
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnchoring carbon layers and oxygen vacancies endow WO3−x/C electrode with high specific capacity and rate performance for supercapacitors†
Juan Xu *,
Chongyang Li,
Lulu Chen,
Zhongyang Li and
Pibin Bing
*,
Chongyang Li,
Lulu Chen,
Zhongyang Li and
Pibin Bing
Institute of Electric Power, North China University of Water Resources and Electric Power, Zhengzhou, Henan 450000, China. E-mail: xujuan@ncwu.edu.cn
First published on 12th September 2019
Abstract
Herein, novel hierarchical carbon layer-anchored WO3−x/C ultra-long nanowires were developed via a facile solvent-thermal treatment and a subsequent rapid carbonization process. The inner anchored carbon layers and abundant oxygen vacancies endowed the WO3−x/C nanowire electrode with high conductivity, as measured with a single nanowire, which greatly enhanced the redox reaction active sites and rate performance. Surprisingly, the WO3−x/C electrode exhibited outstanding specific capacitance of 1032.16 F g−1 at the current density of 1 A g−1 in a 2 M H2SO4 electrolyte and maintained the specific capacitance of 660 F g−1 when the current density increased to 50 A g−1. Significantly, the constructed WO3−x/C//WO3−x/C symmetric supercapacitors achieved specific capacitance of 243.84 F g−1 at the current density of 0.5 A g−1 and maintained the capacitance retention of 94.29% after 5000 charging/discharging cycles at the current density of 4 A g−1. These excellent electrochemical performances resulted from the fascinating structure of the WO3−x/C nanowires, showing a great potential for future energy storage applications.
1. Introduction
Over the past decades, electrochemical supercapacitors (ESs) have gradually stood out from other electrochemical energy storage devices, profiting from their distinguishing advantages of high power density, rational energy density and long-term cycle stability.1–3 As is well-established, the electrochemical performance of ESs is heavily dependent on their electrode materials.4–7 Apart from commercial carbon and conducting polymer materials, transition metal oxides have received considerable attention owing to their good electrochemical reversibility and high specific capacitance in aqueous electrolytes.8–11 Nanostructured tungsten-based materials (WO3 and W18O49) are distinctly appealing in this regard owing to their multiple oxidation states, high electronic conductivity (10 × 10−6 S cm−1), small radii and suitable crystalline phases for fast ion insertion and superior electrochemical performance.12–15To induce more active sites during the redox reaction process, strategies for the nanostructures of different dimensionalities such as nanoparticles, nanowires, nanotubes, and nanosheets as well as 3D porous structures have been reported.16–20 Among them, nanowires are considered to be the most promising structures due to their sufficient interfacial contact areas with the electrolyte as well as the easy percolation of the nanowire network. Furthermore, the electrical conductivity can be improved by compositing/doping WO3−x with conducting alien species.21–23 Recently, Gong et al. reported the growth of carbon-encapsulated WO3−x nanowires via a facile hydrothermal and annealing treatment, achieving areal specific capacitance of 786.8 mF cm−2 at the current density of 20 mA cm−2, but their rate properties were not satisfactory.24 Tian et al. reported the production of W18O49 nanofibers by the addition of ethanol and electrodeposition of a polyaniline film, displaying specific capacitance of 440 F g−1 at the current density of 1 A g−1. However, they could not display a long cycling life.14 In all, all these reported structures cannot satisfy the commercial requirements of high specific capacitance, high rate properties and long lifespan. The major obstacle is that all these reported carbon post-coated methods yield WO3−x nanowires with only surface-coated carbon materials rather than being confined to a single nanowire and thus, the inner nanowire matrix fails to contact with the exterior conductive carbon materials.
Herein, the structure of hierarchical carbon layer-anchored ultra-long nanowires was proposed as a promising platform to achieve high electronic conductivity and superior electrochemical performance. We projected that the WO3–ethylenediamine (WO3–EDA) inorganic–organic hybrid nanowire framework with a unique structure and high specific surface area can provide large contact areas with the electrolyte. After a facile rapid thermal treatment, the anchored carbon layers could be uniformly and tightly distributed in the inside of the WO3−x nanowires through oxygen bridges, which not only significantly contributed to the enhanced conductivity but also helped enable an outstanding rate performance. Significantly, the reduction of carbon to WO3 resulted in the formation of a large amount of oxygen vacancies in WO3−x to achieve promising conductivity. Notably, the as-obtained carbon layer-anchored ultra-long WO3−x nanowire (WO3−x/C)-based electrode exhibited specific capacitance of 1032.16 F g−1 at the current density of 1 A g−1. Amazingly, when the current density was increased to 50 A g−1, the specific capacitance of 660 F g−1 could still be maintained. This extraordinary performance is superior to previously reported values for WO3−x-based electrodes, thus demonstrating the advantage of the carbon layer-anchored nanowire configuration.
2. Experimental section
2.1. Material preparation
The precursor of organic–inorganic hybrid WO3–EDA nanowires was obtained via a facile and low-cost solvent-thermal treatment. In a typical experiment, 1 g WO3 was first slowly added to 75 mL ethylenediamine (EDA) to form a uniform and yellow solution. Subsequently, the solution was transferred to a Teflon-lined stainless steel autoclave and then heated in an electric oven at 180 °C for 12 h, followed by naturally cooling to room temperature. The product was collected by centrifugation, washed thoroughly with deionized water and ethanol several times and then dried in vacuum at 60 °C overnight. Finally, the hierarchical carbon layer-anchored WO3−x nanowires were achieved by the rapid pyrolysis of the as-obtained organic–inorganic hybrid nanowire precursor at 600 °C for 2 h under an Ar flow. The pure WO3 nanowires were produced by annealing the precursor at 600 °C for 2 h under an Air flow.2.2. Material characterization
X-ray powder diffraction (XRD) patterns were recorded using a Philips X'Pert Pro (PANanalytical B.V., Netherlands) Super diffractometer with Cu Kα radiation (λ = 1.54118 Å). SEM images were obtained via field-emission scanning electron microscopy (FE-SEM, FEI Nova NanoSEM 450) equipped with an X-ray energy dispersive spectrometer (EDS). Transmission electron microscopy (TEM, FEI Tecnai G20), Raman scattering (InVo-RENISHAW), Fourier transform infrared spectroscopy (FT-IR, Bruker Vertex 80 V) and X-ray photoelectron spectroscopy (XPS, Kratos AXIS Ultra DLD-600 W) were also performed. The thermogravimetric-differential analysis (TG-DSC) was performed on Perkin Elmer Diamond TG-DTA at a temperature ramping rate of 10 °C min−1 under air. A four-point probe on a Si template was then designed and fabricated to measure the conductivity of a single hierarchical WO3−x/C nanowire and pure WO3 nanowire.2.3. Electrochemical measurements
Electrodes were prepared by mixing the collected samples (80%), Super P (10%) and polyvinylidene fluoride (10%) in N-methyl,2-pyrrolidone to form a slurry. The slurry was stirred uniformly and pasted on the carbon current collector. The collector was cut into a square shape with 1 cm × 1 cm dimensions for electrodes and placed into a vacuum oven (80 °C) overnight to remove moisture and residual hydrocarbons. The three-electrode electrochemical cell was fabricated with a platinum foil as the counter electrode, a saturated calomel electrode as the reference electrode and the hierarchical WO3−x/C nanowire electrode as the working electrode. The symmetric supercapacitor was measured with a two-electrode configuration, including two slices of electrode material with the same size (1 × 1 cm2) and the same mass loading (1.8 mg), a cellulose membrane as the separator, and 2 M H2SO4 solution as the electrolyte. All the electrochemical measurements in both the three-electrode and two-electrode systems were performed on a CHI660D electrochemical workstation.3. Results and discussion
The above-mentioned organic–inorganic hybrid WO3–EDA nanowires were acquired in high yields via a facile hydrothermal treatment with a small quantity of WO3 as the raw material and EDA as the solvent. As shown in Fig. 1, EDA plays a vital role in the production of the hierarchical organic–inorganic hybrid nanowires. In the WO3–EDA hybrid nanowires, the organic EDA molecules intercalated into the inorganic WO3 framework on a sub-nanometer-scale.25 The inorganic units are connected by H-bonding of N–H⋯O–W.26–28 Following the facile rapid pyrolysis of these hybrid WO3–EDA nanowires in a pure Ar atmosphere, we finally achieved uniform thin carbon layer-anchored WO3−x nanowires owing to the instant carbonization of EDA and confined effects. In addition, the formation of abundant oxygen vacancies was induced because of the reduction of carbon to WO3.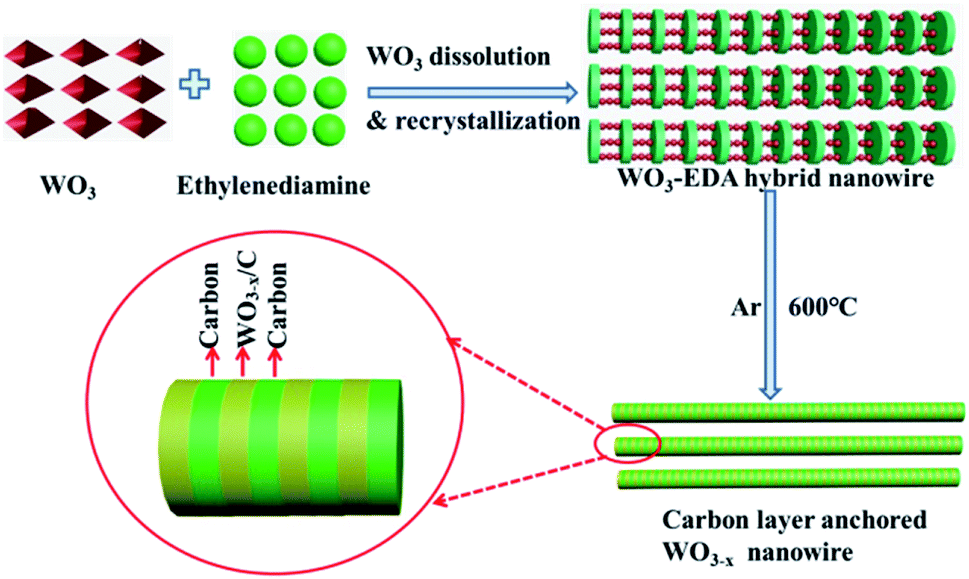 | ||
| Fig. 1 The formation process of hierarchical carbon layer-anchored ultra-long WO3−x nanowires from the organic–inorganic hybrid WO3–EDA precursor. | ||
Fig. 2a and b show the typical scanning electron microscopy (SEM) images of the hierarchical WO3−x/C nanowires. Fig. 2a shows the hierarchical WO3−x/C nanowires with a uniform length of 10–20 μm. Obviously, the nanowires exhibited a diameter of 50–100 nm. The nanowires possessed the same morphology as that of the WO3–EDA hybrid precursor, which is characterized in Fig. S1.† The presence of EDA molecules in the WO3–EDA hybrid precursor is confirmed by the FTIR spectrum in Fig. 2c, which displays the ν-OH stretching mode at 3433 cm−1 and the ν-CH2 bending mode at 2960 cm−1.26 The sharp peaks at around 1620 cm−1 and 3245 cm−1 correspond to the δ-NH2 and ν-NH2 stretching modes, indicating that the interlayer organic molecules were intercalated as ammonium cations rather than neutral amines. In addition, the absorption peaks at around 590 cm−1 and 850 cm−1 can be attributed to the ν-(O–W–O) lattice vibration modes in WO3.26 These results gave significant evidence that the EDA molecules intercalated into the WO3 lattice. Moreover, the XRD pattern in Fig. 2d clearly shows the presence of three level diffraction peaks at 2θ = 11.73°, 15.94°, and 19.13° (i.e., d1 = 0.71 nm, d2 = 0.54 nm, and d3 = 0.44 nm), respectively, which are owing to the intercalation of the EDA molecules into the WO3 nanowires. Furthermore, the other peaks well-matched with the previous results for monoclinic WO3/EDA (JCPDS Card no. 43-1034).26 Fig. 2e shows the XRD pattern of the final carbon layer-anchored WO3−x/C nanowire network, which can be indexed to monoclinic WO3−x (JCPDS Card no. 36-0103). The Raman spectra in Fig. 2f further confirm the presence of carbon in the WO3−x nanowire network.29 The absorption peaks at around 688.2 cm−1 and 804.4 cm−1 can be attributed to the δ-(O–W–O) lattice vibration mode and ν-(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) lattice vibration mode in WO3, respectively.30 Moreover, the two broad peaks at 1360 cm−1 and 1578 cm−1 correspond to the D band and G band, which indicate the existence of carbon coating layers originating from the disordered and ordered graphitic carbon, respectively. The ID/IG value was calculated to be about 1.05, which indicated a reasonable degree of graphitization.31–34
O) lattice vibration mode in WO3, respectively.30 Moreover, the two broad peaks at 1360 cm−1 and 1578 cm−1 correspond to the D band and G band, which indicate the existence of carbon coating layers originating from the disordered and ordered graphitic carbon, respectively. The ID/IG value was calculated to be about 1.05, which indicated a reasonable degree of graphitization.31–34
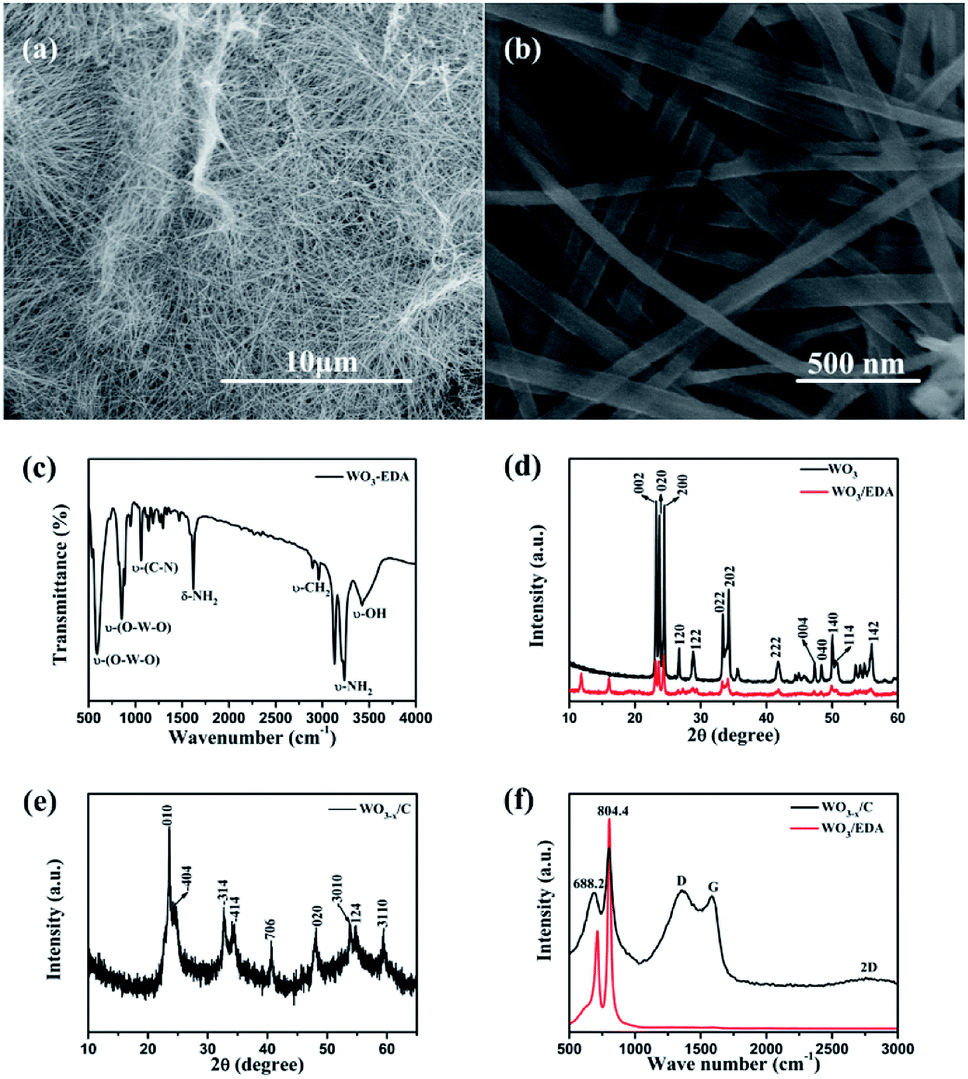 | ||
| Fig. 2 (a and b) SEM images of different magnifications of hierarchical WO3−x/C nanowires. (c) FTIR spectrum of the WO3–EDA organic–inorganic hybrid precursor. (d) XRD patterns of the raw material WO3 and the WO3–EDA precursor. (e) XRD pattern of the hierarchical WO3−x/C nanowires. (f) Raman spectra of the WO3–EDA hybrid precursor and hierarchical WO3−x/C nanowires. | ||
Fig. 3 further confirms the crystalline structure and element distribution of the WO3−x/C nanowires. The diameter of the WO3−x/C nanowires was about 20 nm. Obviously, Fig. 3a shows the carbon layers tightly anchored on the inside of the WO3−x layers, which is consistent with the description in Fig. 1. In addition, the lattice fringes of the WO3 layers and carbon layers are very clear in Fig. 3b. Fig. 3c shows the TEM image and X-ray energy dispersive spectroscopy (EDS) maps of a single nanowire, confirming the uniform distribution of W, O, C and N.
 | ||
| Fig. 3 (a and b) TEM images and (c) EDS mapping results of the carbon layer-anchored WO3−x nanowires. | ||
The X-ray photoelectron spectroscopy (XPS) results acquired from the pure WO3 nanowires (Fig. S2†) and WO3−x/C nanowires are shown in Fig. 4a. The presence of N1s in the XPS spectra of the WO3−x/C nanowires clearly proved the insertion of EDA into the WO3 nanowires. Moreover, the ratio of O to W was less than 3, indicating the generation of oxygen vacancies in the hierarchical carbon layer-anchored WO3−x nanowires. The fine XPS spectra of N1s are shown in Fig. S3.† The two fine XPS peaks of W of pure WO3 (Fig. 4b) at 36.03 eV and 38.48 eV correspond to 4f7/2 and 4f5/2 of W6+, which are in agreement with the XRD pattern. However, the binding energy was lower than that for the corresponding fine spectra of W4f7/2 and W4f5/2 in the WO3−x/C nanowires by 0.17 eV.10,14,24,25 The shifts in the binding energies can provide valuable information. As previously reported, this evidences the introduction of oxygen vacancies and the perturbation of the electronic environment after incorporating a small amount of N anions with different electronegativities. The C content in the hierarchical WO3−x/C nanowires was measured to be 3.5 wt% according to the TG curve in Fig. 4c. According to the calculation in Fig. 4d, the electrical conductance is 1.64 × e−3 1/Ω for a single hierarchical WO3−x/C nanowire, which is approximately three orders of magnitude higher than that of a single pure WO3 nanowire with 1.00 × e−6 1/Ω. This is consistent with the prominent role of the anchoring of carbon layers, which can indicate the excellent electrochemical performance of the WO3−x/C nanowire electrodes.
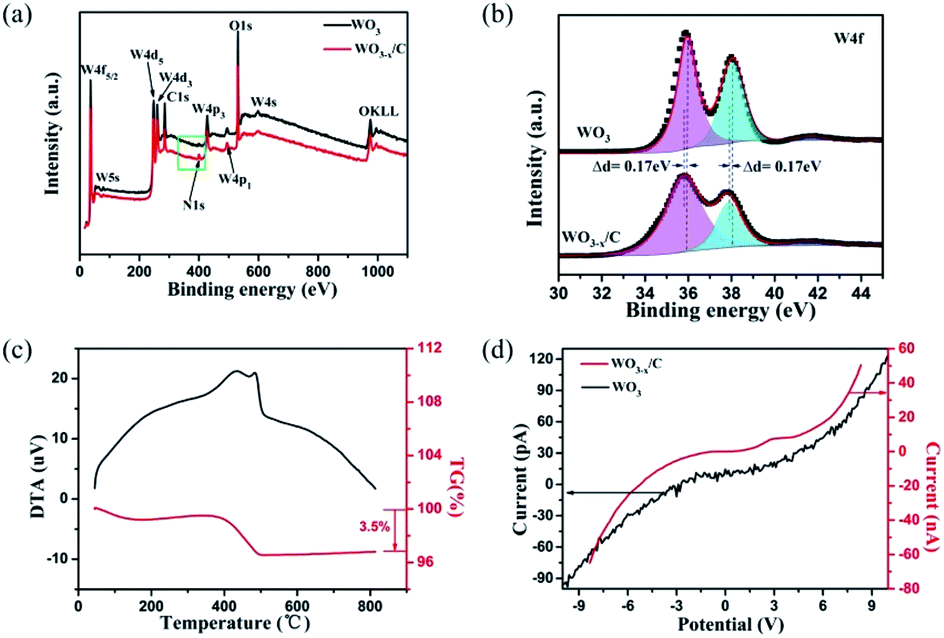 | ||
| Fig. 4 (a) Survey XPS of pure WO3 nanowires and WO3−x/C nanowires. (b) High-resolution W4f spectra of WO3 nanowires and WO3−x/C nanowires. (c) TGA and DSC profiles of the WO3−x/C nanowires. (d) Electrical conductivity of the WO3 nanowires and WO3−x/C nanowires. | ||
The electrochemical performance of the as-prepared WO3−x/C nanowire electrode in comparison with that of the pure WO3 nanowire electrode was evaluated using three-electrode cell configuration in 2 M H2SO4 electrolyte. The results are clearly given in Fig. 5a–f. Notably, the WO3−x/C nanowire electrode delivered larger CV curve areas and wider voltage window than the pure WO3 nanowire electrode at the same scan rate and current density (Fig. 5a–d), which indicated its enhanced specific capacitance and good redox reaction kinetics. Obviously, broad peaks were detected in the CV curves of the two samples, which corresponded to the slightly sloping plateaus in the GCD curves at different current densities, indicating redox-active WO3−x with reversible Faradaic reactions as the dominant charge storage mechanism.35–38 The voltage window was 0.9 eV for the WO3−x/C electrode, which was larger than that for the pure WO3 electrode (0.6 eV) (Fig. 5c and d). Prominently, this was ascribed to the introduction of oxygen vacancies in the WO3−x/C nanowires.25,26,28,29 The gravimetric specific capacitances were calculated from the GCD curves and the result was plotted vs. mass current density (Fig. 5e). Ultra-high specific capacitance of 1032.16 F g−1 was acquired at 1 A g−1 for the WO3−x/C electrode, which was much higher than that for the pure WO3 electrode (308.15 F g−1) at 1 A g−1. When the current density increased to 2, 5, 10, 20, and 50 A g−1, the specific capacitances gradually reduced to 896.96, 886.61, 835.67, 765.33, and 660 F g−1, respectively. Obviously, the pure WO3 electrode exhibited a much poorer rate performance (Fig. 5e). The cyclic performance of the WO3−x/C electrode was much better than that of the pure WO3 electrode (Fig. 5f), and the capacitance retention of 100% could be maintained after 5000 cycles. This significant electrochemical performance was attributed to the carbon layer-anchored nanowire structure and the introduction of numerous oxygen vacancies. In fact, the excellent structure with high electrical conductivity makes a substantial contribution to the high specific capacitance of the WO3−x/C electrode, and this 1D nanowire morphology offers more electro-active sites, which are easily accessible by the electrolyte ions during the redox reactions even at high rates.
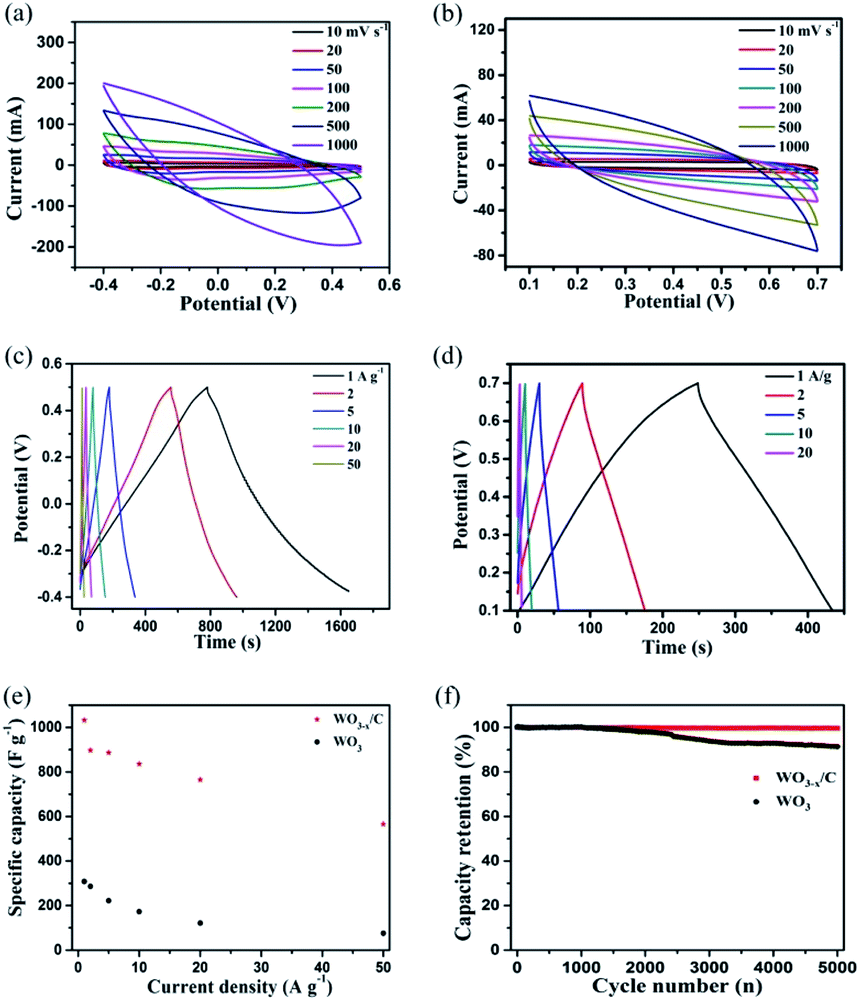 | ||
| Fig. 5 (a and b) Typical CV curves at different scan rates and (c–e) GCD profiles at different current densities and corresponding rate performance of the WO3−x/C nanowire electrode and pure WO3 nanowire electrode in the three-electrode configuration. (f) Cycle stability of the hierarchical WO3−x/C nanowire and pure WO3 nanowire electrodes for 5000 cycles. | ||
Fig. 6 shows the electrochemical performance of the as-assembled hierarchical WO3−x/C//WO3−x/C symmetric supercapacitor (SSC). A schematic of the SSC device is clearly shown in Fig. 6a. The CV profiles (Fig. 6b) of the SSC device measured at different scan rates implied a very stable electrochemical performance with symmetric rectangular curves at the voltage window of 1.2 V. Obviously, the GCD curves (Fig. 6c) of the device obtained at a series of current densities ranging from 0.5 A g−1 to 5 A g−1 exhibited a nearly perfect symmetric triangular shape with a prominent charge/discharge capacitive performance, which corresponded to the coulombic efficiency of nearly 100% at every charge/discharge cycle. The gravimetric capacitances were calculated using the results from the GCD profiles and plotted versus current density (Fig. 6d). The devices showed outstanding specific capacitance of 243.84 F g−1 at the current density of 0.5 A g−1. When the current density increased to 1, 2, 4, and 5 A g−1, the specific capacitances gradually decreased to 194.64, 150.32, 100.97, and 59.46 F g−1, respectively. The cell also demonstrated outstanding capacitance retention of 94.29% even after 5000 charge/discharge cycles at the high current density of 4 A g−1 (Fig. 6e). These results are considerably superior to that for previously reported WO3-based symmetric supercapacitors.10,12,24,29 The prominent electrochemical performance of the developed SSC device implied that the hierarchical carbon layer-anchored WO3−x/C nanowire electrode is a promising candidate for practical applications in next-generation energy storage devices, presenting a new solution of carbon layers and oxygen vacancies integrated into metal oxides in the process of material preparation.
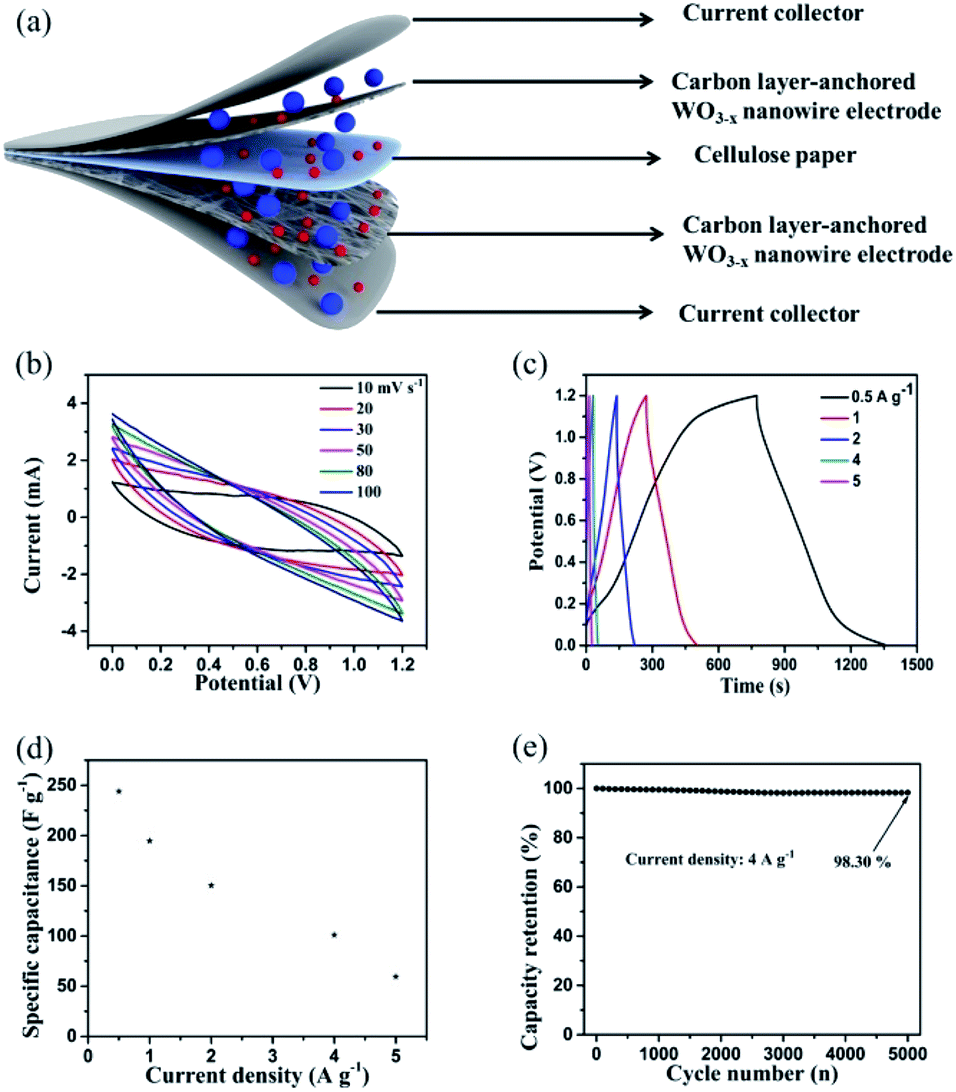 | ||
| Fig. 6 Electrochemical properties of the assembled hierarchical WO3−x/C//WO3−x/C symmetric supercapacitor (SSC): (a) schematic illustration of the SSC device, where the blue spheres represent (SO4)2− and the red spheres represent H+. (b) CV curves at different scan rates. (c and d) GCD curves and specific capacitance plots at various current densities, respectively. (e) Cyclic stability at the current density of 4 A g−1. | ||
4. Conclusions
In summary, WO3−x/C ultra-long nanowires with in situ anchored carbon layers were successfully achieved via a facile hydrothermal treatment and a subsequent rapid carbonization process; they were also employed as promising electrode materials for supercapacitors. The hierarchical WO3−x/C nanowire electrode achieved outstanding specific capacitance of 1032.16 F g−1 at the current density of 1 A g−1 owing to its enhanced electrochemical active sites and improved conductivity due to the introduction of abundant oxygen vacancies and inner anchored carbon layers. Amazingly, the assembled WO3−x/C//WO3−x/C symmetric supercapacitors exhibited an excellent electrochemical performance of 243.84 F g−1 at the current density of 0.5 A g−1 and maintained the specific capacitance retention of 94.29% even after 5000 charging/discharging cycles. This synthetic strategy exhibits a new protocol to develop carbonization-engineered metal oxides as electrode materials for applications in supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Doctoral Research Grant funded by the North China University of Water Resources and Electric Power.Notes and references
- P. Zhao, M. Yao, H. Ren, N. Wang and S. Komarneni, Appl. Surf. Sci., 2019, 463, 931–938 CrossRef CAS.
- L. Lim, Y. Liu, W. Liu, R. Tjandra, L. Rasenthiram, Z. Chen and A. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 39576–39583 CrossRef CAS PubMed.
- Z. Liu, H. Zhang, Q. Yang and Y. Chen, Electrochim. Acta, 2018, 287, 149–157 CrossRef CAS.
- H. J. Tang, J. Y. Wang, H. J. Yin, H. J. Zhao, D. Wang and Z. Y. Tang, Adv. Mater., 2015, 27, 1117–1123 CrossRef CAS PubMed.
- A. Namdarian, A. G. Tabrizi, A. Maseleno, A. Mohammadi and S. E. Moosavifard, Int. J. Hydrogen Energy, 2018, 43, 17780–17787 CrossRef CAS.
- Y. Z. Chao, S. B. Chen, H. Q. Chen, X. J. Hu, Y. Ma, W. S. Gao and Y. X. Bai, Electrochim. Acta, 2018, 276, 118–124 CrossRef CAS.
- H. Z. Chi, Y. Q. Wu, Y. K. Shen, C. Zhang, Q. Xiong and H. Qin, Electrochim. Acta, 2018, 289, 158–167 CrossRef CAS.
- R. Farzana, R. Rajarao, B. R. Bhat and V. Sahajwalla, J. Ind. Eng. Chem., 2018, 65, 387–396 CrossRef CAS.
- A. A. B. Hamra, H. N. Lim, S. M. Hafiz, S. Kamaruzaman, S. A. Rashid, R. Yunus, M. Altarawneh, Z. T. Jiang and N. M. Huang, Electrochim. Acta, 2018, 285, 9–15 CrossRef CAS.
- G. Wang, Y. Yang, D. Han and Y. Li, Nano Today, 2017, 13, 23–39 CrossRef CAS.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed.
- J. Xu, T. T. Ding, J. Wang, J. Zhang, S. Wang, C. Q. Chen, Y. Y. Fang, Z. H. Wu, K. F. Huo and J. N. Dai, Electrochim. Acta, 2015, 174, 728–734 CrossRef CAS.
- P. A. Shinde, A. C. Lokhande, A. M. Patil and C. D. Lokhande, J. Alloys Compd., 2019, 770, 1130–1137 CrossRef CAS.
- Y. Tian, S. Cong, W. Su, H. Chen, Q. Li, F. Geng and Z. Zhao, Nano Lett., 2014, 14, 2150–2156 CrossRef CAS PubMed.
- Z. Shao, X. Fan, X. Liu, Z. Yang, L. Wang, Z. Chen and W. Zhang, J. Alloys Compd., 2018, 765, 489–496 CrossRef CAS.
- B. K. Urhan and U. Demir, Electrochim. Acta, 2019, 302, 109–118 CrossRef CAS.
- J. Li, G. Zhang, N. Chen, X. Nie, B. Ji and L. Qu, ACS Appl. Mater. Interfaces, 2017, 9, 24840–24845 CrossRef CAS PubMed.
- X. Li, Y. Tang, J. Song, W. Yang, M. Wang, C. Zhu, W. Zhao, J. Zheng and Y. Lin, Carbon, 2018, 129, 236–244 CrossRef CAS.
- Z. Gao, J. Wang, Z. Li, W. Yang, B. Wang, M. Hou, Y. He, Q. Liu, T. Mann, P. Yang, M. Zhang and L. Liu, Chem. Mater., 2011, 23, 3509–3516 CrossRef CAS.
- D. Ruan, R. Lin, K. Jiang, X. Yu, Y. Zhu, Y. Fu, Z. Wang, H. Yan and W. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 29699–29706 CrossRef CAS PubMed.
- R. M. Tamgadge and A. Shukla, Electrochim. Acta, 2018, 289, 342–353 CrossRef CAS.
- S. Xie, Z. Bi, Y. Chen, X. He, X. Guo, X. Gao and X. Li, Appl. Surf. Sci., 2018, 459, 774–781 CrossRef CAS.
- D. V. Zhuzhelskii, E. G. Tolstopjatova, S. N. Eliseeva, A. V. Ivanov, S. Miao and V. V. Kondratiev, Electrochim. Acta, 2019, 299, 182–190 CrossRef CAS.
- C. Yao, B. Wei, H. Li, G. Wang, Q. Han, H. Ma and Q. Gong, J. Mater. Chem. A, 2017, 5, 56–61 RSC.
- J. Xu, Z. Liao, J. Zhang, B. Gao, P. K. Chu and K. Huo, J. Mater. Chem. A, 2018, 6, 6916–6921 RSC.
- W. Li, F. Xia, J. Qu, P. Li, D. H. Chen, Z. Chen, Y. Yu, Y. Lu, R. A. Caruso and W. G. Song, Nano Res., 2014, 7, 903–916 CrossRef CAS.
- Z. Hu, L. Xu, Y. Yang, H. Yao, H. Zhu, B. Hu and S. Yu, Chem. Sci., 2016, 7, 4276–4283 RSC.
- R. Wu, J. Zhang, Y. Shi, D. Liu and B. Zhang, J. Am. Chem. Soc., 2015, 137, 6983–6986 CrossRef CAS PubMed.
- H. Zheng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. Kalantar-zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef CAS.
- J. Zhao, Y. Tian, Z. Wang, S. Cong, D. Zhou, Q. Zhang, M. Yang, W. Zhang, F. Geng and Z. Zhao, Angew. Chem., Int. Ed., 2016, 55, 7161–7165 CrossRef CAS PubMed.
- M. S. Kim, E. Lim, S. Kim, C. Jo, J. Chun and J. Lee, Adv. Funct. Mater., 2017, 27, 1603921 CrossRef.
- Y. Zhong, X. H. Xia, S. J. Deng, J. Y. Zhan, R. Y. Fang, Y. Xia, X. L. Wang, Q. Zhang and J. P. Tu, Adv. Energy Mater., 2017, 8, 1701110 CrossRef.
- W. X. Liu, R. L. Yin, X. L. Xu, L. Zhang, W. H. Shi and X. H. Cao, Adv. Sci., 2019, 1802373 CrossRef PubMed.
- X. L. Xu, W. H. Shi, P. Li, S. F. Ye, C. Z. Ye, H. J. Ye, T. M. Lu, A. A. Zheng, J. X. Zhu, L. X. Xu, M. Q. Zhong and X. H. Cao, Chem. Mater., 2017, 29, 6058–6065 CrossRef CAS.
- Y. An, Y. Yang, Z. Hu, B. Guo, X. Wang, X. Yang, Q. Zhang and H. Wu, J. Power Sources, 2017, 337, 45–53 CrossRef CAS.
- A. D. Jagadale, V. S. Kumbhar, D. S. Dhawale and C. D. Lokhande, Electrochim. Acta, 2013, 98, 32–38 CrossRef CAS.
- Q. Liao, N. Li, S. Jin, G. Yang and C. Wang, ACS Nano, 2015, 9, 5310–5317 CrossRef CAS PubMed.
- X. L. Xu, W. H. Shi, W. X. Liu, S. F. Ye, R. L. Yin, L. Zhang, L. X. Xu, M. H. Chen, M. Q. Zhong and X. h. Cao, J. Mater. Chem. A, 2017, 6, 24086 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03886h |
| This journal is © The Royal Society of Chemistry 2019 |