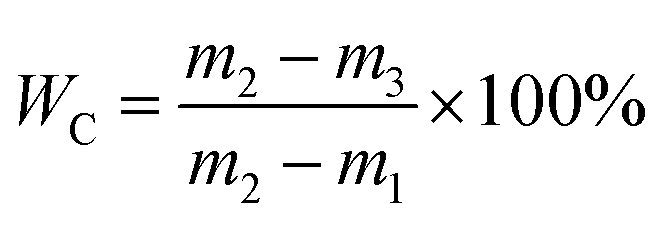DOI:
10.1039/C9RA03781K
(Paper)
RSC Adv., 2019,
9, 27593-27601
Enhanced dewaterability of waste activated sludge by a combined use of permanganate and peroxymonosulfate
Received
20th May 2019
, Accepted 21st August 2019
First published on 2nd September 2019
Abstract
Ever-increasing efforts have been made to develop rapid and practical conditioning methods of sludge dewatering. This study demonstrated an innovative combination of potassium permanganate (KMnO4) and peroxymonosulfate (PMS) for sludge dewatering. The combined use of KMnO4 and PMS (KMnO4/PMS) showed its superiority in improving sludge dewaterability over the separate use of KMnO4 or PMS. By dosing 4 mmol g−1 VSS KMnO4 and 3 mmol g−1 VSS PMS, the dewaterability of waste activated sludge (WAS) significantly enhanced as capillary suction time (CST) decreased from 73.65 s to 24.65 s while the water content of dewatered sludge cake (WC) decreased from 78.96% to 70.47%. Apart from CST and WC, the KMnO4/PMS process could also affect negative zeta potential, sludge flocs size and the concentrations of protein and polysaccharide in extracellular polymeric substances (EPS). The enhanced sludge dewaterability and changes of the physicochemical characteristics of the WAS samples during the KMnO4/PMS process were actually ascribed to sulfate radicals (SO4˙−) and hydroxyl radicals (HO˙) in situ generated via PMS activation by manganese oxides (MnOx) in the states of MnO2 and Mn3O4 transferred from KMnO4 oxidation, which was verified by transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX) techniques and radical scavenging experiments. Moreover, the Fourier transform infrared spectroscopy (FTIR) analysis further confirmed that the in situ generated SO4˙− and HO˙ could improve sludge dewaterability. Thus, the KMnO4/PMS process could be considered as a promising conditioning method of sludge dewatering.
1. Introduction
The management of large quantities of waste activated sludge (WAS) produced from activated sludge processes has posed great challenges to municipal wastewater treatment plants (WWTPs) due to great economic and environmental burden.1–4 As reported, the treatment and disposal of excess sludge usually accounts for up to 60% of the total operating cost in WWTPs.2 Consequently, how to substantially cut down this kind of cost has become a significant issue, and many researchers and practitioners have tried their best to tackle this problem. WAS has a high water content in the form of free and bound water (over 90%).5 And sludge dewatering has been generally considered as a promising strategy in reducing sludge volume and cost of sludge transport and ultimate disposal.3,4 However, the sludge dewatering process is usually inhibited due to the presence of extracellular polymeric substances (EPS) that can retain a large amount of water, especially bound water.6,7 Hence, increasing efforts have been focused on development of efficient sludge dewatering techniques, which can promote the release of bound water from EPS. So far, thermal, ultrasonication, freezing and thawing, and chemical oxidation have been investigated to improve sludge dewaterability.7–12 Among all the developed methods, advanced oxidation processes (AOPs) have recently shown their superiority in sludge disintegration and dewatering.
AOPs usually rely on the produced highly reactive radical species, such as hydroxyl radicals (HO˙) and sulfate radicals (SO4˙−) to disrupt WAS and degrade EPS, thereby enhancing sludge dewaterability.2,13,14 It has been reported that the Fenton (combination of H2O2 and Fe2+) and Fenton-like (e.g., simultaneous addition of H2O2 and Fe3+) processes based on HO˙ production could efficiently improve the dewaterability of WAS.1,7 However, only under strongly acidic conditions (pH = 2–5) can these processes generate sufficient amounts of HO˙ for better sludge dewaterability, which significantly hindered the sludge dewatering process and subsequent disposal.15–17 Compared to HO˙ (E0 = 1.8–2.7 V), SO4˙− owns a higher redox potential (E0 = 2.5–3.1 V), a longer life time (30–40 μs) and a wider working pH range (pH 4–9).4,18 Therefore, pretty much attention has been recently paid to the SO4˙−-based AOPs as new alternative methods of sludge conditioning. SO4˙− is usually generated through the activation of peroxydisulfate (PDS, S2O82−) and peroxymonosulfate (PMS, HSO5−) by ultraviolet (UV), heat, base and (transition) metal catalysts.19–25
Our recent study investigated α-MnO2 with different morphologies (nanoparticles, nanoflowers and nanorods) synthesized via a facile hydrothermal method as activators of PMS for ciprofloxacin (CIP) degradation, and found that α-MnO2 nanoflowers achieved much higher degradation efficiency of CIP than the other three MnO2 (α-MnO2 nanorods, α-MnO2 nanoparticles and commercial MnO2), which would be attributed to its higher catalytic activity of PMS and larger quantities of SO4˙− produced from its activation of PMS.26 Interestingly, Cui et al. recently studied chemical oxidation of benzene and trichloroethylene (TCE) by an innovative combined use of permanganate (KMnO4) and PMS (KMnO4/PMS), and also found that it was mainly colloidal and amorphous MnO2 in situ generated from KMnO4 oxidation that activated PMS to trigger powerful SO4˙−-mediated oxidation of both benzene and TCE.27 As known to us all, both KMnO4 and PMS have been widely used as powerful oxidants to destruct organic pollutants in water and wastewater. Recently, the two powerful oxidants were also applied to enhance the filterability of waste activated sludge.28,29 Wu et al. reported that KMnO4 could efficiently disintegrate the excess sludge with soluble chemical oxygen demand (SCOD) increasing by 3473%, and that the optimal KMnO4/sludge solid mass ratio was 0.1 with a stable disintegration degree (DDCOD) of about 34% while suitable reaction time was 30 min.28 Niu et al. found that PMS oxidation could effectively break sludge particles, and that EPS increased significantly and transferred to slime layer after PMS treatment.29 Yang et al. also reported that PMS could facilitate the disintegration of WAS and the biodegradability of organics could be enhanced after treatment by PMS.30 Although it has been proven that KMnO4 and PMS could disintegrate WAS and enhance sludge dewaterability, the synergistic effect of both oxidants on the dewaterability of WAS still remains unknown.
In this paper, we proposed a combined use of KMnO4 and PMS (KMnO4/PMS) for sludge dewatering. The dewaterability of WAS during the KMnO4/PMS process by the measurement of variations in capillary suction time (CST) and water content of dewatered sludge cake (WC) was initially investigated. Then, the physicochemical characteristics of WAS (CST, WC, zeta potential, particle size and the concentrations of protein and polysaccharide in EPS) at different dosages of KMnO4 and PMS were further studied. Finally, the mechanism of sludge dewatering by the KMnO4/PMS process was deeply explored.
2. Materials and methods
2.1. Raw sludge and chemicals
The raw sludge was collected from Qige municipal wastewater treatment plant in Hangzhou, China. A sieve with 20 mesh was then used to remove big gravels and debris from the collected sludge samples. The supernatant was finally decanted to acquire the denser sludge samples used in this study after plain sedimentation. The acquired denser sludge samples were stored at 4 °C in a refrigerator prior to use, and all the experiments were completed within 48 h. The basic characteristics of the sludge samples were measured and shown in Table 1.
Table 1 Basic characteristics of sludge samples
| Parameter |
Unit |
Value |
| pH |
— |
6.85 ± 0.05 |
| Water content |
% |
96.205 ± 0.005 |
| Volatile suspended solids (VSS) |
g L−1 |
30.364 ± 0.394 |
| Total suspended solids (TSS) |
g L−1 |
39.133 ± 0.951 |
| VSS/TSS |
% |
77.355 ± 0.875 |
| CST |
s |
73.65 ± 1.45 |
All the chemicals were used without further purification. Peroxymonosulfate (Oxone®, KHSO5·0.5 KHSO4·0.5 K2SO4, KHSO5 ≥47%) was provided by Aladdin Co., Ltd. (Shanghai, China). Permanganate (KMnO4, ≥99.5%), sodium hydroxide (NaOH, ≥96.0%), and sulfuric acid (H2SO4, 95.0–98.0%) were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
2.2. Experimental procedures
All the sludge dewatering experiments were conducted in an Erlenmeyer flask with a volume of 500 mL. In each test, 300 mL sludge sample was added and then mixed with a stoichiometric amount of PMS. When PMS was completely dissolved under thorough stirring, H2SO4 or NaOH was used to adjust the solution pH to 7. Finally, the reaction was immediately initiated by dosing a calculated amount of KMnO4 in a water bath apparatus (300 rpm), and was ceased after 120 min. All the experiments were duplicated at least, and the average values with standard deviations were presented.
2.3. Analytical methods
2.3.1. CST and WC determination. The dewaterability of WAS is commonly evaluated by CST and WC. CST was measured using a CST instrument (Type 304, Triton Ltd., UK) equipped with an 18 mm diameter funnel and a standard Whatman no. 17 chromatography-grade paper. To determine WC, the sludge samples were pretreated by a vacuum filtration method. Briefly, 50 mL sludge sample was poured into a standard Buchner funnel and filtered at a constant vacuum pressure for 15 min. The water content of the vacuum-filtered sludge cake trapped by the filter paper was measured according to the standard method, and calculated according to eqn (1):| |
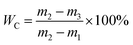 | (1) |
where m1 is the weight of the filter paper, m2 is the weight of the vacuum-filtered sludge cake, and m3 is the weight of the vacuum-filtered sludge cake completely dried at 105 °C.
2.3.2. EPS extraction and analysis. EPS were extracted using a heat extraction method described by Yu et al.6 Briefly, 50 mL sludge sample was centrifuged at 4000g for 5 min at 4 °C to acquire the supernatant as soluble EPS (S-EPS). The sludge pellets were then diluted with a 0.05 w/v % NaCl solution that was pre-heated to 70 °C. The diluted sludge solution was immediately sheared by a vortex mixer for 1 min and centrifuged at 4000g for 10 min at 4 °C to collect the supernatant as the loosely bound EPS (LB-EPS). The residual sludge pellets were resuspended in a 0.05 w/v % NaCl solution and heated to 60 °C in a water bath for 30 min. At short notice, the resuspended sludge solution was centrifuged at 4000g for 15 min at 4 °C, and the supernatant was collected as the tightly bound EPS (TB-EPS).Before the analyses of protein and polysaccharide (assumed as dominant components of EPS), the EPS samples were pretreated with an optimal amount of Na2SO3 to quench the residual colored KMnO4. The pretreated EPS sample was then filtered through a 0.45 μm cellulose acetate membrane. The protein content was quantified by the modified Lowry–Folin method using bovine serum albumin as the standard.31 The polysaccharide content was determined by the phenol-sulfuric acid method using glucose as the standard.32
2.3.3. Other analyses. The zeta potential of the WAS samples was analyzed using a Zetasizer Nano (Nano-ZS90, Malvern Instrument Ltd., UK). The particle size distribution was measured by a laser particle size analyzer (LAP-W2000H) using ultrapure water as the dispersant. The Fourier transform infrared spectroscopy (FTIR) analysis of sludge samples was recorded in a wavenumber range of 4000–400 cm−1 by a Nicolet 6700 spectrometer (Thermo, USA).Given the difficulty in separating the formed solids from the conditioned WAS samples, the extracted EPS solution was used and treated with the KMnO4/PMS process to monitor whether manganese oxides (MnOx) formed after KMnO4 oxidation of the organics in EPS. Briefly, after the extracted EPS solution was treated with the KMnO4/PMS process, the reaction solution was filtered through a 0.45 μm cellulose acetate membrane to collect the brown-red solids. Then, the collected solids were rinsed alternately several times using ethanol and ultrapure water, and vacuum-dried at 60 °C overnight. Finally, the dried solid samples were scanned by transmission electron microscopy (TEM) to capture the microscale crystallization and structure and energy dispersive X-ray spectroscopy (EDX) to analyze the elemental composition.
3. Results and discussion
3.1. Feasibility of sludge dewatering with the KMnO4/PMS process
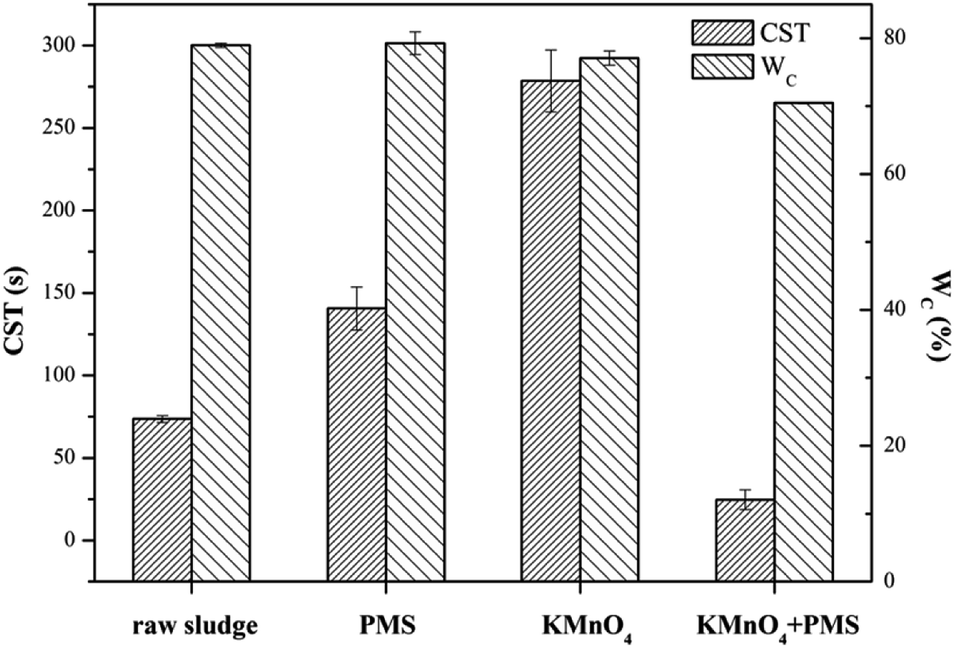
Fig. 1 compared the dewaterability of the WAS samples conditioned by KMnO4 alone, PMS alone, and a combination of KMnO4 and PMS (KMnO4/PMS), respectively. As observed, the CST increased from 73.65 s to 140.60 s and 278.55 s, respectively when the WAS samples were conditioned by 7 mmol g−1 VSS PMS and KMnO4 alone, respectively, which indicated that neither PMS alone nor KMnO4 alone could efficiently improve the sludge dewaterability, and that the two individuals even worsened the dewaterability. The deterioration of sludge dewaterability was likely resulted from the progressive cell lysis and further releasing of intracellular biopolymers under the influence of KMnO4 or PMS when the intracellular biopolymers could not be degraded effectively in current condition.33 Lee et al. also found that the PMS conditioning could deteriorate the dewaterability after the WAS samples were treated with different dosages of PMS at room temperature.5 Surprisingly, a combined use of 4 mmol g−1 VSS KMnO4 and 3 mmol g−1 VSS PMS instantly decreased the CST from 73.65 s to 24.65 s, and apparently showed its synergistic effect on significantly improving the sludge dewaterability. Besides, the decrease of WC (from 78.96% to 70.47%) also indicated the synergistic effect after the WAS samples were conditioned by the KMnO4/PMS process while the WC values almost remained unchanged after the samples were treated with PMS alone and KMnO4 alone (79.25% and 77.07%, respectively). The synergistic effect of the KMnO4/PMS process on the WAS dewaterability was interpreted with the decrease of both CST and WC, which also implied the generation of SO4˙− and HO˙ during the KMnO4/PMS process as mentioned in Section 1.
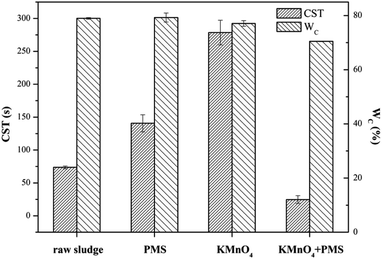 |
| | Fig. 1 Dewaterability change in the WAS samples conditioned by different methods (experimental conditions: [KMnO4] = [PMS] = 7 mmol g−1 VSS, [KMnO4] + [PMS] = 4 mmol g−1 VSS + 3 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
3.2. Effect of the KMnO4/PMS process on the physicochemical characteristics of the WAS samples
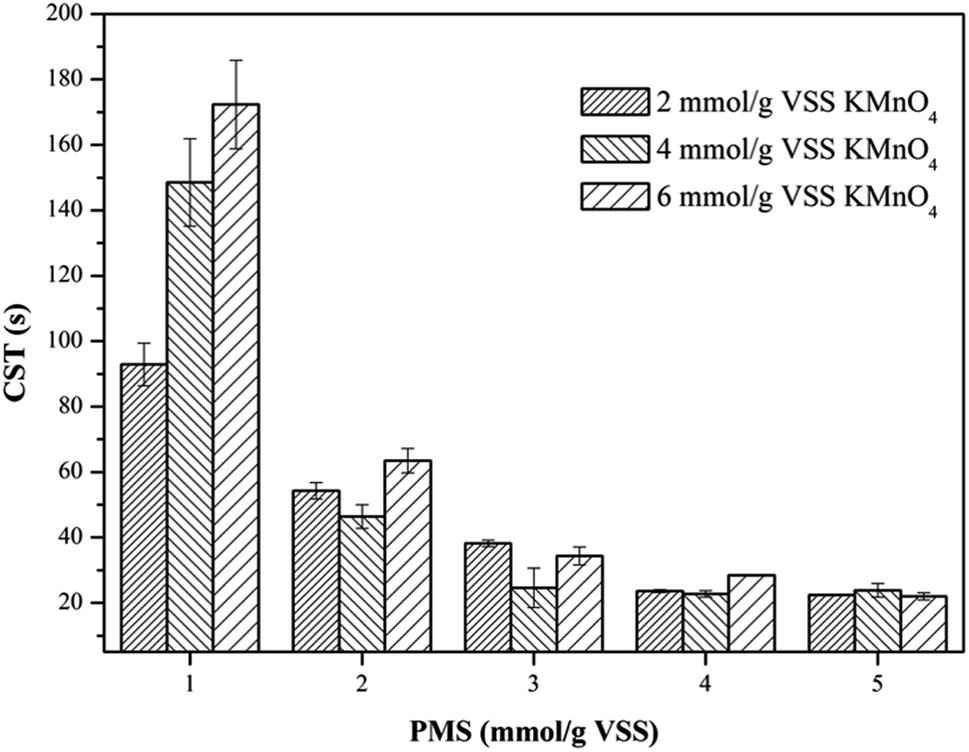
3.2.1. Capillary suction time (CST). The dewaterability of the WAS samples were further investigated by simultaneously dosing different amounts of KMnO4 and PMS. As shown in Fig. 2, when PMS dosage was fixed at 1 mmol g−1 VSS, the increase of KMnO4 dosage from 2 mmol g−1 VSS to 6 mmol g−1 VSS did not decrease the CST, and instead jeopardized the dewaterability, which indicated the absence of the synergistic effect of the KMnO4/PMS process at a low PMS dosage. This might be because the in situ generated SO4˙− and HO˙ during the KMnO4/PMS process was too few to improve the dewaterability of the WAS samples when PMS dosage was too low. However, increasing PMS dosage to 2 mmol g−1 VSS efficiently enhanced the dewaterability, and the CST decreased from 73.65 s to 54.30 s, 46.45 s and 63.45 s, respectively with increasing KMnO4 dosage from 2 mmol g−1 VSS to 6 mmol g−1 VSS. When PMS dosage continuously increased to 5 mmol g−1 VSS, the CST decreased to 22.45 s, 23.85 s and 22.00 s, respectively as KMnO4 dosage increased from 2 mmol g−1 VSS to 6 mmol g−1 VSS. This is because the higher PMS dosage, the more SO4˙− and HO˙ generated to achieve better sludge dewaterability.34 The similar results were also reported by Liu et al. when the Fe2+-activated PMS was used for sludge conditioning.4 Remarkably, the CST values did not decrease all the way, and instead decreased firstly and then increased with sustainably increasing KMnO4 dosage, which might be ascribed to the consumption of the generated SO4˙− and HO˙ by KMnO4 with a too high dosage.35 Liu et al. also observed the similar results when using the magnetic Fe3O4–MnO2 core–shell nanocomposites-activated PMS for 4-chlorophenol degradation.36
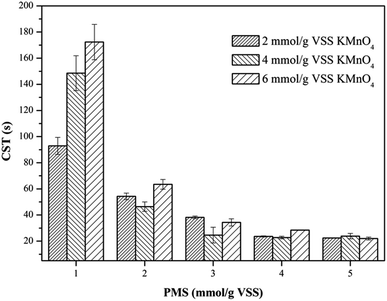 |
| | Fig. 2 Effect of the KMnO4/PMS process on CST of the WAS samples (experimental conditions: [KMnO4] = 2–6 mmol g−1 VSS, [PMS] = 1–5 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
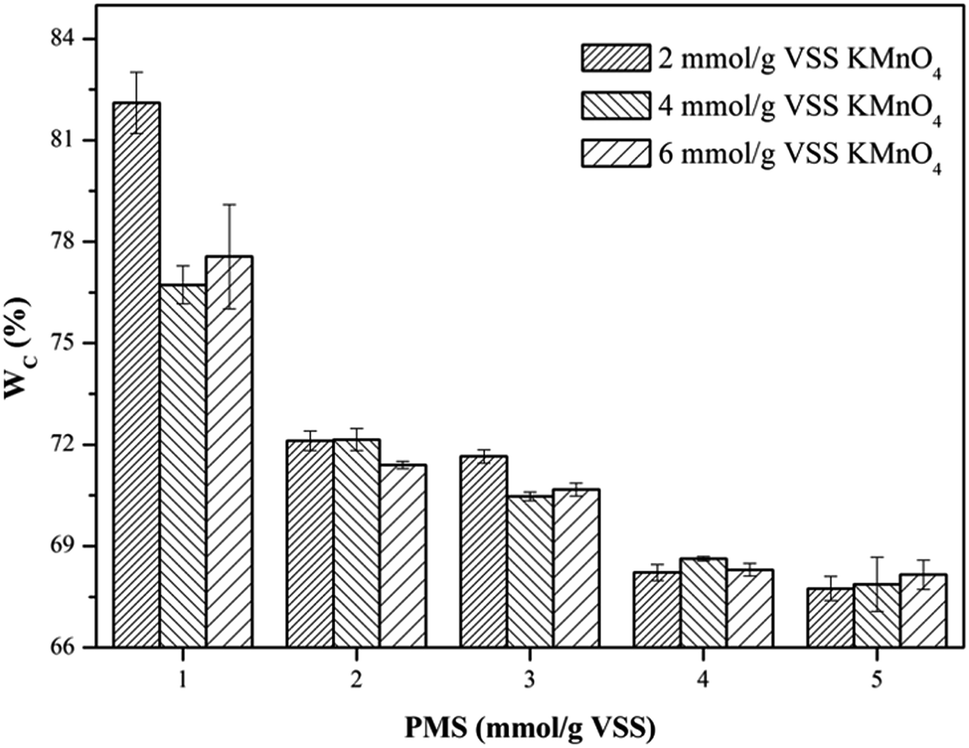
3.2.2. Water content of dewatered sludge cake (WC). The profiles of WC of the WAS samples after conditioning of the KMnO4/PMS process with different KMnO4 and PMS dosages were depicted in Fig. 3. As can be seen, the WC changes in the WAS samples were quite similar to the CST changes discussed in Section 3.2.1. By dosing 1 mmol g−1 VSS PMS, the WC after conditioning with 2 mmol g−1 VSS, 4 mmol g−1 VSS and 6 mmol g−1 VSS KMnO4, respectively (82.11%, 76.73% and 77.56%) almost remained unchanged, compared to the WC of the raw sludge (78.96%). This suggested that the synergistic effect of the KMnO4/PMS process with 1 mmol g−1 VSS PMS would not work. However, the WC of the WAS samples would continue to decrease with the ever-increasing PMS dosages, which was also attributed to more SO4˙− and HO˙ generated from the KMnO4/PMS process. The WC after conditioning could decrease to about 67.5%, and by 11.46% compared to the WC of the raw sludge, which was higher than the variation in the WC of the WAS samples conditioned by the Fenton-like process (8.7%).1
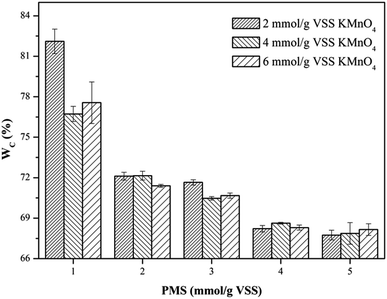 |
| | Fig. 3 Effect of the KMnO4/PMS process on WC of the WAS samples (experimental conditions: [KMnO4] = 2–6 mmol g−1 VSS, [PMS] = 1–5 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
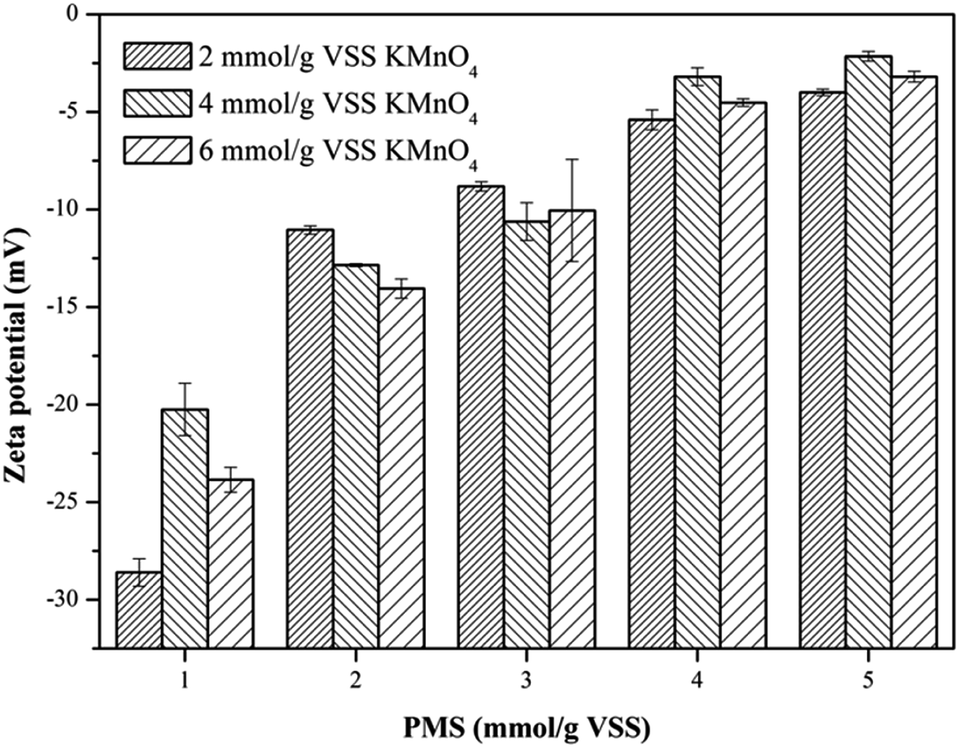
3.2.3. Zeta potential. To deeply investigate the effect of the KMnO4/PMS process on the WAS dewaterability, the zeta potentials of the WAS samples before and after conditioning were also measured. The unconditioned raw sludge was negatively charged with the zeta potential of −17.7 mV, which was due to the ionization of the carboxyl and amino in the protein of EPS.37 It can be obviously observed from Fig. 4 that the zeta potentials of the WAS samples were affected by dosing either KMnO4 or PMS. The zeta potential tended to increase to 0 mV with increasing KMnO4 or PMS dosage. By simultaneously dosing 4 mmol g−1 VSS KMnO4 and 5 mmol g−1 VSS PMS, the zeta potential increased to −2.15 mV. The increase in the zeta potential suggested the ever-increasing sludge dewaterability, which quite matched with the changes of the CST and WC. Zhen et al. also reported that the sludge dewaterability improved as the zeta potential increased in the Fe2+-activated persulfate process.38 According to the DLVO theory, the aggregation of sludge flocs is mainly controlled by the surface charge. The decrease in the negative surface charge of sludge flocs would reduce the electrostatic repulsion and bring about the increment of interaction energy, which led to the aggregation of sludge flocs and finally enhanced sludge dewaterability. Therefore, it can be concluded that sludge flocs are easier for agglomeration and sedimentation, and thus dewatering as the zeta potential increases.39
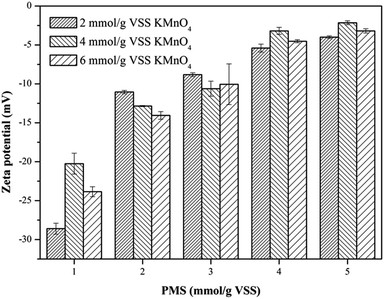 |
| | Fig. 4 Effect of the KMnO4/PMS process on zeta potential of the WAS samples (experimental conditions: [KMnO4] = 2–6 mmol g−1 VSS, [PMS] = 1–5 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
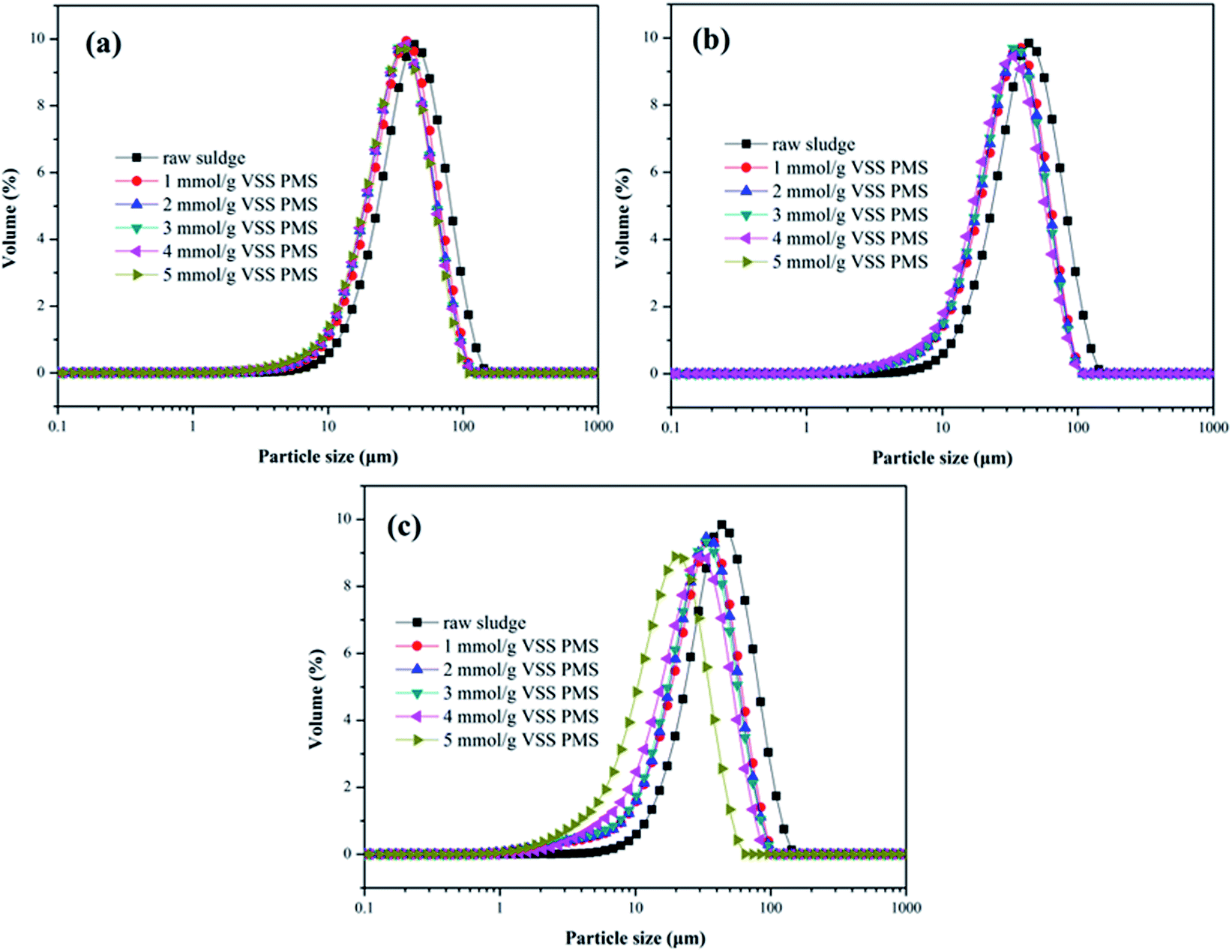
3.2.4. Particle size distribution. As depicted in Fig. 5, effect of the KMnO4/PMS process on particle size distribution of the WAS samples was also investigated. The D50 value of the unconditioned sludge flocs was determined to be 39.2 μm. When the WAS samples were conditioned with 2 mmol g−1 VSS KMnO4 and 1 mmol g−1 VSS PMS, the D50 value decreased to 32.95 μm. With PMS dosage increasing to 5 mmol g−1 VSS, the D50 value also decreased to 30.08 μm. By dosing 4 mmol g−1 VSS and 6 mmol g−1 VSS KMnO4, the D50 values of the conditioned sludge flocs decreased from 30.42 μm and 28.35 μm to 25.78 μm and 24.44 μm, respectively with increasing PMS dosage. Therefore, increasing both KMnO4 and PMS dosages could reduce the size of the conditioned sludge flocs. The decrease of the sludge flocs size suggested that the KMnO4/PMS process could efficiently break the sludge flocs to enhance the sludge dewaterability as the flocs breakage could provide more passages for the removal of water in the inner of sludge flocs.40 It was also reported by Higgins and Novak that the sludge dewaterability was adversely impacted by the flocs in the range of 1–100 μm and decreased as the quantities of flocs in this size range increased.41
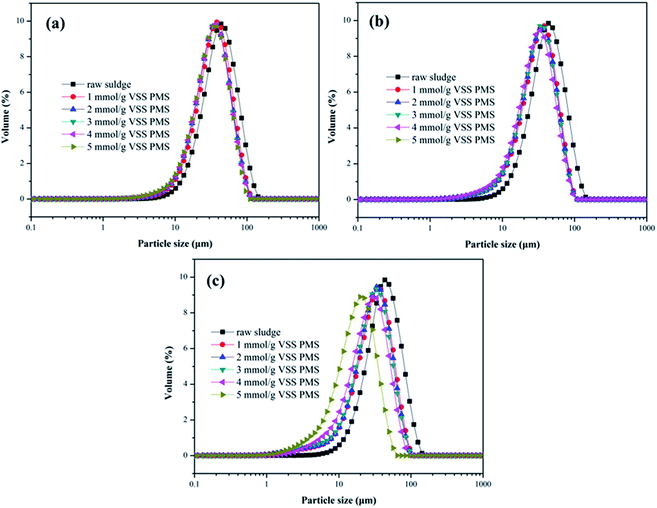 |
| | Fig. 5 Effect of the KMnO4/PMS process on particle size distribution of the WAS samples: (a) 2 mmol g−1 VSS KMnO4; (b) 4 mmol g−1 VSS KMnO4; (c) 6 mmol g−1 VSS KMnO4 (experimental conditions: [PMS] = 1–5 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
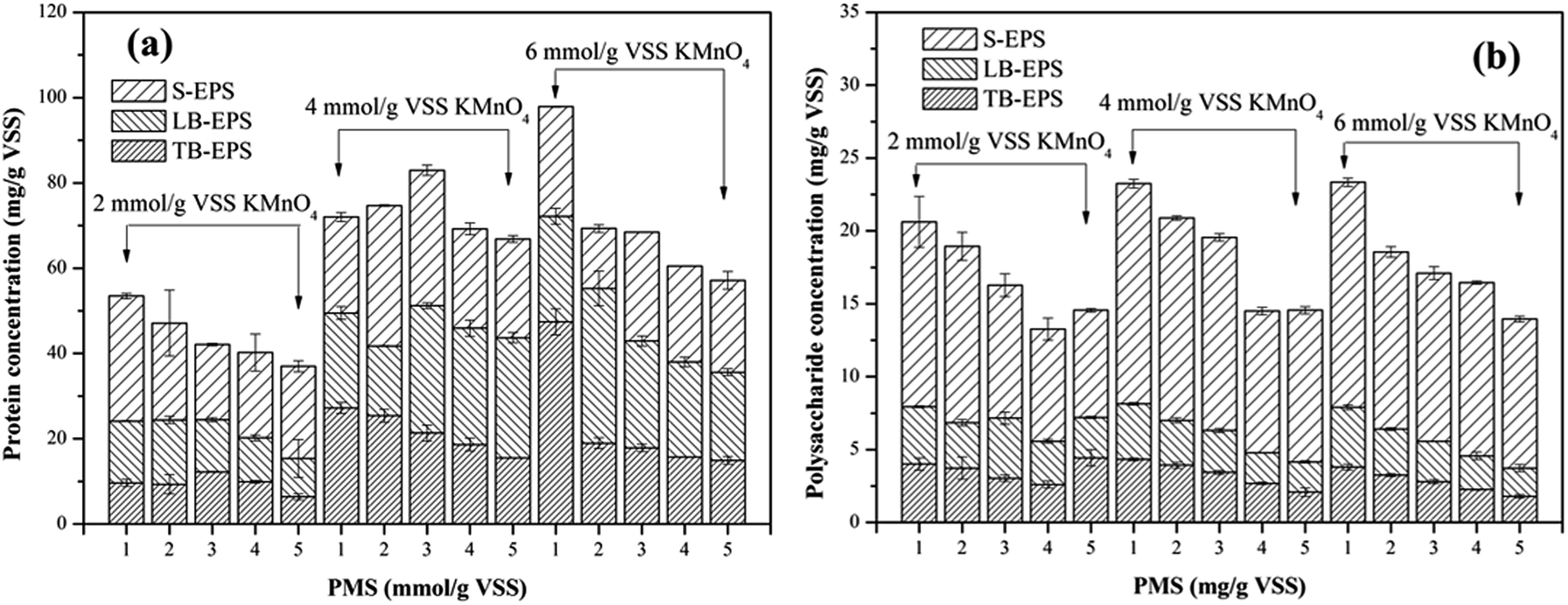
3.2.5. Extracellular polymeric substances (EPS). EPS can have significant effects on the compressibility of sludge and the ability to bind water molecules of sludge flocs, and thus the dewaterability of sludge.42 Since EPS are mainly composed of protein and polysaccharide, variations in the concentrations of protein and polysaccharide in different EPS fractions (S-EPS, LB-EPS and TB-EPS) were monitored to reveal why the KMnO4/PMS process could improve sludge dewaterability. As illustrated in Fig. 6a, with the increase of PMS dosage, the protein content in total EPS tended to decrease. By dosing 2 mmol g−1 VSS KMnO4, the protein content in total EPS decreased from 53.46 mg g−1 VSS to 36.99 mg g−1 VSS while the protein content in S-EPS increased from 21.57 mg g−1 VSS to 29.31 mg g−1 VSS before decreasing to 21.59 mg g−1 VSS, as PMS dosage increased. Moreover, the protein content in LB-EPS decreased from 25.21 mg g−1 VSS to 9.01 mg g−1 VSS while the protein content in TB-EPS decreased from 9.68 mg g−1 VSS to 6.39 mg g−1 VSS. Similar changes of the protein content in total EPS, S-EPS, LB-EPS and TB-EPS could also be observed when 4 mmol g−1 VSS and 6 mmol g−1 VSS KMnO4 were dosed. In contrast to Fig. 6a and b showed that changes of the polysaccharide and protein content in EPS resembled each other.
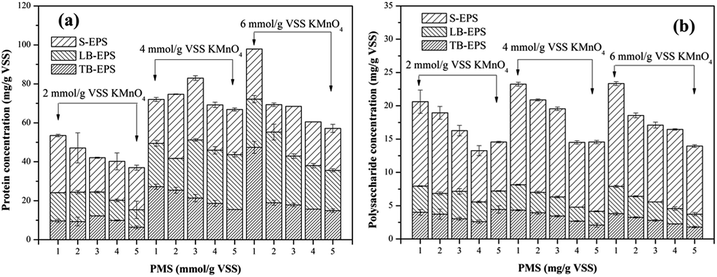 |
| | Fig. 6 Effect of the KMnO4/PMS process on EPS of the WAS samples: (a) protein; (b) polysaccharide (experimental conditions: [KMnO4] = 2–6 mmol g−1 VSS, [PMS] = 1–5 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
The dissolved organics (protein and polysaccharide) in S-EPS increased before decreasing because TB-EPS were dissolved and transferred to S-EPS after the sludge flocs were destructed by SO4˙− and HO˙ generated in the KMnO4/PMS process.29 Nevertheless, more SO4˙− and HO˙ were generated to degrade the organics in S-EPS when PMS dosage continued to increase, so the protein and polysaccharide content in S-EPS tended to decrease. In addition, the slump of the protein and polysaccharide content in S-EPS, LB-EPS and TB-EPS accounted for SO4˙− and HO˙ generation in the KMnO4/PMS process and its high reactivity toward S-EPS, LB-EPS, and even TB-EPS.4
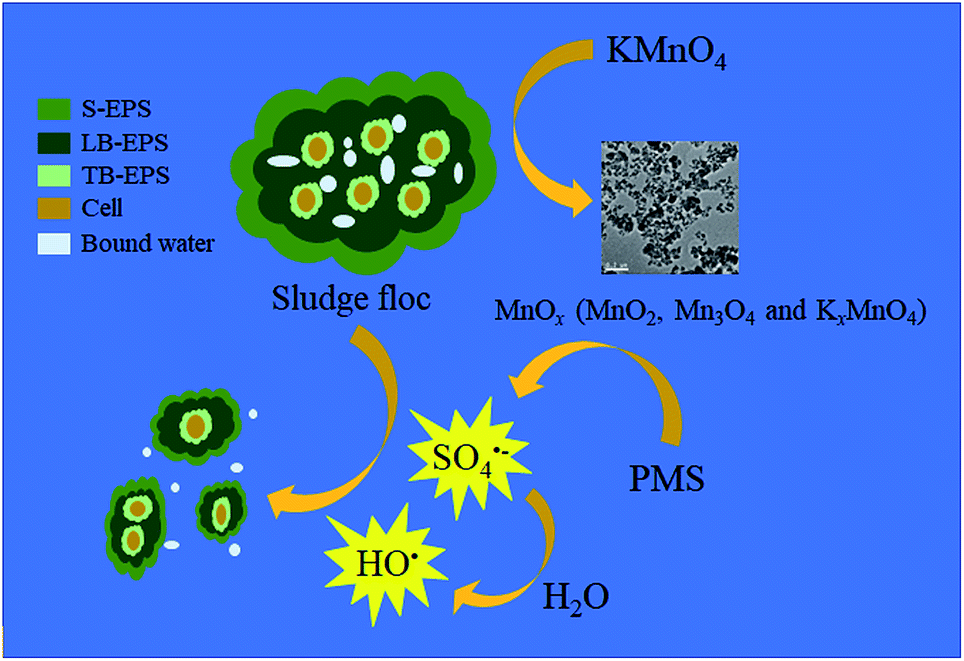
3.3. Mechanism of WAS dewatering by the KMnO4/PMS process
Due to the versatility and efficiency, KMnO4 was deemed as an important precursor for the fabrication of manganese oxide nanomaterials through chemical reduction, including manganese dioxides, tetraoxides and oxyhydroxides.43 Wu et al. used KMnO4 as a conditioner for disintegration of excess activated sludge, and found that after sludge oxidation KMnO4 mainly transferred to the states of MnO2 and Mn3O4 while some compounds of KxMnO4 also existed in the solids.28 Due to the multivalent nature of Mn, MnO2 and Mn3O4 have been proven to possess a high catalytic activity toward PMS for SO4˙− generation.44,45 Thus, in the KMnO4/PMS process, manganese oxides (MnOx) in the state of MnO2 and Mn3O4 might be generated from the reduction of KMnO4, then MnO2 and Mn3O4 further activated PMS to produce SO4˙− and HO˙.
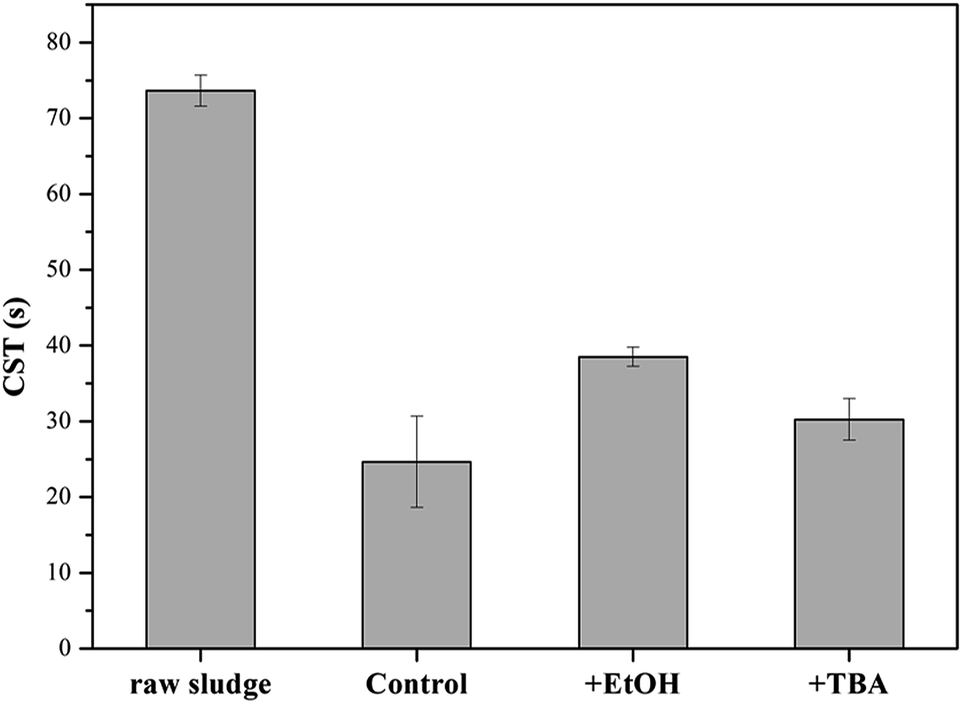
In order to verify the hypothesis, the formed solids were separated from the KMnO4/PMS process. And the samples were observed through TEM and EDX. From Fig. 7a–d, it could be clearly seen that some nanoscale solids did form in the KMnO4/PMS process. Moreover, the EDX analysis further confirmed that the formed solids were mainly composed of element Mn, O and K (Fig. 7e), and the weight percentages of Mn, O and K were recorded to be 20.9%, 74.26% and 4.82%. It suggested that KMnO4 was disintegrate into MnOx under the influence of EPS. According to the previous study, the solids might also be MnOx in the states of MnO2, Mn3O4 and KxMnO4.28 Meanwhile, scavenging experiments using ethanol (EtOH) and tert-butyl alcohol (TBA) as radical scavengers were conducted to confirm the formation of SO4˙− and HO˙ in the KMnO4/PMS process. As can be seen in Fig. 8, after extra addition of 300 mmol g−1 VSS EtOH and TBA, the CST decreased from 73.65 s to 38.50 s and 30.25 s, respectively while decreasing from 73.65 to 24.65 s without addition of any radical scavenger. Moreover, the reduction value of CST with the addition of EtOH was higher than that with the addition of TBA. Such difference in CST value drop by the two radical scavengers implied that both SO4˙− and HO˙ were generated in the KMnO4/PMS process because EtOH can effectively quench both SO4˙− and HO˙ while TBA reacts much faster with HO˙ than SO4˙−.25
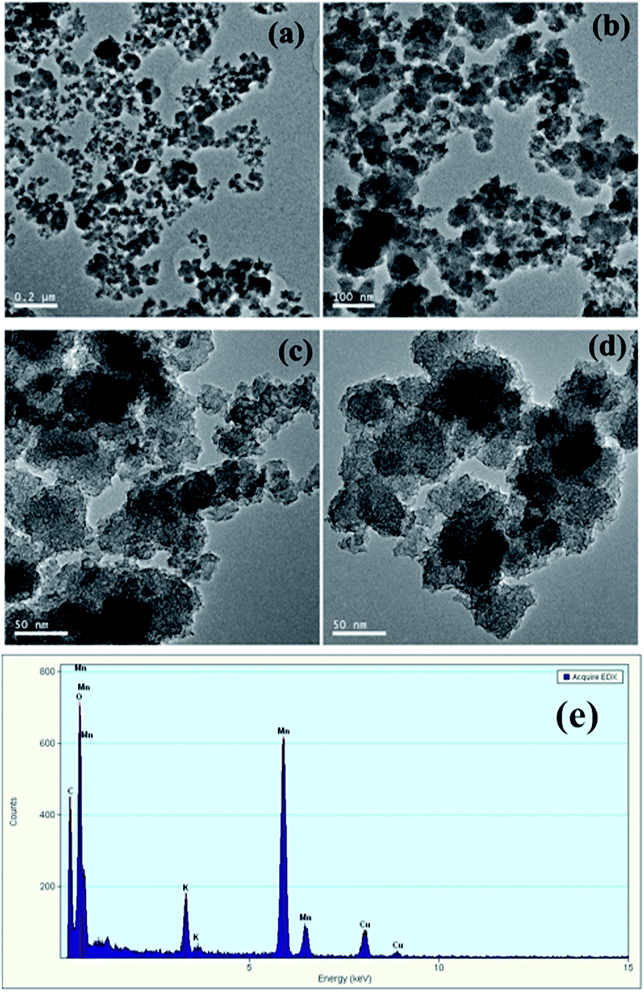 |
| | Fig. 7 TEM image of the solid samples (a–d) and EDX spectra of the solid samples (e). | |
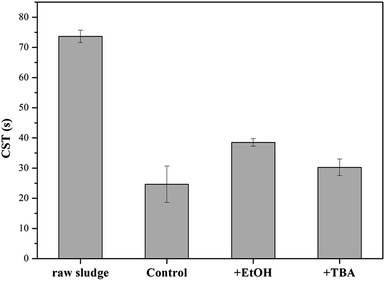 |
| | Fig. 8 Effect of EtOH and TBA on the dewaterability of the WAS samples by the KMnO4/PMS process (experimental conditions: [KMnO4] + [PMS] = 4 mmol g−1 VSS + 3 mmol g−1 VSS, [EtOH] = [TBA] = 300 mmol g−1 VSS, pH0 = 7, T = 25 °C). | |
Based on both the results above and current studies, a possible mechanism was proposed. As depicted in Fig. 9, Firstly, KMnO4 transformed into MnO2, Mn3O4 and KxMnO4 after oxidizing the organics in EPS. Then, the in situ generated MnO2 and Mn3O4 efficiently activated PMS to generate SO4˙− and HO˙ (eqn (2)–(5)). In detail, HSO5− (the species of PMS) initially integrated with ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) MnIV and MnIII located at the surface of MnO2 and Mn3O4 by surface hydroxyl groups (eqn (6)). Then, MnIV and MnIII would be reduced to MnIII and MnII, respectively with the generation of SO5˙− (eqn (7)). Meanwhile, the formed MnIII and MnII could be oxidized to MnIV and MnIII with the formation of SO4˙− (eqn (8) and (9)). Furthermore, HO˙ could also be produced through the reaction between SO4˙− and H2O (eqn (5)).46 Finally, SO4˙− and HO˙ degraded the organics in EPS to break the sludge flocs so that huge quantities of bound water were released and became free water, thus significantly enhancing sludge dewaterability.
MnIV and MnIII located at the surface of MnO2 and Mn3O4 by surface hydroxyl groups (eqn (6)). Then, MnIV and MnIII would be reduced to MnIII and MnII, respectively with the generation of SO5˙− (eqn (7)). Meanwhile, the formed MnIII and MnII could be oxidized to MnIV and MnIII with the formation of SO4˙− (eqn (8) and (9)). Furthermore, HO˙ could also be produced through the reaction between SO4˙− and H2O (eqn (5)).46 Finally, SO4˙− and HO˙ degraded the organics in EPS to break the sludge flocs so that huge quantities of bound water were released and became free water, thus significantly enhancing sludge dewaterability.
| | |
2Mn3O4 + HSO5− → 3Mn2O3 + SO4˙− + H+
| (2) |
| | |
2MnO2 + HSO5− → SO5˙− + OH− + Mn2O3
| (3) |
| | |
Mn2O3 + HSO5− → SO4˙− + 2MnO2 + H+
| (4) |
| | |
SO4˙− + H2O → HO˙ + H+ + SO42−
| (5) |
| | |
MnIV(III) – OH + HSO5− → MnIV(III) − (O)OSO3− + H2O
| (6) |
| | |
MnIV(III) − (O)SO3− + H2O → MnIII(II) − OH + SO5˙− + H+
| (7) |
| | |
MnIII(II) − OH + HSO5− → MnIII(II) − (O)OSO−3 + H2O
| (8) |
| | |
MnIII(II) − (O)OSO3− + H2O → MnIV(III) − OH + SO4˙− + OH−
| (9) |
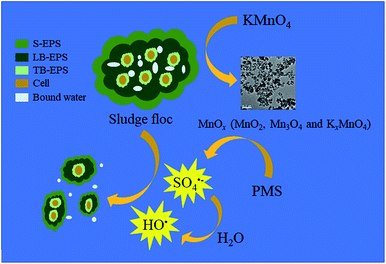 |
| | Fig. 9 Schematic diagram of PMS activation by KMnO4 during the sludge dewatering process. | |
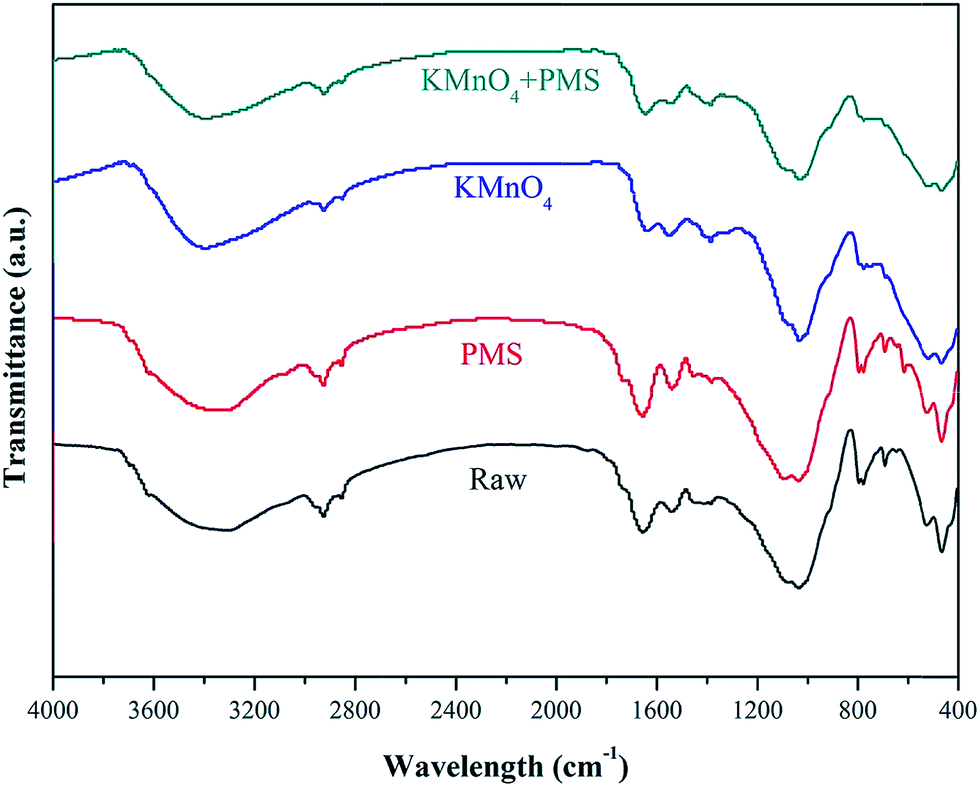
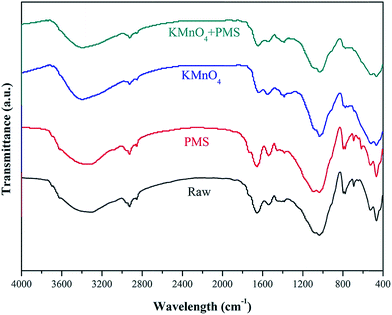
FTIR spectrometer was also used to observed the variations of WAS functional groups after conditioning in different conditions. As illustrated in Fig. 10, the broad absorption band from 3200 to 3400 cm−1 could be attributed to the stretching vibrations of O–H group of polymeric substances.47 The peaks at 2925 and 2852 cm−1 were asymmetric and symmetric vibration of CH2 of aliphatic structures and lipids, respectively.48 The absorption band between 1640 to 1660 cm−1 was associated with the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N, and C–N stretching vibration and N–H deformation vibration at 1550–1560 cm−1.49 The typical band located at 1040–1070 cm−1 was due to the stretching vibration of OH.49 Besides, some peaks (<1000 cm−1) monitored in the “fingerprint” region could be attributed to the phosphate or sulfur functional groups, which is the functional groups for the production of nucleic acids.48 Compared with conditioning by KMnO4 or PMS alone, it can be clearly seen that the intensity of absorption bands of WAS decreased obviously after treatment by the KMnO4/PMS process, implying that SO4˙− and HO˙ generated in the KMnO4/PMS process could effectively degrade the proteins and polysaccharides in EPS and then resulted in the release of bound water and the improvement of sludge filterability.
O and C–N, and C–N stretching vibration and N–H deformation vibration at 1550–1560 cm−1.49 The typical band located at 1040–1070 cm−1 was due to the stretching vibration of OH.49 Besides, some peaks (<1000 cm−1) monitored in the “fingerprint” region could be attributed to the phosphate or sulfur functional groups, which is the functional groups for the production of nucleic acids.48 Compared with conditioning by KMnO4 or PMS alone, it can be clearly seen that the intensity of absorption bands of WAS decreased obviously after treatment by the KMnO4/PMS process, implying that SO4˙− and HO˙ generated in the KMnO4/PMS process could effectively degrade the proteins and polysaccharides in EPS and then resulted in the release of bound water and the improvement of sludge filterability.
 |
| | Fig. 10 FTIR spectra of the WAS samples treated at different conditions. | |
4. Conclusion
This paper investigated the application of a combined use of KMnO4 and PMS (KMnO4/PMS) for sludge dewatering. The major conclusions can be drawn as followed:
(1) Compared with the separate use of KMnO4 or PMS, the combined use of KMnO4 and PMS (KMnO4/PMS) could significantly improve sludge dewaterability. After conditioning with the KMnO4/PMS process, CST of the WAS samples decreased from 73.65 s to 24.65 s. However, the dewaterability of WAS would be worsened by conditioning with KMnO4 alone or PMS alone because the CST value increased significantly.
(2) The KMnO4/PMS process exerted significant effects on the physicochemical characteristics of the WAS samples. When the WAS samples were treated by the KMnO4/PMS process, both CST and WC would decrease, the zeta potential was less negative, and the particle size would also decrease as the sludge flocs were broken into smaller fragments. Besides, the concentrations of protein and polysaccharide in EPS were also reduced due to the synergistic effect of KMnO4 and PMS.
(3) The combined use of TEM and EDX techniques verified the generation of MnOx in the states of MnO2, Mn3O4 and KxMnO4 after KMnO4 oxidation. The radical scavenging experiments confirmed that the in situ generated MnO2 and Mn3O4 could activate PMS to generate SO4˙− and HO˙, which were responsible for significantly enhanced sludge dewaterability. Moreover, the FTIR analysis further indicated that SO4˙− and HO˙ generated in the KMnO4/PMS process could improve sludge dewaterability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Fund of State Key Laboratory of Pollution Control and Resource Reuse (PCRRF17023).
References
- D.-Q. He, L.-F. Wang, H. Jiang and H.-Q. Yu, Chem. Eng. J., 2015, 272, 128–134 CrossRef CAS.
- M. S. Kim, K. M. Lee, H. E. Kim, H. J. Lee, C. Lee and C. Lee, Environ. Sci. Technol., 2016, 50, 7106–7115 CrossRef CAS PubMed.
- H. Yuan, N. Zhu and L. Song, Bioresour. Technol., 2010, 101, 4285–4290 CrossRef CAS.
- J. Liu, Q. Yang, D. Wang, X. Li, Y. Zhong, X. Li, Y. Deng, L. Wang, K. Yi and G. Zeng, Bioresour. Technol., 2016, 206, 134–140 CrossRef CAS.
- K.-M. Lee, M. S. Kim and C. Lee, Sustainable Environ. Res., 2016, 26, 177–183 CrossRef CAS.
- W. Yu, J. Yang, S. Tao, Y. Shi, J. Yu, Y. Lv, S. Liang, K. Xiao, B. Liu, H. Hou, J. Hu and X. Wu, Water Res., 2017, 126, 342–350 CrossRef CAS PubMed.
- R. Mo, S. Huang, W. Dai, J. Liang and S. Sun, Chem. Eng. J., 2015, 269, 391–398 CrossRef CAS.
- C. Bougrier, J. P. Delgenès and H. Carrère, Chem. Eng. J., 2008, 139, 236–244 CrossRef CAS.
- X. Feng, J. Deng, H. Lei, T. Bai, Q. Fan and Z. Li, Bioresour. Technol., 2009, 100, 1074–1081 CrossRef CAS PubMed.
- K. Hu, J. Q. Jiang, Q. L. Zhao, D. J. Lee, K. Wang and W. Qiu, Water Res., 2011, 45, 5969–5976 CrossRef CAS PubMed.
- C.-J. Cheng and P. K. A. Hong, Bioresour. Technol., 2013, 142, 69–76 CrossRef CAS PubMed.
- G. Y. Zhen, X. Q. Lu, Y. Y. Li and Y. C. Zhao, Bioresour. Technol., 2013, 136, 654–663 CrossRef CAS PubMed.
- P. Shi, R. Su, S. Zhu, M. Zhu, D. Li and S. Xu, J. Hazard. Mater., 2012, 229–230, 331–339 CrossRef CAS PubMed.
- J. Yu, K. Xiao, J. Yang, W. Yu, K. Pei, Y. Zhu, J. Wang, S. Liang, J. Hu, H. Hou and B. Liu, ACS Sustainable Chem. Eng., 2019, 7, 324–331 CrossRef CAS.
- E. Neyens, J. Baeyens, M. Weemaes and B. De heyder, J. Hazard. Mater., 2003, 98, 91–106 CrossRef CAS.
- H. Zhang, J. Yang, W. Yu, S. Luo, L. Peng, X. Shen, Y. Shi, S. Zhang, J. Song, N. Ye, Y. Li, C. Yang and S. Liang, Water Res., 2014, 59, 239–247 CrossRef CAS PubMed.
- Y. Li, X. Yuan, D. Wang, H. Wang, Z. Wu, L. Jiang, D. Mo, G. Yang, R. Guan and G. Zeng, Bioresour. Technol., 2018, 262, 294–301 CrossRef CAS PubMed.
- K. Xiao, Y. Chen, X. Jiang, Q. Yang, W. Y. Seow, W. Zhu and Y. Zhou, Water Res., 2017, 109, 13–23 CrossRef CAS PubMed.
- T. K. Lau, C. Wei and N. J. D. Graham, Environ. Sci. Technol., 2007, 41, 613 CrossRef CAS PubMed.
- G. Ying-Hong, M. Jun, L. Xu-Chun, F. Jing-Yun and C. Li-Wei, Environ. Sci. Technol., 2011, 45, 9308 CrossRef PubMed.
- C. Tan, N. Gao, Y. Deng, N. An and J. Deng, Chem. Eng. J., 2012, 203, 294–300 CrossRef CAS.
- J. Deng, Y. Shao, N. Gao, Y. Deng, S. Zhou and X. Hu, Chem. Eng. J., 2013, 228, 765–771 CrossRef CAS.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- C. Qi, X. Liu, J. Ma, C. Lin, X. Li and H. Zhang, Chemosphere, 2016, 151, 280–288 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- J. Deng, Y. Ge, C. Tan, H. Wang, Q. Li, S. Zhou and K. Zhang, Chem. Eng. J., 2017, 330, 1390–1400 CrossRef CAS.
- J. Cui, L. Zhang, B. Xi, J. Zhang and X. Mao, Chem. Eng. J., 2017, 313, 815–825 CrossRef CAS.
- C. Wu, G. Zhang, P. Zhang and C.-C. Chang, Chem. Eng. J., 2014, 240, 420–425 CrossRef CAS.
- T. Niu, Z. Zhou, W. Ren, L.-M. Jiang, B. Li, H. Wei, J. Li and L. Wang, Int. Biodeterior. Biodegrad., 2016, 106, 170–177 CrossRef CAS.
- J. Yang, X. Liu, D. Wang, Q. Xu, Q. Yang, G. Zeng, X. Li, Y. Liu, J. Gong and J. Ye, Water Res., 2019, 148, 239–249 CrossRef CAS PubMed.
- B. Fr/olund, T. Griebe and P. H. Nielsen, Appl. Microbiol. Biotechnol., 1995, 43, 755–761 CrossRef.
- M. DuBois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- K. Xiao, Y. Chen, X. Jiang, V. K. Tyagi and Y. Zhou, Water Res., 2016, 105, 470–478 CrossRef CAS PubMed.
- C. Cai, H. Zhang, X. Zhong and L. Hou, J. Hazard. Mater., 2015, 283, 70–79 CrossRef CAS PubMed.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- J. Liu, Z. Zhao, P. Shao and F. Cui, Chem. Eng. J., 2015, 262, 854–861 CrossRef CAS.
- J. Liu, Y. Wei, K. Li, J. Tong, Y. Wang and R. Jia, Water Res., 2016, 90, 225–234 CrossRef CAS PubMed.
- G. Zhen, X. Lu, B. Wang, Y. Zhao, X. Chai, D. Niu, A. Zhao, Y. Li, Y. Song and X. Cao, Bioresour. Technol., 2012, 124, 29–36 CrossRef CAS PubMed.
- H. Gharibi, M. H. Sowlat, A. H. Mahvi, M. Keshavarz, M. H. Safari, S. Lotfi, M. Bahram Abadi and A. Alijanzadeh, Chemosphere, 2013, 90, 1487–1494 CrossRef CAS PubMed.
- G. Zhen, X. Lu, Y. Li, Y. Zhao, B. Wang, Y. Song, X. Chai, D. Niu and X. Cao, Bioresour. Technol., 2012, 119, 7–14 CrossRef CAS PubMed.
- M. J. Higgins and J. T. Novak, Water Environ. Res., 1997, 69, 215–224 CrossRef CAS.
- D. Mowla, H. N. Tran and D. G. Allen, Biomass Bioenergy, 2013, 58, 365–378 CrossRef CAS.
- K. A. M. Ahmed, Journal of Taibah University for Science, 2016, 10, 412–429 CrossRef.
- E. Saputra, S. Muhammad, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Environ. Sci. Technol., 2013, 47, 5882–5887 CrossRef CAS PubMed.
- E. Saputra, S. Muhammad, H. Sun, H.-M. Ang, M. O. Tadé and S. Wang, J. Colloid Interface Sci., 2013, 407, 467–473 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao and C. Yang, Chem. Eng. J., 2017, 327, 481–489 CrossRef CAS.
- Z. Chen, W. Zhang, D. Wang, T. Ma, R. Bai and D. Yu, Water Res., 2016, 103, 170–181 CrossRef CAS PubMed.
- H.-y. Pei, W.-r. Hu and Q.-h. Liu, J. Hazard. Mater., 2010, 178, 397–403 CrossRef CAS PubMed.
- J. Laurent, M. Casellas, H. Carrere and C. Dagot, Chem. Eng. J., 2011, 166, 841–849 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *c
*c