DOI:
10.1039/C9RA04311J
(Paper)
RSC Adv., 2019,
9, 26126-26135
The synthesis of a 99mTc-labeled tetravalent targeting probe upon isonitrile coordination to 99mTcI for enhanced target uptake in saturable systems†
Received
8th June 2019
, Accepted 11th August 2019
First published on 21st August 2019
Abstract
The presence of excess unlabeled ligands in the injectate hinders the target uptake of 99mTc-labeled targeting vectors. To address the issue, we previously developed a chemical design which provides a 99mTc-labeled trivalent RGD probe upon CN-βAla-Gly-Gly-c(RGDfK) (Lβ) coordination to [99mTc][Tc(CO)3]+ core at pH 6.0. In this study, we extended our coordination mediated synthesis of the trivalent RGD probe to that of a tetravalent one. Our initial attempts reacting Lβ with [99mTc][Tc(CO)3]+ core at pH 8.0 failed to provide [99mTc][Tc(CO)2(Lβ)4]+ due to the formation of multiple side products. A γ-aminobutylic acid (GABA) based isonitrile ligand CN-GABA-Gly-Gly-c(RGDfK) (LG), on the other hand, avoided the side reaction and selectively provided [99mTc][Tc(CO)2(LG)4]+ (99mTc-[LG]4) at pH 8.0. 99mTc-[LG]4 exhibited higher binding affinity to integrin αvβ3 than its unlabeled ligand, and visualized U87MG tumor without tedious post-labeling purification. These results indicate that the metal coordination-mediated syntheses of 99mTc-labeled multivalent probes have been successfully applied to a tetravalent one, which would allow a wider range of choices for designing novel 99mTc-labeled multivalent probes of high in vivo target uptake.
Introduction
Medical applications of metallic radionuclides have been rapidly expanding due to their unique roles in clinical nuclear medicine.1–5 Metallic radionuclides emitting γ photon or β+ particles are used for clinical nuclear imaging based on single photon emission computed tomography (SPECT) or positron emission tomography (PET), respectively, whereas ones emitting β− or α particles are used for internal radiotherapy of malignant tumors. To achieve specific delivery of metallic radionuclides to target tissues such as cancer, peptides or antibody fragments which have high affinity to target molecules are often used as a targeting vector.6 Since these molecules cannot form stable complexes with metallic radionuclides, they need to be modified with proper chelating agents, and carefully designed chelator-vector conjugates (hereafter referred as “ligands”) should be employed for radiolabeling reactions (Fig. 1a).7 Owing to the extremely low concentration of metallic radionuclides (for example, 99mTc < 1 μM (ref. 8)), however, these complexation reactions need to be performed with excess ligands (≈100 μM) to ensure high radiochemical yields in a short reaction time. As a result, the reaction solutions would contain not just the objective radiolabeled compounds but also excess unlabeled ligands (Fig. 1a). Upon the injection of these solutions into subjects, the target uptakes of the radiolabeled compounds would be competitively inhibited by the excess unlabeled ligands due to their similar binding affinities to target molecules (Fig. 1a).9
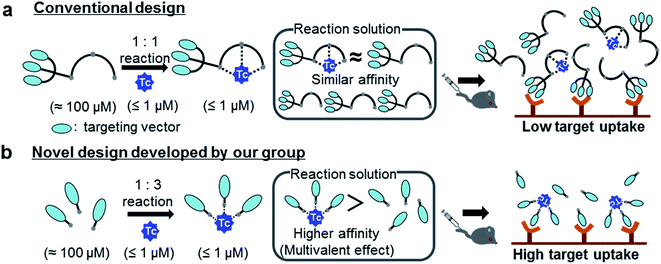 |
| | Fig. 1 Schematic illustration of a conventional and a novel chemical design. (a) The reaction solution, along with the 99mTc-labeled probe, contains excess unlabeled ligands whose binding affinity to a target molecule is similar to that of the 99mTc-labeled probe. Upon injection into subjects without post-labeling purification, the target uptake of the 99mTc-labeled probe is competitively inhibited by the presence of the unlabeled ligands. (b) The 99mTc-labeled trivalent probe would exhibit enhanced target binding affinity compared with its unlabeled ligand due to the multivalent effect. This superior target binding affinity of the 99mTc-labeled probe would enable high in vivo target uptake of the radiotracer without post-labeling purification. Adapted with permission from Mizuno et al., Purification-Free Method for Preparing Technetium-99m-Labeled Multivalent Probes for Enhanced in Vivo Imaging of Saturable Sytems, J. Med. Chem., 2016, 59, 3331–3339. Copyright 2016 American Chemical Society.10 | |
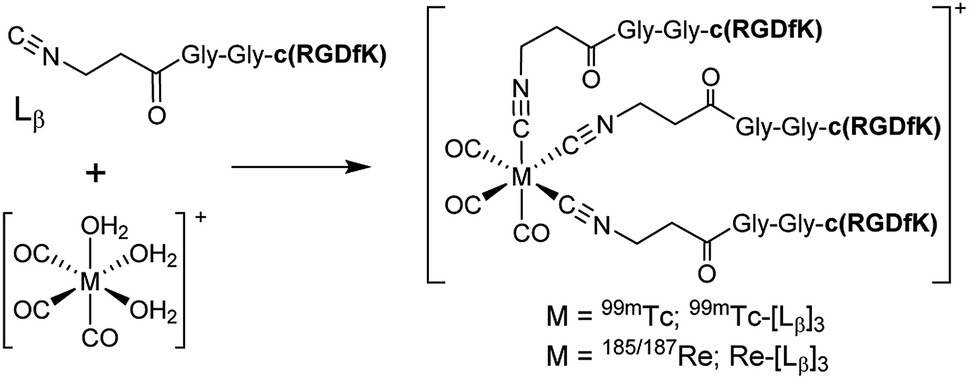
To circumvent the issue, our group has developed a novel chemical design concept of metal coordination mediated multivalency using the combination of [99mTc][TcI(CO)3]+ core and isonitrile ligands (Fig. 1b).10,11 To test the validity of this design concept, we selected c(RGDfK) peptides as a model targeting vector, which has high affinity and specificity to integrin αvβ3.12 [99mTc][TcI(CO)3(OH2)3]+ was firstly prepared as a labeling precursor, whose water molecules were then substituted by CN-βAla-Gly-Gly-c(RGDfK) (Fig. 2, Lβ), a monovalent RGD ligand, at slightly acidic conditions (pH 6.0), providing [99mTc][TcI(CO)3(Lβ)3]+ (Fig. 2, 99mTc-[Lβ]3), a 99mTc-labeled trivalent RGD probe. 99mTc-[Lβ]3 exhibited higher binding affinity to integrin αvβ3 compared with its unlabeled monovalent ligand (Lβ), due to the multivalent effect.13–15 This superior target binding affinity of 99mTc-[Lβ]3 enabled its high in vivo tumor uptake even in the presence of excess unlabeled ligands (Fig. 1b). We also demonstrated the applicability of this design concept to 99mTc-labeled bivalent probes16 and 67Ga-labeled trivalent ones17 using D-penicillamine and Schiff base as a chelating molecule, respectively.
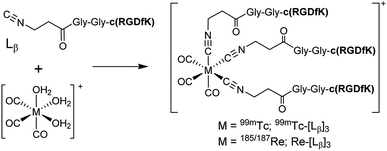 |
| | Fig. 2 Reaction scheme of Lβ and [99mTc][TcI(CO)3(OH2)3]+. A 99mTc-labeled trivalent RGD probe [99mTc][TcI(CO)3(Lβ)3]+ (99mTc-[Lβ]3) is generated from a reaction between [99mTc][TcI(CO)3(OH2)3]+ and Lβ in the molar ratio of 1 to 3. | |
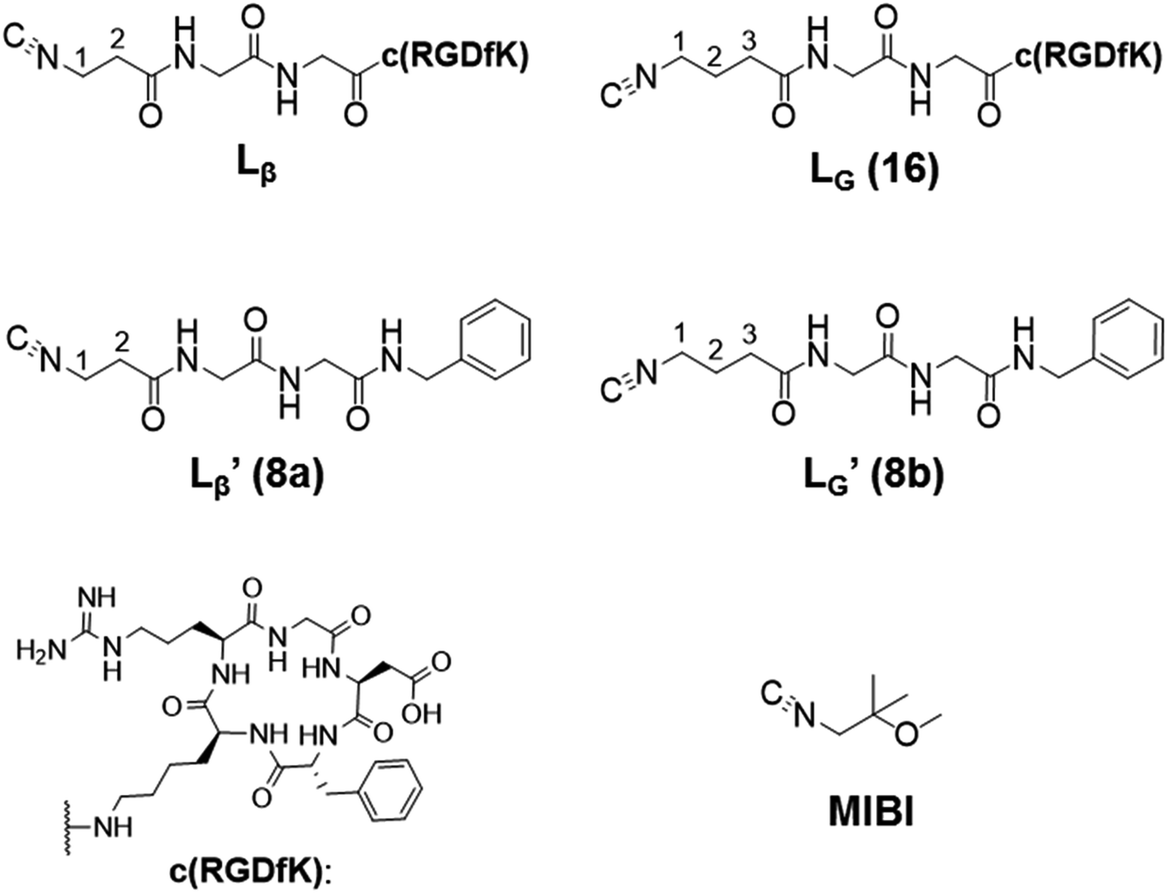
Our design concept would not be restricted to bivalent or trivalent probes but could be applied to other valent probes such as tetravalent or hexavalent ones. For example, it was reported that the reaction of methoxy-isobutyl-isonitrile (Fig. 3, MIBI) with [99mTc][TcI(CO)3(OH2)3]+ at basic conditions generated [99mTc][TcI(CO)2(MIBI)4]+, where one CO molecule in [99mTc][TcI(CO)3(MIBI)3]+ was further substituted by another MIBI molecule.18 This result indicates that the preparation of 99mTc-labeled tetravalent RGD probes would be possible by employing appropriate isonitrile ligands and reaction conditions. However, it is still unknown if any isonitrile ligands could provide 99mTc-labeled tetravalent probes in high radiochemical yields. The aim of this study was thereby to demonstrate whether our design concept of metal coordination mediated multivalency could be extended to 99mTc-labeled tetravalent probes. For this purpose, here we examined the influence of the structure of isonitrile ligands on the formation of their corresponding 99mTc-labeled tetravalent probes.
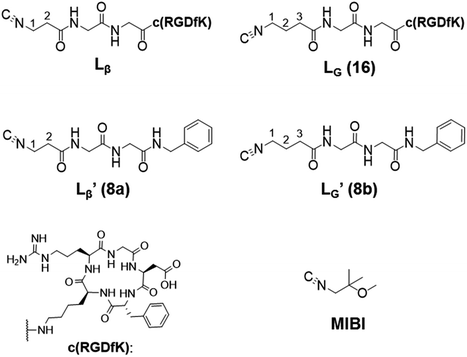 |
| | Fig. 3 The chemical structures of Lβ, LG, 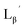 , , 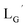 , c(RGDfK), and MIBI. , c(RGDfK), and MIBI. | |
Results and discussion
99mTc-labeling reaction of Lβ and 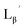
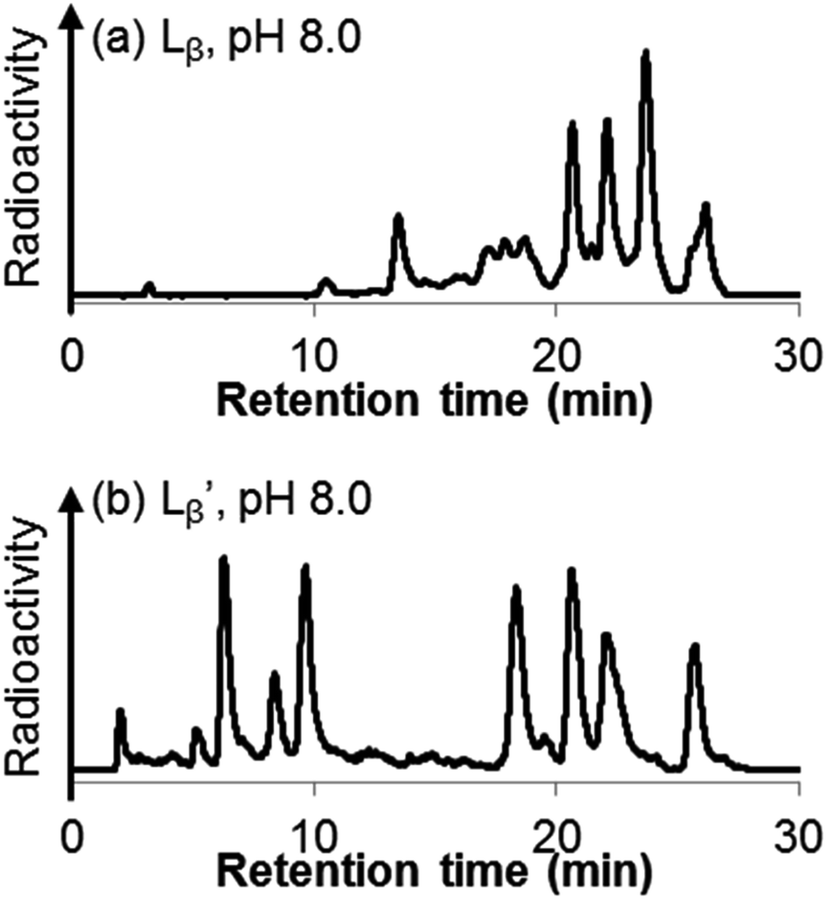
Following the previous report,18 we at first attempted synthesizing [99mTc][TcI(CO)2(Lβ)4]+ by reacting [99mTc][TcI(CO)3(OH2)3]+ with Lβ at pH 8.0. Contrary to our expectation, however, multiple side products were generated at this slightly basic reaction condition (Fig. 4a). Since the formation of multiple side products was not observed in the previous report18 where MIBI was used as a coordinating ligand, we speculated that the presence of RGD peptide might have caused some undesirable side reactions. To verify this speculation, we reacted [99mTc][TcI(CO)3(OH2)3]+ at pH 8.0 with CN-βAla-Gly-Gly-NH-Bn (Fig. 3, 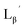 ), where c(RGDfK) in Lβ was replaced with benzylamine. However, as shown in Fig. 4b, the formation of multiple side products was again observed, suggesting that the side reaction observed here was not caused by the presence of RGD peptides but by other moieties in Lβ.
), where c(RGDfK) in Lβ was replaced with benzylamine. However, as shown in Fig. 4b, the formation of multiple side products was again observed, suggesting that the side reaction observed here was not caused by the presence of RGD peptides but by other moieties in Lβ.
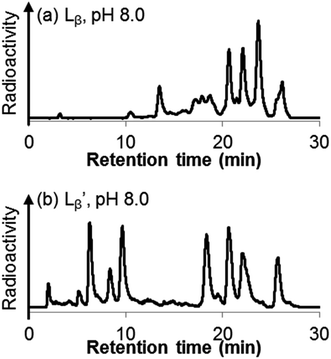 |
| | Fig. 4 HPLC analyses of 99mTc-labeling reactions under the conditions of (a) [Lβ] = 150 μM, 110 °C for 20 min at pH 8.0 (system 11) and (b) 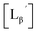 = 1 mM, 100 °C for 30 min at pH 8.0 (system 6). = 1 mM, 100 °C for 30 min at pH 8.0 (system 6). | |
Stabilities of 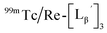 in acidic and basic solutions
in acidic and basic solutions
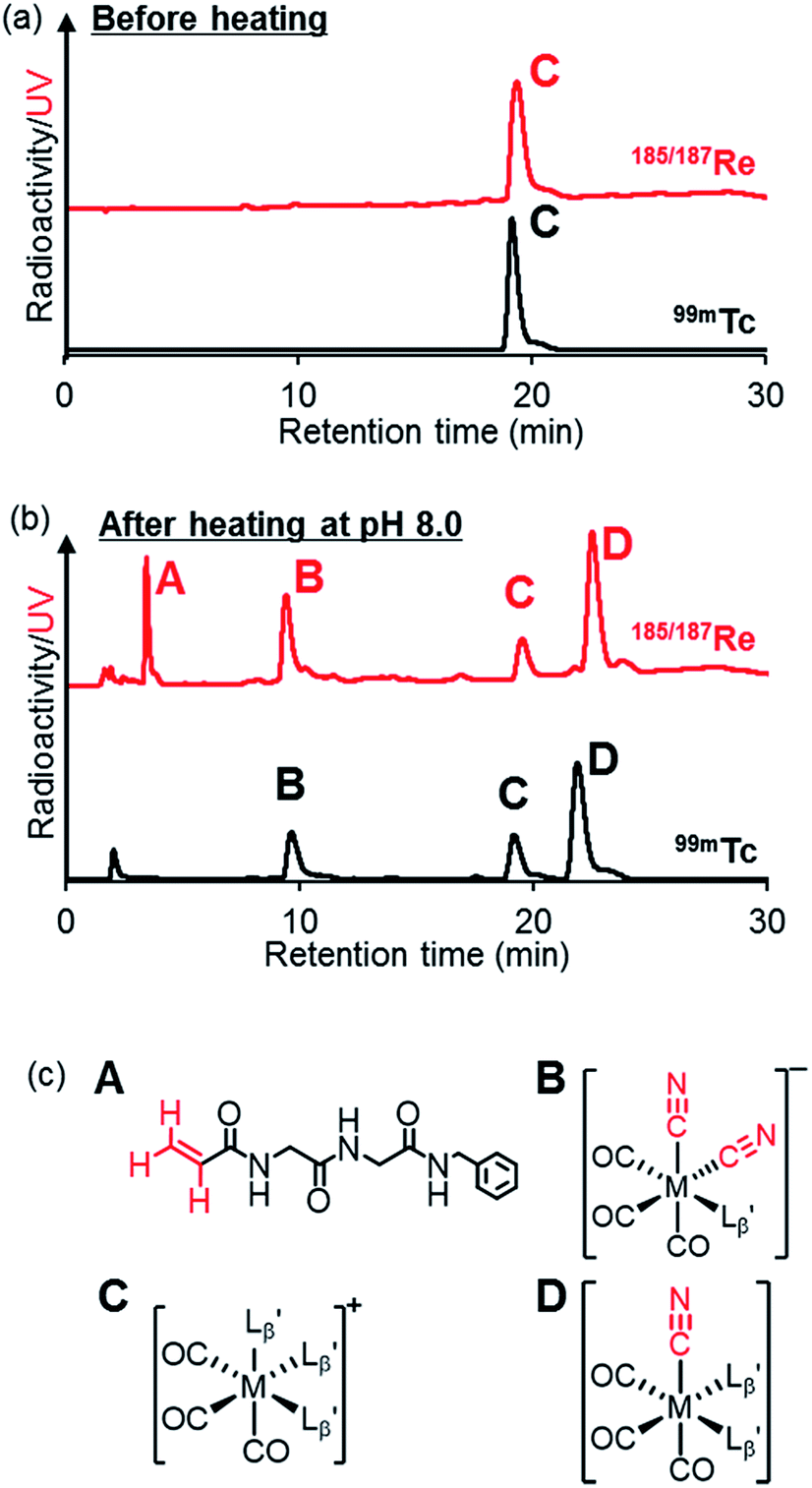
To gain an insight into the mechanism of the side reaction, we evaluated the stabilities of 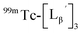 and
and 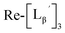 at pH 8.0 (Fig. 5). Since technetium and rhenium share similar coordination chemistry due to their group homology, we employed non-radioactive rhenium analogues to characterize their tracer 99mTc counterparts.
at pH 8.0 (Fig. 5). Since technetium and rhenium share similar coordination chemistry due to their group homology, we employed non-radioactive rhenium analogues to characterize their tracer 99mTc counterparts. 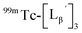 was prepared from a labeling reaction at pH 6.0 and was purified by HPLC.
was prepared from a labeling reaction at pH 6.0 and was purified by HPLC. 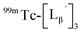 was then dissolved in aqueous solution at pH 8.0 containing
was then dissolved in aqueous solution at pH 8.0 containing 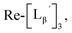 and the solution was heated at 100 °C for 30 min. As shown in Fig. 5b,
and the solution was heated at 100 °C for 30 min. As shown in Fig. 5b, 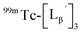 and
and 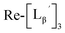 exhibited a similar decomposition pattern upon heating at pH 8.0, whereas they remained intact at pH 6.0 (Fig. S1†). These results indicate that the decomposition of
exhibited a similar decomposition pattern upon heating at pH 8.0, whereas they remained intact at pH 6.0 (Fig. S1†). These results indicate that the decomposition of 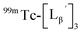 under basic conditions primarily accounts for the generation of the multiple peaks observed in Fig. 4b.
under basic conditions primarily accounts for the generation of the multiple peaks observed in Fig. 4b.
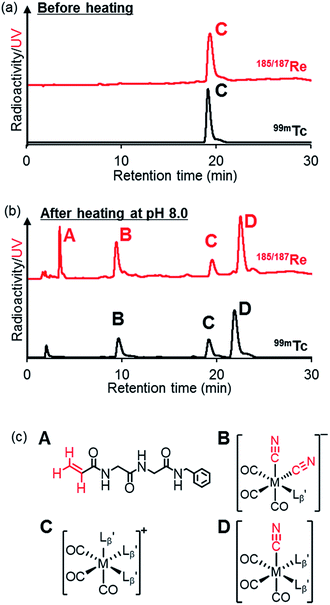 |
| | Fig. 5 The stability of 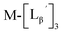 (M = 99mTc and 185/187Re) in basic solution. HPLC analyses (system 6) of (M = 99mTc and 185/187Re) in basic solution. HPLC analyses (system 6) of 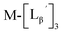 (a) before heating and (b) after heating at 100 °C for 30 min at pH 8.0. (c) The chemical structures of the products generated upon heating and the intact complex (a) before heating and (b) after heating at 100 °C for 30 min at pH 8.0. (c) The chemical structures of the products generated upon heating and the intact complex 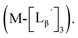 | |
We isolated compounds A, B and D (Fig. 5b) and determined their chemical structures by 1H NMR, MS, and/or IR (Fig. 5c). The NMR spectrum of compound A showed coupling constants unique to geminal (2JHH = 2 Hz) and vicinal (3JHH = 10 and 17 Hz) couplings (Fig. S8b†), which proved the presence of an alkene structure. The IR spectrum of compound A (Fig. S8c†) also verified the presence of alkene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, ν = 1647.88 cm−1) and excluded the presence of isonitrile, whose IR peak should appear at around 2150 cm−1. On the other hand, compounds B and D showed MS spectra (Fig. S7a and S6a,† respectively) unique to rhenium isotopic patterns (the ratio of 185Re/187Re = 37/63). These results along with their m/z values indicated that compounds B and D had structures of
C, ν = 1647.88 cm−1) and excluded the presence of isonitrile, whose IR peak should appear at around 2150 cm−1. On the other hand, compounds B and D showed MS spectra (Fig. S7a and S6a,† respectively) unique to rhenium isotopic patterns (the ratio of 185Re/187Re = 37/63). These results along with their m/z values indicated that compounds B and D had structures of 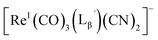 and
and 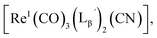 respectively (Fig. 5c).
respectively (Fig. 5c).
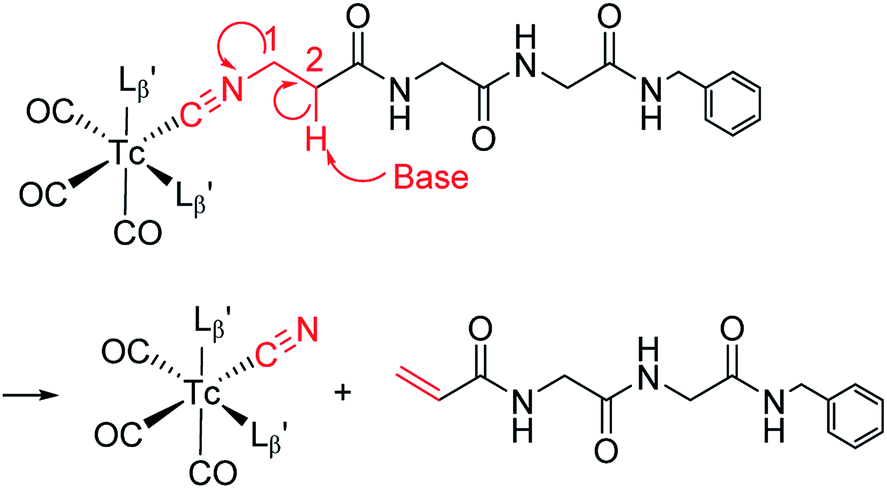
The mechanism of the side reaction
The decomposition reaction of 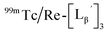 has the following three characteristics: (1) one of the coordinated isonitrile ligands is eliminated from the Tc/Re-complex with leaving an isonitrile ion on the Tc/Re-complex, (2) the eliminated ligand loses the isonitrile group, and its terminal structure is converted to an alkene structure, (3) this reaction proceeds only at high hydroxide ion concentration. These features possess the characteristics of the “bimolecular elimination reaction”.19 We thereby hypothesized that the side reaction observed at basic conditions (Fig. 4) would also proceed via the following three simultaneous events: (1) the abstraction of the 2-proton in
has the following three characteristics: (1) one of the coordinated isonitrile ligands is eliminated from the Tc/Re-complex with leaving an isonitrile ion on the Tc/Re-complex, (2) the eliminated ligand loses the isonitrile group, and its terminal structure is converted to an alkene structure, (3) this reaction proceeds only at high hydroxide ion concentration. These features possess the characteristics of the “bimolecular elimination reaction”.19 We thereby hypothesized that the side reaction observed at basic conditions (Fig. 4) would also proceed via the following three simultaneous events: (1) the abstraction of the 2-proton in 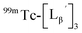 by a hydroxide ion, (2) the departure of
by a hydroxide ion, (2) the departure of 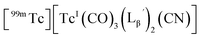 from
from 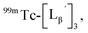 and (3) the formation of an alkene structure (Fig. 6).
and (3) the formation of an alkene structure (Fig. 6).
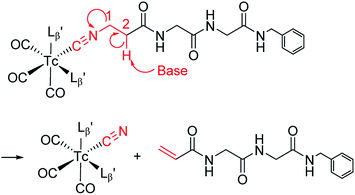 |
| | Fig. 6 A proposed reaction mechanism of the side reaction proceeding at basic conditions. | |
Design and synthesis of 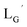 and 99mTc-labeling of
and 99mTc-labeling of 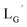
According to the hypothesis proposed above, the high acidity of the 2-proton in 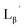 (Fig. 6) would be essential for the side reaction to proceed. In addition, this high acidity of the 2-proton can be partly attributed to the electron withdrawing effect from the neighboring carbonyl group. We thereby newly designed a γ-aminobutyric acid (GABA) based isonitrile ligand (
(Fig. 6) would be essential for the side reaction to proceed. In addition, this high acidity of the 2-proton can be partly attributed to the electron withdrawing effect from the neighboring carbonyl group. We thereby newly designed a γ-aminobutyric acid (GABA) based isonitrile ligand (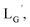 Fig. 3). As the 2-proton in
Fig. 3). As the 2-proton in 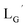 is no longer present next to the carbonyl group (Fig. 3), the 2-proton in
is no longer present next to the carbonyl group (Fig. 3), the 2-proton in 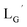 would possess much lower acidity.
would possess much lower acidity.
99mTc-labeling reactions of 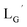 were performed in a procedure similar to that of
were performed in a procedure similar to that of 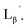 [99mTc][TcI(CO)3(OH2)3]+ was prepared as a labeling precursor, mixed with
[99mTc][TcI(CO)3(OH2)3]+ was prepared as a labeling precursor, mixed with 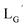 and heated at 100 °C for 30 min at pH 8.0. The HPLC analysis of this reaction solution is presented in Fig. 7c, which shows a completely different chromatographic profile from that in Fig. 4b. As shown in Fig. 7c, the 99mTc-labeling reaction of
and heated at 100 °C for 30 min at pH 8.0. The HPLC analysis of this reaction solution is presented in Fig. 7c, which shows a completely different chromatographic profile from that in Fig. 4b. As shown in Fig. 7c, the 99mTc-labeling reaction of 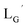 at pH 8.0 generated only two peaks, and the retention time of the major radioactivity peak was similar to that of
at pH 8.0 generated only two peaks, and the retention time of the major radioactivity peak was similar to that of 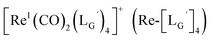 characterized by ESI-MS and IR (Fig. 7b and c). These results supported the validity of our working hypothesis that the abstraction of the 2-proton by a base is the key step for the side reaction. We also identified the minor radioactivity peak as
characterized by ESI-MS and IR (Fig. 7b and c). These results supported the validity of our working hypothesis that the abstraction of the 2-proton by a base is the key step for the side reaction. We also identified the minor radioactivity peak as 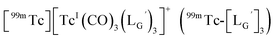 based on its similar HPLC retention time to that of
based on its similar HPLC retention time to that of 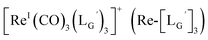 (Fig. 7a and c).
(Fig. 7a and c).
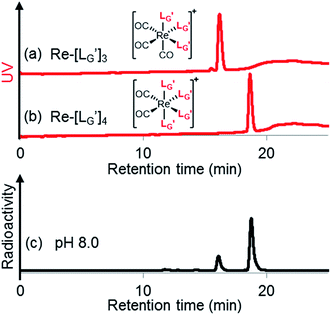 |
| | Fig. 7 HPLC analyses (system 7) of (a) 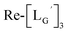 and (b) and (b) 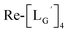 . (c) An HPLC analysis (system 7) of a 99mTc-labeling reaction of . (c) An HPLC analysis (system 7) of a 99mTc-labeling reaction of  under the condition of under the condition of  = 1 mM, 100 °C for 30 min at pH 8.0. = 1 mM, 100 °C for 30 min at pH 8.0. | |
A GABA-based isonitrile ligand with c(RGDfK) peptides
Based on the above findings, we subsequently designed and synthesized a GABA-based isonitrile ligand containing c(RGDfK) peptides (LG, Fig. 3). The synthesis of LG was accomplished by a condensation reaction of CN-GABA-TFP (7b) and H2N-Gly-Gly-c(RGDfK) (15), followed by HPLC purification (Scheme S1†). Its corresponding rhenium complex, [ReI(CO)2(LG)4]+ (Re-[LG]4), was also synthesized as a surrogate compound for its 99mTc counterpart.
As a result of preliminary evaluations on its 99mTc-labelling reactions, we established a reaction condition which reproducibly provides [99mTc][TcI(CO)2(LG)4]+ (99mTc-[LG]4) in over 90% radiochemical yields (Fig. 8). The ligand concentration required to provide 99mTc-[LG]4 (200 μM) was lower than that needed for 99mTc-[Lβ]3 (350 μM), which is advantageous to mitigate the competitive inhibition by unlabeled ligand in vivo.9,20,21 Since isonitrile is more stable in basic solutions than in acidic ones,22 the result that 99mTc-[LG]4 was prepared under lower ligand concentration could be attributed to the improved stability of the isonitrile moiety in LG during the labeling reactions.
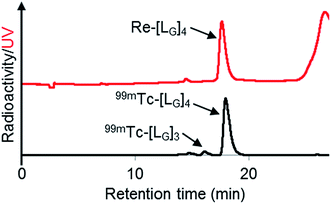 |
| | Fig. 8 HPLC analyses (system 9) of Re-[LG]4 and a 99mTc-labeling reaction performed under the condition of [LG] = 200 μM, 85 °C for 1 h, and 37.5 mM P.B. (pH 8.0). | |
The stability of 99mTc-[LG]4 in an excess histidine solution and murine plasma was evaluated by TLC and HPLC. HPLC analyses were performed after removing plasma protein from the samples. The recovery rates of the radioactivity from plasma samples to supernatant solutions were 96.8 ± 0.4% (n = 3) upon addition of ethanol and protein precipitation. No decomposition of 99mTc-[LG]4 was observed at all even after 6 h incubation in both solutions (Table S1 and Fig. S2†). These results indicate that 99mTc-[LG]4 possesses stability sufficient for in vivo applications.
In vitro binding affinities of RGD derivatives
To examine the differences in integrin αvβ3 binding affinities between monovalent and tetravalent RGD peptides, we calculated the IC50 values of LG and Re-[LG]4 in a competitive binding experiment using [125I]I-c(RGDyV) and U87MG cell as a competed radioligand and an integrin αvβ3 positive cell line, respectively. We included c(RGDyV) into the study as an internal standard. As shown in Table 1 and Fig. S3†, Re-[LG]4 showed much lower IC50 value than those of LG and c(RGDyV), indicating that the metal complex has gained enhanced binding affinity due to the multivalent effect. This superior binding affinity is essential for 99mTc-[LG]4 to achieve high in vivo target uptake when injected into subjects without post-labeling purification.
Table 1 The IC50 values of each compound
| Compound |
IC50, nM |
95% C.I.a |
| C.I. = confidential interval. |
| LG |
107 |
90.2−126 |
| Re-[LG]4 |
2.59 |
1.90−3.54 |
| c(RGDyV) |
34.8 |
27.5−44.0 |
In vivo study of 99mTc-[LG]4
We evaluated the biodistribution of 99mTc-[LG]4 in U87MG-bearing nude mice at 1 h post injection, and these results are summarized in Table 2. There were three different samples tested in this study: (1) HPLC-purified 99mTc-[LG]4 (without the unlabeled ligand (LG)), (2) unpurified 99mTc-[LG]4 (containing 5 nmol of LG) and (3) HPLC-purified 99mTc-[LG]4 with 5 nmol of Re-[LG]4. The second sample represent our chemical design where a 99mTc-labeled multivalent RGD probe prepared from a monovalent ligand is injected into subjects without postlabeling purification (Fig. 1b), whereas the third sample imitates a conventional design where a 99mTc-labeled tetravalent RGD probe prepared from a ligand already possessing tetravalent RGD is injected into subjects without postlabeling purification (Fig. 1a). Though the tumor uptake of 99mTc-[LG]4 was slightly impaired by the presence of 5 nmol of LG in the injectate (5.00 ± 0.84% ID g−1 vs. 3.08 ± 0.43% ID g−1), its tumor uptake value was significantly higher than that of the conventional design (3.08 ± 0.43% ID g−1 vs. 1.81 ± 0.14% ID g−1).
Table 2 Biodistribution at 1 h postinjection in U87MG-bearing micea
| Tissue |
Purified 99mTc-[LG]4 (LG = 0 nmol) |
Un-purified 99mTc-[LG]4 (LG = 5 nmol) |
Purified 99mTc-[LG]4 + Re-[LG]4 (Re-[LG]4 = 5 nmol) |
| The results were expressed as percent injected dose per gram (% ID g−1) ± SD (n = 5 or 6). Statistical analysis was performed using one-way analysis of variance followed by Tukey's multiple-comparison test (* different from “un-purified 99mTc-[LG]4 (LG = 5 nmol)”, p < 0.01). |
| Blood |
0.26 ± 0.04 |
0.58 ± 0.39 |
0.21 ± 0.04 |
| Liver |
1.85 ± 0.23 |
1.29 ± 0.14 |
0.95 ± 0.14 |
| Spleen |
2.16 ± 0.31 |
1.29 ± 0.12 |
0.95 ± 0.09 |
| Kidney |
13.05 ± 0.87 |
13.12 ± 1.87 |
12.28 ± 1.14 |
| Pancreas |
0.93 ± 0.07 |
0.46 ± 0.06 |
0.27 ± 0.05 |
| Heart |
1.10 ± 0.13 |
0.52 ± 0.05 |
0.36 ± 0.07 |
| Lung |
2.25 ± 0.47 |
1.60 ± 0.11 |
0.84 ± 0.25 |
| Stomach |
4.57 ± 2.43 |
4.21 ± 2.01 |
2.36 ± 0.83 |
| Intestine |
4.01 ± 0.89 |
3.34 ± 1.06 |
1.99 ± 0.65 |
| Muscle |
0.74 ± 0.17 |
0.44 ± 0.12 |
0.26 ± 0.23 |
| Tumorb |
5.00 ± 0.84* |
3.08 ± 0.43 |
1.81 ± 0.14* |
| T/Blood |
19.0 ± 2.31 |
6.15 ± 3.03 |
8.81 ± 1.48 |
| T/Liver |
2.72 ± 0.49 |
2.35 ± 0.40 |
1.93 ± 0.22 |
| T/Muscle |
7.10 ± 2.04 |
7.19 ± 2.12 |
9.60 ± 4.11 |
The tumor to blood ratio of un-purified 99mTc-[LG]4 was not higher than that of purified 99mTc-[LG]4 + Re-[LG]4. This is attributed to the higher blood radioactivity level of unpurified 99mTc-[LG]4 (0.58 ± 0.39% ID g−1) than that of purified 99mTc-[LG]4 + Re-[LG]4 (0.21 ± 0.04% ID g−1). Though further study would be needed to elucidate the cause of the higher blood radioactivity level, we are considering the possibility that the difference in the normal tissue uptakes (Table 2) affected the blood clearance of 99mTc-[LG]4, especially in the elimination phase. Because blood clearance in the elimination phase can be affected by redistribution from normal tissues, the higher normal tissue uptakes could cause higher blood radioactivity level in the elimination phase.
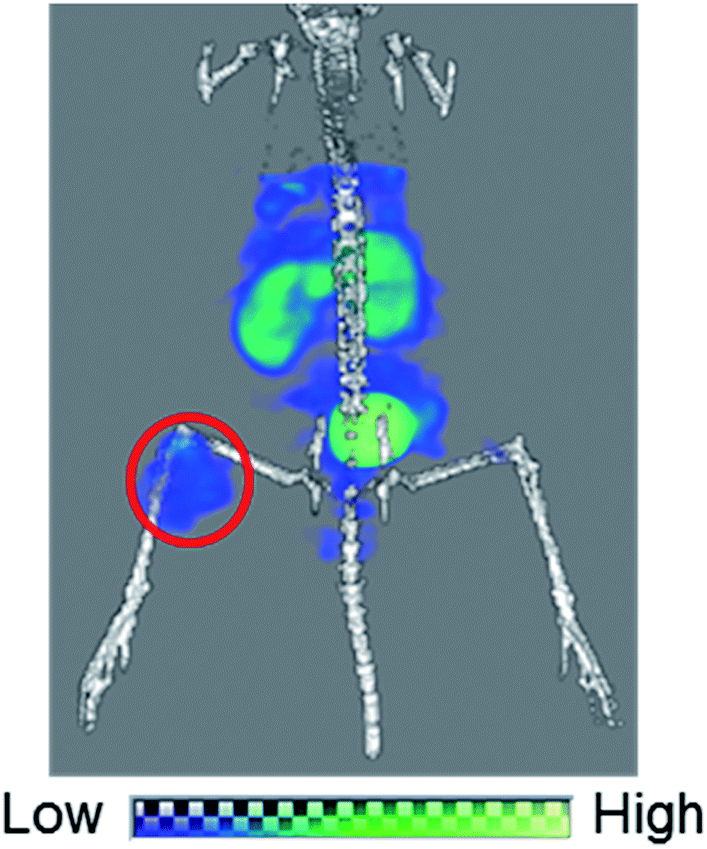
Lastly, we performed a SPECT/CT imaging study using unpurified 99mTc-[LG]4 containing 5 nmol of LG. A representative SPECT/CT image is shown in Fig. 9, which clearly visualized U87MG tumor despite the presence of excess unlabeled ligand in the injectate, owing to the high tumor uptake of 99mTc-[LG]4. These results again demonstrated the utility of our design concept for achieving high in vivo target uptake of 99mTc-labeled probes without tedious post-labeling purification.
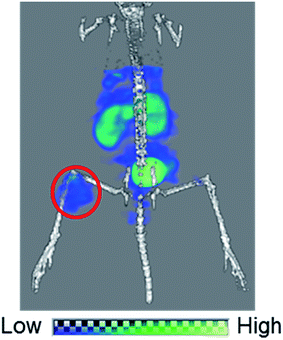 |
| | Fig. 9 A SPECT/CT image of a U87MG tumor-bearing male nude mouse over 30–94 min post i.v. injection of 3.7 MBq of 99mTc-[LG]4 containing 5 nmol of LG. Red circle indicates the U87MG tumor. | |
Conclusion
In this study, we have found a way to prepare a 99mTc-labeled tetravalent RGD probe upon isonitrile ligands coordination to 99mTcI in the molar ratio of 4 to 1. Our initial attempts reacting a βAla-based isonitrile ligand (Lβ) with [99mTc][Tc(CO)3]+ core at pH 8.0 failed to provide [99mTc][Tc(CO)2(Lβ)4]+ due to the bimolecular elimination reaction. GABA-based isonitrile ligands (LG and 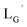 ), on the other hand, avoided the side reaction and provided 99mTc-labeled tetravalent probes from reactions at basic conditions. In the SPECT/CT study, 99mTc-[LG]4 clearly visualized integrin αvβ3 positive tumor without tedious post-labeling purification to remove its excess unlabeled ligand. These results clearly indicate that the metal coordination-mediated synthesis of 99mTc-labeled bivalent and trivalent probes has been successfully extended to that of tetravalent ones, which would lead to a wider range of choices for designing novel 99mTc-labeled multivalent probes of high in vivo target uptake. In addition, because 99mTc-hexakis-isonitriles such as [99mTc][Tc(MIBI)6]+ also possess very high in vivo stability, we are currently working to apply our metal coordination mediated multivalency concept to hexavalent ones, and are going to report the results in the near future elsewhere.
), on the other hand, avoided the side reaction and provided 99mTc-labeled tetravalent probes from reactions at basic conditions. In the SPECT/CT study, 99mTc-[LG]4 clearly visualized integrin αvβ3 positive tumor without tedious post-labeling purification to remove its excess unlabeled ligand. These results clearly indicate that the metal coordination-mediated synthesis of 99mTc-labeled bivalent and trivalent probes has been successfully extended to that of tetravalent ones, which would lead to a wider range of choices for designing novel 99mTc-labeled multivalent probes of high in vivo target uptake. In addition, because 99mTc-hexakis-isonitriles such as [99mTc][Tc(MIBI)6]+ also possess very high in vivo stability, we are currently working to apply our metal coordination mediated multivalency concept to hexavalent ones, and are going to report the results in the near future elsewhere.
Experimental section
General
[185/187Re(CO)3(OH2)3]Br (19),23 Boc-Gly-Gly-OH (1),10 CN-βAla-TFP (7a),10 CN-βAla-Gly-Gly-c(RGDfK) (Lβ)10 and H2N-Gly-Gly-c(RGDfK)·CF3CO2H (15)10 were synthesized according to the previous reports. Technetium-99m as Na99mTcO4 was eluted in saline solution on daily basis from a 99Mo/99mTc generator (FUJIFILM RI Pharma Co., Ltd., Tokyo, Japan). All other reagents were purchased from Wako Pure Chemical Industries, Ltd. (Tokyo, Japan) and were used without further purification. Mass spectrometry was carried out using a JMS-T100LP (JEOL Ltd., Tokyo, Japan) or an Agilent 6130 Series Quadrupole LC/MS spectrometer (Agilent technologies, Tokyo). 1H NMR/13C NMR spectra was recorded on a JEOL ECS-400 (400 MHz/100 MHz, respectively) spectrometer (JEOL Ltd, Tokyo). Infrared (IR) spectra were recorded on a FT/IR-4700 (Jasco Co., Tokyo, Japan).
Boc-Gly-Gly-NH-Bn (2)
Boc-Gly-Gly-OH (1) (180 mg, 0.775 mmol) and benzylamine (77 μL, 0.705 mmol) were dissolved in dichloromethane (4 mL). To this stirred mixture was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC HCl, 270 mg, 1.41 mmol), and the mixture was stirred at room temperature for 1 h. The solvent was removed in vacuo and the residue was dissolved in chloroform (15 mL) and washed with 5% citric acid (10 mL) and 5% NaHCO3 (10 mL). The organic layer was then dried over magnesium sulfate. After removing magnesium sulfate by filter, the organic solvent was removed in vacuo to afford the compound 2 as a white solid (189 mg, 76%). 1H NMR (400 MHz, DMSO-d6): δ 1.37 (s, 9H, tBu), 3.57 (d, 2H, HαGly), 3.74 (d, 2H, HαGly), 4.29 (d, 2H, CH2-phenyl), 7.05 (t, 1H, NH), 7.21–7.33 (m, 5H, Harom), 8.11 (t, 1H, NH), 8.28 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 28.16 (C(CH3)3), 41.96 (CαGly), 42.07 (CH2-phenyl), 43.42 (CαGly), 78.16 (C(CH3)3), 126.73 (Carom), 127.10 (Carom), 128.22 (Carom), 139.24 (Carom), 155.91 (CO), 168.79 (CO), 169.74 (CO). ESI-MS, m/z: 322.18 [M + H]+, found 322.24.
H2N-Gly-Gly-NH-Bn·CF3CO2H (3)
The compound 2 (104 mg, 0.324 mmol) was dissolved in trifluoroacetic acid (TFA, 2.5 mL) and stirred at room temperature for 1 h. After removing TFA in vacuo, the compound 3 was successively used in the following condensation reactions without further purification.
4-Formylaminobutanoic acid (5b)
4-Aminobutanoic acid (2.00 g, 19.4 mmol) was dissolved in formic acid/acetic anhydrate (20 mL/15 mL) and stirred at 95 °C under N2 atmosphere for 30 min. After the solvent was evaporated in vacuo, the residue was washed with diethyl ether to afford the compound 5b as a white solid (1.00 g, 39%). 1H NMR (400 MHz, DMSO-d6): δ 1.62 (m, 2H, CH2CH2CH2), 2.22 (t, 2H, CH2CO), 3.08 (q, 2H, NHCH2), 7.99 (s, 1H, formylamino), 8.02 (s, 1H, NH). ESI-MS, m/z: 130.05 [M − H]−, found 130.09.
2,3,5,6-Tetrafluorophenyl 4-formylaminobutanoate (6b)
The compound 5b (525 mg, 4.00 mmol) and 2,3,5,6-tetrafluoro phenol (TFP, 797 mg, 4.80 mmol) were dissolved in chloroform (8 mL). To this stirred mixture was added EDC HCl (1.53 g, 8.00 mmol), and the mixture was stirred at room temperature for 2 hours. The solvent was removed in vacuo and purified with silica gel column chromatography (hexane/ethyl acetate = 1/1) to afford the compound 6b as a white powder (929 mg, 83%). 1H NMR (400 MHz, DMSO-d6): δ 1.81 (m, 2H, CH2CH2CH2), 2.81 (t, 2H, CH2CO), 3.18 (q, 2H, NHCH2), 7.95 (m, 1H, Harom), 8.03 (s, 1H, formylamino), 8.11 (s, 1H, NH). ESI-MS, m/z: 302.04 [M + Na]+, found 301.97.
2,3,5,6-Tetrafluorophenyl 4-isocyanobutanoate (CN-GABA-TFP) (7b)
The compound 6b (717 mg, 2.57 mmol) and Burgess reagent24 (918 mg, 3.85 mmol) were dissolved in dichloromethane (24 mL) and stirred at 50 °C for 30 minutes. After removing the solvent in vacuo, the residue was purified with silica gel chromatography (hexane/ethyl acetate = 10/1) to afford the compound 7b as a dark yellow oil (404 mg, 60%). 1H NMR (400 MHz, CDCl3): δ 2.16 (br, 2H, CH2CH2CH2), 2.91 (t, 2H, CH2CO), 3.59 (m, 2H, NCH2), 7.02 (m, 1H, Harom). IR (ATR, ν/cm−1): 2149.28 (s, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).
N).
CN-βAla-Gly-Gly-NH-Bn 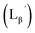 (8a)
(8a)
CN-βAla-TFP (7a) was synthesized as previously reported.10 To a solution of the compound 7a (120 mg, 0.486 mmol) and H2N-Gly-Gly-NH-Bn·CF3CO2H (3) (108.6 mg, 0.324 mmol) in N,N-dimethylformamide (DMF, 2 mL) was added NaHCO3 (40.8 mg, 0.486 mmol). The mixture was stirred at room temperature for 1 h. After removing NaHCO3 by filtration, the solvent was removed in vacuo and the crude compound was purified with silica gel column chromatography (chloroform/methanol = 20/1) to afford the compound 8a as a white solid (70 mg, 72%). 1H NMR (400 MHz, DMSO-d6): δ 2.56 (br, 2H, CH2CH2CO), 3.64 (br, 2H, CH2CH2CO), 3.74 (d, 2H, HαGly), 3.77 (d, 2H, HαGly), 4.29 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, phenyl), 8.23 (t, 1H, NH), 8.29 (t, 1H, NH), 8.37 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 34.47 (CH2CH2CO), 37.47 (t, CH2CH2CO), 41.97 (CαGly), 42.07 (CαGly), 42.23 (CH2-phenyl), 126.76 (Carom), 127.17 (Carom), 128.25 (Carom), 139.32 (Carom), 155.73 (t, CN), 168.78 (CO), 169.15 (2C, CO). ESI-MS, m/z: 325.13 [M + Na]+, found 325.19. IR (ATR, ν/cm−1): 2150.24 (s, CN).
CN-GABA-Gly-Gly-NH-Bn 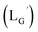 (8b)
(8b)
To a solution of CN-GABA-TFP (7b) (106.8 mg, 0.409 mmol) and H2N-Gly-Gly-NH-Bn·CF3CO2H (3) (100.2 mg, 0.299 mmol) in DMF (2 mL) was added NaHCO3 (75.2 mg, 0.895 mmol). The mixture was stirred at room temperature for 6 h. After removing NaHCO3 by filtration, the solvent was removed in vacuo and the crude product was purified with silica gel column chromatography (chloroform/methanol = 20/1) to afford the compound 8b as a white solid (60 mg, 63%). 1H NMR (400 MHz, DMSO-d6): δ 1.79 (m, 2H, CH2CH2CH2), 2.27 (t, 2H, CH2CH2CH2), 3.50 (m. 2H, CNCH2), 3.72 (d, 2H, HαGly), 3.74 (d, 2H, HαGly), 4.28 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, Harom), 8.23 (t, 1H, NH), 8.28–8.32 (m, 2H, NH). ESI-MS, m/z: 339.14 [M + Na]+, found 339.16. IR (ATR, ν/cm−1): 2147.35 (s, CN).
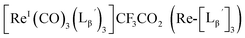 (9)
(9)
[ReI(CO)3(OH2)3]Br (19) (9.7 mg, 0.024 mmol) and 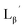 (8a) (65 mg, 0.215 mmol) were dissolved in 0.1 M acetate buffer (pH 6.0, 21.5 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 1 and lyophilized to afford the compound 9 as a white powder (30 mg, 97%). 1H NMR (400 MHz, DMSO-d6): δ 2.70 (t, 2H, CH2CH2CO), 3.76 (d, 2H, HαGly), 3.81 (d, 2H, HαGly), 4.09 (t, 2H, CH2CH2CO), 4.28 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, Harom), 8.27 (t, 1H, NH), 8.36 (t, 1H, NH), 8.43 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 33.89 (CH2CH2CO), 41.06 (CH2CH2CO), 42.02, 42.08, (CαGly, CH2-phenyl), 126.79 (Carom), 127.18 (Carom), 128.26 (Carom), 139.31 (Carom), 158.24 (q, CN), 168.80 (CO), 168.96 (CO), 169.10 (CO), 182.71 (ReCO). ESI-MS, m/z: 1177.35 [M]+, found 1177.56. IR (ATR, ν/cm−1): 2244.74 (w, CN), 2212.92 (m, CN), 2057.67, 1990.18 (s, CO).
(8a) (65 mg, 0.215 mmol) were dissolved in 0.1 M acetate buffer (pH 6.0, 21.5 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 1 and lyophilized to afford the compound 9 as a white powder (30 mg, 97%). 1H NMR (400 MHz, DMSO-d6): δ 2.70 (t, 2H, CH2CH2CO), 3.76 (d, 2H, HαGly), 3.81 (d, 2H, HαGly), 4.09 (t, 2H, CH2CH2CO), 4.28 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, Harom), 8.27 (t, 1H, NH), 8.36 (t, 1H, NH), 8.43 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 33.89 (CH2CH2CO), 41.06 (CH2CH2CO), 42.02, 42.08, (CαGly, CH2-phenyl), 126.79 (Carom), 127.18 (Carom), 128.26 (Carom), 139.31 (Carom), 158.24 (q, CN), 168.80 (CO), 168.96 (CO), 169.10 (CO), 182.71 (ReCO). ESI-MS, m/z: 1177.35 [M]+, found 1177.56. IR (ATR, ν/cm−1): 2244.74 (w, CN), 2212.92 (m, CN), 2057.67, 1990.18 (s, CO).
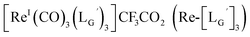 (10)
(10)
[ReI(CO)3(OH2)3]Br (19) (2.6 mg, 6.4 μmol) and 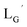 (8b) (20 mg, 63.2 μmol) were dissolved in 0.1 M phosphate buffer (P.B., pH 7.0, 6.32 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 3 and lyophilized to afford the compound 10 as a white powder (8.9 mg, quant.). 1H NMR (400 MHz, DMSO-d6): δ 1.93 (m, 2H, CH2CH2CH2), 2.29 (t, 2H, CH2CH2CH2), 3.75 (d, 4H, HαGly), 3.98 (t, 2H, CNCH2), 4.28 (d, 2H, CH2-phenyl), 7.10–7.33 (m, 5H, Harom), 8.23 (t, 1H, NH), 8.29 (t, 1H, NH), 8.34 (t, 1H, NH). ESI-MS, m/z: 1219.40 [M]+, found 1219.47. IR (ATR, ν/cm−1): 2238.95 (w, CN), 2208.09 (m, CN), 2053.82, 1981.50 (s, CO).
(8b) (20 mg, 63.2 μmol) were dissolved in 0.1 M phosphate buffer (P.B., pH 7.0, 6.32 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 3 and lyophilized to afford the compound 10 as a white powder (8.9 mg, quant.). 1H NMR (400 MHz, DMSO-d6): δ 1.93 (m, 2H, CH2CH2CH2), 2.29 (t, 2H, CH2CH2CH2), 3.75 (d, 4H, HαGly), 3.98 (t, 2H, CNCH2), 4.28 (d, 2H, CH2-phenyl), 7.10–7.33 (m, 5H, Harom), 8.23 (t, 1H, NH), 8.29 (t, 1H, NH), 8.34 (t, 1H, NH). ESI-MS, m/z: 1219.40 [M]+, found 1219.47. IR (ATR, ν/cm−1): 2238.95 (w, CN), 2208.09 (m, CN), 2053.82, 1981.50 (s, CO).
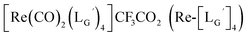 (11)
(11)
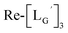 (10) (3.6 mg, 0.0027 mmol) and
(10) (3.6 mg, 0.0027 mmol) and 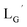 (8b) (4.7 mg, 0.0149 mmol) were dissolved in 0.1 M P.B. (pH 8.0, 1.47 mL). The mixture was heated at 100 °C for 6 h, after which time additional
(8b) (4.7 mg, 0.0149 mmol) were dissolved in 0.1 M P.B. (pH 8.0, 1.47 mL). The mixture was heated at 100 °C for 6 h, after which time additional 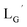 (8b) (4.7 mg, 0.0149 mmol) was added. After heating at 100 °C for another 6 h, the crude product was purified by preparative HPLC using system 3 and lyophilized to afford the compound 11 as a white powder (0.4 mg, 10%). ESI-MS, m/z: 1507.56 [M]+, found 1507.65. IR (ATR, ν/cm−1): 2228.34 (w, CN), 2160.85 (s, CN), 1996.93, 1953.54 (s, CO).
(8b) (4.7 mg, 0.0149 mmol) was added. After heating at 100 °C for another 6 h, the crude product was purified by preparative HPLC using system 3 and lyophilized to afford the compound 11 as a white powder (0.4 mg, 10%). ESI-MS, m/z: 1507.56 [M]+, found 1507.65. IR (ATR, ν/cm−1): 2228.34 (w, CN), 2160.85 (s, CN), 1996.93, 1953.54 (s, CO).
[99mTc][TcI(CO)3(OH2)3]+
[99mTc][TcI(CO)3(OH2)3]+ was prepared according to a reported method25 with slight modifications. Briefly, a kit containing 0.285 mg sodium tetraborate decahydrate (Na2B4O7·10H2O), 0.715 mg sodium carbonate (Na2CO3), 1 mg sodium(+)-tartrate dihydrate (C4H4Na2O6·2H2O), and 0.45 mg sodium boranocarbonate [Na2(H3BCO2)] was purged with N2 atmosphere before adding 300 μL [99mTc]TcO4− (∼333 MBq) to a sealed vial. After the mixture was heated for 14 min at 100 °C, the vial was allowed to stand at room temperature for 3 min. The pH of the solution was then adjusted to 6, 7, or 8 with 1 M AcOH. Ten min after the neutralization, AgNO3 (7.1 mg, 0.042 mmol) was added to the solution to precipitate out Cl− as AgCl. After filtering the solution through 0.45 μm Cosmonice Filter W (NACALAI TESQUE, Inc., Kyoto), the solution was used for labeling reactions with Lβ and LG. For the reactions with 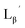 and
and 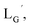 1N HCl was used for neutralization and [99mTc][TcI(CO)3(OH2)3]+ solution was used without the removal of Cl−.
1N HCl was used for neutralization and [99mTc][TcI(CO)3(OH2)3]+ solution was used without the removal of Cl−.
99mTc-labeling reaction of Lβ
A 5 μL of freshly prepared [99mTc][TcI(CO)3(OH2)3]+ (∼5.55 MBq) was added to a vial containing 95 μL of Lβ solution (0.1 M P.B., pH 8.0) to reach the final ligand concentration of 150 μM. The mixture was heated at 110 °C for 20 min under N2 atmosphere in a heating block. The reaction solution was analyzed by HPLC using system 11.
99mTc-labeling reaction of 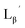
A 20 μL solution of freshly prepared [99mTc][TcI(CO)3(OH2)3]+ (∼22.2 MBq) was added to a vial containing 380 μL of 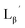 solution (0.1 M P.B., pH 6.0 or 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 6.
solution (0.1 M P.B., pH 6.0 or 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 6.
Stability of 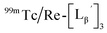 in acidic and basic solutions
in acidic and basic solutions
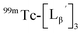 was prepared by the reaction at pH 6.0 which was described above and purified by HPLC (system 6). The eluent of the objective fraction was evaporated in vacuo and the residue was dissolved in 0.1 M P.B. (pH 6.0 or 8.0) containing 40 nmol of
was prepared by the reaction at pH 6.0 which was described above and purified by HPLC (system 6). The eluent of the objective fraction was evaporated in vacuo and the residue was dissolved in 0.1 M P.B. (pH 6.0 or 8.0) containing 40 nmol of 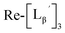 to prepare 111 kBq
to prepare 111 kBq 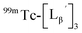 /40 nmol
/40 nmol 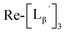 /100 μL sample solutions. The sample of pH 6.0 was analyzed by HPLC (system 6) before heating. Both solutions were then heated at 100 °C for 30 min in a heating block and analyzed by HPLC (system 6) after heating.
/100 μL sample solutions. The sample of pH 6.0 was analyzed by HPLC (system 6) before heating. Both solutions were then heated at 100 °C for 30 min in a heating block and analyzed by HPLC (system 6) after heating.
Characterization of three peaks generated upon heating 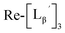
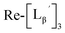 (9) was prepared by the method described above. Twenty nine mg of
(9) was prepared by the method described above. Twenty nine mg of 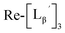 (0.225 mmol) was dissolved in 5 mL of 0.1 M P.B. (pH 8.0) and the mixture was heated at 100 °C for 5 h. The crude product was purified by preparative HPLC using system 2 to afford the compound 12 (1.4 mg, 23% calculated from the Re-complex) as a white solid, the compound 14 (1.8 mg, 13%) as a purple oil and the compound 13 (6.1 mg, 30%) as an orange solid. The spectrum data of the compound 12, 13 and 14 are shown down below.
(0.225 mmol) was dissolved in 5 mL of 0.1 M P.B. (pH 8.0) and the mixture was heated at 100 °C for 5 h. The crude product was purified by preparative HPLC using system 2 to afford the compound 12 (1.4 mg, 23% calculated from the Re-complex) as a white solid, the compound 14 (1.8 mg, 13%) as a purple oil and the compound 13 (6.1 mg, 30%) as an orange solid. The spectrum data of the compound 12, 13 and 14 are shown down below.
The eliminated ligand (compound A in Fig. 5b) (12)
1H NMR (400 MHz, DMSO-d6): δ 3.75 (d, 2H, HαGly), 3.83 (d, 2H, HαGly), 4.28 (d, 2H, CH2-phenyl), 5.61 (dd, 1H, J = 2, 10 Hz, Halkene), 6.08 (dd, 1H, J = 2, 17 Hz, Halkene), 6.31 (dd, 1H, J = 10, 17 Hz, Halkene), 7.21–7.33 (m, 5H, Harom), 8.27 (t, 1H, NH), 8.34 (t, 1H, NH), 8.44 (t, 1H, NH). ESI-MS, m/z: 298.12 [M + Na]+, found 298.10. IR (ATR, ν/cm−1): 1647.88 (s, CC).
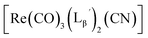 (compound D in Fig. 5b) (13)
(compound D in Fig. 5b) (13)
1H NMR (400 MHz, DMSO-d6): δ 2.69 (t, 2H, CH2CH2CO), 3.75 (d, 2H, HαGly), 3.80 (d, 2H, HαGly), 4.07 (t, 2H, CH2CH2CO), 4.28 (d, 2H, CH2-phenyl), 7.21−7.33 (m, 5H, Harom), 8.26 (t, 1H, NH), 8.30 (t, 1H, NH), 8.44 (t, 1H, NH). ESI-MS, m/z: 924.21 [M + Na]+, found 924.38.
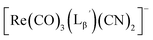 (compound B in Fig. 5b) (14)
(compound B in Fig. 5b) (14)
ESI-MS, m/z: 627.10 [M + 2H]+, found 627.22.
99mTc-labeling reaction of 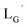
A 20 μL solution of freshly prepared [99mTc][TcI(CO)3(OH2)3]+ (∼22.2 MBq) was added to a vial containing 380 μL of 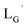 solution (0.1 M P.B., pH 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 7.
solution (0.1 M P.B., pH 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 7.
CN-GABA-Gly-Gly-c(RGDfK) (LG) (16)
To a solution of the compound 15 (13.8 mg, 16.6 μmol) and N,N-diisopropylethylamine (4.9 μL, 28.8 μmol) in DMF (180 μL) was added the compound 7b (4.0 mg, 16 μmol). The reaction mixture was stirred at room temperature for 1 h, after which time diethyl ether was added to the solution to form precipitate. The precipitate was collected and purified with preparative HPLC using system 4, and lyophilized to afford the compound 16 as a white powder (7.0 mg, 52%). ESI-MS, m/z: 813.40 [M + H]+, found 813.49.
[ReI(CO)3(LG)3]CF3CO2 (Re-[LG]3) (17) and [ReI(CO)2(LG)4]CF3CO2 (Re-[LG]4) (18)
LG (16) (6.0 mg, 7.4 μmol) and [ReI(CO)3(OH2)3]Br (19) (0.3 mg, 0.7 μmol) were dissolved in 50 mM P.B. (pH 8.0, 720 μL). The mixture was heated at 110 °C for 2 h, after which time additional LG (1.5 mg, 1.9 μmol) was added to the solution. After heating at 110 °C for another 2 h, the crude was purified with preparative HPLC using system 5 and lyophilized to afford the compounds 17 and 18 as white powders (0.5 mg and 25% for the compound 17, and 0.2 mg and 8% for the compound 18). ESI-MS (17), m/z: 1355.06 [M + H]2+, found 1355.26. ESI-MS (18), m/z: 874.13 [M + 3H]4+, found 874.4.
99mTc-labeling reaction of LG
LG (16) (20 nmol) was dissolved in 75 μL of 50 mM P.B. at pH 8.0. A 25 μL solution of freshly prepared [99mTc][TcI(CO)3(OH2)3]+ (∼27.8 MBq) was added to this LG solution, followed by heating at 85 °C for 1 h under N2 atmosphere. The resulting solution was analyzed by HPLC using system 9.
Stability assessment
The unlabeled ligand in 99mTc-[LG]4 was removed by HPLC using system 9. The radioactive peak was collected and the solvent was removed in vacuo. The residue was reconstituted in 0.001% v/v TritonX-100 in D-PBS(−), and 10 μL of the 99mTc-[LG]4 solution was mixed with 190 μL of histidine solution to reach the final histidine concentration of 10 mM. Similarly 10 μL of the 99mTc-[LG]4 solution was mixed with 190 μL of freshly prepared murine plasma. After incubation at 37 °C for 1 and 6 h, 1 μL of aliquots were drawn and analyzed with radio-TLC (n = 3) (Silica gel 60 F254, Merck Ltd., Tokyo) developed with MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10% w/w AcONH4 in water = 1:1. For HPLC analysis of the histidine sample, the incubation solution was directly analyzed by HPLC using system 9. For HPLC analysis of the plasma sample, 200 μL of EtOH was added to 100 μL of the plasma sample to precipitate the proteins. The sample was centrifuged at 15000g for 5 min at 4 °C. The supernatant was collected and, the pellet was washed with 300 μL of 66% v/v EtOH in D-PBS(−) and centrifuged. This washing step was repeated twice. The supernatants of both centrifugation steps were combined and analyzed by HPLC using system 9. The recovery rate of radioactivity from the plasma sample was determined by following equation, (the radioactivity of the supernatant)/(the original radioactivity of the plasma sample) × 100 (n = 3). The radioactivity was determined using an autowell γ counter (WIZARD 1480, PerkinElmer Japan Co., Ltd., Yokohama, Japan).
:10% w/w AcONH4 in water = 1:1. For HPLC analysis of the histidine sample, the incubation solution was directly analyzed by HPLC using system 9. For HPLC analysis of the plasma sample, 200 μL of EtOH was added to 100 μL of the plasma sample to precipitate the proteins. The sample was centrifuged at 15000g for 5 min at 4 °C. The supernatant was collected and, the pellet was washed with 300 μL of 66% v/v EtOH in D-PBS(−) and centrifuged. This washing step was repeated twice. The supernatants of both centrifugation steps were combined and analyzed by HPLC using system 9. The recovery rate of radioactivity from the plasma sample was determined by following equation, (the radioactivity of the supernatant)/(the original radioactivity of the plasma sample) × 100 (n = 3). The radioactivity was determined using an autowell γ counter (WIZARD 1480, PerkinElmer Japan Co., Ltd., Yokohama, Japan).
Binding affinity to integrin αvβ3
U87MG human glioma cells were grown in Dulbecco's Modified Eagle Medium (Sigma-Aldrich Japan K.K., Tokyo) supplemented with 10% v/v fetal bovine serum (Nippon Biosupply Center, Tokyo), and 1% v/v penicillin–streptomycin (10000 unit–10 mg mL−1, Sigma-Aldrich Japan K.K.) at 37 °C in humidified atmosphere containing 5% CO2. Multiscreen DV filter plates (Merck Millipore, MA) were seeded with 2 × 105 cells in the binding buffer (20 mM Tris, 150 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, 0.1% BSA, pH 7.4) and the mixtures were incubated at 37 °C with [125I]I-c(RGDyV) in the presence of increasing concentrations of c(RGDyV), LG, Re-[LG]3, or Re-[LG]4. The total incubation volume was adjusted to 200 μL. After incubation for 1 h at 37 °C, the plates were filtered, and washed twice with 200 μL of the ice cold binding buffer. The polyvinylidene fluoride (PVDF) filters were collected and the radioactivity was determined using an autowell γ counter. The IC50 values were calculated by fitting the data by nonlinear regression using GraphPad Prism (GraphPad Software, Inc., San Diego, CA). All the binding experiments were carried out with quadruplet samples.
Animal model
All animal procedures were performed in accordance with the institutional guidelines approved by the Chiba University Animal Care Committee and experiments were approved by the Chiba University Animal Care Committee. BALBc nu/nu male mice (Japan SLC, Inc., Shizuoka, Japan) of 6 week-old were xenografted by subcutaneous injection of U87MG human glioblastoma cells (5 × 106 cells per 50 μL of culture medium) into their right hind legs. The mice were subjected to biodistribution studies as well as SPECT/CT imaging studies when the tumor weight reached 0.1–0.5 g.
Biodistribution
Male nude mice bearing tumor xenografts of U87MG were injected via the tail vein with 100 μL (11.1 kBq) of 99mTc-[LG]4 (LG = 0 nmol), 99mTc-[LG]4 (LG = 5 nmol) or 99mTc-[LG]4 (Re-[LG]4 = 5 nmol). 99mTc-[LG]4 (LG = 0 nmol) was prepared by diluting an HPLC-purified 99mTc-[LG]4 solution with D-PBS (−). 99mTc-[LG]4 (Re-[LG]4 = 5 nmol) was prepared by adding Re-[LG]4 to an HPLC-purified 99mTc-[LG]4 solution. Un-purified 99mTc-[LG]4 solution was used for 99mTc-[LG]4 (LG = 5 nmol). The animals were sacrificed and dissected at 1 h after administration. The tissues of interest were removed and weighed, and the radioactivity was determined with an autowell gamma counter. The results are presented as the percentage injected dose per gram (% ID g−1) or percentage injected dose per tissue (% ID). Values were expressed as mean ± SD for a group of 5 or 6 animals.
Small animal SPECT/CT imaging study
SPECT images were acquired over 30–94 min after intravenous administration of 99mTc-[LG]4 (100 μL, 3.7 MBq, LG = 5 nmol) to male BALBc nu/nu mice bearing U87MG xenografts via the tail vein. CT scans were performed before SPECT scans for anatomic reference. The mice were anaesthetized with 1–2% (v/v) isoflurane (DS Pharma Animal Health, Osaka, Japan) and positioned on the animal bed where anesthesia was continuously delivered via a nose cone system. SPECT imaging and X-ray CT imaging were performed by use of small animal SPECT/CT system (Triumph Lab SPECT4/CT, TriFoil Imaging Inc., Chatsworth, CA) equipped with a five pinhole (0.5 mm) collimator. Data acquisition was performed for 64 min at 240 s per projection with stepwise rotation of 16 projections over 360°.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Y. M. was supported by JSPS Research Fellowships for Young Scientists. This work was supported in part by JSPS Research Fellows Program (No. 264896). We thank Prof. Atsushi Nishida (Chiba University) for valuable discussion on the mechanism of the side reaction.
References
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS PubMed.
- B. M. Zeglis, J. L. Houghton, M. J. Evans, N. Viola-Villegas and J. S. Lewis, Inorg. Chem., 2014, 53, 1880–1899 CrossRef CAS PubMed.
- D. Brasse and A. Nonat, Dalton Trans., 2015, 44, 4845–4858 RSC.
- S. Bhattacharyya and M. Dixit, Dalton Trans., 2011, 40, 6112–6128 RSC.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- M. Morais, A. Paulo, L. Gano, I. Santos and J. D. G. Correia, J. Organomet. Chem., 2013, 744, 125–139 CrossRef CAS.
- S. Liu, Adv. Drug Delivery Rev., 2008, 60, 1347–1370 CrossRef CAS PubMed.
- S. Liu and S. Chakraborty, Dalton Trans., 2011, 40, 6077–6086 RSC.
- J. R. Ballinger, Q. J. Nucl. Med. Mol. Imaging, 2002, 46, 224–232 CAS.
- Y. Mizuno, T. Uehara, H. Hanaoka, Y. Endo, C.-W. Jen and Y. Arano, J. Med. Chem., 2016, 59, 3331–3339 CrossRef CAS PubMed.
- Y. Mizuno, T. Uehara, H. Hanaoka, Y. Endo, C.-W. Jen and Y. Arano, J. Med. Chem., 2017, 60, 6768–6769 CrossRef CAS PubMed.
- R. Haubner, H. J. Wester, U. Reuning, R. Senekowitsch-Schmidtke, B. Diefenbach, H. Kessler, G. Stöcklin and M. Schwaiger, J. Nucl. Med., 1999, 40, 1061–1071 CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed.
- S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS PubMed.
- Y. Taira, T. Uehara, M. Tsuchiya, H. Takemori, Y. Mizuno, S. Takahashi, H. Suzuki, H. Hanaoka, H. Akizawa and Y. Arano, Bioconjugate Chem., 2018, 29, 459–466 CrossRef CAS PubMed.
- H. A. Holik, T. Uehara, S. Nemoto, T. Rokugawa, Y. Tomizawa, A. Sakuma, Y. Mizuno, H. Suzuki and Y. Arano, Bioconjugate Chem., 2018, 29, 2909–2919 CrossRef CAS PubMed.
- X. Chen, Y. Guo, Q. Zhang, G. Hao, H. Jia and B. Liu, J. Organomet. Chem., 2008, 693, 1822–1828 CrossRef CAS.
- J. F. Bunnett, Angew. Chem., Int. Ed. Engl., 1962, 1, 225–235 CrossRef CAS.
- W. C. Eckelman, A. G. Jones, A. Duatti and R. C. Reba, Drug Discov. Today, 2013, 18, 984–991 CrossRef CAS PubMed.
- W. C. Eckelman, Eur. J. Nucl. Med. Mol. Imaging, 1995, 22, 249–263 CrossRef CAS PubMed.
- K. Sung and C. C. Chen, Tetrahedron Lett., 2001, 42, 4845–4848 CrossRef CAS.
- N. Lazarova, S. James, J. Babich and J. Zubieta, Inorg. Chem. Commun., 2004, 7, 1023–1026 CrossRef CAS.
- S. M. Creedon, H. K. Crowley and D. G. McCarthy, J. Chem. Soc., Perkin Trans. 1, 1998, 1015–1017 RSC.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135–3136 CrossRef CAS PubMed.
Footnote |
† Electronic supplementary information (ESI) available: HPLC methods, synthesis scheme of the compounds 8a, 8b, 16, stability of 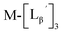 (M = 185/187Re and 99mTc) at pH 6.0, in vitro stability of 99mTc-[LG]4, competitive inhibition curves of the tested compounds, MS, 1H-NMR, 13C-NMR, IR, and/or purity check of key compounds. See DOI: 10.1039/c9ra04311j (M = 185/187Re and 99mTc) at pH 6.0, in vitro stability of 99mTc-[LG]4, competitive inhibition curves of the tested compounds, MS, 1H-NMR, 13C-NMR, IR, and/or purity check of key compounds. See DOI: 10.1039/c9ra04311j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Tomoya Uehara
ab,
Tomoya Uehara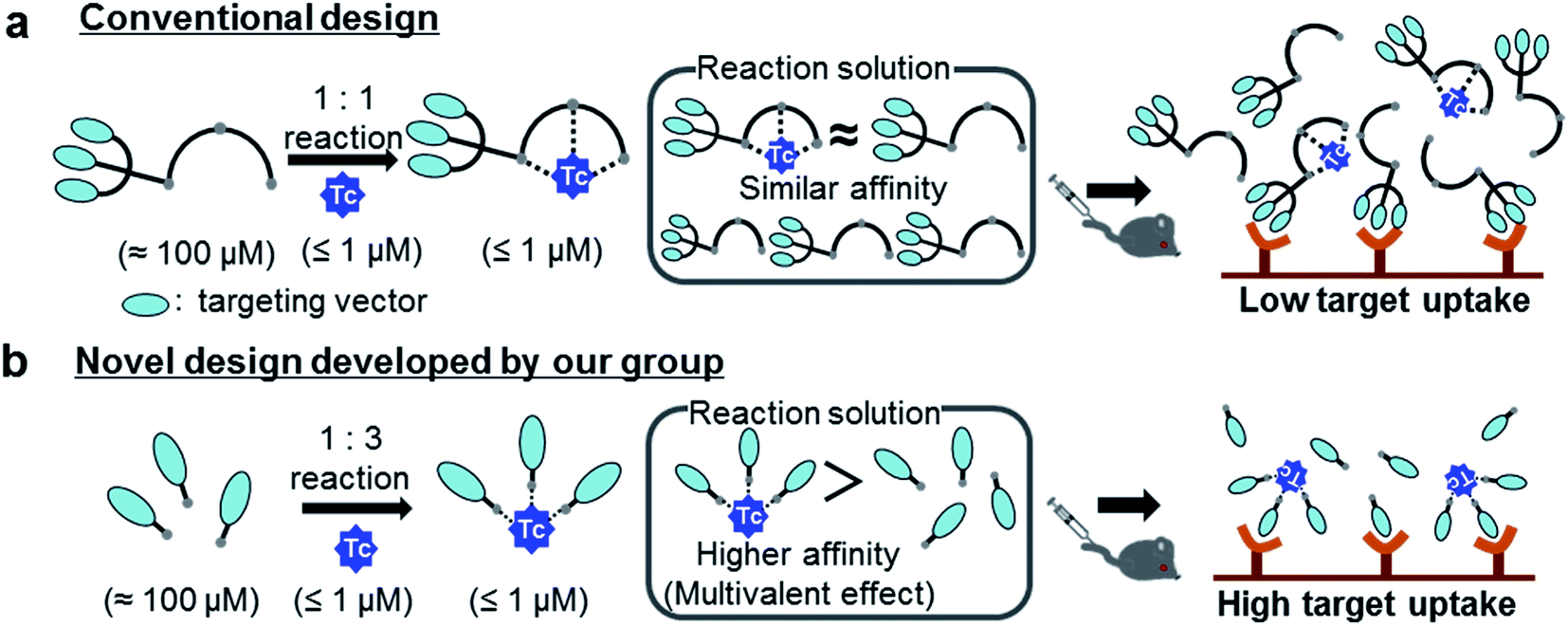
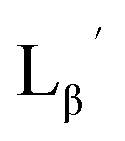 ,
, 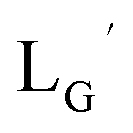 , c(RGDfK), and MIBI.
, c(RGDfK), and MIBI.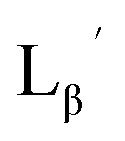
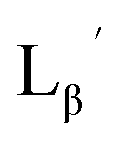 ), where c(RGDfK) in Lβ was replaced with benzylamine. However, as shown in Fig. 4b, the formation of multiple side products was again observed, suggesting that the side reaction observed here was not caused by the presence of RGD peptides but by other moieties in Lβ.
), where c(RGDfK) in Lβ was replaced with benzylamine. However, as shown in Fig. 4b, the formation of multiple side products was again observed, suggesting that the side reaction observed here was not caused by the presence of RGD peptides but by other moieties in Lβ.
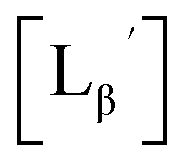 = 1 mM, 100 °C for 30 min at pH 8.0 (system 6).
= 1 mM, 100 °C for 30 min at pH 8.0 (system 6).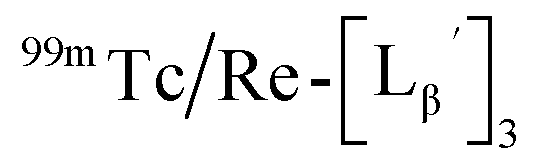
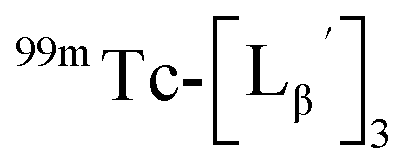 and
and 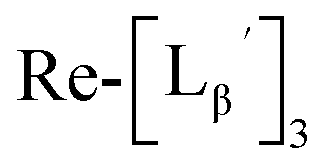 at pH 8.0 (Fig. 5). Since technetium and rhenium share similar coordination chemistry due to their group homology, we employed non-radioactive rhenium analogues to characterize their tracer 99mTc counterparts.
at pH 8.0 (Fig. 5). Since technetium and rhenium share similar coordination chemistry due to their group homology, we employed non-radioactive rhenium analogues to characterize their tracer 99mTc counterparts. 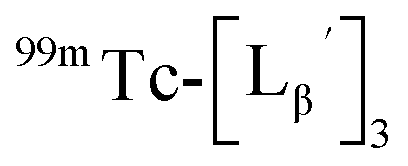 was prepared from a labeling reaction at pH 6.0 and was purified by HPLC.
was prepared from a labeling reaction at pH 6.0 and was purified by HPLC. 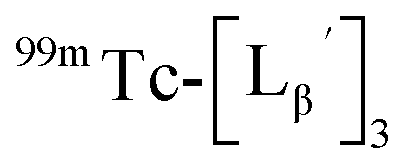 was then dissolved in aqueous solution at pH 8.0 containing
was then dissolved in aqueous solution at pH 8.0 containing 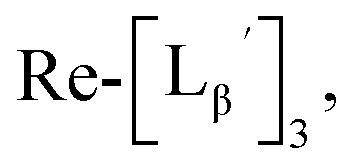 and the solution was heated at 100 °C for 30 min. As shown in Fig. 5b,
and the solution was heated at 100 °C for 30 min. As shown in Fig. 5b, 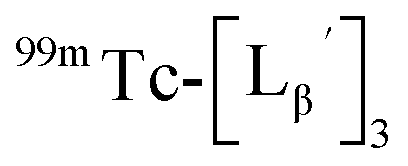 and
and 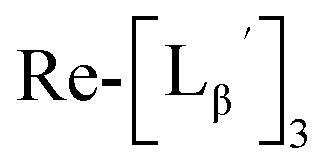 exhibited a similar decomposition pattern upon heating at pH 8.0, whereas they remained intact at pH 6.0 (Fig. S1†). These results indicate that the decomposition of
exhibited a similar decomposition pattern upon heating at pH 8.0, whereas they remained intact at pH 6.0 (Fig. S1†). These results indicate that the decomposition of 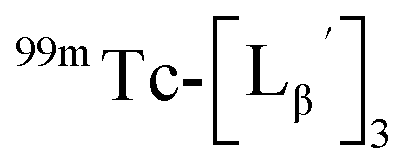 under basic conditions primarily accounts for the generation of the multiple peaks observed in Fig. 4b.
under basic conditions primarily accounts for the generation of the multiple peaks observed in Fig. 4b.
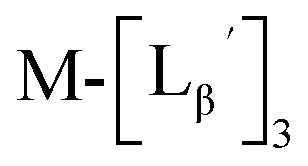 (M = 99mTc and 185/187Re) in basic solution. HPLC analyses (system 6) of
(M = 99mTc and 185/187Re) in basic solution. HPLC analyses (system 6) of 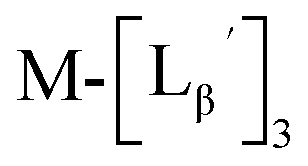 (a) before heating and (b) after heating at 100 °C for 30 min at pH 8.0. (c) The chemical structures of the products generated upon heating and the intact complex
(a) before heating and (b) after heating at 100 °C for 30 min at pH 8.0. (c) The chemical structures of the products generated upon heating and the intact complex 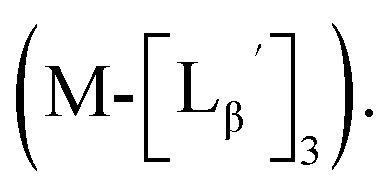
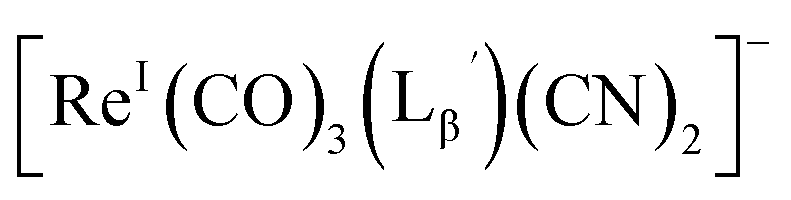 and
and 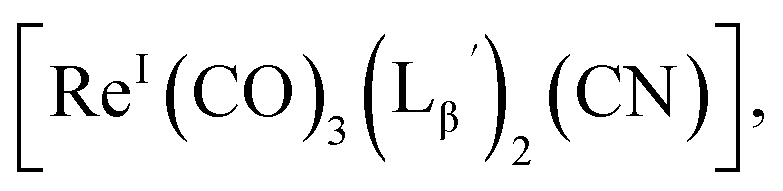 respectively (Fig. 5c).
respectively (Fig. 5c).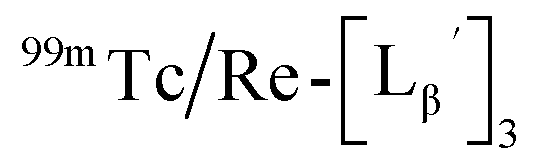 has the following three characteristics: (1) one of the coordinated isonitrile ligands is eliminated from the Tc/Re-complex with leaving an isonitrile ion on the Tc/Re-complex, (2) the eliminated ligand loses the isonitrile group, and its terminal structure is converted to an alkene structure, (3) this reaction proceeds only at high hydroxide ion concentration. These features possess the characteristics of the “bimolecular elimination reaction”.19 We thereby hypothesized that the side reaction observed at basic conditions (Fig. 4) would also proceed via the following three simultaneous events: (1) the abstraction of the 2-proton in
has the following three characteristics: (1) one of the coordinated isonitrile ligands is eliminated from the Tc/Re-complex with leaving an isonitrile ion on the Tc/Re-complex, (2) the eliminated ligand loses the isonitrile group, and its terminal structure is converted to an alkene structure, (3) this reaction proceeds only at high hydroxide ion concentration. These features possess the characteristics of the “bimolecular elimination reaction”.19 We thereby hypothesized that the side reaction observed at basic conditions (Fig. 4) would also proceed via the following three simultaneous events: (1) the abstraction of the 2-proton in 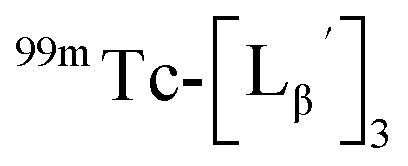 by a hydroxide ion, (2) the departure of
by a hydroxide ion, (2) the departure of 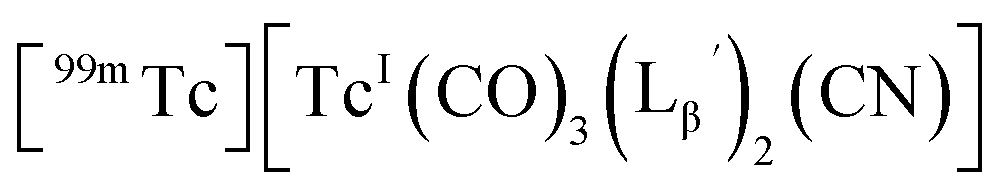 from
from 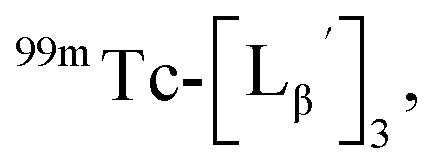 and (3) the formation of an alkene structure (Fig. 6).
and (3) the formation of an alkene structure (Fig. 6).
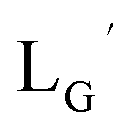
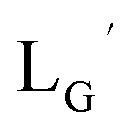
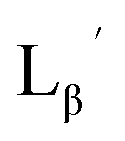 (Fig. 6) would be essential for the side reaction to proceed. In addition, this high acidity of the 2-proton can be partly attributed to the electron withdrawing effect from the neighboring carbonyl group. We thereby newly designed a γ-aminobutyric acid (GABA) based isonitrile ligand (
(Fig. 6) would be essential for the side reaction to proceed. In addition, this high acidity of the 2-proton can be partly attributed to the electron withdrawing effect from the neighboring carbonyl group. We thereby newly designed a γ-aminobutyric acid (GABA) based isonitrile ligand (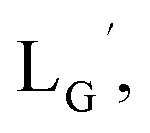 Fig. 3). As the 2-proton in
Fig. 3). As the 2-proton in 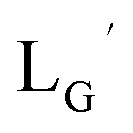 is no longer present next to the carbonyl group (Fig. 3), the 2-proton in
is no longer present next to the carbonyl group (Fig. 3), the 2-proton in 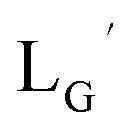 would possess much lower acidity.
would possess much lower acidity.
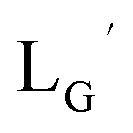 were performed in a procedure similar to that of
were performed in a procedure similar to that of 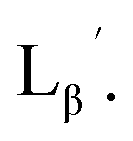 [99mTc][TcI(CO)3(OH2)3]+ was prepared as a labeling precursor, mixed with
[99mTc][TcI(CO)3(OH2)3]+ was prepared as a labeling precursor, mixed with 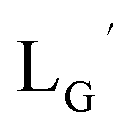 and heated at 100 °C for 30 min at pH 8.0. The HPLC analysis of this reaction solution is presented in Fig. 7c, which shows a completely different chromatographic profile from that in Fig. 4b. As shown in Fig. 7c, the 99mTc-labeling reaction of
and heated at 100 °C for 30 min at pH 8.0. The HPLC analysis of this reaction solution is presented in Fig. 7c, which shows a completely different chromatographic profile from that in Fig. 4b. As shown in Fig. 7c, the 99mTc-labeling reaction of 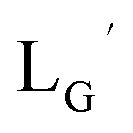 at pH 8.0 generated only two peaks, and the retention time of the major radioactivity peak was similar to that of
at pH 8.0 generated only two peaks, and the retention time of the major radioactivity peak was similar to that of 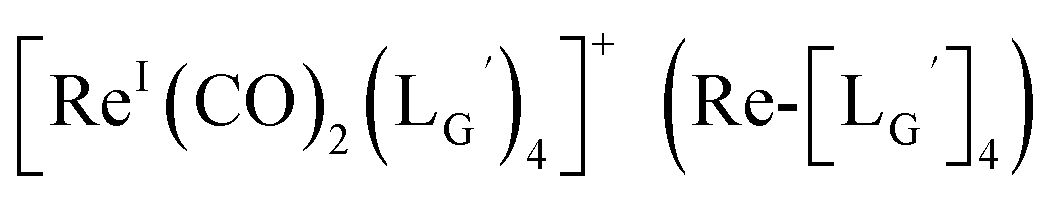 characterized by ESI-MS and IR (Fig. 7b and c). These results supported the validity of our working hypothesis that the abstraction of the 2-proton by a base is the key step for the side reaction. We also identified the minor radioactivity peak as
characterized by ESI-MS and IR (Fig. 7b and c). These results supported the validity of our working hypothesis that the abstraction of the 2-proton by a base is the key step for the side reaction. We also identified the minor radioactivity peak as 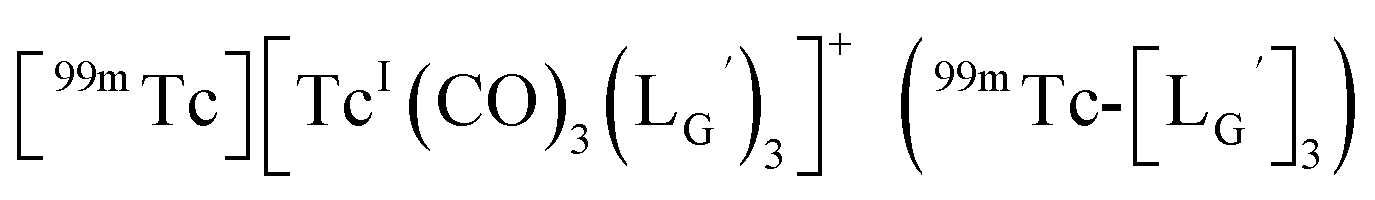 based on its similar HPLC retention time to that of
based on its similar HPLC retention time to that of 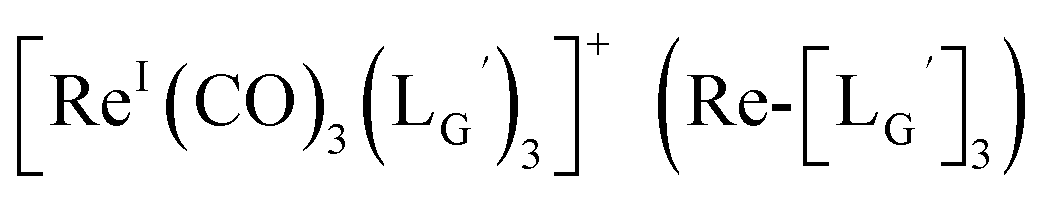 (Fig. 7a and c).
(Fig. 7a and c).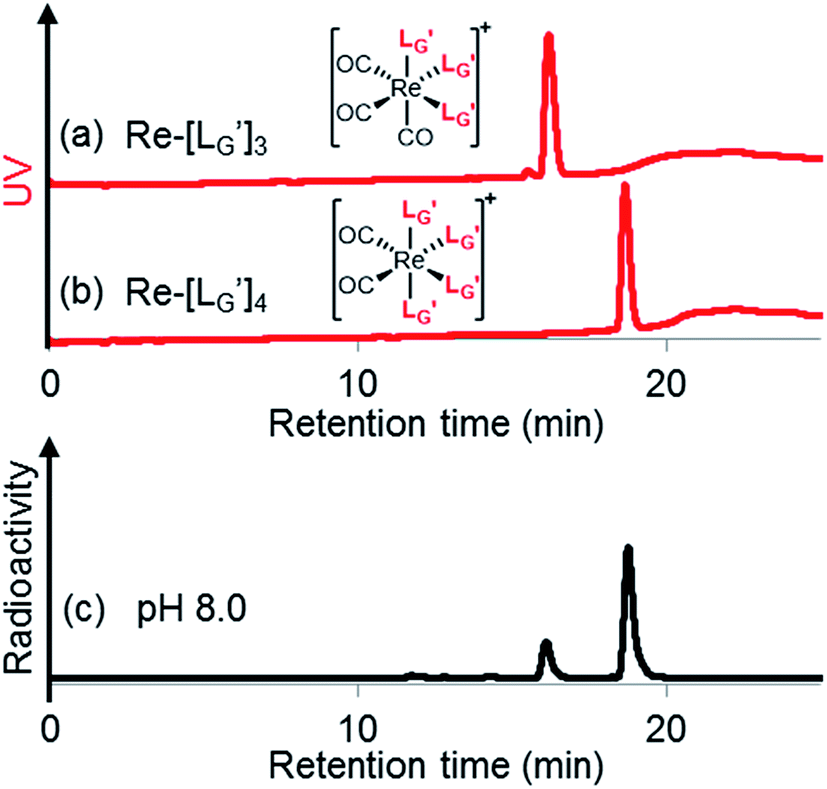
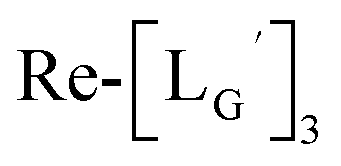 and (b)
and (b) 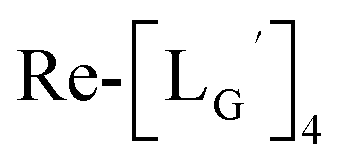 . (c) An HPLC analysis (system 7) of a 99mTc-labeling reaction of
. (c) An HPLC analysis (system 7) of a 99mTc-labeling reaction of 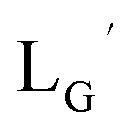 under the condition of
under the condition of 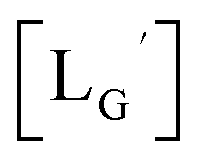 = 1 mM, 100 °C for 30 min at pH 8.0.
= 1 mM, 100 °C for 30 min at pH 8.0.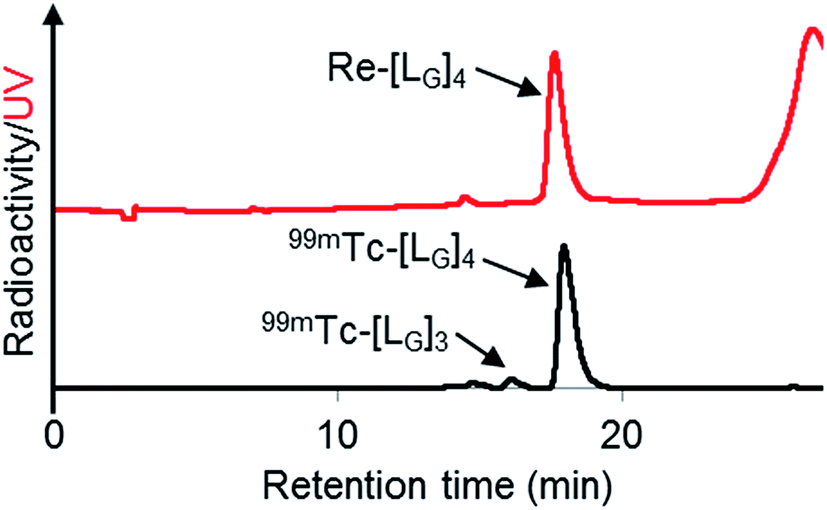
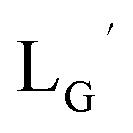 ), on the other hand, avoided the side reaction and provided 99mTc-labeled tetravalent probes from reactions at basic conditions. In the SPECT/CT study, 99mTc-[LG]4 clearly visualized integrin αvβ3 positive tumor without tedious post-labeling purification to remove its excess unlabeled ligand. These results clearly indicate that the metal coordination-mediated synthesis of 99mTc-labeled bivalent and trivalent probes has been successfully extended to that of tetravalent ones, which would lead to a wider range of choices for designing novel 99mTc-labeled multivalent probes of high in vivo target uptake. In addition, because 99mTc-hexakis-isonitriles such as [99mTc][Tc(MIBI)6]+ also possess very high in vivo stability, we are currently working to apply our metal coordination mediated multivalency concept to hexavalent ones, and are going to report the results in the near future elsewhere.
), on the other hand, avoided the side reaction and provided 99mTc-labeled tetravalent probes from reactions at basic conditions. In the SPECT/CT study, 99mTc-[LG]4 clearly visualized integrin αvβ3 positive tumor without tedious post-labeling purification to remove its excess unlabeled ligand. These results clearly indicate that the metal coordination-mediated synthesis of 99mTc-labeled bivalent and trivalent probes has been successfully extended to that of tetravalent ones, which would lead to a wider range of choices for designing novel 99mTc-labeled multivalent probes of high in vivo target uptake. In addition, because 99mTc-hexakis-isonitriles such as [99mTc][Tc(MIBI)6]+ also possess very high in vivo stability, we are currently working to apply our metal coordination mediated multivalency concept to hexavalent ones, and are going to report the results in the near future elsewhere.
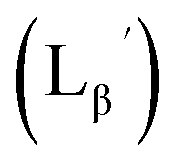
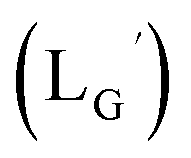
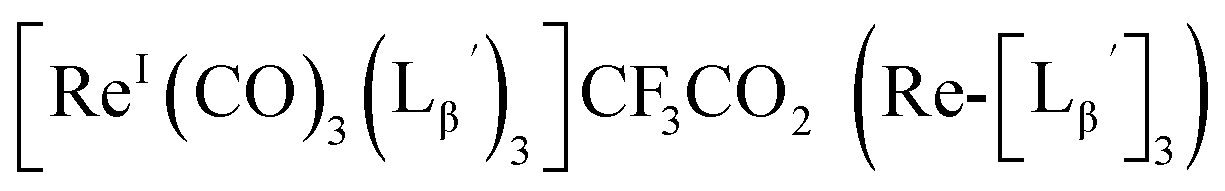
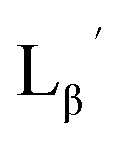 (8a) (65 mg, 0.215 mmol) were dissolved in 0.1 M acetate buffer (pH 6.0, 21.5 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 1 and lyophilized to afford the compound 9 as a white powder (30 mg, 97%). 1H NMR (400 MHz, DMSO-d6): δ 2.70 (t, 2H, CH2CH2CO), 3.76 (d, 2H, HαGly), 3.81 (d, 2H, HαGly), 4.09 (t, 2H, CH2CH2CO), 4.28 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, Harom), 8.27 (t, 1H, NH), 8.36 (t, 1H, NH), 8.43 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 33.89 (CH2CH2CO), 41.06 (CH2CH2CO), 42.02, 42.08, (CαGly, CH2-phenyl), 126.79 (Carom), 127.18 (Carom), 128.26 (Carom), 139.31 (Carom), 158.24 (q, CN), 168.80 (CO), 168.96 (CO), 169.10 (CO), 182.71 (ReCO). ESI-MS, m/z: 1177.35 [M]+, found 1177.56. IR (ATR, ν/cm−1): 2244.74 (w, C
(8a) (65 mg, 0.215 mmol) were dissolved in 0.1 M acetate buffer (pH 6.0, 21.5 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 1 and lyophilized to afford the compound 9 as a white powder (30 mg, 97%). 1H NMR (400 MHz, DMSO-d6): δ 2.70 (t, 2H, CH2CH2CO), 3.76 (d, 2H, HαGly), 3.81 (d, 2H, HαGly), 4.09 (t, 2H, CH2CH2CO), 4.28 (d, 2H, CH2-phenyl), 7.21–7.33 (m, 5H, Harom), 8.27 (t, 1H, NH), 8.36 (t, 1H, NH), 8.43 (t, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 33.89 (CH2CH2CO), 41.06 (CH2CH2CO), 42.02, 42.08, (CαGly, CH2-phenyl), 126.79 (Carom), 127.18 (Carom), 128.26 (Carom), 139.31 (Carom), 158.24 (q, CN), 168.80 (CO), 168.96 (CO), 169.10 (CO), 182.71 (ReCO). ESI-MS, m/z: 1177.35 [M]+, found 1177.56. IR (ATR, ν/cm−1): 2244.74 (w, C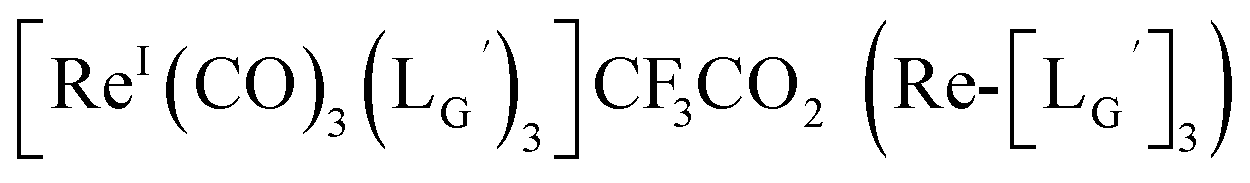
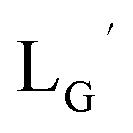 (8b) (20 mg, 63.2 μmol) were dissolved in 0.1 M phosphate buffer (P.B., pH 7.0, 6.32 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 3 and lyophilized to afford the compound 10 as a white powder (8.9 mg, quant.). 1H NMR (400 MHz, DMSO-d6): δ 1.93 (m, 2H, CH2CH2CH2), 2.29 (t, 2H, CH2CH2CH2), 3.75 (d, 4H, HαGly), 3.98 (t, 2H, CNCH2), 4.28 (d, 2H, CH2-phenyl), 7.10–7.33 (m, 5H, Harom), 8.23 (t, 1H, NH), 8.29 (t, 1H, NH), 8.34 (t, 1H, NH). ESI-MS, m/z: 1219.40 [M]+, found 1219.47. IR (ATR, ν/cm−1): 2238.95 (w, C
(8b) (20 mg, 63.2 μmol) were dissolved in 0.1 M phosphate buffer (P.B., pH 7.0, 6.32 mL). After heating at 100 °C for 3 h, the crude was purified by preparative HPLC using system 3 and lyophilized to afford the compound 10 as a white powder (8.9 mg, quant.). 1H NMR (400 MHz, DMSO-d6): δ 1.93 (m, 2H, CH2CH2CH2), 2.29 (t, 2H, CH2CH2CH2), 3.75 (d, 4H, HαGly), 3.98 (t, 2H, CNCH2), 4.28 (d, 2H, CH2-phenyl), 7.10–7.33 (m, 5H, Harom), 8.23 (t, 1H, NH), 8.29 (t, 1H, NH), 8.34 (t, 1H, NH). ESI-MS, m/z: 1219.40 [M]+, found 1219.47. IR (ATR, ν/cm−1): 2238.95 (w, C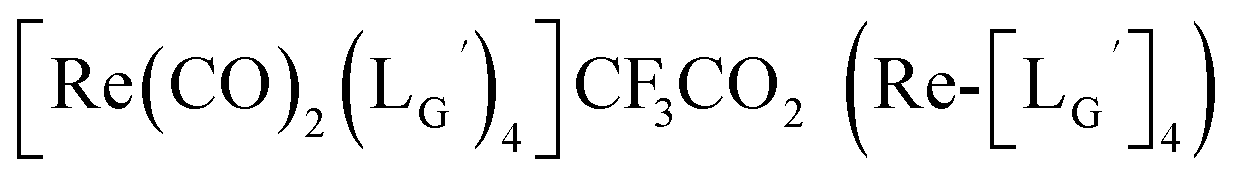
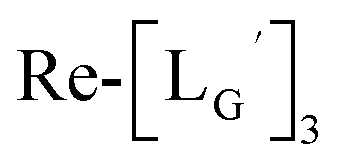 (10) (3.6 mg, 0.0027 mmol) and
(10) (3.6 mg, 0.0027 mmol) and 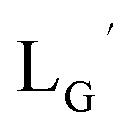 (8b) (4.7 mg, 0.0149 mmol) were dissolved in 0.1 M P.B. (pH 8.0, 1.47 mL). The mixture was heated at 100 °C for 6 h, after which time additional
(8b) (4.7 mg, 0.0149 mmol) were dissolved in 0.1 M P.B. (pH 8.0, 1.47 mL). The mixture was heated at 100 °C for 6 h, after which time additional 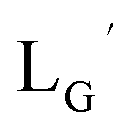 (8b) (4.7 mg, 0.0149 mmol) was added. After heating at 100 °C for another 6 h, the crude product was purified by preparative HPLC using system 3 and lyophilized to afford the compound 11 as a white powder (0.4 mg, 10%). ESI-MS, m/z: 1507.56 [M]+, found 1507.65. IR (ATR, ν/cm−1): 2228.34 (w, C
(8b) (4.7 mg, 0.0149 mmol) was added. After heating at 100 °C for another 6 h, the crude product was purified by preparative HPLC using system 3 and lyophilized to afford the compound 11 as a white powder (0.4 mg, 10%). ESI-MS, m/z: 1507.56 [M]+, found 1507.65. IR (ATR, ν/cm−1): 2228.34 (w, C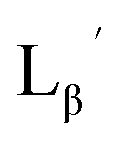 and
and 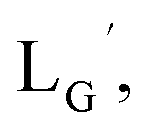 1N HCl was used for neutralization and [99mTc][TcI(CO)3(OH2)3]+ solution was used without the removal of Cl−.
1N HCl was used for neutralization and [99mTc][TcI(CO)3(OH2)3]+ solution was used without the removal of Cl−.
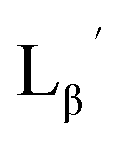
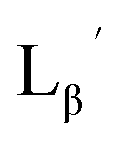 solution (0.1 M P.B., pH 6.0 or 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 6.
solution (0.1 M P.B., pH 6.0 or 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 6.
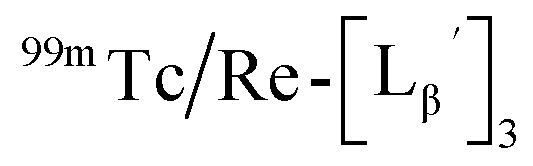
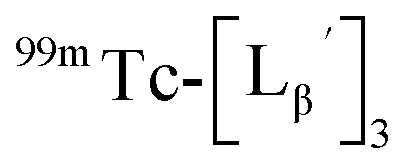 was prepared by the reaction at pH 6.0 which was described above and purified by HPLC (system 6). The eluent of the objective fraction was evaporated in vacuo and the residue was dissolved in 0.1 M P.B. (pH 6.0 or 8.0) containing 40 nmol of
was prepared by the reaction at pH 6.0 which was described above and purified by HPLC (system 6). The eluent of the objective fraction was evaporated in vacuo and the residue was dissolved in 0.1 M P.B. (pH 6.0 or 8.0) containing 40 nmol of 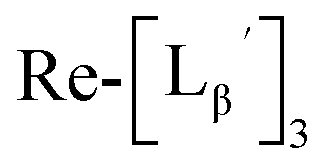 to prepare 111 kBq
to prepare 111 kBq 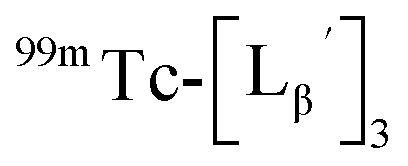 /40 nmol
/40 nmol 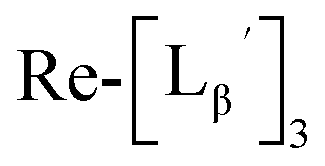 /100 μL sample solutions. The sample of pH 6.0 was analyzed by HPLC (system 6) before heating. Both solutions were then heated at 100 °C for 30 min in a heating block and analyzed by HPLC (system 6) after heating.
/100 μL sample solutions. The sample of pH 6.0 was analyzed by HPLC (system 6) before heating. Both solutions were then heated at 100 °C for 30 min in a heating block and analyzed by HPLC (system 6) after heating.
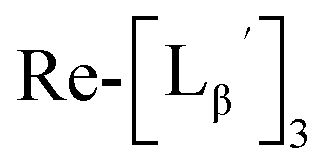
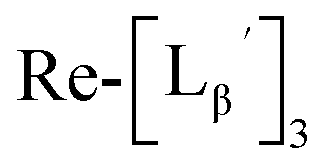 (9) was prepared by the method described above. Twenty nine mg of
(9) was prepared by the method described above. Twenty nine mg of 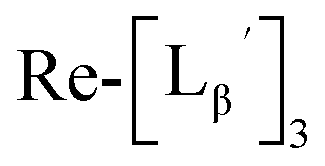 (0.225 mmol) was dissolved in 5 mL of 0.1 M P.B. (pH 8.0) and the mixture was heated at 100 °C for 5 h. The crude product was purified by preparative HPLC using system 2 to afford the compound 12 (1.4 mg, 23% calculated from the Re-complex) as a white solid, the compound 14 (1.8 mg, 13%) as a purple oil and the compound 13 (6.1 mg, 30%) as an orange solid. The spectrum data of the compound 12, 13 and 14 are shown down below.
(0.225 mmol) was dissolved in 5 mL of 0.1 M P.B. (pH 8.0) and the mixture was heated at 100 °C for 5 h. The crude product was purified by preparative HPLC using system 2 to afford the compound 12 (1.4 mg, 23% calculated from the Re-complex) as a white solid, the compound 14 (1.8 mg, 13%) as a purple oil and the compound 13 (6.1 mg, 30%) as an orange solid. The spectrum data of the compound 12, 13 and 14 are shown down below.
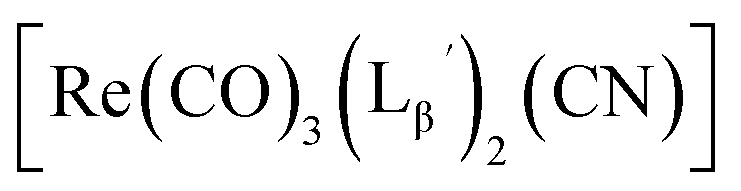
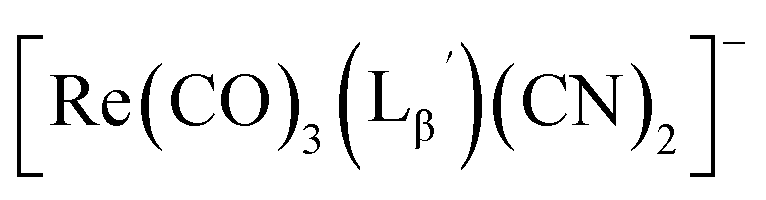
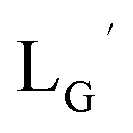
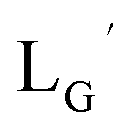 solution (0.1 M P.B., pH 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 7.
solution (0.1 M P.B., pH 8.0) to reach the final ligand concentration of 1 mM. The mixtures were heated at 100 °C for 30 min under N2 atmosphere in a heating block. The reaction solutions were analyzed by HPLC using system 7.
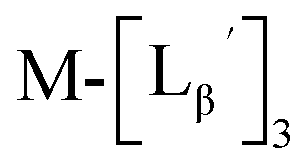 (M = 185/187Re and 99mTc) at pH 6.0, in vitro stability of 99mTc-[LG]4, competitive inhibition curves of the tested compounds, MS, 1H-NMR, 13C-NMR, IR, and/or purity check of key compounds. See DOI: 10.1039/c9ra04311j
(M = 185/187Re and 99mTc) at pH 6.0, in vitro stability of 99mTc-[LG]4, competitive inhibition curves of the tested compounds, MS, 1H-NMR, 13C-NMR, IR, and/or purity check of key compounds. See DOI: 10.1039/c9ra04311j