
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA proposed protocol based on integrative metabonomics analysis for the rapid detection and mechanistic understanding of sulfur fumigation of Chinese herbal medicines†
Dai
Shengyun
ab,
Wang
Yuqi
a,
Wang
Fei
ac,
Mei
Xiaodan
a and
Zhang
Jiayu
 *de
*de
aSchool of Chinese Pharmacy, Beijing University of Chinese Medicine, Beijing 102488, China
bNational Institute of Food and Drug Control, Beijing 100050, China
cDepartment of Pharmacy, People Hospital of Peking University, Beijing 100044, China
dBeijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, China. E-mail: zhangjiayu0615@163.com
eSchool of Pharmacy, Binzhou Medical University, Yantai 264003, China
First published on 2nd October 2019
Abstract
In the current work, Lonicera japonica Flos (FLJ) was selected as a model Chinese herbal medicine (CHM) and a protocol was proposed for the rapid detection of sulfur-fumigated (SF) CHMs. A multiple metabonomics analysis was conducted using HPLC, NIR spectroscopy and a UHPLC-LTQ-Orbitrap mass spectrometer. First, the group discriminatory potential of each technique was respectively investigated based on PCA. Then, the effect of mid-level metabonomics data fusion on sample spatial distribution was evaluated based on data obtained using the above three technologies. Furthermore, based on the acquired HRMS data, 76 markers discriminating SF from non-sulfur-fumigated (NSF) CHMs were observed and 49 of them were eventually characterized. Moreover, NIR absorptions of 18 sulfur-containing markers were identified to be in close correlation with the discriminatory NIR wavebands. In conclusion, the proposed protocol based on integrative metabonomics analysis that we established for the rapid detection and mechanistic explanation of the sulfur fumigation of CHMs was able to achieve variable selection, enhance group separation and reveal the intrinsic mechanism of the sulfur fumigation of CHMs.
1. Introduction
Sulfur fumigation is a traditional storage method for Chinese herbal medicines (CHMs). It was first applied to the processing of Dioscoreae rhizoma, and has been widely misused in various CHMs in the last two decades, such as in the processing of Chrysanthemi Flos, Gastrodia Rhizoma, Radix Paeoniae Alba and Lonicera japonica Flos (FLJ).1–3 It plays an important role in the production and post-harvest handling of CHMs due to its usage in moisturizing, bleaching, retaining freshness and killing parasites.4–6 However, sulfur fumigation induces significant chemical transformations in inherent herbal constituents, resulting in alterations in the bioactivities, pharmacokinetics and toxicities of CHMs.7,8 Besides, it often leads to the production in CHMs of excessive sulfur dioxide (SO2), sulfates, sulfites, heavy metal residues and other detrimental exogenous materials, which exhibit harmful potential toxicities or side effects to human health.9–11Although the use of sulfur fumigation has been officially restricted in China since 2005,12 some illicit herbal farmers and wholesalers still misuse sulfur fumigation during the post-harvest handling and storage of CHMs. Moreover, SO2 residue-based detection standards formulated by many countries and organizations are often ineffective at evaluating the degree of sulfur fumigation because of the high volatility of SO2. Current studies mainly focus on total SO2 residues and neglect the transformations of inherent herbal constituents and corresponding mechanisms.13–16 Therefore, the development of rapid and sensitive approaches based on stable quality-markers (Q-markers), such as sulfur-containing derivatives, to discriminate sulfur-fumigated (SF) CHMs from non-sulfur-fumigated (NSF) CHMs is urgently needed.17
Integrative omics combining and interpreting data from multiple sources have already been adopted to successfully elucidate the mechanisms of human diseases, such as diabetes, obesity and schizophrenia.18,19 Besides, integrative omics analysis has been used to characterize genes in the context of the molecular pathophysiology of the disease and its interacting genes and pathways.20,21 Likewise, multi-omics data collected using various detection technologies such as liquid chromatography combined with mass spectrometry (LC-MS), high-performance liquid chromatography (HPLC) and near infrared (NIR) spectroscopy have been used for the screening and identification of Q-markers for the analysis of CHMs.22,23 Of these technologies, HPLC retains the practicality and principles of LC, while increasing the overall interlaced attributes of sensitivity and resolution, MS has emerged as a powerful tool for quantitative and qualitative analysis of the complex components in CHMs, and NIR spectroscopy is a very rapid and alternative non-destructive method that shows electromagnetic absorption signals in the NIR region associated with specific chemical structures and that can be assigned to specific chemical functional groups and molecular structures. Nevertheless, although each technique has its own powerful capabilities for specific issues, any data set obtained by one single technique cannot capture the complexity of the overall system. Thus, integrative metabonomics analysis based on multiple levels of data fusion and correlation combines the information provided by various analytical technologies so as to achieve much better statistical predictions and interpretations than those obtained from any individual technique.
FLJ, also known as Jin Yin Hua in Chinese, is one of the most well-known CHMs. It is derived from the dried buds or flowers of Lonicera japonica Thunb. and contains various biological ingredients such as organic acids, flavonoids and iridoid glycosides.24–27 Pharmacological investigations indicated that FLJ displays various pharmacological activities, such as hepatoprotective, cytoprotective, anti-microbial, anti-oxidative, anti-viral and anti-inflammatory activities.28–30 FLJ is also used in many food products, such as FLJ tea, a well-known health drink that has been highly praised for thousands of years for clearing away heat and toxic materials and treating exogenous pathogenic wind-heat.31 However, in the last two decades, sulfur fumigation has been frequently misused in post-harvest handling during the drying and storage of FLJ. Therefore, we used FLJ as a study case to present a proposal for a protocol based on integrative metabonomics analysis in order to clarify the inherent chemical transformations of CHMs and to classify the CHMs based on these transformations. SF and non-sulfur-fumigated (NSF) FLJ along with organic acids, flavonoids and iridoid glycosides were used to verify the effectiveness of the established strategy.
2. Materials and methods
2.1 Chemicals, reagents and herbal materials
A total of 22 batches of authenticated NSF FLJ samples were collected from several different areas in China (Table S1†). All of their identities were authenticated using morphological and histological methods to be the dried buds of L. japonica Thunb. according to the monograph of Chinese Pharmacopoeia (version 2015).32 The authenticated specimens were deposited in the Beijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, China. Standard substances including 3-caffeoylquinic acid (3-CQA), 4-CQA, 5-CQA, 3,4-dicaffeoylquinic acid (3,4-DiCQA), 3,5-diCQA, 4,5-diCQA, lonicerin, secologanic acid, swertiamarin and luteolin 7-O-β-glucoside were purchased from Chengdu Bio-purify Phytochemicals Ltd (Sichuan, China) with their purities all greater than 98% (Fig. S1 and Table S2†). HPLC-grade acetonitrile and methanol were provided by Fisher Scientific (Fisher, Fair Lawn, NJ, USA). Formic acid was provided by Aldrich (St. Louis, MO). All of the other chemicals were of analytical grade and obtained commercially from Beijing Chemical Works (Beijing, China). De-ionized water was purified using a Milli-Q Gradient A 10 System (Millipore, Billerica, MA). The 0.22 μm membranes were purchased from Xinjinghua Co. (Shanghai, China).2.2 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) in an ultrasonic bath (40 kHz, Eima Ultrasonics Corp., Germany) for 30 min at room temperature. Then the same solvent was added to compensate for the lost weight during the extraction process. The methanol solution was subjected to centrifugation (10000 g) for 10 minutes, and then filtered through a 0.22 μm microporous membrane before being injected into an LC-MS system for analysis. To ensure the quality of the HPLC and LC-MS-based metabonomics data, pooled quality control (QC) samples were prepared by mixing equal amounts of 37 sample solutions.
:30, v/v) in an ultrasonic bath (40 kHz, Eima Ultrasonics Corp., Germany) for 30 min at room temperature. Then the same solvent was added to compensate for the lost weight during the extraction process. The methanol solution was subjected to centrifugation (10000 g) for 10 minutes, and then filtered through a 0.22 μm microporous membrane before being injected into an LC-MS system for analysis. To ensure the quality of the HPLC and LC-MS-based metabonomics data, pooled quality control (QC) samples were prepared by mixing equal amounts of 37 sample solutions.
NIR, HPLC-DAD and UHPLC-LTQ-Orbitrap MS data were collected from these samples. The conditions for the three methods are listed in the ESI.†
2.3 Primary metabonomics data processing
All of the data were subjected to principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) to interpret the interrelationships between the samples. With respect to PLS-DA, samples were divided into a calibration set for modeling and a validation set for the established model evaluation. The prediction set consisted of samples that were not used for the calibration set.The very high quantities of acquired UHPLC-LTQ-Orbitrap MS raw data were processed with an Xcalibur 2.1 workstation (Thermo Scientific, Germany). The normalization was accomplished using Sieve 2.1 software (Thermo Scientific, USA), which was specifically used to perform background subtraction, component detection and peak alignment. SIMCA-P+ 11.5 (Umetrics, Sweden) and Unscrambler 7.0 (CAMO, Norway) software were utilized to carry out the spectral pre-processing. PCA and PLS were conducted using Matlab version R2009a (The MathWorks, Inc., USA) with Statistical Toolbox and in-house functions. The iToolbox utilized to run synergy interval partial least squares (siPLS) analysis algorithms was downloaded from http://www.models.kvl.dk/ for the NIR wavelength selection.
2.4 Integrative metabonomics analysis
3. Results and discussion
3.1 Basic data sets for the three technologies
Typical NIR, HPLC-DAD and total ion chromatogram (TIC) results of the representative SF and NSF samples are presented in Fig. S2.† After peak alignment and removal of the missing values, 90 features were finally obtained for the HPLC-based metabonomics analysis. Meanwhile, for each of 37 samples, the NIR and MS data sets included 1557 and 5000 variables, respectively.3.2 Primary metabonomics data analysis
Primary data analysis results were obtained respectively from unsupervised PCA and supervised PLS-DA for the three different techniques (Table 1 and Fig. 1).| Technique | PCA | PLS | ||||||
|---|---|---|---|---|---|---|---|---|
| Lvs | R 2(X) | Lvs | R 2(Y) | Q 2 | Q 2-intercept | Permuted R2 value | p-Value | |
| a Lvs: the number of latent variables. | ||||||||
| HPLC-DAD | 3 | 35.7% | 3 | 94% | 63.5% | −0.262 | 0.624 | 1.16 × 10−5 |
| NIR | 5 | 39.0% | 3 | 97.2% | 55.2% | 0.504 | 0.504 | 0.037 |
| LC-MS | 6 | 72.0% | 2 | 94.9% | 82.9% | −0.264 | 0.439 | 3.73 × 10−11 |
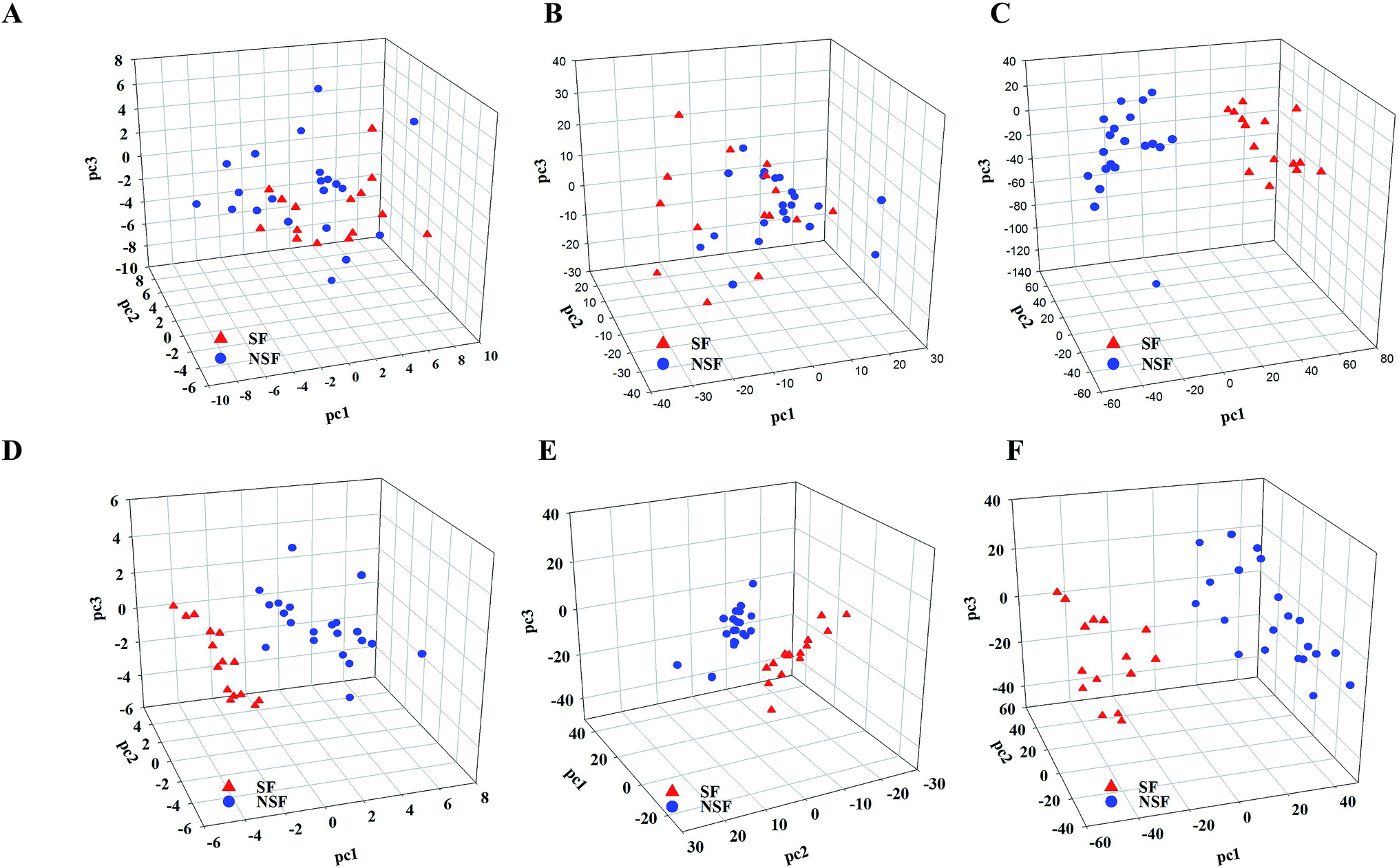 | ||
| Fig. 1 The results of primary metabonomics data fusion analysis. (A–C) PCA for HPLC-DAD, NIR and LC-MS; (D–F) PLS-DA for HPLC-DAD, NIR and LC-MS. | ||
As for PCA, SF and NSF FLJ samples were not explicitly clustered into two groups with regards to HPLC-DAD and NIR analysis (The preprocess method was SG9+2nd, and the results obtained from the other preprocess methods are illustrated in Fig. S3 and Table S3†). While for the LC-MS analysis results, the 37 SF and NSF samples did segregate into two distinct groups. Fig. 1A–C show plots of these scores for these three techniques, respectively. A distinct classification trend could be observed in the LC-MS score plot. However, the results for these samples were nevertheless scattered considerably, with that for sample number 16 attributed to the NSF group being located in the SF group, which indicated that some of the variation in the samples cannot be obtained from the PCA.
Therefore, PLS-DA was performed to improve the group separation. The PLS-DA model resulted in a clear separation of the SF and NSF samples for each of the three different technologies (Fig. 1D–F). As for the HPLC and LC-MS analyses, statistical models were considered to be statistically significant when the corresponding Q2-intercept values (−0.262 and −0.264) for the permutation model were negative. Meanwhile, the permuted R2 values (0.624 and 0.439) were lower than the original R2-values (0.903 and 0.927). Additionally, analysis of variance of the cross-validated predictive residuals (CV-ANOVA) tests were performed to confirm that the SF and NSF groups discriminated by PLS-DA were significantly different. The common practice was to interpret a p value (1.16 × 10−5 and 3.73 × 10−11) dramatically lower than 0.05 as contributing a significant model. As for the NIR analysis (for which the preprocess method was SG9+2nd, and the results obtained from the other preprocess methods are shown in Fig. S4†), the Q2-intercept value was lower than 0.5, indicating the poor predictive capability of PLS-DA here. The poor predictive capability was also verified by the finding of a positive Q2-intercept value from the permutation test. Therefore, the use of the NIR and HPLC technologies did not achieve a satisfactory classification based on PCA while the use of MS did so.
3.3 Integrative metabonomics analysis
| Techniques | PCA | PLS-PCA | ||
|---|---|---|---|---|
| Lvs | R 2(X) | Lvs | R 2(X) | |
| NIR-HPLC | 5 | 93.5% | 5 | 55.2% |
| HPLC-MS | 5 | 71.4% | 6 | 79.4% |
| NIR-MS | 6 | 75.0% | 5 | 66.9% |
| NIR-HPLC-MS | 6 | 74.4% | 5 | 66.7% |
Compared to the primary metabonomics analyses for HPLC and NIR alone, the principal factor total cumulative based on the results of the fusion of NIR and HPLC data was higher, with a value of 93.5%. However, the discrimination was still unsatisfactory. NSF and SF samples were dispersed in the three-dimensional space, indicating that the key information about the discrimination between the two analyses might not be captured (Fig. 2A). HPLC-MS-, NIR-MS- and NIR-HPLC-MS-based metabonomics data fusion generated summarized principal factorial plane results that were similar to the result for MS analysis mentioned above. Besides, many more variables were in the MS data set than in the HPLC data set, and hence the information contained in the MS data set could cover up the limited information of HPLC to some extent. Therefore, the results from MS-HPLC metabonomics data fusion were similar to those from primary metabonomics based on the MS data set (Table 3).
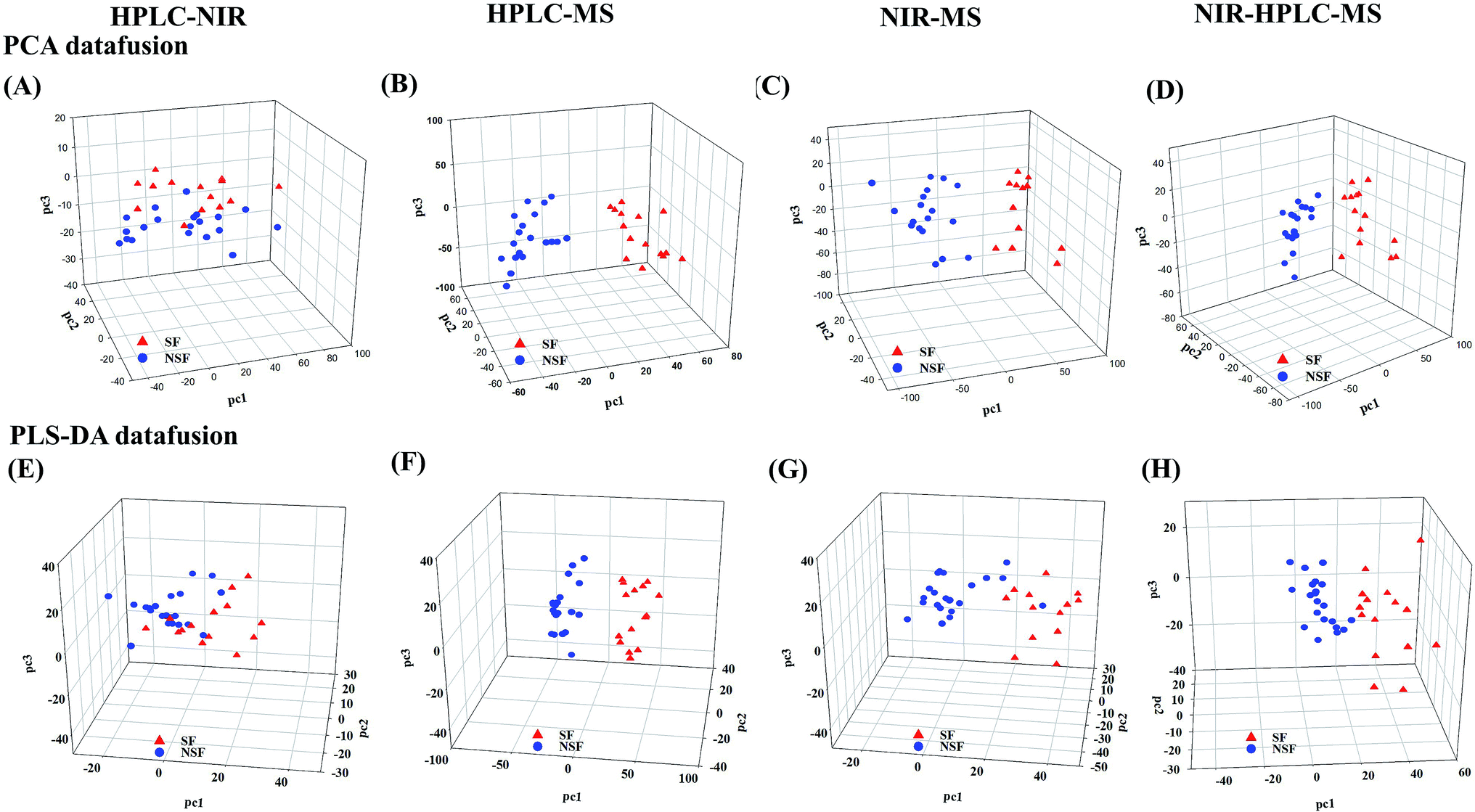 | ||
| Fig. 2 The results of mid-level metabonomics data fusion analysis. (A–D) Mid-level metabonomics data fusion analysis without variable selection for NIR-HPLC, HPLC-MS, NIR-MS and NIR-HPLC-MS. (E–H) Mid-level metabonomics data fusion analysis with variable selection for NIR-HPLC, HPLC-MS, NIR-MS and NIR-HPLC-MS. | ||
| No. | t R | Experimental mass | Formula [M–H]− | MS/MS fragment ions | Identification |
|---|---|---|---|---|---|
| a Identified by comparison with reference standards; CQA, caffeoylquinic acid; DiCQA, dicaffeoylquinic acid; pCoCQA, p-coumaroylcaffeoylquinic acid; CFQA, caffeoylferuloylquinic acid. | |||||
| M1a | 4.47 | 353.0869 | C16H17O9 | MS2[353]: 191, 179, 135 | 3-CQA |
| M2a | 6.91 | 353.0858 | C16H17O9 | MS2[353]: 191, 179, 161 | 5-CQA |
| M3a | 7.73 | 353.0856 | C16H17O9 | MS2[353]: 173, 179, 191, 135 | 4-CQA |
| M4a | 2.14 | 373.1122 | C16H21O10 | MS2[373]: 193, 149, 167, 179, 119 | Swertiamarin |
| M5a | 5.30 | 373.1118 | C16H21O10 | MS2[373]: 211, 167, 149, 193, 179 | Secologanic acid |
| M6 | 7.88 | 373.1118 | C16H21O10 | MS2[373]: 193, 149, 167, 179 | Swertiamarin isomer |
| M7 | 4.23 | 375.1292 | C16H23O10 | MS2[375]: 213, 169, 151 | Loganin acid isomer |
| M8 | 4.84 | 375.1280 | C16H23O10 | MS2[375]: 213, 169, 151, 195 | Loganin acid |
| M9 | 5.84 | 375.1273 | C16H23O10 | MS2[375]: 213, 169, 151 | Loganin acid isomer |
| M10 | 6.63 | 375.1292 | C16H23O10 | MS2[375]: 195, 151 | Loganin acid isomer |
| M11 | 1.81 | 391.1231 | C16H23O11 | MS2[391]: 229, 211, 193, 185, 167, 149 | Secologanic acid hydrate |
| M12 | 2.45 | 391.1255 | C16H23O11 | MS2[391]: 211, 229, 193, 167, 149, 185 | Secologanic acid hydrate |
| M13 | 14.33 | 403.1223 | C17H23O11 | MS2[403]: 371, 223, 179, 121, 91 | Secologanin |
| M14 | 1.63 | 433.0428 | C16H17O12S | MS2[433]: 241, 415, 353, 161, 191, 287 | CQA sulfate |
| M15 | 2.53 | 433.0427 | C16H17O12S | MS2[433]: 415, 387, 353, 241, 353 | CQA sulfate |
| M16 | 2.66 | 433.0433 | C16H17O12S | MS2[433]: 241, 415, 387, 259, 353 | CQA sulfate |
| M17 | 4.62 | 433.0423 | C16H17O12S | MS2[433]: 415.387, 259 | CQA sulfate |
| M18 | 5.01 | 433.0419 | C16H17O12S | MS2[433]: 415, 241, 161, 259, 387 | CQA sulfate |
| M19 | 1.12 | 435.0591 | C16H17O12S | MS2[435]: 353, 191, 179 | CQA sulfite |
| M20 | 3.15 | 437.0720 | C16H21O12S | MS2[437]: 193, 149, 373, 355 | Secologanic acid sulfite |
| M21a | 19.06 | 447.0916 | C21H19O11 | MS2[447]: 285 | Luteolin-7-O-glucoside |
| M22 | 21.06 | 447.0918 | C21H19O11 | MS2[447]: 285 | Luteolin-7-O-glucoside isomer |
| M23 | 1.93 | 455.0822 | C16H23O13S | MS2[455]: 373, 411, 437, 193, 211 | Secologanic acid sulfite |
| M24 | 2.15 | 455.0836 | C16H23O13S | MS2[455]: 373, 437, 411, 193, 211 | Secologanic acid sulfite |
| M25 | 18.22 | 463.0854 | C21H19O12 | MS2[463]: 301, 271, 445 | Hyperoside isomer |
| M26 | 18.73 | 463.0861 | C21H19O12 | MS2[463]: 301, 445, 271 | Hyperoside |
| M27 | 23.05 | 499.1231 | C25H23O11 | MS2[499]: 337, 173, 335, 353 | 4-pCo-1-CQA |
| M28 | 23.49 | 499.1233 | C25H23O11 | MS2[499]: 353, 337, 191, 335, 179 | 5-pCo-3-CQA |
| M29 | 25.21 | 499.1230 | C25H23O11 | MS2[499]: 353, 337, 179, 191 | 3-pCo-4-CQA |
| M30a | 20.36 | 515.1155 | C25H23O11 | MS2[515]: 353, 335, 173, 179 | 3,4-DiCQA |
| M31a | 20.85 | 515.1155 | C25H23O11 | MS2[515]: 353, 191, 179, 335 | 3,5-DiCQA |
| M32a | 22.44 | 515.1163 | C25H23O11 | MS2[515]: 353, 191, 179, 335, 353 | 4,5-DiCQA |
| M33 | 17.34 | 527.0494 | C21H19O14S | MS2[527]: 447, 285, 481 | Luteolin-7-O-glucoside sulfate |
| M34 | 23.82 | 529.1343 | C26H25O12 | MS2[529]: 367, 179, 335, 353, 193 | 3-C-4-FQA |
| M35 | 24.60 | 529.1340 | C26H25O12 | MS2[529]: 353, 367, 191, 179 | 5-C-3-FQA |
| M36 | 25.86 | 529.1335 | C26H25O12 | MS2[529]: 353, 367, 173, 335 | cis-5-C-3-FQA |
| M37 | 8.73 | 543.0431 | C21H19O15S | MS2[543]: 463, 381, 525, 301 | Hyperoside sulfate |
| M38 | 12.76 | 543.0432 | C21H19O15S | MS2[543]: 381, 301, 381, 463 | Hyperoside sulfate |
| M39 | 18.80 | 593.1488 | C27H29O15 | MS2[593]: 285, 447 | Lonicerin isomer |
| M40 | 19.71 | 593.1483 | C27H29O15 | MS2[593]: 285, 447 | Lonicerin isomer |
| M41a | 20.50 | 593.1486 | C27H29O15 | MS2[593]: 285 | Lonicerin |
| M42 | 16.70 | 595.0737 | C25H23O15S | MS2[595]: 549, 577, 415, 241, 259 | DiCQA sulfate |
| M43 | 16.98 | 595.0750 | C25H23O15S | MS2[595]: 549, 577, 415, 301, 397 | DiCQA sulfate |
| M44 | 17.61 | 595.0748 | C25H23O15S | MS2[595]: 577, 549, 415, 433, 241, 259 | DiCQA sulfate |
| M45 | 17.89 | 595.0737 | C25H23O15S | MS2[595]: 577, 549, 415, 433, 241, 259 | DiCQA sulfate |
| M46 | 19.38 | 595.0745 | C25H23O15S | MS2[595]: 577, 549, 415, 433, 259 | DiCQA sulfate |
| M47 | 21.25 | 595.0745 | C25H23O15S | MS2[595]: 577, 415, 549, 433, 259, 241 | DiCQA sulfate |
| M48 | 22.70 | 607.1653 | C28H31O15 | MS2[607]: 299 | Chrysoeriol-7-O-β-D-neohesperidoside |
| M49 | 18.30 | 609.1403 | C27H29O16 | MS2[609]: 301, 300, 271, 255, 179, 591 | Rutin |
Subsequently, we investigated the mid-level metabonomics data fusion with PLS-DA to improve the group separation. The results from NIR-HPLC, HPLC-MS, NIR-MS and NIR-HPLC-MS data fusions were respectively presented after preliminarily screening all of data acquired from an individual platform according to variable importance values (VIP > 1.0). The initial NIR, HPLC and MS data sets included 1557, 90, and 5000 variables, respectively. After the screening based on the VIP scores, 607, 27 and 1843 variables, respectively, were considered to be the most effective variables and hence retained for the subsequent discrimination. So now, a new PCA model could be constructed to enhance the group discrimination of SF and NSF samples based on the generated variables data set.
HPLC-NIR data fusion using the new PCA model yielded much better results than ever before (Fig. 2E), even though 37 batches of FLJ were not distinctly clustered into two groups. Meanwhile, the HPLC-MS data fusion generated much better results without any misclassification, while one misclassification was still found in the NIR-MS data fusion results (no. 16 was still far from the NSF group) (Fig. 2F and G).
Fig. 2H shows the results obtained from NIR-MS-HPLC metabonomics data fusion. Although no. 16 was not correctly allocated into the NSF group, the group discrimination potential was significantly improved when compared with those obtained from metabonomics data fusion without variable selection. Thus it could be seen from the results that metabonomics data fusion with variable selection made greater improvements in class separation than did the metabonomics data fusion without variable selection.
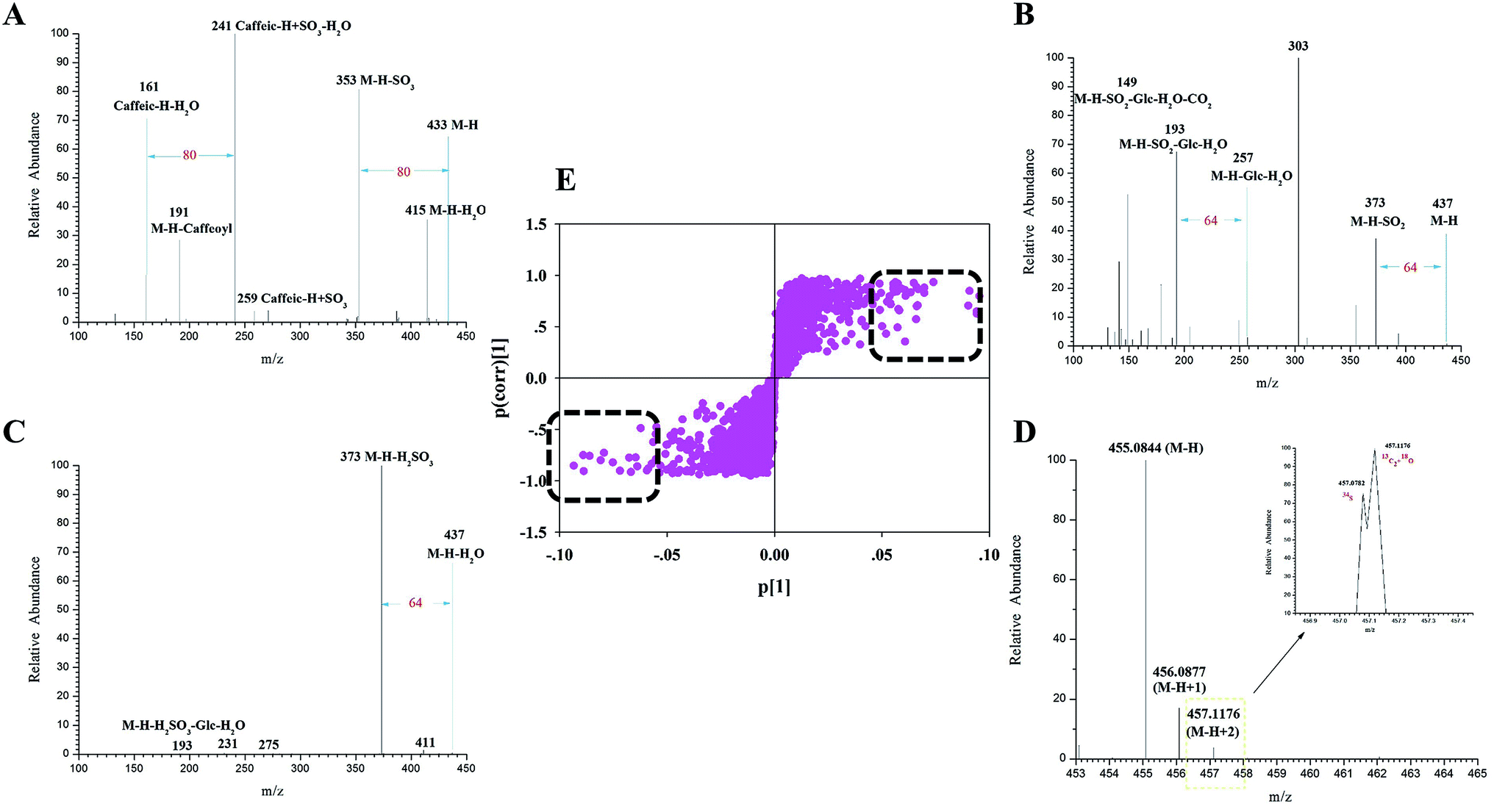 | ||
| Fig. 3 The mass fragmentation behaviors of identified markers. (A) HRMS1 spectrum of M14. (B) ESI-MS2 spectrum of M20. (C) HRMS1 spectrum of M23. (D) ESI-MS2 spectrum of M24. (E) The S-plot of LC-MS metabonomics analysis. | ||
Markers 1, 2 and 3 yielded identical [M–H]− ions at an m/z value of 353.0867 (C16H23O10, mass error within ±5 ppm) in negative ion mode. Their deprotonated molecular ions all generated a series of diagnostic fragment ions including those with m/z values of 191 [M–H–caffeoyl]−, 179 [caffeic–H]− and 173 [M–H–caffeoyl–H2O]−.37 CQAs attributed to three different linkage positions of caffeoyl groups on quinic acid have been reported to display different intensities of their ESI-MS2 base peak ions and predominant product ions. Meanwhile, based on retention times and MSn spectra of the corresponding reference substances and literature data, markers 1–3 were identified to be 5-CQA (Fig. S5†), 3-CQA and 4-CQA, respectively.
Markers 14–18 generated their deprotonated [M–H]− molecular ions each at an m/z of 433.0435 (C16H17O12S, mass error within ±5 ppm). In their ESI-MS2 spectra, the diagnostic product ions were at m/z values of 415 [M–H–H2O]−, 387 [M–H–H2O–CO]−, 353 [M–H–SO3]−, 259 [caffeic–H + SO3]− and 241 [caffeic–H + SO3–H2O]−. The observation of the pair of ions at m/z values of 433 and 353 (Fig. 3A) further confirmed that the sulfate moiety was introduced to the CQA molecule, which has to the best of our knowledge never been reported before. Finally, markers 14–18 were tentatively identified as isomeric CQA sulfate.
Similarly, the ESI-MS2 spectrum of marker 20 (Fig. 3B) showed an m/z signal corresponding to its deprotonated [M–H]− molecular ion at a value of 437.0748 (C16H21O12S, error within ±5 ppm). Moreover, the characteristic product ions at m/z values of 373 [M–H–SO2]−, 193 [M–H–SO2–Glc–H2O]− and 149 [M–H–SO2–Glc–H2O–CO2]− were all observed. Based on the observation of the signals at the m/z values of 193 and 149 coupled with its [M–H]− ion, marker 20 may be concluded to be secologanic acid.38 Meanwhile, the observation of the product ion at the m/z value of 373 confirmed that the sulfite moiety was introduced into the iridoid molecule. Accordingly, marker 20 was tentatively identified as isomeric secologanic acid sulfate.
In addition, a combination of the isotopic pattern combined and chromatography analyses was used for screening sulfur-containing compounds in the complex systems, mainly because the 34S isotopic ion has been shown to be drastically affected by 13C2 and 18O.39 Markers 23 and 24 produced their [M–H]− ions each at an m/z of 455.0822 (C16H23O12S, error within ±5 ppm). And both of them generated a series of fragment ions at m/z values of 437 [M–H–H2O]−, 411 [M–H–CO2]−, 373 [M–H–H2SO3]−, 211 [M–H–H2SO3–Glc]− and 193 [M–H–H2SO3–Glc–H2O]−. Furthermore, they simultaneously produced the isotopic patterns of the 34S ion at an m/z of 457.07822 and of the 13C2 + 18O ion at an m/z of 457.11760. Their characteristic product ions at m/z values of 437 and 373 probably resulted from the occurrence of the sulfite moiety in some of the iridoid molecules. Accordingly, markers 23 and 24 were putatively identified as secologanic acid-sulfite or its isomers (Fig. 3C and D).
Taken together, a total of 49 discriminatory markers (Table S4†) attributed to iridoids, organic acids and flavones were screened and characterized according to the fragmentation behaviors, isotopic patterns and diagnostic product ions obtained using the UHPLC-LTQ-Orbitrap MS coupled with the established integrated strategy. Eighteen of these markers were assigned to sulfate/sulfite derivatives of iridoid and chlorogenic acid, which could be chosen as the characteristic Q-markers for SF FLJ discrimination.40 (Note that Fig. S6† shows a histogram of signal intensities of sulfur derivatives.)
| Preprocessing method | PCA | PLS-DA | |||||
|---|---|---|---|---|---|---|---|
| Lv | R 2(X) | R 2(Y) | Lvs | R 2(X) | R 2(Y) | Q 2 | |
| Baseline | 3 | 0.998 | 0.997 | 3 | 0.997 | 0.501 | 0.326 |
| Spectroscopic transformation | 3 | 0.999 | 0.998 | 3 | 0.999 | 0.390 | 0.248 |
| MSC | 6 | 0.999 | 0.997 | 3 | 0.858 | 0.454 | 0.206 |
| Normalization | 5 | 0.999 | 0.999 | 3 | 0.975 | 0.494 | 0.309 |
| Original | 3 | 0.999 | 0.999 | 3 | 0.999 | 0.497 | 0.249 |
| SG91st | 5 | 0.899 | 0.831 | 4 | 0.845 | 0.827 | 0.601 |
| SG92nd | 5 | 0.621 | 0.381 | 3 | 0.425 | 0.827 | 0.309 |
| SG111st | 4 | 0.693 | 0.578 | 3 | 0.583 | 0.791 | 0.476 |
| SG112nd | 6 | 0.582 | 0.234 | 3 | 0.292 | 0.922 | 0.418 |
| SNV | 4 | 0.997 | 0.996 | 3 | 0.993 | 0.517 | 0.336 |
| WDS | 3 | 0.999 | 0.999 | 3 | 0.999 | 0.538 | 0.159 |
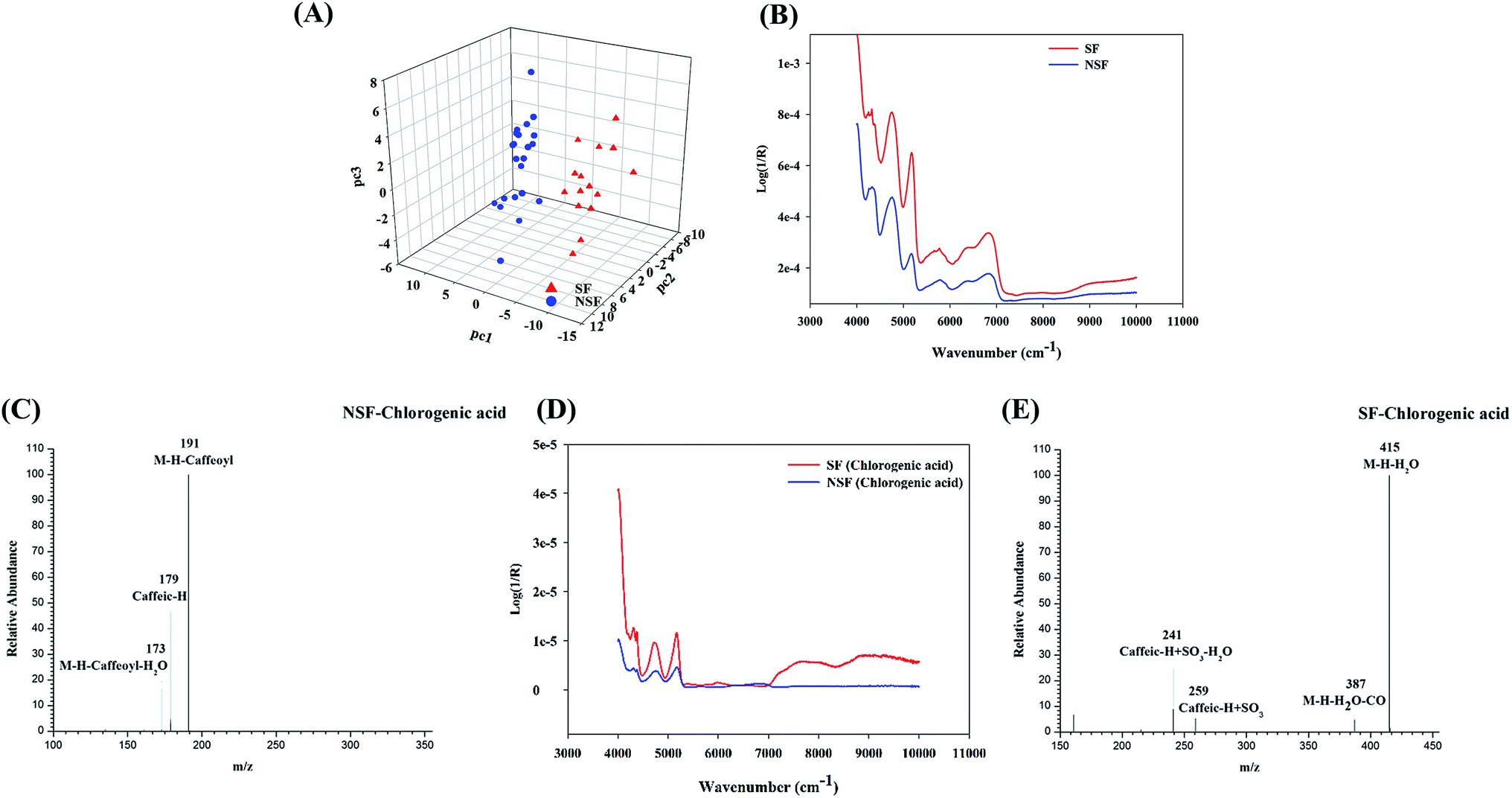 | ||
| Fig. 4 The MOCA for the FLJ. (A) The discriminatory information of preprocess method of SG9+1st; (B) the synchronous 2D-COS auto-peak analysis of the SF and NSF samples; (C) the mass fragmentation behaviors of SF chlorogenic acid; (D) the synchronous 2D-COS auto-peak analysis of the SF and NSF chlorogenic acid samples; (E) the mass fragmentation behaviors of SF chlorogenic acid. | ||
To validate the above-mentioned results, one of the main representative chemical constituents, namely chlorogenic acid (5-CQA), was subjected to sulfur fumigation and analyzed using the same methods. The autocorrelation curves of the SF and NSF chlorogenic acid samples (Fig. 4D) were derived from their respective 2D-COS spectra. Obvious differences between the SF and NSF chlorogenic acid samples in the wavebands between 5000 and 5200 cm−1 were observed, which was also in accordance with the wavebands screened using the siPLS model. The subsequent LC-HRMS analysis of an SF chlorogenic acid mixture also indicated the presence of newly generated constituents (Fig. 4C and E) except for the prototype drug during the process of sulfur fumigation. Through analyzing the fragment ions of the S-derivatives, it was found that SO3 (79.9568) and H2SO3 (81.9725) were the characteristic neutral losses of organic sulfates or sulfites. The assignments of these newly emerged peaks were confirmed to be the rudimentary sulfate derivatives of chlorogenic acid based on the HRMS data, which indicated a mass 79.95 Da (SO3) more than that of standard reference. It also indicated that the results of NIR were reliable and credible in the discrimination of SF FLJ.
3.4 Proposed protocol for the rapid detection and mechanistic explanation of the sulfur fumigation of CHMs using SF FLJ as a study case
Evaluating the quality of CHMs and judging their authenticity are two major challenges. Sulfur fumigation has attracted increasing attention due to its alteration of CHM quality resulting from its damage to bioactive components, generating excessive sulfur dioxide residue and especially changing the chemically active ingredients in CHMs.NIR, HPLC and LC-MS were proposed to be used to evaluate the quality of CHMs. However, the variations during sulfur fumigation were much more complicated than expected. Furthermore, the amount of data obtained based on one single method was still limited, making it difficult to expound on the mechanism of sulfur fumigation. To experimentally support our inference, we selected FLJ as a model herb in this study. With the development of a few high-throughput strategies, integrative metabonomics analysis was applied to integrate the multiple interactions of NIR spectra, HPLC chromatograms and HRMS data. The results aimed to reveal whether the herb underwent sulfur fumigation and to illuminate the inherent mechanism of the NIR judgment method by associating NIR with HPLC and UHPLC-MS analyses. Our established approach was applied to rapidly discriminate SF FLJ among many unknown samples, and is expected to be greatly beneficial for guaranteeing CHM quality.
To perform the analysis of the sulfur fumigation of CHMs, the process of sulfur fumigation was first simulated in the laboratory. According to the characteristics of each analytical technology, optimum analytical conditions were adopted and the corresponding high-quality data of SF and NSF CHM samples were obtained. All of these experiments provided the foundation for subsequent data analysis, which is illustrated in Fig. 5.
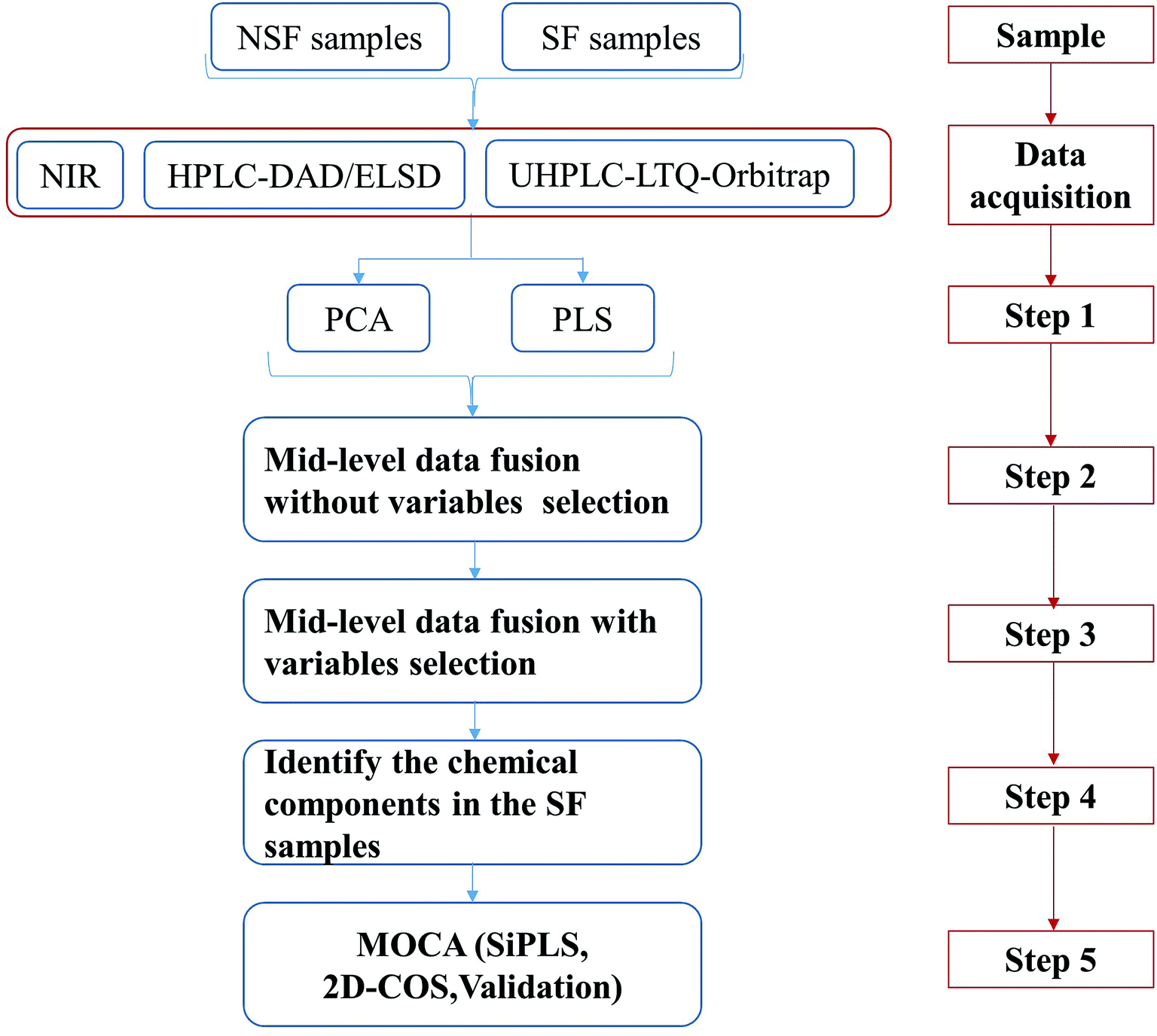 | ||
| Fig. 5 A suggested protocol for the rapid discrimination of SF CHMs and an explanatory mechanism. | ||
Step 1: Performing PCA and PLS-DA for the single technology.
This step was focused on the analytical capability of each single technology and whether the SF and NSF CHMs could be distinguished. Our study demonstrated that NIR spectroscopy based on a data preprocessing method (SG9+2nd) with a multivariate calibration approach such as PCA and PLS-DA was the appropriate tool to discriminate SF from NSF FLJ samples. The chemical constituents in FLJ samples displayed strong ultraviolet absorption, which was observed with HPLC-DAD at 330 nm, 238 nm, 254 nm and 280 nm. Peak areas (≥150 mAU) were selected separately through the data fusion of the four wavelengths and then analyzed by performing PCA and PLS-DA. In addition, a UHPLC-LTQ-Orbitrap high-resolution MS was employed to comprehensively and dynamically profile the chemical constituents in FLJ. The derivative content during the sulfur fumigation process was not abundant enough, i.e., the signals of sulfur-containing analytes would have been drowned out by the contribution of inherent constituents.
Step 2 and Step 3: Performing the integrative metabonomics analysis, such as mid-level metabonomics data fusion analysis without/with variable selection.
In our previous study, the data fusion of NIR- and HRMS-based metabonomics-like analysis was successfully applied to accomplishing the discrimination of SF Ophiopogon Radix.17 Herein, we combined three kinds of analytical techniques including NIR, HPLC and UHPLC-HRMS to obtain the dimensional information of SF samples, and investigated two types of mid-level metabonomics data fusion strategies as illustrated in Step 2 and Step 3. For that, informative features of the raw data from a single instrument were separately extracted using their own protocol from sample preparation to data preprocessing.
The comparison between the unique model and the metabonomics data fusion model is illustrated in Fig. 6. No single analytical platform could be utilized to accurately discriminate the SF samples based on PCA score plots. HPLC and NIR led to classification without rhyme or reason and HRMS could not correctly discriminate one of the SF samples (no. 16). Thus, we believed that utilizing mid-level metabonomics data fusion without variable selection to obtain more accurate characteristics of the samples might be a much better choice. As a result, the potential to discriminate between of NSF and SF samples was actually improved with no. 16 still in the wrong class, and the results were worse than the individual application of MS. Mid-level fusion with variable selection was employed and clearly improved the class separation, as samples were correctly classified and less scattered (Fig. 6K). Taking the classification into account, the fusion of NIR and HRMS data, accomplished with high accuracy, provided the best model (Fig. 6I and J). Moreover, variables that were selected before classification generated better classification results than those obtained when all variables were used. Overall, the proposed metabonomics data fusion approach demonstrated an ability to effectively discriminate key information from raw analysis data.
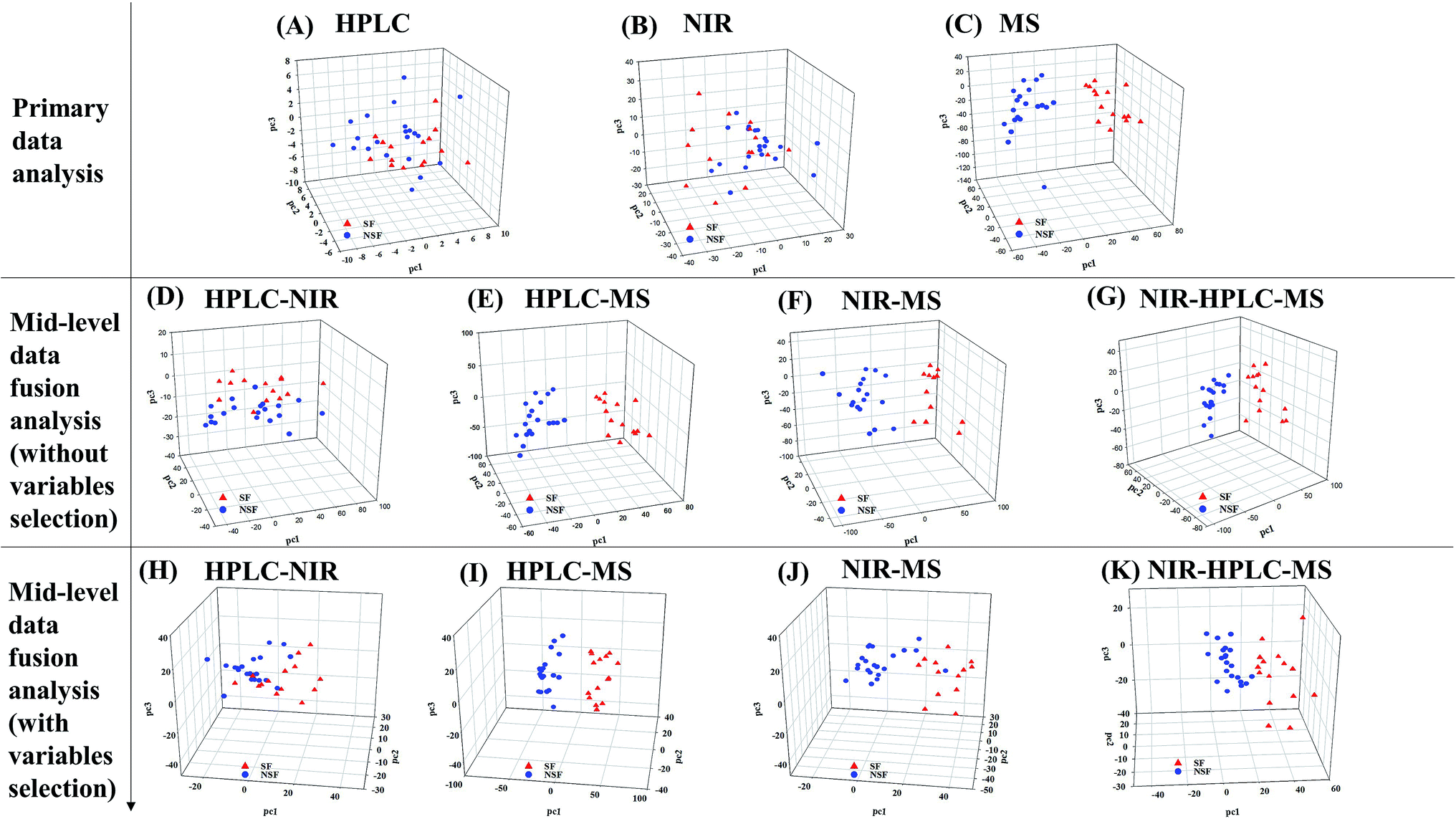 | ||
| Fig. 6 Comparison between the unique model and the metabonomics data fusion model. (A–C) PCA for HPLC-DAD, NIR and LC-MS. (D–G) Result for HPLC-NIR, HPLC-MS, NIR-MS and NIR-HPLC-MS data fusion without variable selection analysis. (H–K) Result for HPLC-NIR, HPLC-MS, NIR-MS and NIR-HPLC-MS data fusion with variable selection analysis. | ||
The results demonstrated that the mid-level metabonomics data fusion methods were much better than all of the primary analyses, which meant that the information obtained from individual techniques was in fact insufficient. The results from both kinds of mid-level fusion strategies accomplished the effective discrimination of SF FLJ samples.
Step 4: Identifying the discriminatory markers attributed to group separation.
LTQ-Orbitrap high-resolution MS has been one of the most powerful approaches used for the rapid identification of multiple constituents in CHMs.42,43 It has been used to combine high trapping capacity and multiple data acquisition of linear ion traps to generate a large amount of information from MS1 and MSn data. In this study, a highly sensitive and effective strategy was utilized for rapidly screening and identifying SF FLJ by using PIL-DE acquisition based on a hybrid LTQ-Orbitrap mass spectrometer to accomplish the overall acquisition of data sets, which helped allow for a search of a greater number of potential active compounds especially for the sulfur-containing constituents. As a result, 49 markers including iridoids, organic acids and the sulfur-containing derivatives were positively or tentatively identified.
Step 5: Application of MOCA for deriving mechanistic explanations of the sulfur fumigation process and the corresponding method validation.
An NIR spectrum was constructed from different wavebands, but not every waveband displayed a special discrimination ability. Therefore, siPLS analysis was employed to screen the potential wavebands that presented the significant differences between SF and NSF samples. In step 4 mentioned above, some sulfur-containing constituents were identified that would explain the potential NIR wavebands. Chlorogenic acid was selected as the example to validate whether the new sulfur-containing derivatives were produced after the sulfur fumigation process.
4. Conclusions
Our work indicated that the proposed protocol for the rapid detection of SF CHMs is also beneficial for revealing the intrinsic mechanism of sulfur fumigation and boosting the ability to discriminate SF from NSF FLJ samples, and could serve as an example for future research on rapidly detecting other SF CHMs. Integrative metabonomics analysis was also found to be beneficial for evaluating the quality of and rapidly detecting CHMs. Our work also suggests a future trend of integrating multiple metabonomics datasets from different technologies to achieve a sound evaluation.Conflicts of interest
All authors declare that they have no conflict of interest.Acknowledgements
This work has been financially supported by National Natural Science Foundation of China (No. 81303206), Beijing National Natural Science Foundation (No. 7173265) and Beijing Nova Program (Z171100001117029).References
- C. Kang, C. J. Lai, D. Zhao, T. Zhou, D. H. Liu, C. Lv, S. Wang, L. Kang, J. Yang and Z. L. Zhan, J. Hazard. Mater., 2017, 340, 221–230 CrossRef CAS.
- M. Kong, H. H. Liu, J. Wu, M. Q. Shen, Z. G. Wang, S. M. Duan, Y. B. Zhang, H. Zhu and S. L. Li, J. Ethnopharmacol., 2017, 212, 95–105 CrossRef.
- A. L. Guo, L. M. Chen, X. Q. Liu, Q. W. Zhang, H. M. Gao, Z. M. Wang, X. Wei and Z. Z. Wang, Molecules, 2014, 19, 16640–16655 CrossRef PubMed.
- W. L. T. Kan, B. Ma and G. Lin, Front. Pharmacol., 2011, 2, 84 Search PubMed.
- X. Jiang and L. F. Huang, Sulfur fumigation, Phytomedicine, 2013, 20, 97–105 CrossRef CAS.
- M. Q. Carter, M. H. Chapman, F. Gabler and M. T. Brandl, Food Microbiol., 2015, 49, 189–196 CrossRef CAS.
- L. Zhang, H. Shen, J. Xu, J. D. Xu, Z. L. Li, J. Wu, Y. T. Zou, L. F. Liu and S. L. Li, Food Chem., 2018, 246, 202–210 CrossRef CAS.
- S. M. Duan, J. Xu, Y. J. Bai, Y. Ding, M. Kong, H. H. Liu, X. Y. Li, Q. S. Zhang, H. B. Chen and L. F. Liu, Food Chem., 2016, 192, 119–124 CrossRef CAS.
- B. Ma, W. L. T. Kan, Z. He, S. L. Li and L. Ge, J. Ethnopharmacol., 2017, 195, 222–230 CrossRef CAS.
- R. J. Li, X. J. Kou, J. J. Tian, Z. Q. Meng, Z. W. Cai, F. Q. Cheng and C. Dong, Chemosphere, 2014, 112, 296–304 CrossRef CAS.
- A. Agar, V. Küçükatay, P. Yargicoglu, S. Bilmen, S. Gümüşlü and G. Yücel, Diabetes Metab., 2000, 26, 140–144 CAS.
- Pharmacopoeia of People's Republic of China, Part L, China Medical Science and Technology Press, Beijing, 2005 Search PubMed.
- X. Jin, L. Y. Zhu, H. Shen, J. Xu, S. L. Li, X. B. Jia, H. Cai, B. C. Cai and R. Yan, Food Chem., 2012, 135, 1141–1147 CrossRef CAS PubMed.
- M. Kong, H. H. Liu, J. Xu, C. R. Wang, M. Lu, X. N. Wang, Y. B. Li and S. L. Li, J. Pharm. Biomed. Anal., 2014, 98, 424–433 CrossRef CAS.
- X. Chen, X. Li, X. Mao, H. Huang, T. Wang, Z. Qu, M. Jing and W. Gao, Food Chem., 2017, 224, 224–232 CrossRef CAS.
- Y. J. Bai, J. D. Xu, M. Kong, Q. Gao, L. F. Liu and S. L. Li, Food Res. Int., 2015, 76, 387–394 CrossRef CAS.
- S. Y. Dai, Z. Z. Lin, B. Xu, Y. Q. Wang, X. Y. Shi, J. Y. Zhang and Y. J. Qiao, Talanta, 2018, 189, 641–648 CrossRef CAS.
- K. J. Karczewski and M. P. Snyder, Nat. Rev. Genet., 2018, 19, 299–310 CrossRef CAS.
- M. S. Kwon, Y. Kim, S. Lee, J. Namkung, T. Yun, S. G. Yi, S. Han, M. Kang, W. K. Sun and J. Y. Jang, BMC Genomics, 2015, 16, S4 CrossRef.
- M. D. Ritchie, E. R. Holzinger, L. Ruowang, S. A. Pendergrass and K. Dokyoon, Nat. Rev. Genet., 2015, 16, 85–97 CrossRef CAS.
- K. Anshul, M. Wouter, E. Jason, B. Misha, Y. Angela, H.-M. Alireza, K. Pouya, Z. Zhizhuo, W. Jianrong and Z. Michael J, Nature, 2015, 518, 317–330 CrossRef.
- Y. J. Bai, J. D. Xu, M. Kong, Q. Gao, L. F. Liu and S. L. Li, Food Res. Int., 2015, 76, 387–394 CrossRef CAS PubMed.
- W. Z. Yang, Y. B. Zhang, W. Y. Wu, L. Q. Huang, D. A. Guo and C. X. Liu, Acta Pharm. Sin. B, 2017, 7, 439–446 CrossRef.
- Y. Kashiwada, Y. Omichi, S. I. Kurimoto, H. Shibata, Y. Miyake, T. Kirimoto and Y. Takaishi, Phytochemistry, 2013, 96, 423–429 CrossRef CAS.
- Y. Yu, C. G. Zhu, S. J. Wang, W. X. Song, Y. C. Yang and S. J. Gong, J. Nat. Prod., 2013, 76, 2226–2233 CrossRef CAS.
- S. Qiu, W. Z. Yang, X. J. Shi, C. L. Yao, M. Yang, L. Xuan, B. H. Jiang, W. Y. Wu and D. A. Guo, Anal. Chim. Acta, 2015, 893, 65–76 CrossRef CAS.
- L. Y. Lin, P. P. Wang, Z. Y. Du, W. C. Wang, Q. F. Cong, C. P. Zheng, C. Jin, D. Kan and C. H. Shao, Int. J. Biol. Macromol., 2016, 88, 130–137 CrossRef CAS.
- M. H. Han, W. S. Lee, A. Nagappan, S. H. Hong, J. H. Jung, C. Park, H. J. Kim, G. Y. Kim, G. Kim and J. M. Jung, Phytother. Res., 2016, 30, 1824–1832 CrossRef CAS.
- Z. X. Liu, Z. Cheng, Q. J. He, B. Lin, P. Y. Gao, L. Z. Li, Q. B. Liu and S. J. Song, Fitoterapia, 2016, 110, 44–51 CrossRef CAS.
- C. Park, W. S. Lee, M. H. Han, K. S. Song, S. H. Hong, A. Nagappan, G. Y. Kim, G. S. Kim, J. M. Jung and C. H. Ryu, Phytother. Res., 2017, 32, 504–513 CrossRef.
- X. Shang, H. Pan, M. Li, X. Miao and H. Ding, J. Ethnopharmacol., 2011, 138, 1–21 CrossRef CAS.
- Chinese Pharmacopoeia Commission, Chinese Pharmacopoeia, China Medical Science Press, Beijing, 2015, vol. 1 Search PubMed.
- S. Y. Dai, Z. P. Shang, F. Wang, Y. Cao, X. Y. Shi, Z. Z. Lin, Z. Wang, N. Li, J. Q. Lu and Y. J. Qiao, Sci. Rep., 2017, 7, 9971 CrossRef PubMed.
- J. Y. Zhan, P. Yao, C. W. Bi, K. Y. Zheng, W. L. Zhang, J. P. Chen, T. T. Dong, Z. R. Su and K. W. Tsim, Phytomedicine, 2014, 21, 1318–1324 CrossRef CAS.
- A. Biancolillo, R. Bucci, A. L. Magrì, A. D. Magrì and F. Marini, Anal. Chim. Acta, 2014, 820, 23–31 CrossRef CAS.
- J. Y. Zhang, Z. J. Wang, Q. Zhang, F. Wang, Q. Ma, Z. Z. Lin, J. Q. Lu and Y. J. Qiao, Talanta, 2014, 124, 111–122 CrossRef CAS.
- J. Y. Zhang, Q. Zhang, N. Li, Z. J. Wang, J. Q. Lu and Y. J. Qiao, Talanta, 2013, 104, 1–9 CrossRef CAS.
- X. Sun, X. B. Cui, H. M. Wen, C. X. Shan, X. Z. Wang, A. Kang, C. Chai and W. Li, J. Pharm. Biomed. Anal., 2017, 141, 19–31 CrossRef CAS.
- M. Yang, Z. Zhou and D. A. Guo, Anal. Chim. Acta, 2015, 894, 44–53 CrossRef CAS.
- X. Zhang, Z. Ning, D. Ji, Y. Chen, C. Mao and T. Lu, J. Sep. Sci., 2015, 38, 3825–3831 CrossRef CAS PubMed.
- R. F. Goddu, in Near-infrared spectrophotometry, Advances in Analytical Chemistry and Instrumentation, ed. C. N. Reilly, 2nd edn, 1960, pp. 347–424 Search PubMed.
- S. Qiu, W. Z. Yang, X. J. Shi, C. L. Yao, M. Yang, X. Liu, B. H. Jiang, W. Y. Wu and D. A. Guo, Anal. Chim. Acta, 2015, 893, 65–76 CrossRef CAS.
- T. H. Wang, J. Zhang, X. H. Qiu, J. Q. Bai, Y. H. Gao and W. Xu, Molecules, 2015, 21, E40 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05032a |
| This journal is © The Royal Society of Chemistry 2019 |