
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sarcoehrenbergilides D–F: cytotoxic cembrene diterpenoids from the soft coral Sarcophyton ehrenbergi†
Mohamed-Elamir F. Hegazy ab,
Tarik A. Mohamedb,
Abdelsamed I. Elshamycd,
Ahmed R. Hamedb,
Mahmoud A. A. Ibrahime,
Shinji Ohtaf,
Akemi Umeyamad,
Paul W. Paré*g and
Thomas Efferth*a
ab,
Tarik A. Mohamedb,
Abdelsamed I. Elshamycd,
Ahmed R. Hamedb,
Mahmoud A. A. Ibrahime,
Shinji Ohtaf,
Akemi Umeyamad,
Paul W. Paré*g and
Thomas Efferth*a
aDepartment of Pharmaceutical Biology, Institute of Pharmacy and Biochemistry, Johannes Gutenberg University, Staudinger Weg 5, 55128 Mainz, Germany
bChemistry of Medicinal Plants Department, National Research Centre, El-Tahrir St., Dokki, Giza 12622, Egypt. E-mail: elamir77@live.com; tarik.nrc83@yahoo.com; n1ragab2004@yahoo.com
cNatural Compound Chemistry Department, National Research Centre, El-Tahrir St., Dokki, Giza 12622, Egypt. E-mail: elshamynrc@yahoo.com
dFaculty of Pharmaceutical Sciences, Tokushima Bunri University, Yamashiro-cho, Tokushima, 770-8514, Japan. E-mail: umeyama@ph.bunri-u.ac.jp
eComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia 61519, Egypt. E-mail: m.ibrahim@compchem.net
fGraduate School of Biosphere Science, Hiroshima University, 1-7-1 Kagamiyama, Higashi-Hiroshima 739-8521, Japan. E-mail: ohta@hiroshima-u.ac.jp
gDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409, USA
First published on 29th August 2019
Abstract
A solvent extract of the soft coral Sarcophyton ehrenbergi afforded cembrene diterpenoids, sarcoehrenbergilid D–F (1–3). Chemical structures were established by modern spectroscopic techniques with absolute stereochemistries determined by circular dichroism (CD) and time-dependent density functional theory electronic CD calculations (TDDFT-ECD). Cytotoxicity activities for 1–3 were evaluated against three human cancer cell lines: lung (A549), colon (Caco-2) and liver (HepG2).
1. Introduction
Soft coral of the genus Sarcophyton (subclass Octocorallia; order Alcyonaceae; family Alcyoniidae) contain a diversity of cyclic diterpenes that usually contain ethers, lactones or furanes around a cembrane framework.1,2 These cembrane diterpenoids exhibit a wide range of structural diversity and biological activity.3–10 Cembranoids, the main metabolites identified in the genus Sarcophyton have been shown to serve as an effective chemical defense against natural predators of coral.11The leather coral Sarcophyton ehrenbergi (von Marenzeller, 1886) produces diverse metabolites with distinct chemical structures as well as promising biological activities.8,12–17 Additionally, prostaglandins (PGs) that regulate a broad range of physiological activities, have been isolated from S. ehrenbergi.18,19
The Red Sea contains a high endemic biota including approximately 50 genera of hermatypic soft coral.20 While Red Sea marine invertebrates have been historically under-reported within the scientific literature, intensive investigation of Red Sea marine life has occurred over the past ten years.8,21–23 To continue efforts to identify new marine metabolites from Red Sea soft coral,6–8,22–24 herein we report three cembrene diterpenoids isolated from S. ehrenbergi (Fig. 1). Absolute stereochemistry of the newly reported compounds was determined by time-dependent density functional theory-electronic circular dichroism (TDDFT-ECD) calculations. All isolated metabolites were probed against three human cancer cell lines.
 | ||
| Fig. 1 Structures of metabolites 1–3. | ||
2. Results and discussion
Freshly collected S. ehrenbergi were rapidly frozen by placing in a −20 °C chamber and kept frozen till time of extraction. The chromatographic separation of the methylene chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (1:1) extract yielded three cembrene diterpenoids derivatives (Fig. 1).
:methanol (1:1) extract yielded three cembrene diterpenoids derivatives (Fig. 1).
Compound 1 was isolated as a white powder with an optical rotation of [α]25D +10.1 (c 0.02, CHCl3). The molecular formula C21H32O5 was determined by high-resolution electron ionization mass spectrum (HREIMS) (m/z 346.2127 [M − H2O]+, calcd 346.2149).
The IR spectrum showed absorption bands at νmax 3450 cm−1 and 1754 cm−1 for hydroxyl and keto groups, respectively. The 13C NMR and distortion less enhancement by polarization transfer (DEPT) spectrum showed 21 carbon signals, classified as five methyls, six methylenes, four methines and six quaternary carbons (Table 1). Additionally, four oxygenated carbons at δC 76.2 (dC), 78.0 (dC), 78.5 (dC) and 78.1 (sC), four olefinic carbon signals at δC 119.5, 121.8, 147.0 and 163.0. These functionalities were obtained by 1H NMR analysis: oxygenated proton signals at δH 3.57 (brd; J = 10.0 Hz), δH 3.14 (brd, J = 5.0 Hz), δH 5.45 (d; J = 10.0 Hz); four methyl singlets at δH 2.02 s, 1.83 s, 1.11 s and 1.03, as well as, one methyl of a methoxy group at δH 3.20 s; olefinic signal at δH 5.14 (d; J = 10.0 Hz) signed for a tri-substituted double bond (Table 1). 1D and 2D NMR spectroscopic data comparison (Table 1) closely corresponded to those of previously isolated metabolites from Sarcophyton species as well as a previously isolated skeleton by Hegazy et al., 2017 (ref. 5–13, 22 and 23) (Fig. 2).
| No. | 1b | 2c | 3c | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a J values (Hz) in parentheses, obtained at 500 and 125 MHz for 1H and 13C NMR, respectively.b Recorded in CDCl3.c Recorded in CDCl3–CD3OD (9:1). |
||||||
| 1 | — | 163.0 | — | 164.4 | — | 163.9 |
| 2 | 5.45 d (10.00) | 78.1 | 5.54 d (9.5) | 81.0 | 5.38 brd (10.00) | 80.2 |
| 3 | 5.14 d (10.00) | 119.5 | 4.99 d (9.00) | 119.4 | 5.10 brd (10.00) | 119.5 |
| 4 | — | 147.0 | — | 141.6 | — | 144.4 |
| 5 | 1.87 m, 2.37 brd (14.00) | 34.6 | 2.11 m, 2.37 m | 41.0 | 1.85 m, 1.62 m | 37.00 |
| 6 | 1.30 m, 2.21 m | 28.7 | 2.04 m, 2.18 dd (6.50, 8.00) | 27.8 | 1.98 m; 1.58 m | 24.7 |
| 7 | 3.14 brd (5.00) | 73.5 | 3.38 brd (10.50) | 78.3 | 3.09 dd (7.5, 2.5) | 84.0 |
| 8 | — | 78.5 | — | 74.5 | — | 70.0 |
| 9 | 1.43 m; 2.00 m | 37.0 | 1.51 m; 1.79 m | 43.1 | 1.90 m, 1.59 m | 40.4 |
| 10 | 1.51 m; 1.85 m | 28.2 | 1.47 m; 1.85 m | 28.9 | 1.58 m, 1.51 m | 23.7 |
| 11 | 3.57 brd (10.00) | 76.2 | 3.16 d (7.50) | 80.0 | 3.29 brd (10.00) | 80.2 |
| 12 | — | 78.0 | — | 80.1 | — | 73.1 |
| 13 | 1.62 m; 1.78 m | 31.0 | 1.49 m, 1.96 m | 34.7 | 2.35 m, 2.24 m | 36.3 |
| 14 | 2.43 brt (12.20), 2.57 m | 20.8 | 1.99 m, 2.41 m | 20.8 | 2.05 m; 2.53 m | 20.3 |
| 15 | — | 121.8 | — | 122.3 | — | 123.1 |
| 16 | — | 174.0 | — | 176.0 | — | 175.5 |
| 17 | 1.83 s | 7.8 | 1.83 s | 8.8 | 1.85 s | 8.9 |
| 18 | 2.02 s | 20.8 | 1.91 brs | 17.1 | 1.83 brs | 16.7 |
| 19 | 1.11 s | 13.6 | 1.39 s | 20.7 | 1.17 s | 20.3 |
| 20 | 1.03 s | 17.0 | 1.03 s | 17.6 | 1.16 s | 23.3 |
| 21 | 3.20 s | 48.3 | ||||
 | ||
Fig. 2 Selected 1H–1H COSY ( ) and HMBC ( ) and HMBC ( ) correlations of 1–3. ) correlations of 1–3. | ||
The signal at δH 5.45 (d; J = 10.0 Hz) correlated with a proton signal at δH 5.14 (d, J = 10.0 Hz) and quaternary olefinic carbons at δC 147.0 and δC 163.0 in DQF-COSY and HMBC (Fig. 2), respectively, allowed for the assignments of H-2, H-3, C-4 and C-1, respectively.8–10,23–25 Correlations in the HMBC spectrum showed several informative connections: H-3 to carbon signals at δC 13.6 (q, olefinic) δC 34.6 (t), allowed for the assignment of H-18 (δH 2.02, s) and H-5 (δH 2.37, brd, J = 14.0), respectively; methyl signal δH 1.83 (s) to C-1 and carbon signal at δC 174.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) attributed to H-17 and C-16, respectively as well as supporting the location of C-1/C-2 lactone ring; methyl singlet at δH 1.11 to δC 73.5 (C-7), δC 37.0 and 78.5 allowed for the location of H3-19 (δC 13.6), C-9 and C-8, respectively; the oxygenated broad doublet at δH 3.57 (δC 79.0) to C-9 and C-20, was assigned to H-11. The assignment of H-7, H2-6 and C-5 was detected through the correlation of the oxygenated methine signal at δH 3.14 (brd, J = 5.0) to a methylene multiplet at δH 1.30/2.21 and a carbon signal at δC 34.6 in DQF-COSY and HMBC, respectively. Additionally, a correlation was detected in DQF-COSY between H-13 (δH 1.78, m) and H-14 (δH 2.43, brt, J = 12.2) as well as to C-20 in HMBC analyses (Fig. 2). An HMBC correlation established the site of a methoxy group (δH 3.20 s, δC 48.3 q) at C-8.
O) attributed to H-17 and C-16, respectively as well as supporting the location of C-1/C-2 lactone ring; methyl singlet at δH 1.11 to δC 73.5 (C-7), δC 37.0 and 78.5 allowed for the location of H3-19 (δC 13.6), C-9 and C-8, respectively; the oxygenated broad doublet at δH 3.57 (δC 79.0) to C-9 and C-20, was assigned to H-11. The assignment of H-7, H2-6 and C-5 was detected through the correlation of the oxygenated methine signal at δH 3.14 (brd, J = 5.0) to a methylene multiplet at δH 1.30/2.21 and a carbon signal at δC 34.6 in DQF-COSY and HMBC, respectively. Additionally, a correlation was detected in DQF-COSY between H-13 (δH 1.78, m) and H-14 (δH 2.43, brt, J = 12.2) as well as to C-20 in HMBC analyses (Fig. 2). An HMBC correlation established the site of a methoxy group (δH 3.20 s, δC 48.3 q) at C-8.
The planar structure assignment of 1 and the C-7/C-12 ether linkage were proposed by 1D, 2D NMR and HREIMS data. The data comparison with those of sarcoehrenbergilid A, as previously reported,23 suggested that 1 and sarcoehrenbergilid A,23 differ only in stereochemistry.
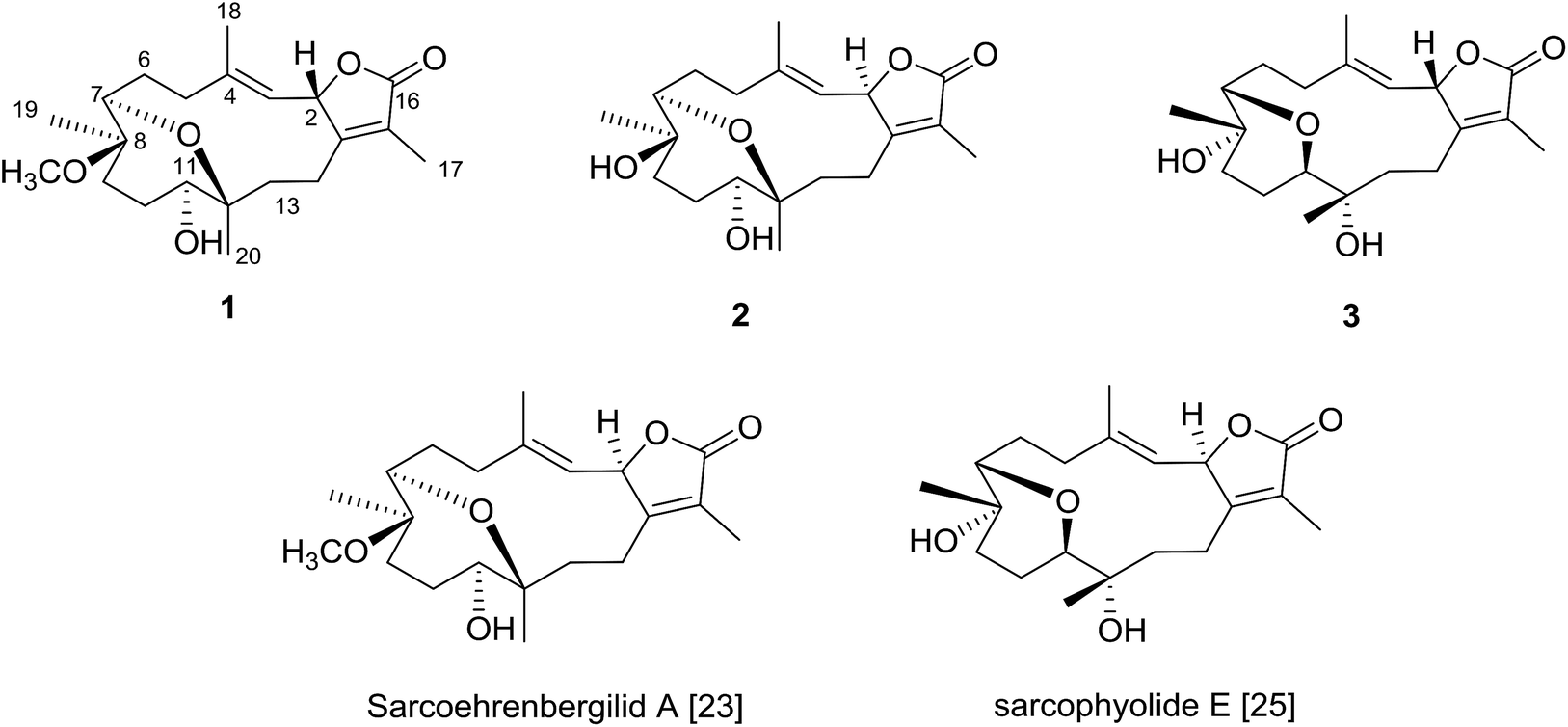
The NOESY spectrum revealed that a γ-lactone at H-2 (δH 5.45, d, J = 10.0 Hz) correlated with CH3-18 (δH 2.02, s); a vicinal coupling with H-3 established a trans configuration and a β-orientation for H-2.8 NOSEY correlations were observed between three methyl groups with alpha protons (e.g., CH3-20 with H-10a, CH3-19 with H-6a/H-10a, and CH3-17 with H-14a) (Fig. 3). H-7 and H-11 was assigned to a β-configuration based on NOSEY correlations with H-5b and H-14b, respectively. Absolute configuration was established by experimental and TDDFT-simulated ECD spectra. All possible conformations of 1 within energy window of 10 kcal mol−1 were generated and optimized at B3LYP/6-31G* level of theory. The first 50 excitation states were then computed based on time-dependent density-functional theory (TDDFT) at B3LYP/6-31G* level in methanol by the PCM model. The generated TDDFT-ECD spectra were Boltzmann-weighted and compared to the experimental spectrum (Fig. 4). The TDDFT-simulated ECD spectrum was in a good agreement with the corresponding experimental ECD spectra (Fig. 4). This comparison revealed the absolute configuration and therefore 1 was assigned as 2S,16:7S,12S-diepoxy-11R-hydroxy-8R-methoxy-16-keto-cembra-1Z,3E-diene (sarcoehrenbergilid D).
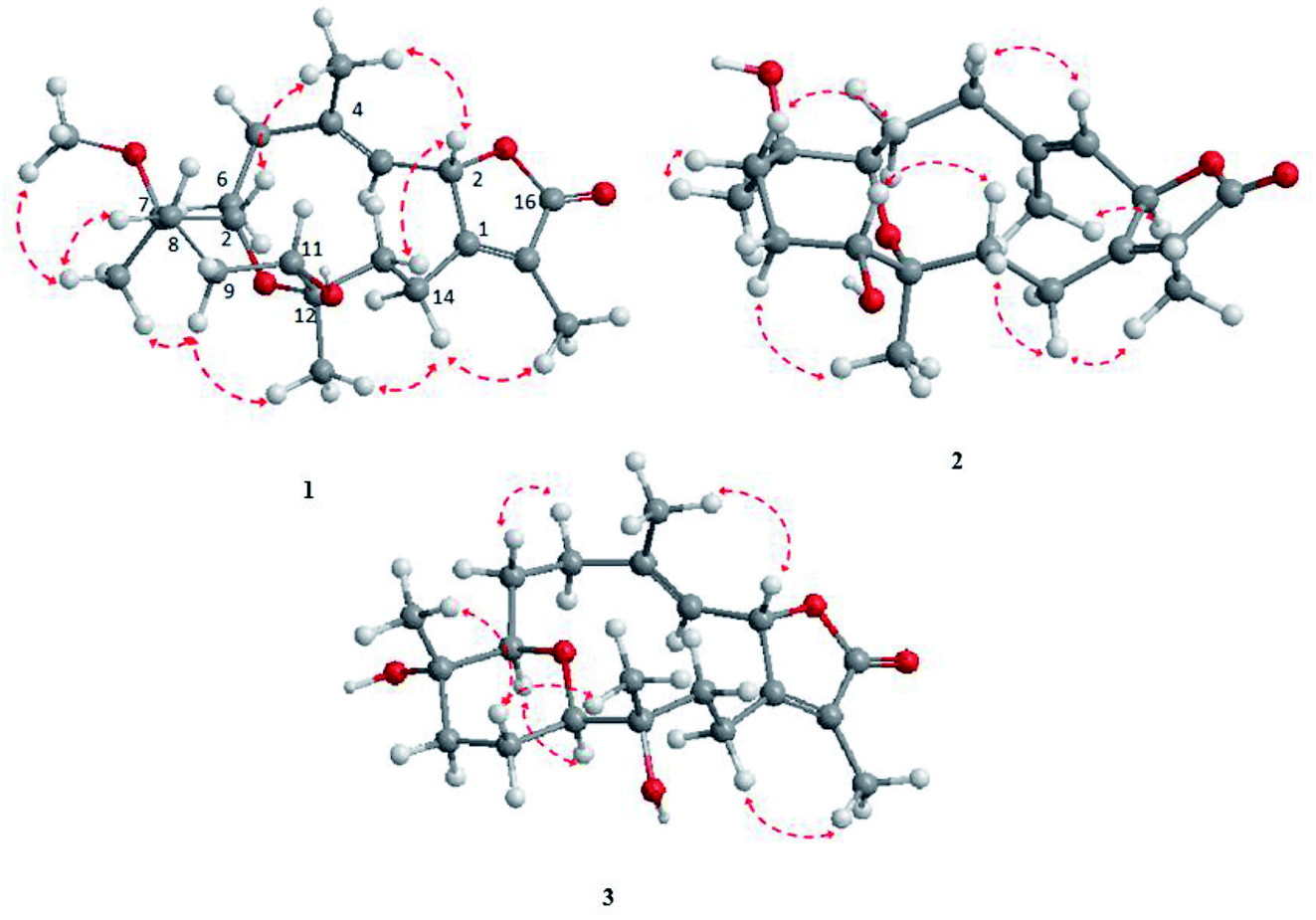 | ||
| Fig. 3 Selected NOESY correlations for 1–3. | ||
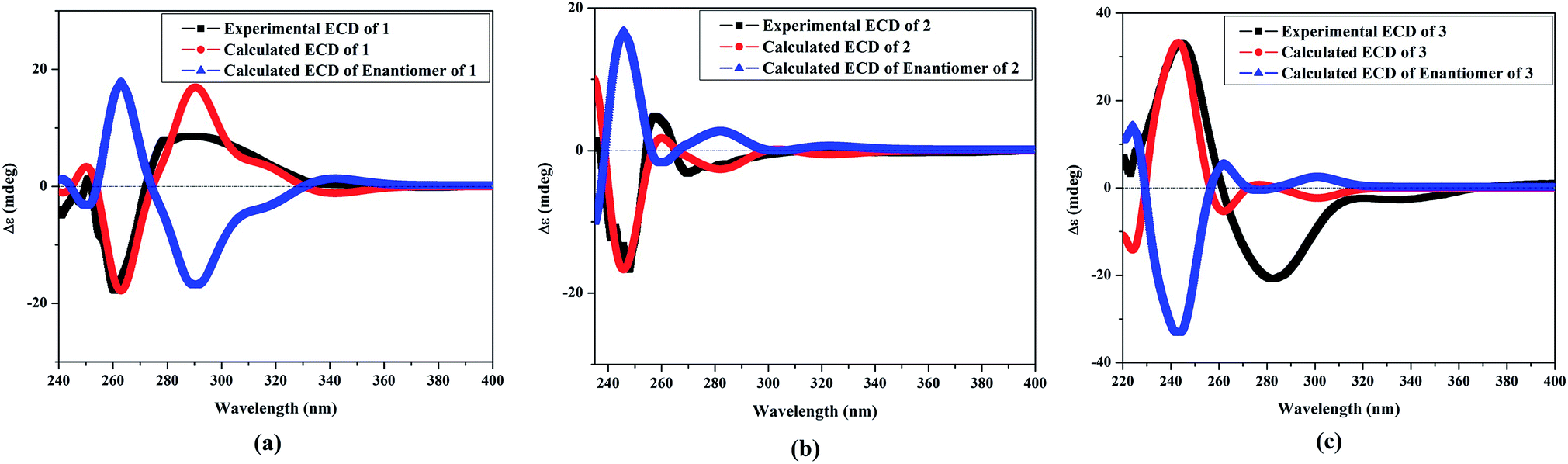 | ||
| Fig. 4 Experimental electronic circular dichroism (ECD in MeOH): (a) 1 compared with the TDDFT-simulated ECD spectra of 2S,7S,12S-diepoxy-11R-hydroxy-8R-methoxy-16-keto-cembra-1Z,3E-diene and 2R,7R,12R-diepoxy-11S-hydroxy-8S-methoxy-16-keto-cembra-1E,3Z-diene; (b) 2 compared with the TDDFT-simulated ECD spectra of 2R,7S,12S-diepoxy-11R-hydroxy-8R-methoxy-16-keto-cembra-1Z,3E-diene and 2S,7R,12R-diepoxy-11S-hydroxy-8S-methoxy-16-keto-cembra-1E,3Z-diene; and (c) 3 compared with the TDDFT-simulated ECD spectra of 2S,7R,11R-diepoxy-12S-hydroxy-8S-methoxy-16-keto-cembra-1Z,3E-diene and 2R,7S,11S-diepoxy-12R-hydroxy-8R-methoxy-16-keto-cembra-1E,3Z-diene. | ||
Compound 2 was isolated as a white powder with a negative optical rotation of [α]25D = −5.4 (c 0.03, CHCl3). The molecular formula (C20H30O5) was detected by high resolution electron ionization (HREIMS) spectrum (m/z 350.2094 [M]+, calcd 350.2093). HREIMS analysis exhibited a molecular ion peak at m/z 350.2094 [M]+ (calcd) The IR spectrum showed characteristic bands at νmax 3445 cm−1 and 1747 cm−1 for hydroxyl and keto groups, respectively. The 13C NMR spectrum revealed twenty carbon signals (Table 1) classified by DEPT as six quaternary, four methines, six methylenes and four methyls carbons. 1D and 2D NMR spectroscopic data were quite close to sarcoehrenbergilid A,23 a formerly isolated diterpenoid from S. ehrenbergi except for an absence of methoxyl groups. For 2 there is an upfield carbon signal at δC 74.5 and a downfield methyl signal at δC 20.7 for C-8 and CH3-19, respectively.
Stereochemistry was established based on coupling constants and NOESY experiments (Fig. 3). NOESY correlation indicated that 2 has the same relative stereochemistry as sarcoehrenbergilid A.23 To determine absolute configuration, TDDFT-ECD calculations were performed on the 2R,7S,8R,11R,12S- and 2S,7R,8S,11S,12R-enantiomers. The final Boltzmann-weighted TDDFT-ECD spectra were then compared to the corresponding experimental ECD curve (Fig. 4). According to the data depicted in Fig. 4, the 2R,7S,8R,11R,12S-enantiomer reproduced all the transitions of the experimental ECD spectrum. Therefore, 2 was assigned as 2R,16:7S,12S-diepoxy-11R-hydroxy-8R-methoxy-16-keto-cembra-1Z,3E-diene (sarcoehrenbergilid E). Compound 3 was isolated as a colorless oil with a negative optical rotation of [α]25D = −10.8 (c 0.01, CHCl3). The molecular formula of C20H30O5 was detected by high resolution electron ionization (HREIMS) analysis (m/z 332.1993 [M − H2O+], calcd 332.1998).
The IR spectrum showed characteristic bands at νmax 3445 cm−1 and 1747 cm−1 for hydroxyl and keto groups, respectively. The 13C NMR spectrum (Table 1) showed 20 carbon resonances classified by DEPT analysis as four methyls, six methylenes, four methines and six quaternary carbons. The 1D (1H, 13C) as well as 2D NMR (1H–1H COSY, HSQC, and HMBC) (Fig. 2) spectroscopic data closely matches a previously reported cemberene compound.26 The NOESY correlation (Fig. 3) as well as the 1H and 13C NMR analyses indicated that 3 is a C-2 epimer of the previously isolated sarcophyolide E26 through the clear difference in downfield shift of H-3 (δH 5.10, d, J = 10.0). Additionally, several carbon signals showed downfield chemical shift in comparison of sarcophyolide E: δC 37.0/36.2 (C-5), 73.1/71.8 (C-12), 123.1/121.7 (C-15), and 175.5/174.9 (C-17), respectively. The carbon signals at δC 163.9 (C-1) and 36.3 (C-13) showed upfield chemical shift in comparison with sarcophyolide E [δC 165.8 (C-1) and 37.3 (C-13)].
The relative configuration for 3 was established based on coupling constants and NOESY experiments (Fig. 3). A NOE correlation between H-7 (δH 3.09 dd, J = 7.5, 2.5) and H-11 (δH 3.29 brd, J = 10.0) established an alpha linkage for the ether bridge between C-7 and C-11. The NOE correlations between H-3 and the γ-lactone-(H-2) as well as vicinal coupling constant indicated a trans-geometry for H-2 and H-3 of the olefinic bond (Fig. 3). As expected, the experimental ECD for 3 and published compound, sarcophyolide E,26 showed inverted direction for positive and negative cotton effect as well as optical rotation (Fig. 4). These data indicated that 3 is the C-2 epimer of sarcophyolides E. Thus, 3 was confirmed to be 2S,16:7R,11R-diepoxy-12S-hydroxy-8S-methoxy-16-keto-cembra-1Z,3E-diene (sarcoehrenbergilid F).
Isolated metabolites 1–3 were tested for cytotoxic activity toward lung (A549), colon (Caco-2) and liver (HepG2) human cancer cell lines based on an MTT reduction assay (Fig. 5). Compounds 1–3 showed most potent activity toward A549 cells with IC25 values of 23.3, 27.3, and 25.4 μM, respectively. Compound 2 and 3 showed weaker activity toward liver (HepG2) human cancer cell lines with IC25 values of 22.6 and 31.8 μM, respectively. The treated human colon cancer cells (Caco-2) cell viability was over 100% for all the isolated compounds (IC25 > 100 μM). Since primary necrosis is not easily differentiated from secondary necrosis that occurs with apoptosis,27 the mode of action will not be considered. To differentiate these distinct biological events requires apoptotic assays accompanying necrosis measurements. A combined necrosis/apoptotic time-course will be presented in a subsequent study to elaborate on mode of action.
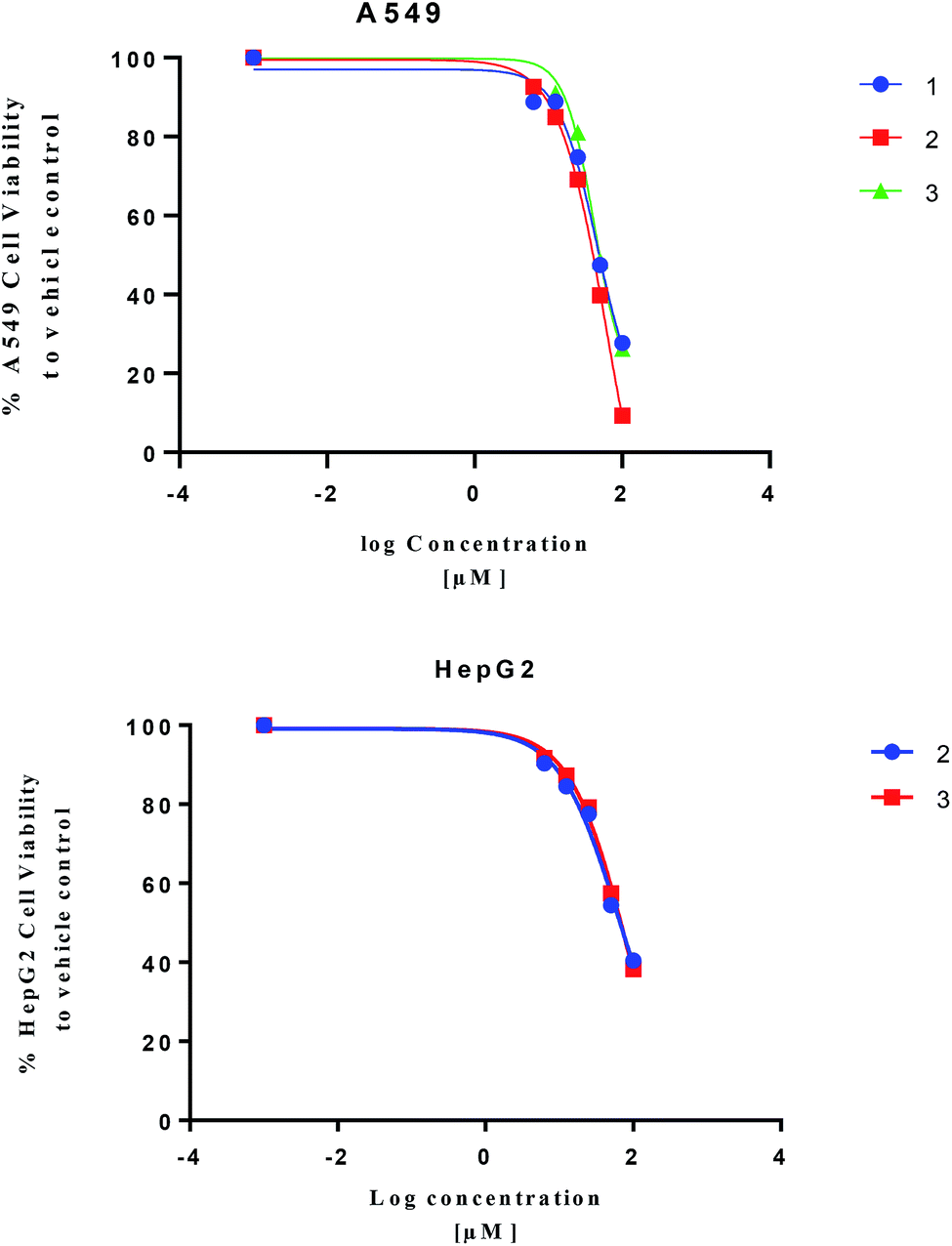 | ||
| Fig. 5 Cytotoxicity assay of 1–3 based on MTT-reduction assay. | ||
3. Experimental section
3.1. General experimental procedures
Circular dichroism was measured on JASCO 810 spectropolarimeter. HREIMS data were collected on a JEOL JMS-700 instrument (Tokyo, Japan). NMR spectra were recorded on a Bruker 500 NMR spectrometer (Japan). JASCO P-2200 polarimeter and JASCO FT/IR-6300 spectrometer was used for optical rotation and infrared measurements, respectively.Normal-phase silica gel 60 (230–400) column chromatography (CC) as well as aluminum TLC plates (silica gel 60 F254) (Merck, Darmstadt, Germany) were used for purification and monitoring spotting, respectively. A H2SO4:MeOH (1:9) spraying reagent was used for spot visualization after heating. HPLC purification was performed using Shimadzu HPLC-RID-10A with YMC-Pack ODS-A analytical (250 × 4.6 mm i.d.) and preparative (250 × 20 mm i.d.) columns (YMC, Tokyo, Japan) for separation.
3.2. Animal material
Sarcophyton ehrenbergi coral was collected from the Red Sea on the Egyptian coast at Hurghada, in March 2016 and identified by Dr M. Al-Hammady. A voucher specimen (03RS27/1) was deposited in the National Institute of Oceanography and Fisheries, marine biological station, Hurghada, Egypt.3.3. Extraction and isolation
Sliced frozen soft coral (2 kg, total wet weight) were extracted with CH2Cl2:MeOH (1:1, v/v) at room temperature (3 L × 4 times). Isolation protocol was performed as described previously by Hegazy et al., 2017 (ref. 23) to afford 1 (5.5 mg), 2 (4 mg) and 3 (6 mg).
3.4. Biological activity
3.5. Computational functional theory calculations
Conformational analysis was performed using Omega2 software30 to obtain the possible conformers for 1–3 within energy window value of 10 kcal mol−1. All resulting conformers were optimized at B3LYP/6-31G* level of theory using Gaussian09 software.30 Frequency calculations were then performed on the optimized structures to ensure the nature of the local minima as well as to estimate the Gibbs free energies. Time-dependent density functional theory (TDDFT) calculations with incorporating a polarizable continuum model (PCM) using methanol as a solvent were carried out at the B3LYP/6-31G* level of theory to calculate the first fifty excitation states. Electronic circular dichroism (ECD) spectra were finally generated using SpecDis 1.71 (SpecDis 2017 (ref. 31 and 32)) by applying Gaussian band shapes with sigma = 0.20–30 eV. The generated ECD spectra were Boltzmann-averaged.4. Conclusions
Cembrene diterpenoids (1–3) were isolated and identified from the S. ehrenbergi soft coral. The isolated compounds were tested against three human cancer cell lines, which resulted in 2 being the most potent compound against lung A549 cancer cell. The absolute stereochemistry of 1–3 were confirmed by comparing experimental and TDDFT-simulated ECD spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Alexander von Humboldt Foundation (Georg Förster Research Fellowship) to MEFH, National Research Centre, Egypt and the Welch Foundation (D-1478). Dr Elshamy gratefully acknowledges the Takeda Science Foundation, Japan for the financial support. Computational work was completed in part with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants No. 5480 & 7972.Notes and references
- B. Yang, X.-F. Zhou, X.-P. Lin, J. Liu, Y. Peng, X.-W. Yang and Y. Liu, Cembrane diterpenes chemistry and biological properties, Curr. Org. Chem., 2012, 16, 1512–1539 CrossRef CAS.
- M. Kobayashi, T. Nakagawa and H. Mitsuhashi, Marine terpenes and terpenoids. I. Structures of four cembrane-type diterpenes: sarcophytol-A, sarcophytol-A acetate sarcophytol-B, and sarcophytonin-A, from the soft coral, Sarcophyton glaucum, Chem. Pharm. Bull., 1979, 27, 2382–2387 CrossRef CAS.
- B. Yang, J. Liu, J. Wang, S. Liao and Y. Liu, Cytotoxic cembrane diterpenoids, Handbook of anticancer drugs from marine origin, ed. S.-K. Kim, Springer International Publishing, Switzerland, 2015 Search PubMed.
- R. Abou El-Ezz, S. Ahmed, M. Radwan, N. Ayoub, M. Afifi, S. Ross, P. Szymanski, H. Fahmy and S. Khalifa, Bioactive cembranoids from the Red Sea soft coral Sarcophyton glaucum, Tetrahedron Lett., 2013, 54, 989–992 CrossRef CAS.
- L. Liang and Y. Guo, Terpenes from the soft corals of the genus Sarcophyton: chemistry and biological activities, Chem. Biodiversity, 2013, 10, 2161–2196 CrossRef CAS PubMed.
- M. E. Hegazy, A. A. El-Beih, A. Y. Moustafa, A. A. Hamdy, M. A. Alhammady, R. M. Selim, M. Abdel-Rehim and P. W. Paré, Cytotoxic cembranoids from the Red Sea soft coral Sarcophyton glaucum, Nat. Prod. Commun., 2011, 6, 1809–1812 CrossRef CAS PubMed.
- M. E. Hegazy, T. A. Mohamed, F. A. Abdel-Latif, M. Alsaid, A. A. Shahat and P. W. Paré, Trochelioid A and B, new cembranoid diterpenes from the Red Sea soft coral Sarcophyton trocheliophorum, Phytochem. Lett., 2013, 6, 383–386 CrossRef CAS.
- A. Elkhateeb, A. El-Beih, A. Gamal-Eldeen, M. Alhammady, S. Ohta, P. Paré and M. E. Hegazy, New terpenes from the Egyptian soft coral Sarcophyton ehrenbergi, Mar. Drugs, 2014, 12, 1977–1986 CrossRef PubMed.
- X.-H. Yan, Z.-Y. Li and Y.-W. Guo, Further new cembranoid diterpenes from the Hainan soft coral Sarcophyton latum, Helv. Chim. Acta, 2007, 90, 1574 CrossRef CAS.
- X.-H. Yan, L.-Y. Feng and Y.-W. Guo, Further new cembrane diterpenes from the Hainan soft coral Sarcophyton latum, Chin. J. Chem., 2008, 26, 150 CrossRef CAS.
- M. E. Hegazy, T. A. Mohamed, M. A. Alhammady, A. M. Shaheen, E. H. Reda, A. I. Elshamy, M. Aziz and P. W. Paré, Molecular architecture and biomedical leads of terpenes from Red Sea marine invertebrate, Mar. Drugs, 2015, 13, 3154–3181 CrossRef CAS PubMed.
- B. F. Bowden, J. C. Coll, W. Hicks, R. Kazlauskas and S. J. Mitchell, Studies of Australian soft corals. X. The isolation of epoxyisoneocembrene-A from Sinulariagrayi and isoneocembrene-A from Sarcophyton ehrenberg, Aust. J. Chem., 1978, 31, 2707–2712 CrossRef CAS.
- G. M. König and A. D. Wright, New cembranoid diterpenes from the soft coral Sarcophyton ehrenbergi, J. Nat. Prod., 1998, 61, 494–496 CrossRef.
- K. H. Shaker, M. Mueller, M. A. Ghani, H. M. Dahse and K. Seifert, Terpenes from the soft corals Litophyton arboreum and Sarcophyton ehrenbergi, Chem. Biodiversity, 2010, 7, 2007–2015 CrossRef CAS PubMed.
- S.-Y. Cheng, S.-K. Wang, S.-F. Chiou, C.-H. Hsu, C.-F. Dai, M. Y. Chiang and C.-Y. Duh, Cembranoids from the octocoral Sarcophyton ehrenbergi, J. Nat. Prod., 2010, 73, 197–203 CrossRef CAS PubMed.
- S.-K. Wang, M.-K. Hsieh and C.-Y. Duh, Three new cembranoids from the Taiwanese soft coral Sarcophyton ehrenbergi, Mar. Drugs, 2012, 10, 1433–1444 CrossRef CAS PubMed.
- S.-K. Wang, M.-K. Hsieh and C.-Y. Duh, New diterpenoids from soft coral Sarcophyton ehrenbergi, Mar. Drugs, 2013, 11, 4318–4327 CrossRef PubMed.
- Z. B. Cheng, Y. L. Deng, C. Q. Fan, Q. H. Han, S. L. Lin, G. H. Tang, H. B. Luo and S. Yin, Prostaglandin Derivatives: Nonaromatic Phosphodiesterase-4 Inhibitors from the Soft Coral Sarcophyton ehrenbergi, J. Nat. Prod., 2014, 77, 1928–1936 CrossRef CAS PubMed.
- V. C. Sekhar, C. B. Rao, H. Ramana, M. M. K. Kuwar and D. V. Rao, New Prostaglandins from the soft coral Sarcophyton ehrenbergi marengeller of Andaman and Nicobar Islands of Indian ocean, Asian J. Chem., 2010, 22, 5353–5358 CAS.
- A. J. Edwards and S. M. Head, Key Environments-Red Sea, Pergamon Press, Oxford, UK, 1987, p. 440 Search PubMed.
- M. A. Farag, D. A. Al-Mahdy, A. Meyer, H. Westphal and L. A. Wessjohann, Metabolomics reveals biotic and abiotic elicitor effects on the soft coral Sarcophyton ehrenbergi terpenoid content, Sci. Rep., 2017, 7, 648 CrossRef PubMed.
- M. A. Farag, A. Porzel, M. A. Al-Hammady, M. E. Hegazy, A. Meyer, T. A. Mohamed, H. Westphal and L. A. Wessjohann, Soft Corals Biodiversity in the Egyptian Red Sea: A Comparative MS and NMR Metabolomics Approach of Wild and Aquarium Grown Species, J. Proteome Res., 2016, 15, 1274–1287 CrossRef CAS PubMed.
- M. E. Hegazy, A. I. Elshamy, T. A. Mohamed, A. R. Hamed, M. A. Ibrahim, S. Ohta and P. Paré, Cembrene diterpenoids with ether linkages from Sarcophyton ehrenbergi: an anti-proliferation and molecular-docking assessment, Mar. Drugs, 2017, 15, 192 CrossRef PubMed.
- M. E. Hegazy, T. A. Mohamed, A. I. Elshamy, M. A. Alhammady, S. Ohta and P. Paré, Casbane diterpenes from red sea coral Sinularia polydactyla, Molecules, 2016, 21, 308 CrossRef PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- Z. Xi, W. Bie, W. Chen, D. Liu, L. V. Ofwegen, P. Proksch and W. Lin, Sarcophyolides B–E, new cembranoids from the soft coral Sarcophyton elegans, Mar. Drugs, 2013, 11(9), 3186–3196 CrossRef PubMed.
- F. K. Chan, K. Moriwaki and M. J. De Rosa, Detection of necrosis by release of lactate dehydrogenase activity, Methods Mol. Biol., 2013, 979, 65–70 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. Dutta, S. Bandyopadhyay, C. Mandal and M. Chatterjee, Development of a modified MTT assay for screening antimonial resistant field isolates of Indian visceral leishmaniasis, Parasitol. Int., 2005, 54, 119–122 CrossRef CAS PubMed.
- OpenEye Scientific Software, OMEGA2.5.1.4, Santa Fe, NM, USA Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel; G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, USA, 2009 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pescitelli, SpecDis, Berlin, Germany, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S21: HR-ESI-MS, 1D, and 2D NMR spectra of compounds 1–3. See DOI: 10.1039/c9ra04158c |
| This journal is © The Royal Society of Chemistry 2019 |