
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium(II)-catalyzed synthesis of indenones through the cyclization of benzenecarbaldehydes with internal alkynes†
Jajula
Kashanna
 *a,
Rathod
Aravind Kumar
b and
Ravada
Kishore
c
*a,
Rathod
Aravind Kumar
b and
Ravada
Kishore
c
aDepartment of Chemistry, Rajiv Gandhi University of Knowledge Technologies, Basar 504107, India. E-mail: jajulakashanna@yahoo.co.in
bOrganic Synthesis & Process Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India
cDepartment of Chemistry, GITAM (Deemed to be University), Visakhapatnam 530045, India
First published on 2nd October 2019
Abstract
The palladium(II)-catalyzed carbocyclization of benzenecarbaldehydes with internal alkynes to afford 2,3-disubstituted indenones was reported. The annulation reaction proceeded through the transmetalation of Pd(II) with an aromatic aldehyde and the insertion of internal alkynes, followed by cyclization via the intramolecular nucleophilic addition of intermediate organopalladium(II) species to the aldehyde group. This reaction proceeded in moderate to good yields with high regioselectivity.
Introduction
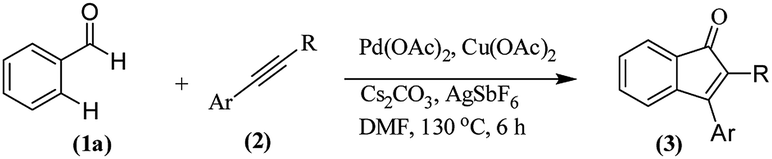
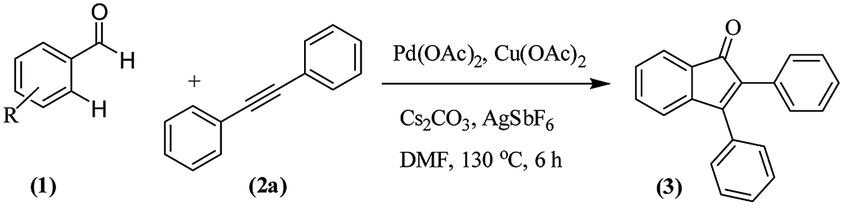
The transition metal-catalyzed oxidative C–H activation/carbocyclization of inactivated arene C–H bonds has evolved as a highly efficient route for a wide variety of building blocks.1,2 In this respect, the direct embodiment of inactivated arene groups in the C–H functionalization reaction is the most excellent synthetic strategy that induces additional features. Mostly, carbocyclization is catalyzed by rhodium3 or palladium4 complexes. Palladium4a–e-catalyzed carbocyclization of C–H bonds has received enormous attention owing to high efficiency, broad scope and good functional group tolerance. Pd-catalyzed oxidative coupling of arenes with alkynes can result in a broad spectrum of homo and heterocycles such as napthalenes,4a carbolines,4b phenanthrenes,4c isoquinolones,4d and benzazepines.4e Some of these methodologies have been successfully applied to the synthesis of natural products.4f–h Inspite of their success, it is essential to explore the C–H activation of inactivated arenes expedited by directing groups, which not only coordinate to metals but also participate in the subsequent reactions to provide novel tandem transformations. This approach serves not only to broaden the scope of palladium-catalyzed C–H functionalization but also to achieve molecular complexity.Indenones, which are an important class of carbocyclic compounds, form core moieties of many natural products.5 They are used as estrogen binding receptors,5a pharmaceuticals5a–e and fungicides.5f Furthermore, they find applications in materials science and photochromic chemistry.5g They also serve as useful precursors in the synthesis of a variety of biomolecules such as the C-nor-D-homosteroid ring system,5h gibberellins5i and indenes.5j Liebeskind and South first reported the preparation of indenones from o-diiodo benzene and different alkynes employing nickel carbonyl as the catalyst.6 Later, other transition metals such as iron,7 palladium8 and rhodium9 (Scheme 1, eqn (1)) were utilized as catalysts for the syntheses of indenones using preactivated aldehydes, nitriles, amides, esters or halides as the starting materials;6–9 among these synthesis methods, Pd-catalyzed annulation of alkynes with ortho-functionalized aryl-carbonyl and aryl-nitrile compounds forms a powerful synthetic strategy.8 In recent times, the direct assembly of indenones via rhodium,10 rhenium,11 cobalt12 and ruthenium13-catalyzed carbocyclization of benzoyl chlorides, benzimides, benzamides, azomethines, arylnitrones, benzaldehydes, nitrostyrenes14a and carboximides14b with alkynes and benzaldehydes with haloiodoketones14c,d (Scheme 1, eqn (2) and (4)) has been reported. However, these methods have certain drawbacks, which include harsh reaction conditions and the requirement of activated or preactivated arenes normally obtained from limited natural resources. Therefore, to overcome these problems, it is necessary to develop novel, convenient and efficient approaches and readily available starting materials for the synthesis of indenones. With this stated objective in the pursuance of our consistent efforts on the development of new synthetic methodologies,15 the direct alkynylation of the aromatic rings of benzenecarbaldehydes with the corresponding diaryl alkynes in the presence of a catalytic amount of palladium acetate (Scheme 1, eqn (5)) was explored and reported.
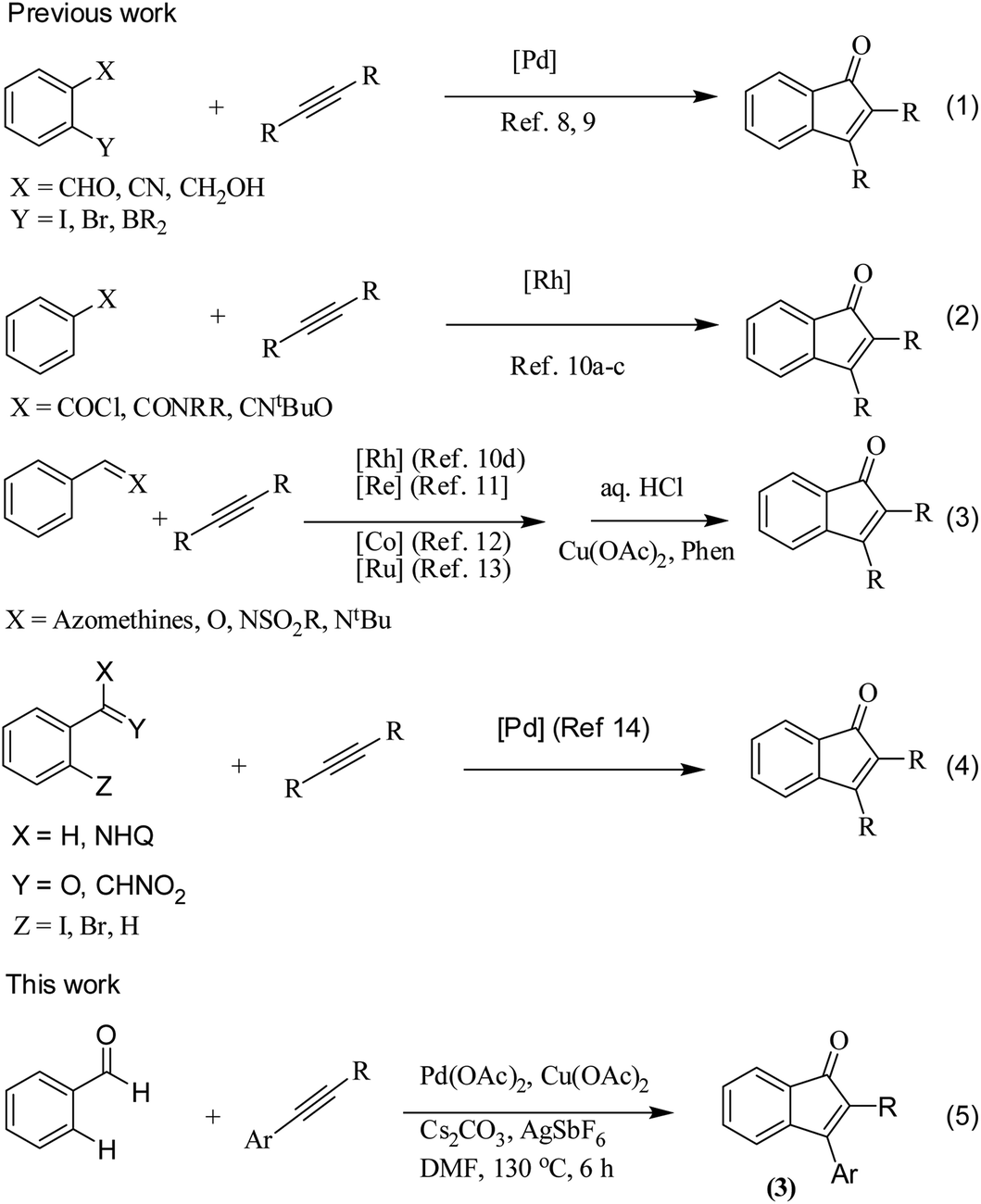 | ||
| Scheme 1 Representative metal-catalyzed synthesis of indenones via C–H activation/carbocyclization. | ||
Results and discussion
We focused our studies on the metal-catalyzed direct C–H activation/carbocyclization of inactivated arenes and found that the palladium complexes possessed catalytic activity for the direct C–H activation/carbocyclization of benzenecarbaldehydes with diaryl alkynes. The reaction of the aldehyde (1a) with diphenylacetylene (2a) in the presence of palladium acetate (10.0 mol%), copper acetate (2.0 equiv.) and cesium carbonate (2.0 equiv.) in DMF (2 mL) at 130 °C for 6 h under a nitrogen atmosphere proceeded to give indenone 3aa. Magnificently, we obtained 41% yield of the desired product (Table 1, entry 1). The yield of 3aa improved to 69% when AgSbF6 was used as the additive. The structure of 3aa was established on the basis of its 1H, 13C NMR and ESI-HRMS results; this structure has been reported in the literature and was obtained by a different route.16|
|
|||||
|---|---|---|---|---|---|
| Entrya | Oxidant | Additive | Base | Solvent | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1a) (0.5 mmol), alkyne (2a) (0.6 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), oxidant (1 equiv.), additive (20 mol%), base(1 equiv.), solvent (2 mL). b Isolated yield after column chromatography. | |||||
| 1 | Cu(OAc)2 | — | Cs2CO3 | DMF | 41 |
| 2 | Cu(OAc)2 | — | Cs2CO3 | DMF | 21 |
| 3 | Cu(OAc)2 | — | — | DMF | Trace |
| 4 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | DMF | 69 |
| 5 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | DCE | 32 |
| 6 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | Dioxane | 39 |
| 7 | Cu2O | AgSbF6 | Cs2CO3 | DMF | Trace |
| 8 | CuO | AgSbF6 | Cs2CO3 | DMF | Trace |
| 9 | CuI | AgSbF6 | Cs2CO3 | DMF | Trace |
| 10 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | Toluene | 13 |
| 11 | Ag2CO3 | — | — | DMF | 31 |
| 12 | Ag2CO3 | AgSbF6 | Cs2CO3 | DMF | 63 |
| 13 | AgOAc | AgSbF6 | Cs2CO3 | DMF | 56 |
| 14 | AgF | AgSbF6 | Cs2CO3 | DMF | 48 |
| 15 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | DMF | 49 |
| 16 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | DMF | 66 |
| 17 | Cu(OAc)2 | AgSbF6 | Cs2CO3 | DMF | 29 |
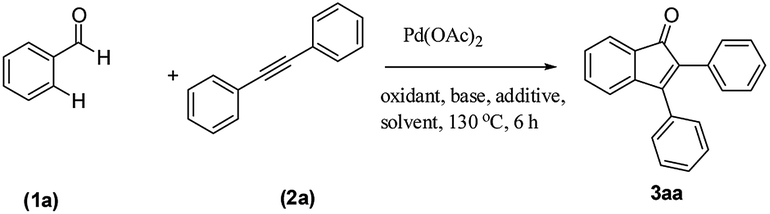
Intrigued by the above optimistic result, the effect of bases, oxidants, additives, solvents and temperatures on the reaction yield was further evaluated and some representative results are shown in Table 1. Various salts including Cu(OAc)2, Cu2O and CuO were examined; Cu(OAc)2 was found to be the optimal choice for the carboannulation reaction (Table 1, entries 1 and 4). In the case of Cu2O and CuO, the yield was <10% (Table 1, entries 8 and 9). The Cu(OAc)2 loading was also evaluated, and the use of 0.5 equiv. of Cu(OAc)2 led to a significant decrease in the yield (21%, entry 2). The use of Cu(OAc)2 without Cs2CO3 produced only traces of the product (entry 3). However, the replacement of Cu(OAc)2 with silver salts such as Ag2CO3, Ag2O, AgOAc and AgF almost resulted in the same yields (entries 11–14). Other bases such as Li2CO3, Na2CO3, NaOAc, NaOtBu, Na3PO4, K2CO3, KOAc and KOtBu were not suitable for this conversion. A survey of the role of solvents revealed that DMF was the optimal candidate (entries 4–10) as the solvent. To probe the process further, the screening of additives disclosed AgSbF6 as the most favored one to push the reaction forward, affording the desired product 3aa in 69% yield (entries 4 and 12). Decreasing the reaction temperature to 100 °C resulted in a lower yield of 49% (entry 15), and no distinct change was detected by raising the reaction temperature (entry 16). When the reaction time was decreased to 3 h, a lower yield of 29% was obtained (entry 17). It may be noted that carboannulation did not proceed in the absence of Pd(OAc)2 and the use of AgSbF6 was critical for achieving a high yield.
With the optimized conditions in hand, we further checked for the substrate scope and generality by varying the structures of the alkynes. As shown in Table 2, various valuable indenones can be conveniently and efficiently obtained in moderate to good yields with high regioselectivity by this novel palladium-catalyzed carboannulation reaction, indicating that this method is general and practically useful. In general, both electron-rich and electron-deficient aromatic diaryl alkynes were suitable for this method, and a wide range of groups such as methyl, fluoro, chloro, methoxy, trifluoromethyl, and hydrogen were tolerated under the reaction conditions (3aa–3am). We further checked the generality of both electron-rich and electron-deficient aromatic disubstituted diaryl alkynes. In general, electron-rich disubstituted diaryl alkynes underwent carboannulation smoothly and gave moderate yields, whereas electron-deficient aromatic disubstituted diaryl alkynes were not suitable for this conversion. Unsymmetrical alkynes also underwent carbocyclization smoothly and gave moderate yields with high regioselectivity. Besides the fact that no traces of regioisomers were detected by in situ1H NMR analysis in all the annulations, the results demonstrated that this novel Pd-catalyzed carboannulation reaction exhibited high regioselectivity.
|
|
|||
|---|---|---|---|
| Entrya | Alkyne (2) | Product (3) | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1) (0.074 g, 0.5 mmol), alkyne (2) (0.6 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), Cu(OAc)2 (0.090 g, 0.5 mmol), Cs2CO3 (0.162 g, 0.5 mmol), AgSbF6 (0.034 g, 0.1 mmol), DMF (2 mL). b Isolated yield after column chromatography. | |||
| 1 |
|
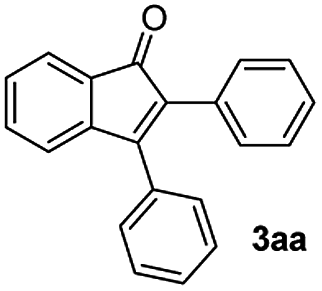
|
69 |
| 2 |
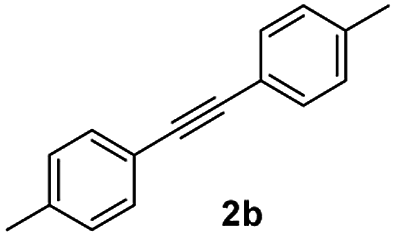
|
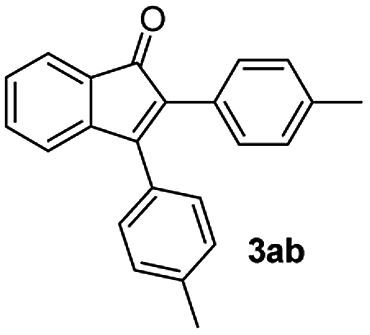
|
71 |
| 3 |
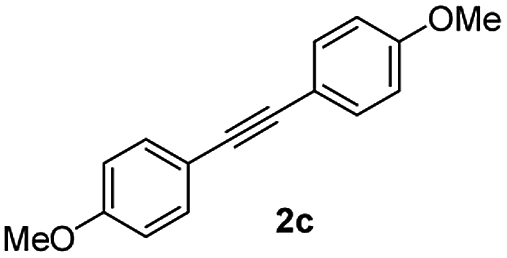
|
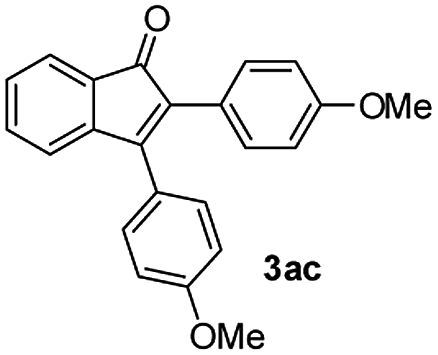
|
73 |
| 4 |
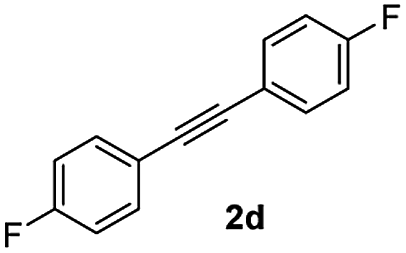
|
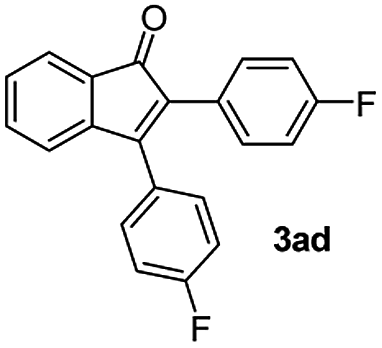
|
59 |
| 5 |
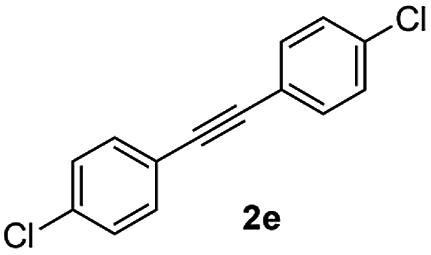
|
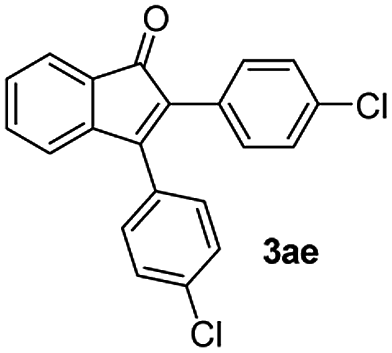
|
55 |
| 6 |
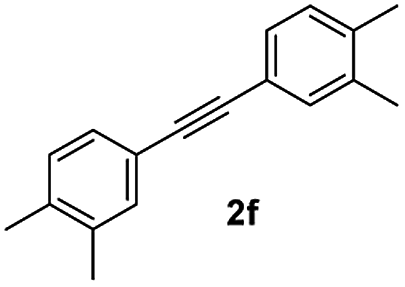
|
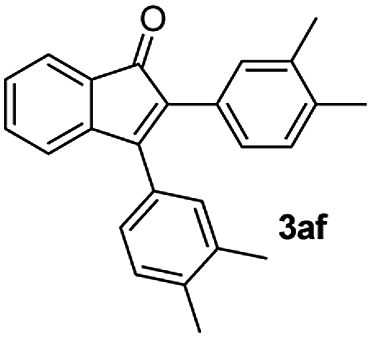
|
77 |
| 7 |
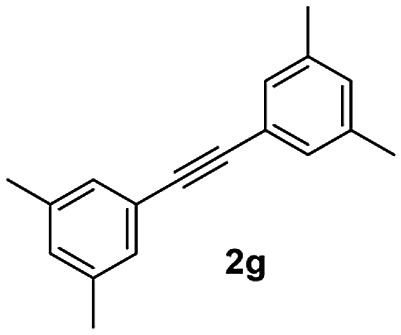
|
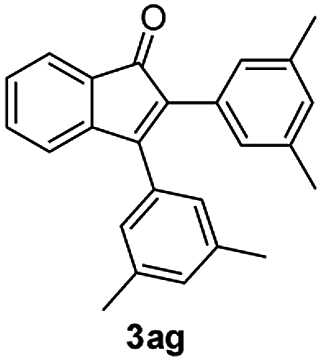
|
75 |
| 8 |
|
|
78 |
| 9 |
|
|
75 |
| 10 |
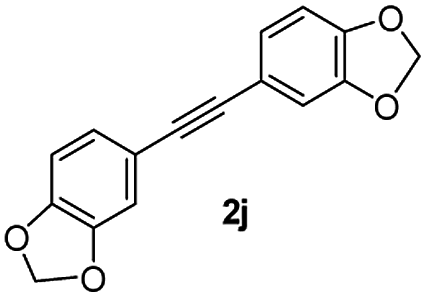
|
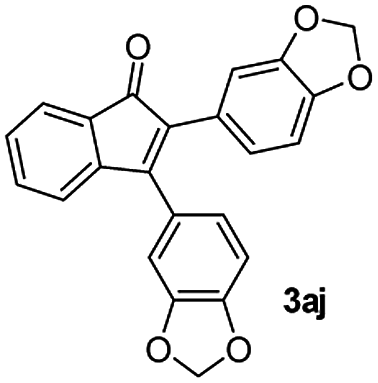
|
59 |
| 11 |
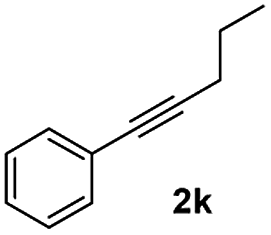
|
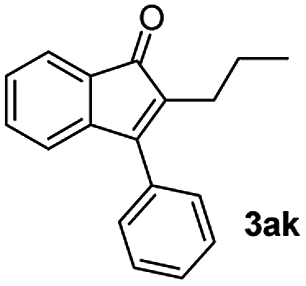
|
45 |
| 12 |
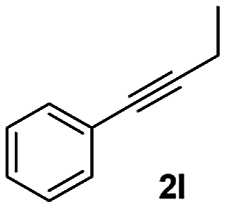
|
|
40 |
We further checked for the substrate scope and generality by varying the structures of benzaldehyde, as shown in Table 3. In general, both electron-rich and electron-deficient compounds were used to prepare different indenones following the above method. The electron-withdrawing groups containing benzenecarbaldehydes underwent C–H annulations smoothly and gave good yields, whereas the electron-rich benzenecarbaldehydes gave low yields of indenones and required a comparatively longer time.
|
|
|||
|---|---|---|---|
| Entrya | Benzaldehyde (1) | Product (3) | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1) (0.5 mmol), alkyne (2) (0.6 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), Cu(OAc)2 (0.090 g, 0.5 mmol), Cs2CO3 (0.162 g, 0.5 mmol), AgSbF6 (0.034 g, 0.1 mmol), DMF (2 mL). b Isolated yield after column chromatography. | |||
| 1 |
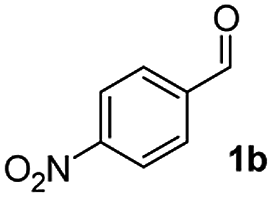
|
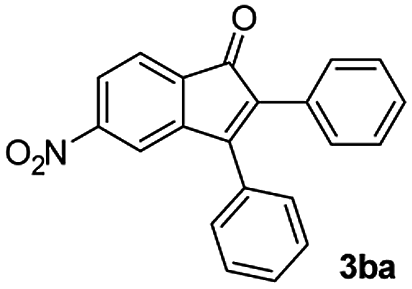
|
43 |
| 2 |
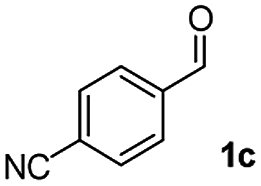
|
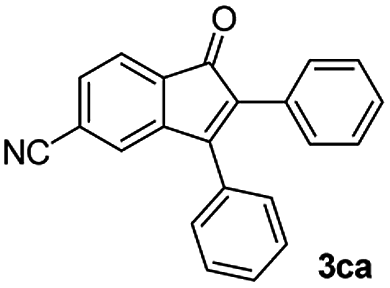
|
49 |
| 3 |
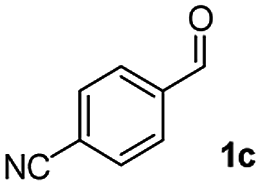
|
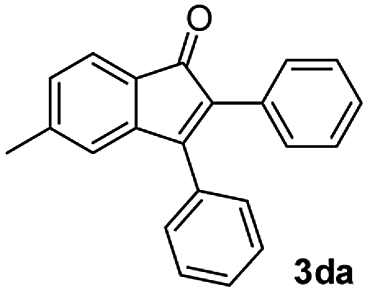
|
55 |
| 4 |
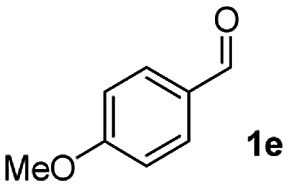
|
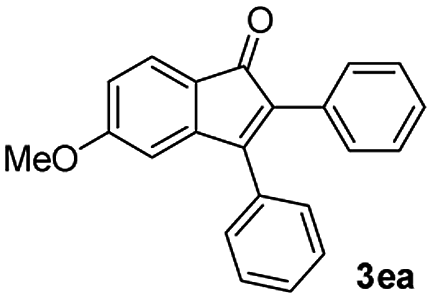
|
51 |
| 5 |
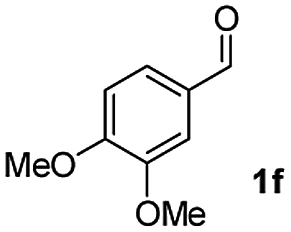
|
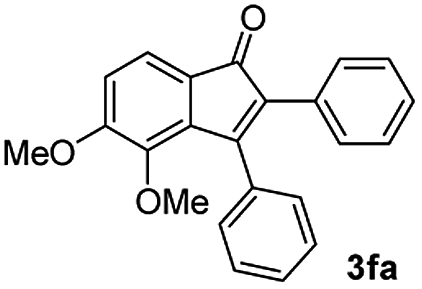
|
55 |
| 6 |
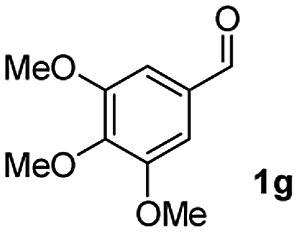
|
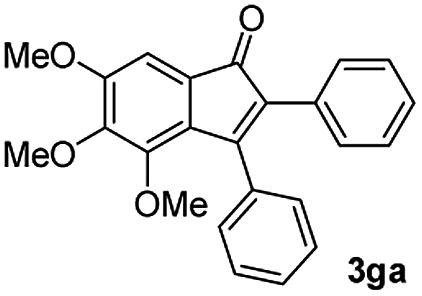
|
57 |
| 7 |
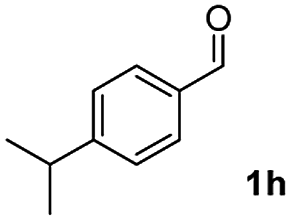
|
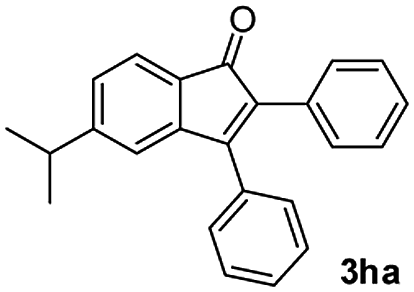
|
53 |
Plausible pathway
On the basis of the current information,8,17 a plausible mechanism for conversion is shown in Scheme 2. The formation of indenone 3aa apparently started with the palladation of an aromatic aldehyde by Pd(OAc)2 to yield the palladium complex 4. Syn-addition of 4 to the triple bond of diphenylacetylene 2 yielded the palladium species 5. This was followed by intramolecular cyclization, which would lead to 6; it further underwent reductive elimination to yield the indenone 3aa. AgSbF6 may be speculated to facilitate the removal of an acetate anion by exchange with [SbF6]−, which could propel the forward reaction. In case of unsymmetrical alkynes, the syn addition of complex 4 to alkyne 2 occurred in such a way that the alkyne carbon bearing the bulky group coupled with the complex 4 to give 5 by possibly avoiding steric hindrance when annulating with the carbaldehyde carbon.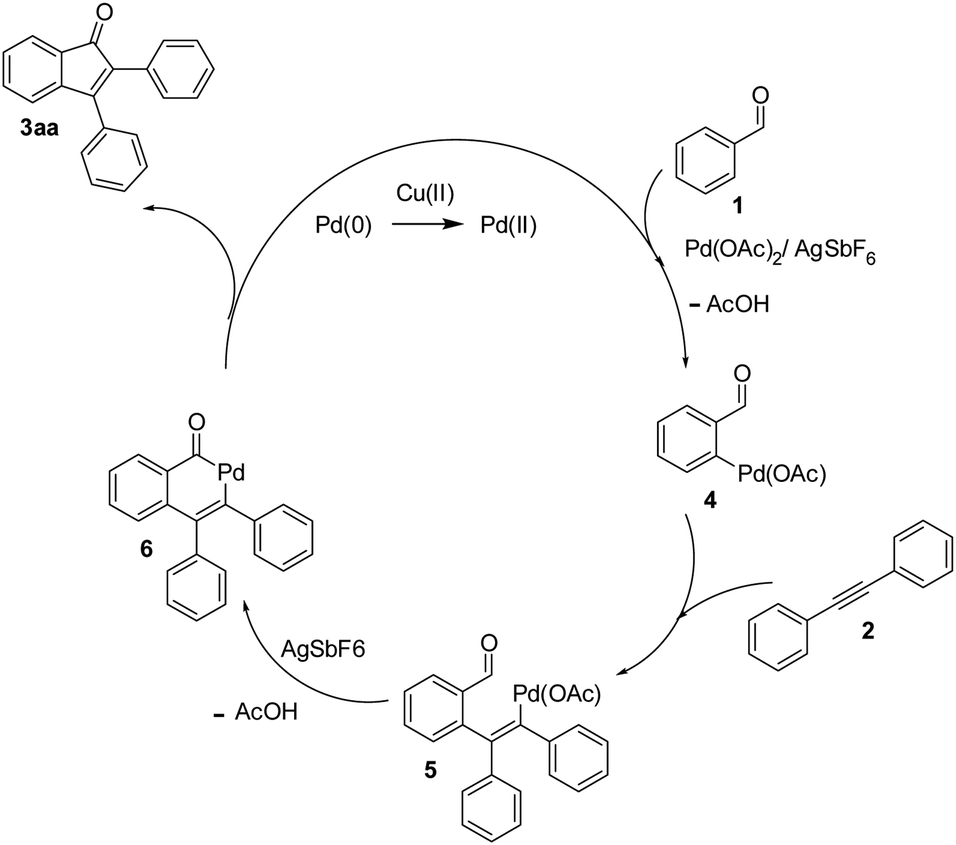 | ||
| Scheme 2 A plausible pathway for the formation of 3aa. | ||
Finally, to express the practical applicability of this protocol on a preparative scale, some reactions were carried out on the gram scale (20 mmol) using the following combinations of substrates: 1a with 2a and 1a with 2g. As per our expectations, the reactions proceeded smoothly to afford the target compounds in high yields as obtained in similar reactions on the milligram scale (entries 1 and 7, Table 2), which demonstrated the practical utility of this method.
Conclusions
In conclusion, we developed one-pot palladium-catalyzed carboannulation between benzaldehyde and internal alkynes for the synthesis of indenones. The reaction involved challenging nucleophilic addition of an organopalladium species to benzaldehyde. With this protocol, generally, high regioselective addition across unsymmetrically substituted alkynes was achievable.Experimental procedure
Materials and method
Solvents were dried according to known methods as appropriate. 1H, 13C spectra (1H, 400 MHz; 13C, 100 MHz) were recorded using a 400 MHz spectrometer in CDCl3 with shifts referenced to SiMe4 (δ = 0). IR spectra were recorded on an FTIR spectrophotometer. Elemental analyses were carried out on a CHN analyzer. Mass spectra were recorded using LC-MS and HRMS (ESI-TOF analyzer) equipment. Column chromatography was performed on silica gel (250–400 mesh) using ethyl acetate (EtOAc)/hexane mixture.General procedure for the synthesis of indenones
Benzaldehyde (0.053 g, 0.5 mmol), Pd(OAc)2 (0.011 g, 0.05 mmol), Cu(OAc)2 (0.090 g, 0.5 mmol), AgSbF6 (0.034 g, 0.1 mmol), Cs2CO3 (0.162 g, 0.5 mmol), DMF (2 mL) and diaryl acetylene (0.6 mmol) were charged into an oven-dried 25 mL round bottomed flask under nitrogen. The mixture was stirred at 130 °C for 6 h. After cooling to room temperature and extracting with EtOAc (3 × 25 mL). The combined ethyl acetate extract was washed with brine (75 mL), dried over anh. Na2SO4 and filtered. The solvent was removed under vacuum and the resulting crude product was purified by silica gel chromatography using hexane/ethyl acetate mixture to afford compounds 3aa–3al. The details on the yields for all the compounds are presented in Tables 2 and 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the desired product 3aa as a red solid. IR (KBr, cm−1) 2911, 1704, 1605, 1447; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 5.4 Hz, 1H), 7.42–7.35 (m, 6H), 7.30–7.21 (m, 6H), 7.17 (d, J = 5.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 196.4, 153.3, 145.2, 133.2, 132.7, 132.4, 130.7, 129.9, 129.2, 128.9, 128.7, 128.5, 128.0, 127.7, 122.9, 121.2; (HRMS (ESI) Calcd for C21H15O [M + H]: m/z 283.1124. Found: 283.1123).
:1) to afford the desired product 3ab as a red solid. IR (KBr, cm−1) 2914, 1703, 1601, 1450; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.3 Hz, 1H), 7.39 (td, J = 7.3 and 1.2 Hz, 3H), 7.27–7.15 (m, 5H), 7.09 (d, J = 8.4 Hz, 2H), 2.42 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.7, 154.7, 145.5, 139.3, 137.5, 133.2, 132.0, 130.9, 129.9, 129.8, 129.4, 128.8, 128.7, 128.4, 127.9, 122.7, 121.1, 21.5, 21.3; (HRMS (ESI) Calcd for C23H19O [M + H]: m/z 311.1436. Found: 311.136)
:1) to afford the desired product 3ac as a red solid. IR (KBr, cm−1) 2911, 1701, 1605, 1439; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.0 Hz, 1H), 7.39 (d, J = 8.1 Hz, 3H), 7.36–7.29 (m, 3H), 7.22 (d, J = 8.1 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.1 Hz, 2H), 3.88 (s, 3H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.0, 159.1, 133.2, 131.2, 131.0, 130.2, 128.6, 125.1, 122.7, 120.9, 114.2, 113.6, 55.3, 55.2; (HRMS (ESI) Calcd for C23H19O3 [M + H]: m/z 343.1335. Found: 343.1335).
:1) to afford the desired product 3ad as a red solid. IR (KBr, cm−1) 2989, 1714, 1608, 1440; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 7.0 Hz, 1H), 7.44 (t, J = 8.1 and 1.0 Hz, 3H), 7.41–7.30 (m, 3H), 7.19 (t, J = 8.1 and 1.0 Hz, 3H), 7.15–6.90 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 196.2, 164.1, 162.1 (d, JC–F = 244. 0 Hz), 154.1 (d, JC–F = 244. 0 Hz),, 144.8 (d, JC–F = 8. 0 Hz),, 133.6, 131.7 (d, JC–F = 8. 0 Hz), 131.5, 130.5 (d, JC–F = 8. 0 Hz), 129.2, 128.4, 126.5 (d, JC–F = 8. 0 Hz),, 123.4, 121.1, 116.3, 116.1, 115.4, 115.2; (HRMS (ESI) Calcd for C21H12F2O2 [M + H]: m/z 319.0935. Found: 319.0959).
:1) to afford the desired product 3ae as a red solid. IR (KBr, cm−1) 2915, 1700, 1605, 1446; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.0 Hz, 1H), 7.43–7.36 (m, 3H), 7.34–7.29 (m, 3H), 7.26–7.23 (m, 2H), 7.20–7.18 (m, 2H), 7.11 (d, J = 7.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 196.1, 154.3, 144.6, 135.3, 134.1, 133.6, 131.4, 131.3, 130.9, 130.4, 129.9, 129.4, 128.7, 128.4, 123.3, 121.1; (HRMS (ESI) Calcd for C21H12Cl2O2 [M + H]: m/z 351.0235. Found: 351.0247).
:1) to afford the desired product 3af as a red solid. IR (KBr, cm−1) 2915, 1704, 1600, 1446; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.0 Hz, 1H), 7.34 (dt, J = 5.6 and 0.8 Hz, 1H), 7.27 (dt, J = 5.7, 0.6 Hz, 1H), 7.22 (brs, 1H), 7.16 (brs, 2H), 7.15 (brs, 1H), 7.09 (d, J = 5.8 Hz, 1H), 7.00 (d, J = 5.8 Hz, 1H), 6.95 (d, J = 5.8 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H), 2.24 (s, 3H), 2.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.0, 154.8, 145.6, 137.9, 136.8, 136.19, 136.11, 133.2, 132.0, 131.0, 130.9, 130.3, 129.9, 129.3, 129.2, 128.6, 128.4, 127.3, 122.6, 121.1, 19.8, 19.6; HRMS (ESI) Calcd For C25H22ONa [M + Na]: m/z 361.1568. Found: 361.1570.
:1) to afford the desired product 3ag as a red solid. IR (KBr, cm−1) 2915, 1709, 1600, 1457; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 2.6 Hz, 1H), 7.37 (dt, J = 5.9, 0.9 Hz, 1H), 7.28 (dt, J = 5.8, 0.6 Hz, 1H), 7.14 (d, J = 5.8 Hz, 1H), 7.05 (brs, 1H), 7.00 (brs, 2H), 6.91 (brs, 2H), 6.89 (brs, 1H), 2.31 (s, 6H), 2.23 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 196.9, 156.4, 145.6, 138.8, 138.1, 137.2, 136.6, 133.3, 132.7, 132.3, 130.8, 130.6, 129.4, 128.7, 127.6, 126.0, 122.7, 121.2, 21.34, 21.32; HRMS (ESI) Calcd for C25H22ONa [M + Na]: m/z 361.1568. Found: 361.1567.
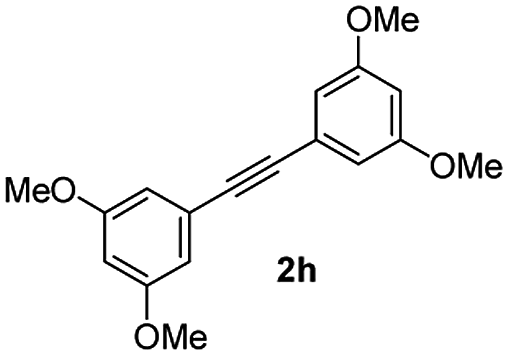
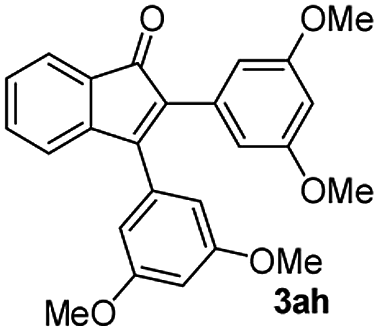
:5) to afford the desired product 3ah as a red solid. IR (KBr, cm−1) 2913, 1709, 1594, 1452; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.6 Hz, 1H), 7.37 (dt, J = 0.9, 5.9 Hz, 1H), 7.28 (dt, J = 0.6, 5.8 Hz, 1H), 7.17 (d, J = 5.8 Hz, 1H), 6.52 (d, J = 1.8 Hz, 2H), 6.49 (t, J = 1.8 Hz, 1H), 6.47 (d, J = 1.8 Hz, 2H), 6.36 (t, J = 1.8 Hz, 1H), 3.71 (s, 6H), 3.65 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 196.2, 161.0, 160.2, 155.6, 145.0, 134.5, 133.5, 132.4, 132.2, 130.6, 129.0, 122.9, 121.4, 107.8, 106.3, 101.4, 100.8, 55.4, 55.2; HRMS (ESI) Calcd for C25H22O5Na [M + Na]: m/z 425.1365. Found: 425.1362.
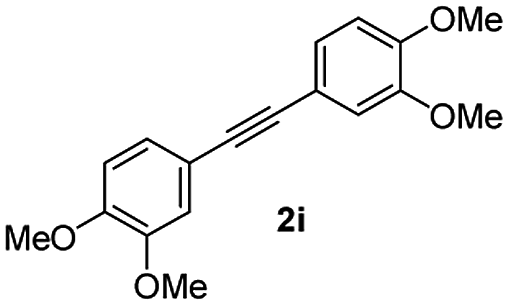
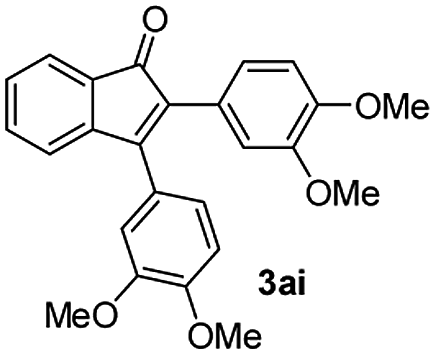
:5) to afford the desired product 3ai as a red solid. IR (KBr, cm−1) 2915, 1693, 1632, 1452; 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 5.4 Hz, 1H), 7.39 (dt, J = 6.0, 0.8 Hz, 1H), 7.29 (dt, J = 5.9, 0.6 Hz, 1H), 7.21 (d, J = 5.8 Hz, 1H), 7.08 (dd, J = 6.6, and 1.5 Hz, 1H), 6.95 (d, J = 6.7 Hz, 2H), 6.88 (dd, J = 9.9 Hz and J = 1.5 Hz, 2H), 6.81 (d, J = 6.7 Hz, 1H), 3.95 (s, 3H), 3.88 (s, 3H), 3.73 (s, 3H), 3.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.8, 153.9, 149.9, 149.1, 148.7, 148.4, 145.3, 133.3, 131.3, 131.0, 128.7, 125.4, 123.6, 123.0, 122.7, 121.6, 120.9, 113.1, 111.9, 111.3, 110.9, 55.9, 55.8, 55.6; HRMS (ESI) Calcd for C25H22O5Na [M + Na]: m/z 425.1365. Found: 425.1363.
:1) to afford the desired product as a red solid. IR (KBr, cm−1) 2914, 1699, 1630, 1499; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.0 Hz, 1H), 7.38 (dt, J = 7.5, 1.1 Hz, 1H), 7.28 (dt, J = 7.7, 0.9 Hz, 1H), 7.17 (d, J = 7.2 Hz, 1H), 6.95 (dd, J ∼8.1, J = 1.7 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.84 (dd, J ∼8.4 Hz and J = 1.6 Hz, 2H), 6.79–6.77 (m, 2H), 6.05 (s, 2H), 5.96 (s, 2H);13C NMR (100 MHz, CDCl3) δ 196.5, 154.1, 148.5, 148.0, 147.4, 147.3, 145.2, 133.4, 131.6, 130.8, 128.8, 126.4, 124.6, 124.2, 122.9, 121.1, 110.2, 108.9, 108.8, 108.4, 101.4, 101.0; HRMS (ESI) Calcd for C23H15O5 [M + H]: m/z 371.0919. Found: 371.0917.
:1) to afford the desired product 3ak as a red solid. IR (KBr, cm−1) 2914, 1703, 1615, 1447; 1H NMR (400 MHz, CDCl3) δ 7.59–7.41 v(m, 6H), 7.39–7.18 (m, 2H), 7.12 (d, J = 7.0 Hz, 1H), 2.72–2.31 (m, 2H), 1.56–1.48 (m, 2H), 0.90 (t, J = 7.0 and 2.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 198.3, 155.2, 145.9, 135.3, 133.1, 132.9, 130.9, 129.0, 128.1, 122.4, 120.5, 120.5, 25.3, 22.6, 14.2; HRMS (ESI) Calcd for C17H15O [M + H]: m/z 235.1124. Found: 235.1100.
:1) to afford the desired product 3aa as a red solid. IR (KBr, cm−1) 2911, 1704, 1605, 1447; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 5.4 Hz, 1H), 7.42–7.35 (m, 6H), 7.30–7.21 (m, 6H), 7.17 (d, J = 5.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 196.4, 153.3, 145.2, 133.2, 132.7, 132.4, 130.7, 129.9, 129.2, 128.9, 128.7, 128.5, 128.0, 127.7, 122.9, 121.2; (HRMS (ESI) Calcd for C21H15O [M + H]: m/z 283.1124. Found: 283.1123).
:1) to afford the desired product 3ab as a red solid. IR (KBr, cm−1) 2914, 1703, 1601, 1450; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.3 Hz, 1H), 7.39 (td, J = 7.3 and 1.2 Hz, 3H), 7.27–7.15 (m, 5H), 7.09 (d, J = 8.4 Hz, 2H), 2.42 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.7, 154.7, 145.5, 139.3, 137.5, 133.2, 132.0, 130.9, 129.9, 129.8, 129.4, 128.8, 128.7, 128.4, 127.9, 122.7, 121.1, 21.5, 21.3; (HRMS (ESI) Calcd for C23H19O [M + H]: m/z 311.1436. Found: 311.136)
:1) to afford the desired product 3ac as a red solid. IR (KBr, cm−1) 2911, 1701, 1605, 1439; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.0 Hz, 1H), 7.39 (d, J = 8.1 Hz, 3H), 7.36–7.29 (m, 3H), 7.22 (d, J = 8.1 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.1 Hz, 2H), 3.88 (s, 3H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.0, 159.1, 133.2, 131.2, 131.0, 130.2, 128.6, 125.1, 122.7, 120.9, 114.2, 113.6, 55.3, 55.2; (HRMS (ESI) Calcd for C23H19O3 [M + H]: m/z 343.1335. Found: 343.1335).
:1) to afford the desired product 3ad as a red solid. IR (KBr, cm−1) 2989, 1714, 1608, 1440; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 7.0 Hz, 1H), 7.44 (t, J = 8.1 and 1.0 Hz, 3H), 7.41–7.30 (m, 3H), 7.19 (t, J = 8.1 and 1.0 Hz, 3H), 7.15–6.90 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 196.2, 164.1, 162.1 (d, JC–F = 244. 0 Hz), 154.1 (d, JC–F = 244. 0 Hz),, 144.8 (d, JC–F = 8. 0 Hz),, 133.6, 131.7 (d, JC–F = 8. 0 Hz), 131.5, 130.5 (d, JC–F = 8. 0 Hz), 129.2, 128.4, 126.5 (d, JC–F = 8. 0 Hz),, 123.4, 121.1, 116.3, 116.1, 115.4, 115.2; (HRMS (ESI) Calcd for C21H12F2O2 [M + H]: m/z 319.0935. Found: 319.0959).
:1) to afford the desired product 3ae as a red solid. IR (KBr, cm−1) 2915, 1700, 1605, 1446; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.0 Hz, 1H), 7.43–7.36 (m, 3H), 7.34–7.29 (m, 3H), 7.26–7.23 (m, 2H), 7.20–7.18 (m, 2H), 7.11 (d, J = 7.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 196.1, 154.3, 144.6, 135.3, 134.1, 133.6, 131.4, 131.3, 130.9, 130.4, 129.9, 129.4, 128.7, 128.4, 123.3, 121.1; (HRMS (ESI) Calcd for C21H12Cl2O2 [M + H]: m/z 351.0235. Found: 351.0247).
:1) to afford the desired product 3af as a red solid. IR (KBr, cm−1) 2915, 1704, 1600, 1446; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.0 Hz, 1H), 7.34 (dt, J = 5.6 and 0.8 Hz, 1H), 7.27 (dt, J = 5.7, 0.6 Hz, 1H), 7.22 (brs, 1H), 7.16 (brs, 2H), 7.15 (brs, 1H), 7.09 (d, J = 5.8 Hz, 1H), 7.00 (d, J = 5.8 Hz, 1H), 6.95 (d, J = 5.8 Hz, 1H), 2.32 (s, 3H), 2.28 (s, 3H), 2.24 (s, 3H), 2.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.0, 154.8, 145.6, 137.9, 136.8, 136.19, 136.11, 133.2, 132.0, 131.0, 130.9, 130.3, 129.9, 129.3, 129.2, 128.6, 128.4, 127.3, 122.6, 121.1, 19.8, 19.6; HRMS (ESI) Calcd For C25H22ONa [M + Na]: m/z 361.1568. Found: 361.1570.
:1) to afford the desired product 3ag as a red solid. IR (KBr, cm−1) 2915, 1709, 1600, 1457; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 2.6 Hz, 1H), 7.37 (dt, J = 5.9, 0.9 Hz, 1H), 7.28 (dt, J = 5.8, 0.6 Hz, 1H), 7.14 (d, J = 5.8 Hz, 1H), 7.05 (brs, 1H), 7.00 (brs, 2H), 6.91 (brs, 2H), 6.89 (brs, 1H), 2.31 (s, 6H), 2.23 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 196.9, 156.4, 145.6, 138.8, 138.1, 137.2, 136.6, 133.3, 132.7, 132.3, 130.8, 130.6, 129.4, 128.7, 127.6, 126.0, 122.7, 121.2, 21.34, 21.32; HRMS (ESI) Calcd for C25H22ONa [M + Na]: m/z 361.1568. Found: 361.1567.
:5) to afford the desired product 3ah as a red solid. IR (KBr, cm−1) 2913, 1709, 1594, 1452; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.6 Hz, 1H), 7.37 (dt, J = 0.9, 5.9 Hz, 1H), 7.28 (dt, J = 0.6, 5.8 Hz, 1H), 7.17 (d, J = 5.8 Hz, 1H), 6.52 (d, J = 1.8 Hz, 2H), 6.49 (t, J = 1.8 Hz, 1H), 6.47 (d, J = 1.8 Hz, 2H), 6.36 (t, J = 1.8 Hz, 1H), 3.71 (s, 6H), 3.65 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 196.2, 161.0, 160.2, 155.6, 145.0, 134.5, 133.5, 132.4, 132.2, 130.6, 129.0, 122.9, 121.4, 107.8, 106.3, 101.4, 100.8, 55.4, 55.2; HRMS (ESI) Calcd for C25H22O5Na [M + Na]: m/z 425.1365. Found: 425.1362.
:5) to afford the desired product 3ai as a red solid. IR (KBr, cm−1) 2915, 1693, 1632, 1452; 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 5.4 Hz, 1H), 7.39 (dt, J = 6.0, 0.8 Hz, 1H), 7.29 (dt, J = 5.9, 0.6 Hz, 1H), 7.21 (d, J = 5.8 Hz, 1H), 7.08 (dd, J = 6.6, and 1.5 Hz, 1H), 6.95 (d, J = 6.7 Hz, 2H), 6.88 (dd, J = 9.9 Hz and J = 1.5 Hz, 2H), 6.81 (d, J = 6.7 Hz, 1H), 3.95 (s, 3H), 3.88 (s, 3H), 3.73 (s, 3H), 3.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.8, 153.9, 149.9, 149.1, 148.7, 148.4, 145.3, 133.3, 131.3, 131.0, 128.7, 125.4, 123.6, 123.0, 122.7, 121.6, 120.9, 113.1, 111.9, 111.3, 110.9, 55.9, 55.8, 55.6; HRMS (ESI) Calcd for C25H22O5Na [M + Na]: m/z 425.1365. Found: 425.1363.
:1) to afford the desired product as a red solid. IR (KBr, cm−1) 2914, 1699, 1630, 1499; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.0 Hz, 1H), 7.38 (dt, J = 7.5, 1.1 Hz, 1H), 7.28 (dt, J = 7.7, 0.9 Hz, 1H), 7.17 (d, J = 7.2 Hz, 1H), 6.95 (dd, J ∼8.1, J = 1.7 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.84 (dd, J ∼8.4 Hz and J = 1.6 Hz, 2H), 6.79–6.77 (m, 2H), 6.05 (s, 2H), 5.96 (s, 2H);13C NMR (100 MHz, CDCl3) δ 196.5, 154.1, 148.5, 148.0, 147.4, 147.3, 145.2, 133.4, 131.6, 130.8, 128.8, 126.4, 124.6, 124.2, 122.9, 121.1, 110.2, 108.9, 108.8, 108.4, 101.4, 101.0; HRMS (ESI) Calcd for C23H15O5 [M + H]: m/z 371.0919. Found: 371.0917.
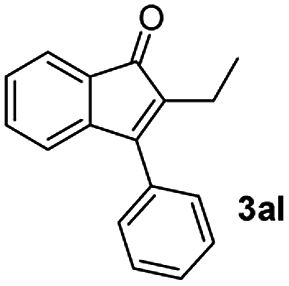
:1) to afford the desired product 3ak as a red solid. IR (KBr, cm−1) 2914, 1703, 1615, 1447; 1H NMR (400 MHz, CDCl3) δ 7.59–7.41 v(m, 6H), 7.39–7.18 (m, 2H), 7.12 (d, J = 7.0 Hz, 1H), 2.72–2.31 (m, 2H), 1.56–1.48 (m, 2H), 0.90 (t, J = 7.0 and 2.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 198.3, 155.2, 145.9, 135.3, 133.1, 132.9, 130.9, 129.0, 128.1, 122.4, 120.5, 120.5, 25.3, 22.6, 14.2; HRMS (ESI) Calcd for C17H15O [M + H]: m/z 235.1124. Found: 235.1100.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank UGC-DSK (New Delhi) for the Dr D. S. Kothari fellowship. JK also thank to Prof. K. C. Kumaraswamy for his expert suggestions. RK acknowledges to the DST for the financial support under the early career research award scheme (project no. ECR/2018/000637). We also thank Director CSIR-IICT for the support (IICT/Pubs/2019/352).Notes and references
- (a) F. Kakiuchiand and S. Murai, Acc. Chem. Res., 2002, 35(10), 826–834 CrossRef; (b) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106(11), 4644–4680 CrossRef CAS; (c) C.-H. Jun, E.-A. Jo and J.-W. Park, Eur. J. Org. Chem., 2007, 1869–1881 CrossRef CAS; (d) D. Kalyani and M. S. Sanford, Top. Organomet. Chem., 2007, 24, 85–116 CrossRef CAS; (e) L. Ackermann, Top. Organomet. Chem., 2007, 24, 35–60 CrossRef CAS; (f) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41(2), 222–234 CrossRef CAS; (g) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110(2), 624–655 CrossRef CAS; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110(2), 1147–1169 CrossRef CAS; (i) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114(3), 1783–1826 CrossRef CAS.
- (a) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102(5), 1731–1770 CrossRef CAS; (b) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345(9–10), 1077–1101 CrossRef CAS; (c) K. Godula and D. Sames, Science, 2006, 312(5770), 67–72 CrossRef CAS; (d) F. Kakiuchi, Top. Organomet. Chem., 2007, 24, 1–33 CrossRef CAS; (e) T. Satoh and M. Miura, Top. Organomet. Chem., 2007, 24, 61–84 CrossRef CAS; (f) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (g) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107(1), 174–238 CrossRef CAS; (h) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41(8), 1013–1025 CrossRef CAS; (i) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15(24), 5874–5883 CrossRef CAS; (j) M. C. Willis, Chem. Rev., 2010, 110(2), 725–748 CrossRef CAS; (k) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110(2), 1147–1169 CrossRef CAS; (l) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC; (m) R. Jazzar, J. Hitce, A. Renaudat, J. S. Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16(9), 2654–2672 CrossRef CAS; (n) L. Ackermann, Chem. Rev., 2011, 111(3), 1315–1345 CrossRef CAS PubMed.
- (a) S. Mochida, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2010, 39(7), 744–746 CrossRef CAS; (b) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132(30), 10565–10569 CrossRef CAS; (c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133(8), 2350–2353 CrossRef CAS; (d) F. W. Patureau, T. Besset and F. Glorius, Angew. Chem., Int. Ed., 2011, 50(5), 1064–1067 CrossRef CAS; (e) C. Zhu and J. R. Falck, Chem. Commun., 2012, 48, 1674–1676 RSC; (f) F. Wang, G. Song, Z. Du and X. J. Li, J. Org. Chem., 2011, 76(8), 2926–2932 CrossRef CAS; (g) J. Willwacher, S. Rakshit and F. Glorius, Org. Biomol. Chem., 2011, 9, 4736–4740 RSC.
- (a) Y.-T. Wu, K.-H. Huang, C.-C. Shin and T.-C. Wu, Chem.–Eur. J., 2008, 14(22), 6697–6703 CrossRef CAS; (b) S. Ding, Z. Shi and N. Jiao, Org. Lett., 2010, 12(7), 1540–1543 CrossRef CAS; (c) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132(40), 14006–14008 CrossRef CAS; (d) H. Zhong, D. Yang, S. Wang and J. Huang, Chem. Commun., 2012, 48, 3236–3238 RSC; (e) L. Wang, J. Huang, S. Peng, H. Liu, X. Jiang and J. Wang, Angew. Chem., Int. Ed., 2013, 52(6), 1768–1772 CrossRef CAS; (f) H. Yokoyama, K. Otaya, H. Kobayashi, M. Miyazawa, S. Yamaguchi and H. Hirai, Org. Lett., 2000, 2(16), 2427–2429 CrossRef CAS; (g) X. Xie, X. Lu, Y. Liu and W. Xu, J. Org. Chem., 2001, 66(20), 6545–6550 CrossRef CAS; (h) A. L. Bowie and D. Trauner, J. Org. Chem., 2009, 74(4), 1581–1586 CrossRef CAS; (i) K. Anderson, K. Calo, T. Pfaffeneder, A. J. P. White and A. G. M. Barrett, Org. Lett., 2011, 13(21), 5748–5750 CrossRef CAS; (j) G. Ji, Y. Duan, S. Zhang and Y. Yang, Catal. Today, 2019, 330(11), 101–108 CrossRef CAS.
- (a) G. M. Anstead, J. L. Ensign, C. S. Peterson and J. A. Katzenellenbogen, J. Org. Chem., 1989, 54(7), 1485–1491 CrossRef CAS; (b) R. E. McDevitt, M. S. Malamas, E. S. Manas, R. J. Unwalla, Z. B. Xu, C. P. Miller and H. A. Harris, Bioorg. Med. Chem. Lett., 2005, 15(12), 3137–3142 CrossRef CAS; (c) H. Gao, J. A. Katzenellenbogen, R. Garg and C. Hansch, Chem. Rev., 1999, 99(3), 723–744 CrossRef CAS PubMed; (d) J. H. Ahn, M. S. Shin, S. H. Jung, S. K. Kang, K. R. Kim, S. D. Rhee, W. H. Jung, S. D. Yang, S. J. Kim, J. R. Woo, J. H. Lee, H. G. Cheon and S. S. Kim, J. Med. Chem., 2006, 49(15), 4781–4784 CrossRef CAS PubMed; (e) W. M. Clark, A. M. Tickner-Eldridge, G. Kris Huang, L. N. Pridgen, M. A. Olsen, R. J. Mills, I. Lantos and N. H. Baine, J. Am. Chem. Soc., 1998, 120(18), 4550–4551 CrossRef CAS; (f) G. P. Jourdan, B. A. Dreikorn, R. E. Hackler, H. R. Hall and W. R. Arnold, in Synthesis and Chemistry of Agrochemicals II, ACS Symposium Series, American Chemical Society, Washington, DC, 1991, p. 566 Search PubMed; (g) E. F. Ullmm and W. A. J. Henderson, J. Am. Chem. Soc., 1966, 88(21), 4942–4960 CrossRef; (h) A. Chattejee and S. Banerjee, Tetrahedron, 1970, 26(11), 2599–2608 CrossRef; (i) H. O. Hose and J. K. Larson, J. Org. Chem., 1968, 33(1), 448–451 CrossRef; (j) E. N. Alesso, D. G. Tombari, A. F. Ibanez, G. Y. MoltrasioIdesias and J. M. Aguirre, Can. J. Chem., 1991, 69, 1166–1170 CrossRef CAS; (k) B. VenkatRamulu and G. Satyanarayana, RSC Adv., 2015, 5, 70972–70976 RSC.
- L. S. Liebeskind and M. S. South, J. Org. Chem., 1980, 45(26), 5426–5429 CrossRef CAS.
- I. R. Butler, Can. J. Chem., 1990, 68, 1979–1987 CrossRef CAS.
- (a) W. Tao, L. J. Silverberg, A. L. Rheingold and R. F. Heck, Organometallics, 1989, 8(11), 2550–2559 CrossRef CAS; (b) R. C. Larock, Q. Tian and A. A. Pletnev, J. Am. Chem. Soc., 1999, 121(13), 3238–3239 CrossRef CAS; (c) A. A. Pletnev, Q. Tian and R. C. Larock, J. Org. Chem., 2002, 67(26), 9276–9287 CrossRef CAS; (d) H. Tsukamoto and Y. Kondo, Org. Lett., 2007, 9(21), 4227–4230 CrossRef CAS; (e) J. Zhang, F. Yang and Y. Wu, Appl. Organomet. Chem., 2011, 25(9), 675–679 CAS; (f) F. Yang, J. Zhang and Y. Wu, Tetrahedron, 2011, 67(16), 2969–2973 CrossRef CAS; (g) N. Wu, A. Messinis, A. S. Batsanov, Z. Yang, A. Whiting and T. B. Marder, Chem. Commun., 2012, 48, 9986–9988 RSC; (h) X. Chen, Q. He, Y. Xie and C. Yang, Org. Biomol. Chem., 2013, 11, 2582–2585 RSC; (i) R. C. Larock and M. J. Doty, J. Org. Chem., 1993, 58(17), 4579–4583 CrossRef CAS; (j) M. L. N. Rao and R. J. Dhanorkar, Tetrahedron, 2014, 70(43), 8067–8078 CrossRef CAS; (k) J. Feng, G. Lu, M. Lv and C. Cai, J. Org. Chem., 2014, 79(21), 10561–10567 CrossRef CAS.
- (a) A. Padwa, K. E. Krumpe, Y. Gareau and U. Chiacchio, J. Org. Chem., 1991, 56(7), 2523–2530 CrossRef CAS; (b) T. Miura and M. Murakami, Org. Lett., 2005, 7(15), 3339–3341 CrossRef CAS; (c) Y. Harada, J. Nakanishi, H. Fujihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2007, 129(17), 5766–5771 CrossRef CAS; (d) T. Morimoto, K. Yamasaki, A. Hirano, K. Tsutsumi, N. Kagawa, K. Kakiuchi, Y. Harada, Y. Fukumoto, N. Chataniand and T. Nishioka, Org. Lett., 2009, 11(8), 1777–1780 CrossRef CAS.
- (a) K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, J. Org. Chem., 1996, 61(20), 6941–6946 CrossRef CAS; (b) B. J. Li, H. Y. Wang, Q. L. Zhu and Z. J. Shi, Angew. Chem., Int. Ed., 2012, 51(16), 3948–3952 CrossRef CAS; (c) Z. Qi, M. Wang and X. Li, Org. Lett., 2013, 15(21), 5440–5443 CrossRef CAS; (d) Y. Chen, F. Wang, W. Zhen and X. Li, Adv. Synth. Catal., 2013, 355(2–3), 353–359 CAS; (e) S. Chen, J. Yu, Y. Jiang, F. Chen and J. Cheng, Org. Lett., 2013, 15(18), 4754–4757 CrossRef CAS PubMed.
- Y. Kuninobu, T. Matsuki and K. Takai, Org. Lett., 2010, 12(13), 2948–2950 CrossRef CAS.
- K. Gao and N. Yoshikai, Chem. Commun., 2012, 48, 4305–4307 RSC.
- P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14(21), 5506–5509 CrossRef CAS.
- (a) J. Kashanna, K. Nagaraju and K. C. Kumara Swamy, Tetrahedron Lett., 2016, 57(14), 1576–1581 CrossRef CAS; (b) A. Ilies, Y. Arslanoglu, T. Matsubara and E. Nakamura, Asian J. Org. Chem., 2018, 7(7), 1327–1329 CrossRef; (c) K. Ramesh and G. Satyanarayana, Eur. J. Org. Chem., 2018, 4135–4146 CrossRef CAS; (d) B. Suchand and G. Satyanarayana, J. Org. Chem., 2017, 82(1), 372–381 CrossRef CAS.
- (a) J. Kashanna, R. A. Kumar, R. Kishore and D. N. Kumar, Chem. Biodiversity, 2018, 15, e1800277 CrossRef PubMed; (b) R. A. Kumar, N. Salvanna, B. Das and J. Kashanna, World Res. J. Appl. Med. Chem., 2019, 4(1), 14–18 CrossRef; (c) A. S. Kumar, R. A. Kumar, E. P. Reddy, V. Satyanarayana, J. Kashanna, B. J. M. Reddy, B. V. Subba Reddy and J. S. Yadav, Nat. Prod. Commun., 2018, 13(5), 599 Search PubMed.
- (a) K. J. Jens and E. Weiss, Chem. Ber., 1984, 117(7), 2469–2478 CrossRef CAS; (b) W. H. Watson and A. Nagl, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2444–2445 CrossRef; (c) M. Hussain, N. T. Hung, R. A. Khera, A. Villinger and P. Langer, Tetrahedron Lett., 2011, 52(2), 184–187 CrossRef CAS.
- H. Ohmiya, Y. Makida, D. Li, M. Tanabe and M. J. Sawamura, J. Am. Chem. Soc., 2010, 132(2), 879–889 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03921j |
| This journal is © The Royal Society of Chemistry 2019 |