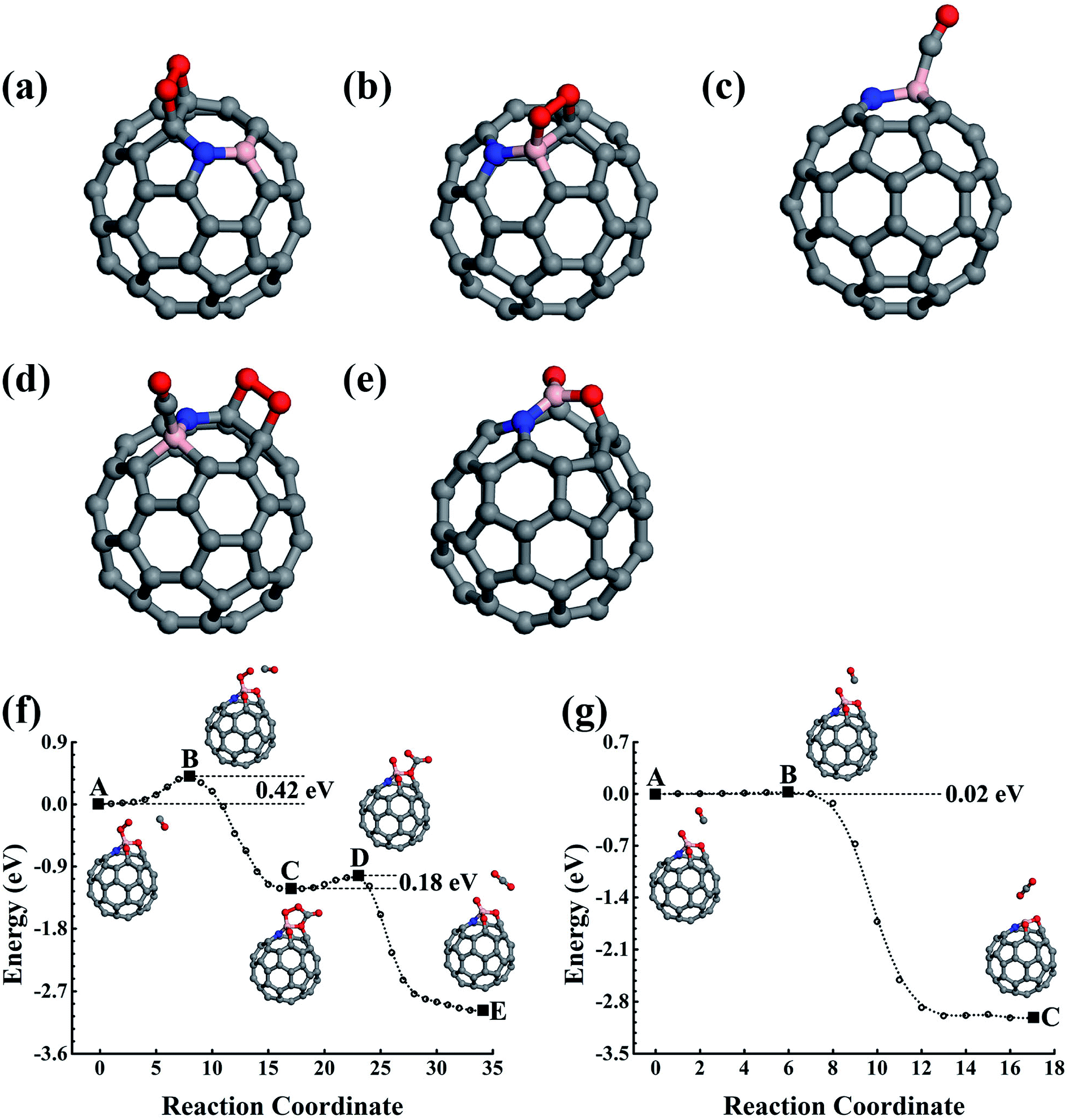 | ||
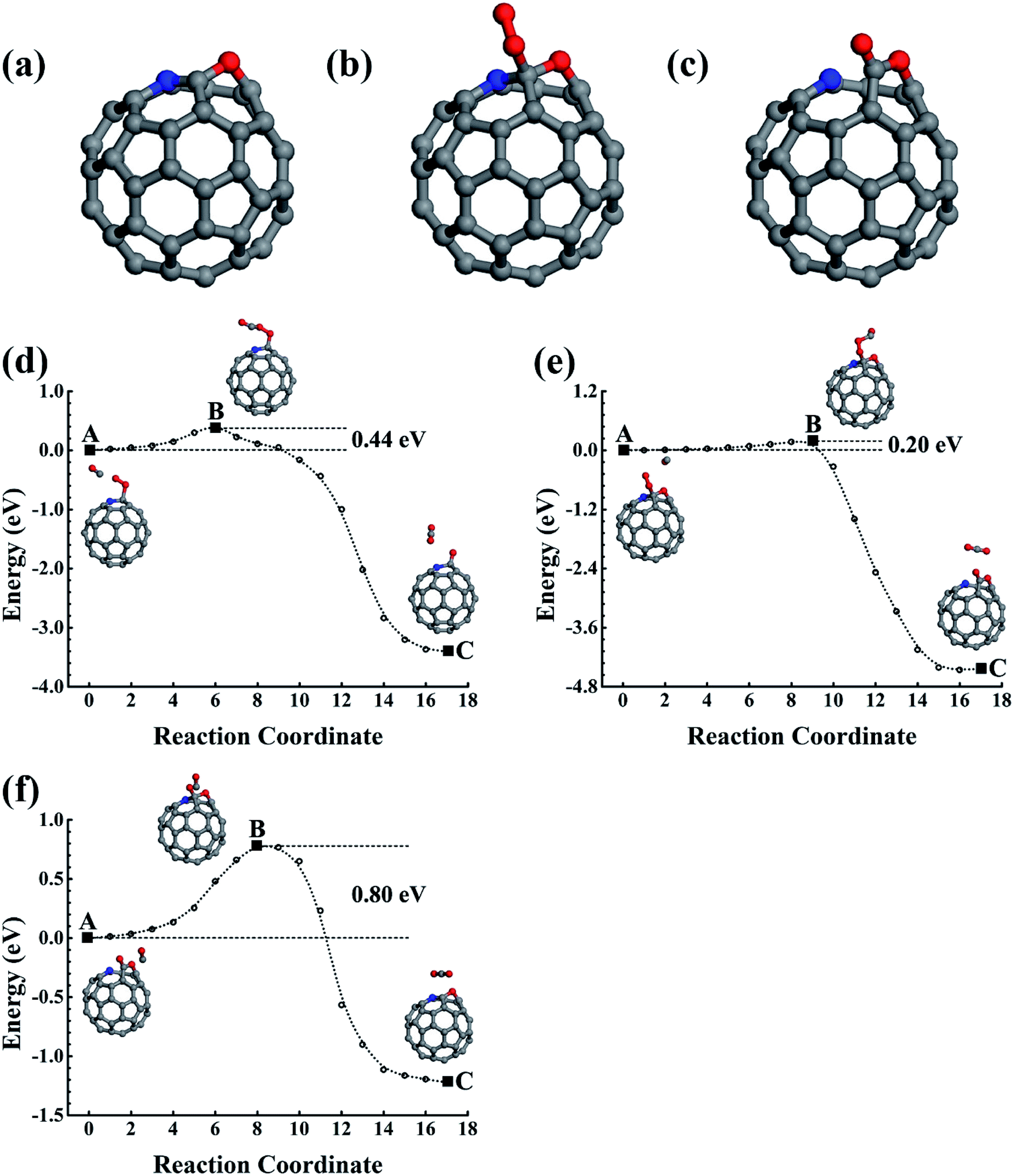
| Fig. 4 The structures of O2 adsorption (a) and (b), CO adsorption (c), and CO and O2 co-adsorption (d) on C58BN. (e) The structure of C58BNO2 oxide. (f) and (g) The reaction pathways for CO oxidization catalyzed by C58BNO2 oxide. C, B, N, and O are represented by the grey, pink, blue, and red balls. | ||
3.3 B3N and BN3 doped fullerene
In addition to the smallest stoichiometric BN-pair doping, we also estimated the nonstoichiometric B–N doping in fullerene. The B3N(BN3), with the N(B) being surrounded by three B(N) atoms, were used as examples to illustrate the B- and N-rich nonstoichiometric co-doping. The doping configurations are shown in Fig. 5a and b, which are confirmed by our structural optimizations as being the lowest energy doping configurations. We also performed 5 ps FPMD simulations at 1000 K for the lowest energy doping configurations of C56B3N and C56BN3, which suggested that they were stable.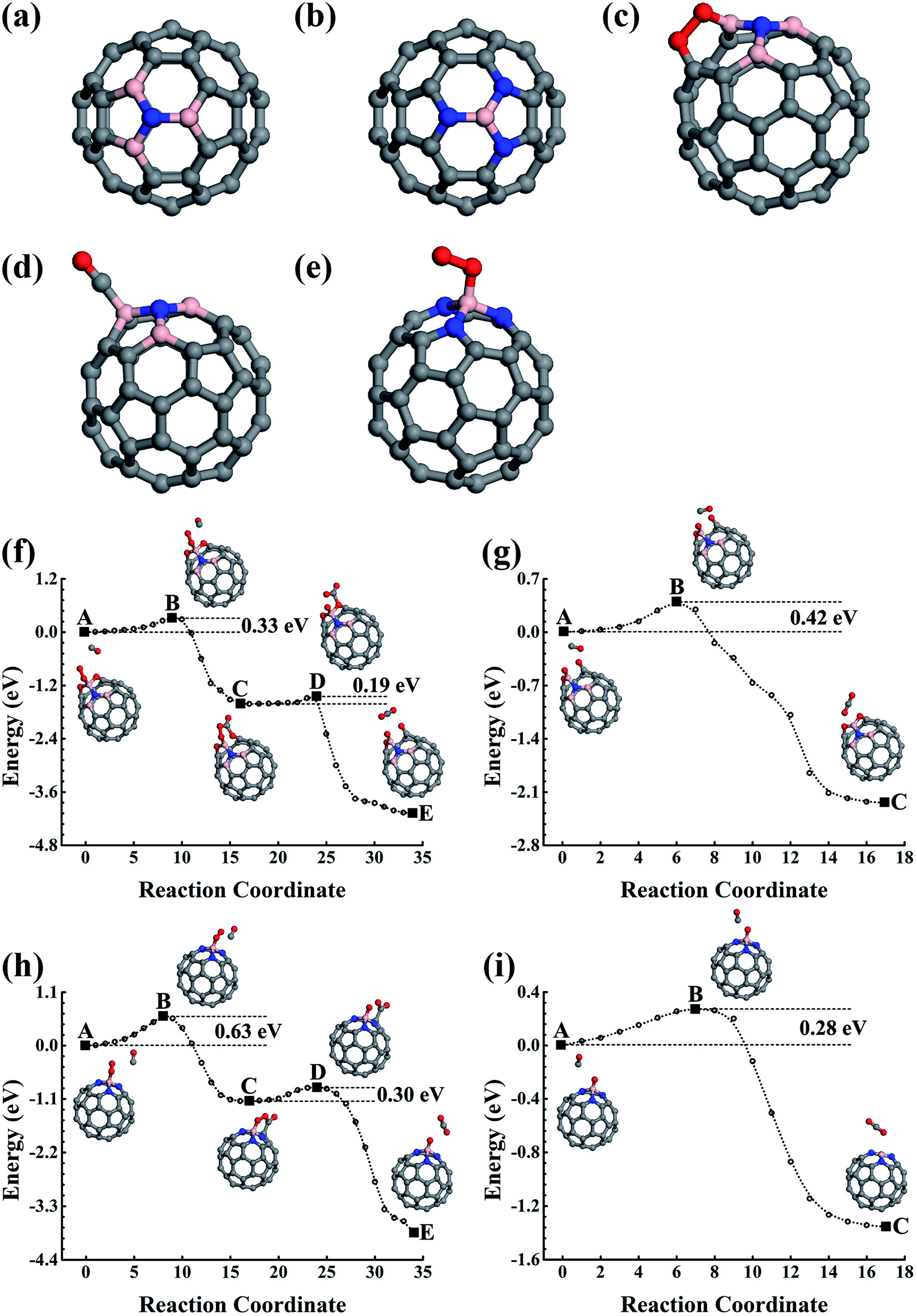 | ||
| Fig. 5 (a)–(e) Structures of B3N and BN3 doped fullerenes and the corresponding gas molecule adsorption configurations on them. (f) and (g) The reaction pathways of CO oxidizations catalyzed by the C56B3N oxide. (h) and (i) Reactions catalyzed by C56BN3. C, B, N, and O are represented by the grey, pink, blue, and red balls. | ||
4. Conclusions
Using elastic band method calculations based on first-principles studies, we have carefully studied low-concentration B and N doped and their co-doped fullerenes. Both C59B and C59N show good catalytic properties for catalyzing CO oxidization, which could also be capable of being oxidized during the first few catalytic reaction cycles. The corresponding oxides were found to have superior catalytic properties, which could be repeatedly used as a catalyst showing durable properties as well. The CO and O2 could be co-adsorbed onto C59B. The co-adsorption configurations require 0.48 or 0.44 eV activation energies to make the CO oxidization occur and produce a C59BO oxide simultaneously, which could be further oxidized in the successive CO oxidization processes to obtain a C59BO3 oxide. In addition, the single O2 adsorption on C59B can also oxidize it with a 0.24 eV barrier. Providing it is used in CO-rich conditions, C59N could retain its structure as a recycling catalyst for CO oxidization. A 0.44 eV rate determining barrier is found. Otherwise, after oxidizing the CO, the produced atomic O atom can overcome the 0.20 eV to oxidize the C59N. Both the C59B and C59N oxides could be used as durable recycling catalysts, which are found to have 0.59 and 0.80 eV rate determining energy barriers, respectively. Interestingly, the co-doping of B and N in low concentration doping fullerene may have superior catalytic properties. The BN-pair doped fullerene would also be oxidized. The produced C58BN oxide can help to oxidize CO with a 0.42 eV rate determining barrier. For the nonstoichiometric B3N and BN3 species, their doping in fullerene is also found to have superior catalytic properties. The B atoms either at the doping domain edge in B3N or at the doping center in BN3 are active sites. C56B3N can be oxidized during catalyzing CO oxidizations. The resulting C56B3N oxide has a rate determining barrier of 0.42 eV for catalyzing successive CO oxidizations. In C56BN3, the central B is protected by its surrounding N atoms against oxidization to remain as a recycling catalytic active site for CO oxidization, which is found to have a 0.63 eV rate determining energy barrier.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC) (Grant No. 11674129).References
- H. Falsig, B. Hvolbœk, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4835 CrossRef CAS PubMed.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2002, 124, 14770 CrossRef CAS PubMed.
- H.-T. Chen, J.-G. Chang, S.-P. Ju and H.-L. Chen, J. Comput. Chem., 2010, 31, 258 CAS.
- J. L. C. Fajín, M. N. D. S. Cordeiro and J. R. B. Gomes, J. Phys. Chem. C, 2008, 112, 17291 CrossRef.
- J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398 CrossRef CAS PubMed.
- H.-L. Chen, C.-H. Su and H.-T. Chen, Chem. Phys. Lett., 2012, 536, 100 CrossRef CAS.
- J. Zhang, H. Jin, M. B. Sullivan, F. C. H. Lim and P. Wu, Phys. Chem. Chem. Phys., 2009, 11, 1441 RSC.
- H.-Y. Su, M.-M. Yang, X.-H. Bao and W.-X. Li, J. Phys. Chem. C, 2008, 112, 17303 CrossRef CAS.
- X.-Q. Gong, Z.-P. Liu, R. Raval and P. Hu, J. Am. Chem. Soc., 2004, 126, 8 CrossRef CAS.
- J. T. Hirvi, T.-J. J. Kinnunen, M. Suvanto, T. A. Pakkanen and J. K. Nørskov, J. Chem. Phys., 2010, 133, 084704 CrossRef.
- S.-H. Oh and G. B. Hoflund, J. Catal., 2007, 245, 35 CrossRef CAS.
- H.-T. Chen, J. Phys. Chem. C, 2012, 116, 6239 CrossRef CAS.
- H.-T. Chen and J.-G. Chang, J. Phys. Chem. C, 2011, 115, 14745 CrossRef CAS.
- L.-C. Hsu, M.-K. Tsai, Y.-H. Lu and H.-T. Chen, J. Phys. Chem. C, 2012, 117, 433 CrossRef.
- M. Huang and S. Fabris, J. Phys. Chem. C, 2008, 112, 8643 CrossRef CAS.
- Z.-P. Liu, X.-Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef.
- C.-M. Wang, K.-N. Fan and Z.-P. Liu, J. Am. Chem. Soc., 2007, 129, 2642 CrossRef CAS.
- G. Chen, S. J. Li, Y. Su, V. Wang, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. C, 2011, 115, 20168 CrossRef CAS.
- X. Fan, J. Li and G. Chen, RSC Adv., 2017, 7, 17417 RSC.
- D. W. Yuan, Z. R. Liu and J. H. Chen, J. Chem. Phys., 2011, 134, 054704 CrossRef CAS.
- D. W. Yuan and Z. Zeng, J. Chem. Phys., 2004, 120, 6574 CrossRef CAS.
- J. Zhang, Z. Fu, Z. Yang and S. F. Li, Phys. Lett. A, 2012, 376, 3235 CrossRef CAS.
- Y.-P. Xie and X.-G. Gong, J. Chem. Phys., 2010, 132, 244302 CrossRef.
- X. Chen, Phys. Chem. Chem. Phys., 2015, 17, 29340 RSC.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207 CrossRef CAS.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023 RSC.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110 CrossRef CAS.
- M. R. Manaa, Solid State Commun., 2004, 129, 379 CrossRef CAS.
- M. R. Manaa, D. W. Sprehn and H. A. Ichord, J. Am. Chem. Soc., 2002, 124, 13990 CrossRef CAS.
- M. R. Manaa, H. A. Ichord and D. W. Sprehn, Chem. Phys. Lett., 2003, 378, 449 CrossRef.
- X. B. Hu, Y. T. Wu and Z. B. Zhang, J. Mater. Chem., 2012, 22, 15198 RSC.
- I. H. Lin, Y. H. Lu and H.-T. Chen, Phys. Chem. Chem. Phys., 2016, 18, 12093 RSC.
- I.-H. Lin, Y.-H. Lu and H.-T. Chen, J. Comput. Chem., 2017, 38, 2041 CrossRef CAS.
- R. Krishnan, S.-Y. Wu and H.-T. Chen, Carbon, 2018, 132, 257 CrossRef CAS.
- M. D. Esrafili, R. Mohammad-Valipour, S. M. Mousavi-Khoshdel and P. Nematollahi, ChemPhysChem, 2015, 16, 3719 CrossRef CAS PubMed.
- Y. Tang, W. Chen, Z. Shen, S. Chang, M. Zhao and X. Dai, Carbon, 2017, 111, 448 CrossRef CAS.
- Y. Tang, Z. Liu, X. Dai, Z. Yang, W. Chen, D. Ma and Z. Lu, Appl. Surf. Sci., 2014, 308, 402 CrossRef CAS.
- S. Sinthika, E. M. Kumar and R. Thapa, J. Mater. Chem. A, 2014, 2, 12812 RSC.
- S. Nigam and M. Chiranjib, ACS Nano, 2008, 2, 1422 CrossRef CAS.
- G. Kress and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef.
- G. Kress and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- Q. Z. Li, J. J. Zheng, J. S. Dang and X. Zhao, ChemPhysChem, 2015, 16, 390 CrossRef CAS.
- Y. Wang, M. Jiao, W. Song and Z. Wu, Carbon, 2017, 114, 393 CrossRef CAS.
- F. Gao, G. Zhao, S. Yang and J. Spivey, J. Am. Chem. Soc., 2013, 135, 3315 CrossRef CAS.
- C. Ricca, F. Labat, C. Zavala, N. Russo, C. Adamo, G. Merino and E. Sicilia, J. Comput. Chem., 2018, 39, 637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02172h |
| This journal is © The Royal Society of Chemistry 2019 |