
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant-free synthesis of porous Au by a urea complex†
Qiang Zhangb,
Biao Zangb and
Shaozhen Wang *a
*a
aAnHui Provincial Engineering Research Center for Polysaccharide Drugs, Institute of Synthesis and Application of Medical Materials, Department of Pharmacy, Wannan Medical College, Wuhu 241002, P. R. China. E-mail: wangshaozhen@wnmc.edu.cn
bAnHui Key Laboratory of Chemo/Biosensing, College of Chemistry and Materials Science, AnHui Normal University, WuHu, 241000, P. R. China
First published on 26th July 2019
Abstract
We report a facile surfactant-free synthetic method to obtain porous and hollow Au nanoparticles using only urea and HAuCl4·4H2O as precursors at 200 °C. The formation mechanism was investigated through X-ray diffraction, scanning electron microscopy, and high-resolution transmission electron microscopy. Moreover, the prepared Au nanocrystals present superior catalytic performance for the reduction of 4-nitrophenol in comparison with solid Au nanoparticles. The catalytic efficiency of porous and hollow Au was nearly 6 times higher than that of Au nanoparticles. Furthermore, the porous and hollow Au maintained excellent stability even after 10 catalytic cycles. Therefore, the as-synthesized porous Au nanoparticles will have potential applications in organic catalysis, biosensing, drug delivery, water pollutant removal, and so on.
1. Introduction
Noble metal nanoparticles still attract considerable attention due to their chemical stability, versatility, and considerable potential applications in sensing,1 medical application,2–4 catalysis,5–7 etc. To improve their application, noble metal nanostructures with diverse shapes have been synthesized, which include nanorods,8,9 nanocages,10 nanostars,11 and porous structures.12,13 Among these structures, porous nanoparticles are particularly attractive because of their larger internal cavity and specific surface area.14Various chemical or physical synthesis methods have been developed for preparing metal nanocrystals with a porous feature, including chemical etching,15 the template method,16,17 and seed-mediated growth.18 As a typical example, Wang et al. prepared porous Pd nanoparticles by a seed-mediated method in an aqueous solution using cetyltrimethylammonium chloride (CTAC) as a stabilizing agent.18 Surfactants, such as poly(vinylpyrrolidone) (PVP) or cetyltrimethylammonium bromide (CTAB), are key for the formation of porous structures in most of the reported syntheses.19 However, the existence of surfactants on the surface of the nanoparticles becomes a huge obstacle in decreasing the active sites for the catalytic reaction.20,21 Moreover, the removal of surfactants is not only a tedious process but also can lead to an agglomeration of particles, which will also decrease their catalytic activity. The surfactant-free synthetic strategy, which will not introduce the stabilizer during the growth procedure, is thus highly attractive. Up to now, there are some studies on the surfactant-free synthesis of solid Au nanoparticles.22–24 For example, Lai et al. prepared homogeneous Au nanoparticles encapsulated by polythiophene analogues.25 However, the growth of the porous and hollow Au nanoparticles is still rarely reported.26 Such an architecture will undoubtedly increase the catalytic sites and thus enhance the catalytic performances.
Herein, we report the surfactant-free synthesis of porous and hollow Au (PHAuNPs) via a incinerate precursors, namely, Gold–Urea Complex (GUC) precursors. These complexes were formed by mixing urea with HAuCl4·4H2O. The precursors were reduced and grown into PHAuNPs under calcination at 200 °C. We found that PHAuNPs show spherical morphology with a size of 50 nm. It was noticed that the nanostructure shows a hollow feature formed by the nanoparticles with a gap of 1 nm externally. The component nanoparticles had a size of around 5 nm. Furthermore, the catalytic performance of the PHAuNPs was assessed by employing the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) as a model reaction. The results revealed that the catalytic activity of PHAuNPs was 6 times higher than that of the commercial solid gold nanoparticles. Moreover, the PHAuNPs also exhibited highly improved stability during the catalytic examinations.
2. Experimental
2.1 Synthesis of Gold–Urea Complex (GUC) precursors
In our proposed synthesis, HAuCl4 (5.89 mM, 1.0 mL) was stirred with urea (0.2 g, 10 mL) at room temperature for 5 min. Then, the mixture was transferred into a round-bottom flask with a reflux condenser to maintain the fast stirring at 100 °C for 10 min. The precipitate obtained was washed with deionized water and ethanol three times and collected for further characterization.2.2 Synthesis of porous and hollow Au nanoparticles (PHAuNPs) and porous and solid Au nanoparticles (PSAuNPs)
Briefly, GUC was diluted with 1 mL ethanol and converted to a neat ampulla via ultrasonication for 2 min. The ampulla was then incinerated at 160 °C or 200 °C. The products obtained were subsequently washed with deionized water and ethanol three times and preserved for further application.2.3 Catalytic reduction of 4-NP
Initially, 5 μL of 4-NP (60 mM) diluted with 2.5 mL of deionized water was added into the standard quartz cuvettes at room temperature, followed by the rapid addition of 150 μL of freshly prepared ice-cold NaBH4 solution (1.0 M). Then, the PHAuNPs (0.5 mg mL−1, 100 μL) and the commercial gold nanoparticles (0.5 mg mL−1, 100 μL) were added into cuvettes sequentially to compare the catalytic activity. The reaction process was monitored by measuring the UV-vis absorption spectra with a time interval of 80 s. The stabilities of the catalysts were tested by a ten-run recycling experiment. After each reaction, 5 μL of 4-NP (60 mM) was directly added into the reaction mixture for the second to tenth runs.2.4 Chemicals and materials
HAuCl4·3H2O, sodium borohydride (NaBH4), and 4-nitrophenol (4-NP) were obtained from Aldrich. Urea and ethanol were purchased from Shantou Xilong and Fuchen TianJin, respectively. The commercial solid gold nanoparticles were purchased from XFNANO (Size: 50 nm, Sodium citrate as a reducing agent). All the aqueous solutions were prepared with twice-distilled water, obtained from a water purifying machine (Hokee, 18.25 MΩ cm−1).2.5 Characterization
SEM images were obtained using a scanning electron microscope (SEM, Hitachi S-4800 Japan). TEM images were captured on a transmission electron microscope (Tecnai G20 S-Twin, Holland). The UV-vis absorbance of 4-NP was monitored by a lambda 35 spectrophotometer. FT-IR spectra were obtained on a Hitachi FTIR-8400S using KBr pellets. Elemental Analyzer (Vario EL cube, Germany) was used to detect the elements (C, H, and N). The surface elemental composition was obtained by X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific, UK) using Al Ka X-ray radiation (1486.8 eV) for excitation. X-ray diffraction (XRD) patterns were collected on a Germany Bruker D8 ADVANCE diffractometer equipped with Cu Kα radiation (λ = 1.5418 Å). Au was determined by an inductive coupled plasma atomic emission spectrometer (ICP, Atomscan Advantage, Thermo Jarrell Ash Corporation).3. Results and discussion
We obtained porous Au nanoparticles with different detailed structures under different temperatures. The products were characterized by transmission electron microscopy (TEM). Interestingly, the Au nanocrystals show a porous structure with a solid center when it was obtained at a low temperature of 160 °C (Fig. 1a). However, the morphology changes into a hollow architecture when the growth temperature was increased to a high temperature of 200 °C (Fig. 1b). It should be noted that the Au nanostructures still possess a porous feature when grown in this condition. Furthermore, the magnified TEM images clearly show that both types of Au nanoparticles are composed of particles of 5 nm. | ||
| Fig. 1 TEM image of the PSAuNPs obtained at 160 °C (a); and PHAuNPs obtained at 200 °C (b). | ||
The absorption spectra of the aqueous solutions of HAuCl4, GUC, and PHAuNPs were monitored at 292 nm, 260 nm, and 575 nm, respectively (Fig. S1†). We can find a blue shift in the absorption of GUC compared with that of HAuCl4, which may be caused by the formation of a complex between HAuCl4 and urea. Furthermore, inductive coupled plasma atomic emission spectrometer and elemental analyzer were used to analyze the element percentage of the sample (Table 1), from which the chemical formula of GUC was determined to be Au(N2H4CO)·H2O. Furthermore, the infrared spectra of GUC(I), urea(II), and PHAuNPs (III) were obtained (Fig. S2†). The spectra reveal that after an interaction between urea and gold chloride, the absorption peak in the spectrum of GUC shows a little shift from 1681 cm−1 to 1629 cm−1 compared to that in the spectrum of urea. Moreover, a characteristic band of terminal isocyanate at 2171 cm−1 and of a coordinated amide group at 3490–3400 cm−1 was detected. Additionally, the band at 574 cm−1 was attributed to ν(Au–N). However, the spectrum of PHAuNPs shows no characteristic bands of coordinated urea because of the high-temperature calcination.
| Element | Au | N | C | H |
|---|---|---|---|---|
| Actual value/wt% | 69.67 | 10.08 | 3.61 | 2.2 |
| Theory value/wt% | 71.63 | 10.18 | 4.36 | 2.18 |
The crystal structure of the products obtained from different calcination times was investigated by powder X-ray diffraction (XRD). As shown in Fig. 2, the lower blue line is the JCPDS data of the standard Au (card number 65–8601). The GUC (Fig. 2b, I) has no characteristic diffraction peak, indicating that Au nanocrystals were not formed. After GUC was calcined at 200 °C for about 20 min, the products turn brownish gray (Fig. 2a, II) and the characteristic peaks of Au appear in the spectra (Fig. 2b, II). Further, the characteristic peaks of gold in the purple-red products (Fig. 2a, III) become sharp as the calcination time was increased to 40 min (Fig. 2b, III). Moreover, the intensity of the peak observed at 35.49° decreased. Further, the XRD characteristic peaks of the black products (Fig. 2a, IV), obtained after calcination for 50 min (Fig. 2b, IV) were consistent with the standard diffraction peaks of Au at 38.19°, 44.43° and 64.69°, which correspond to (111), (200), (220), respectively. Therefore, this proved that the GUC was reduced to gold through a high-temperature reaction.
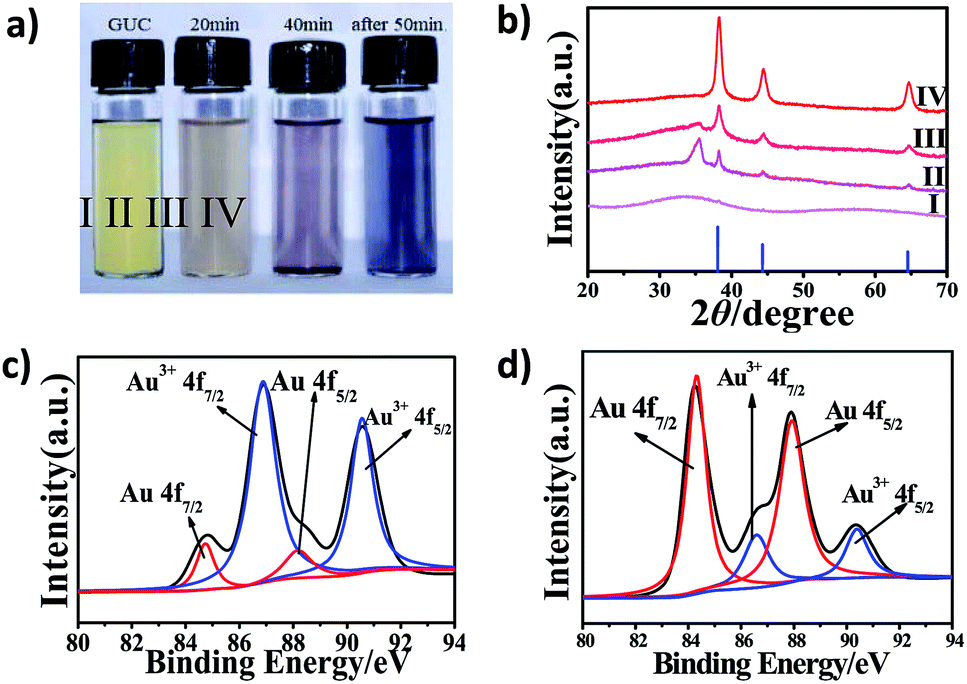 | ||
| Fig. 2 (a) The color change with time gradient calcination. (b) XRD patterns of products under different calcination time (I) GUC (origin), (II) 20 min, (III) 40 min, (IV) 60 min. XPS spectra of Au 4f of the as-prepared GUC (c) and PHAuNPs (d) deposited on the silica wafer. | ||
X-ray photoelectron spectroscopy (XPS) was further employed to characterize the chemical state of the GUC and PHAuNPs. The Au 4f XPS spectrum of the GUC displays two strong peaks at 86.89 and 90.56 eV (Fig. 2c), which was assigned to Au3+ 4f7/2 and Au3+ 4f5/2, respectively. This result indicates that Au3+ is the dominant species at this stage. Further, the N 1s peaks observed at 398.88 eV (Fig. S5a†) was assigned to C–N bonds. The C 1s at 284.83 eV, 285.98 eV, and 288.18 eV (Fig. S5b†) correspond to C–C bonds, C–O bonds, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.27 The survey spectrum again confirms that the GUC was composed of Au, N, C, and O. Thus, these results establish the metal–urea complex structure of GUC. After calcination at 200 °C, the XPS spectrum of the obtained product show two peaks at 84.32 and 87.77 eV, undoubtedly suggesting the Au0 chemical state (Fig. 2d). The above survey result demonstrates that the reactive metal ions were reduced from their positively charged state during the thermal annealing process.
O bonds, respectively.27 The survey spectrum again confirms that the GUC was composed of Au, N, C, and O. Thus, these results establish the metal–urea complex structure of GUC. After calcination at 200 °C, the XPS spectrum of the obtained product show two peaks at 84.32 and 87.77 eV, undoubtedly suggesting the Au0 chemical state (Fig. 2d). The above survey result demonstrates that the reactive metal ions were reduced from their positively charged state during the thermal annealing process.
We also investigated the growth process in this study. Initially, GUC was slightly yellow in color (Fig. S3a†). The corresponding samples exhibited a spherical morphology and a good size dispersion of ∼50 nm diameter, as characterized at low magnification under SEM imaging (Fig. S3b and c†). A single nanoparticle of GUC was focused and amplified under a transmission electron microscope, where it was observed that the GUC changed from a solid spherical structure to a porous and hollow structure in a short time, which was due to the strong electron beams (Fig. S3e†).
As mentioned above, the GUC changed into porous Au nanoparticles by the strong electron beams under the high magnified TEM. Hence, we captured the beam irradiation time-dependent TEM images. Fig. S4a† shows the original low magnification TEM image of GUC (fast-focus shooting). The GUC dispersed homogeneously, with the increase in time. In addition, an increasingly large number of cavities were observed inside the GUC. Meanwhile, the solid parts also began to produce porous structures, and the nanoparticles started to fuse and attach to each other (Fig. S4b to d†). This indicates that the complexes will be reduced to PHAuNPs by giving high energy gradually. Hence, the GUC was calcined to obtain PHAuNPs. After the evaporation of GUC/ethanol dispersion, the color of the products changed from yellow to brownish gray, then turned purple-red, and finally turned black (Fig. 2a).
We further explored the mechanism of the reaction, which reveals that the GUC shape changes on heating under different conditions. The amorphous GUC shows a good dispersion of 50 nm (Fig. 3b). After incinerating at 160 °C, the PSAuNPs were formed with sizes of around 5 nm and a gap of 1 nm both internally and externally (Fig. 3d). When the PHAuNPs were obtained on calcination at 200 °C for about 30 to 50 min (Fig. 3f), the nanostructure maintained the original size of 50 nm, while the nanoparticles of 5 nm size were arranged with a gap of 1 nm externally to form the hollow structure. We speculate that the reason for the temperature-dependent morphological change was the rapid thermal decomposition of urea to NH3 and CO2 at 200 °C. First, the shell of the PHAuNPs was formed due to the fast heating. Urea and ethanol acted together as weak reducing agents, and the gas generated by urea pyrolysis because of the surface tension induces gold particles to move to the sphere surface to form the porous hollow structure. However, urea cannot pyrolyze rapidly at 160 °C. The whole particle was heated uniformly and decomposed and reduced slowly, and the internal gas could not be released quickly to induce the assembly of reduced gold particles to move to the sphere surface. As a result, the PSAuNPs were formed. Both PHAuNPs and PSAuNPs will eventually fusion and deform when heated after 50 min. The small particles overlapped and fused with each other. Finally, larger particles with a diameter of about 20 nm were formed embedded in the polyhedron (Fig. 3h).
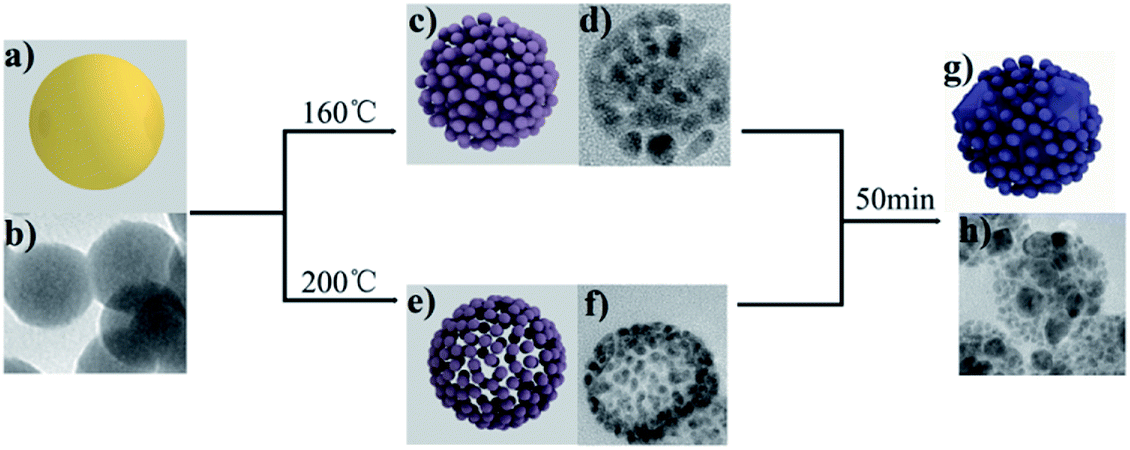 | ||
| Fig. 3 (a) The schematic diagram and (b) TEM image of GUC; (c) the schematic diagram and (d) TEM image of PSAuNPs obtained at 160 °C; (e) the schematic diagram and (f) TEM image of PHAuNPs obtained at 200 °C; (g) the schematic diagram and (h) TEM images of Au calcined after 50 min. | ||
The catalytic properties of PHAuNPs were evaluated via the reduction reaction of 4-NP to 4-AP in the presence of excess NaBH4, which was monitored by collecting ultraviolet-visible absorption (UV-vis) spectra.28 When NaBH4 was added to 4-NP, a yellow solution was obtained, and the time-dependent absorption spectra were measured immediately. The spectra show a maximum absorption peak at 400 nm, which was the absorption peak of 4-nitrophenolate. Further, the yellow solution without catalysts was placed indoors for 18 hours before characterizing it using a UV-vis spectrophotometer. The red line showed no significant change (Fig. S5†), indicating that 4-nitrophenol reduced slowly. After the addition of PHAuNPs, the absorption peak at 400 nm decreases rapidly, and a new absorption peak appeared at 300 nm. This was mainly due to the catalytic reduction of 4-NP to 4-AP (Fig. 4a). As the reaction proceeded, the maximum absorption peak (400 nm) of 4-NP decreased rapidly and disappeared completely after the reaction was carried out for just 240 s. Since the amount of NaBH4 was in excess, this reduction reaction was assumed to follow pseudo first-order kinetics, and the rate constant was thus utilized to evaluate the reaction rate. Fig. 4b shows the linear fit between the reaction time and −ln(Ct/C0) for the catalytic reduction of 4-NP using PHAuNPs as the catalyst. The kinetic rate constant Kapp, calculated directly from the slope, was estimated to be 12.1 × 10−3 s−1.
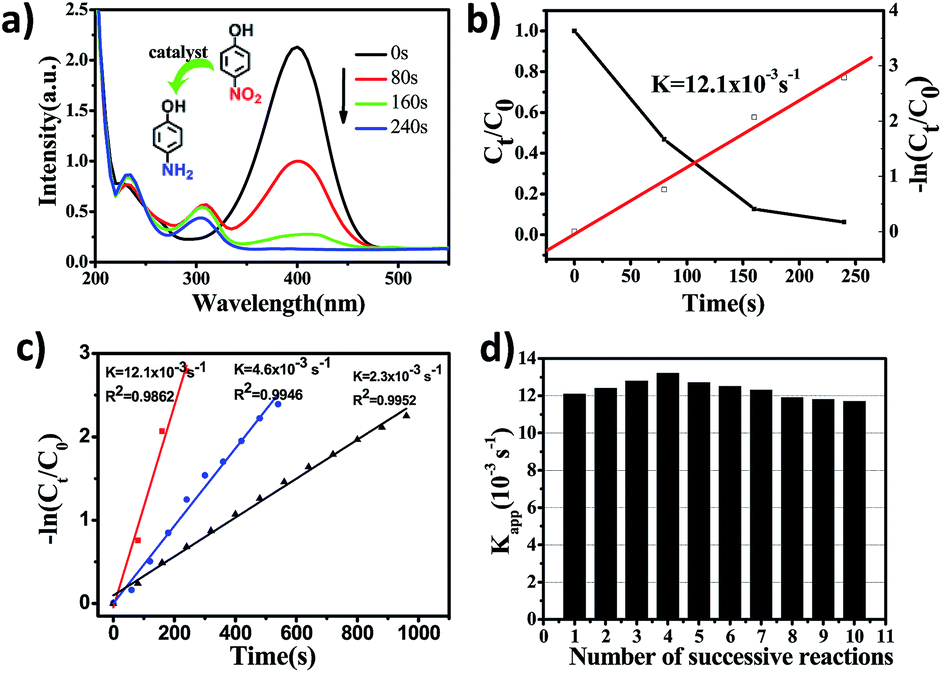 | ||
| Fig. 4 (a) Time-dependent absorption spectra of the reduction of 4-NP to 4-AP catalyzed by PHAuNPs. (b) The black and white squares stand for the Ct/C0 and −ln(Ct/C0) versus reaction time, respectively, for the reduction of 4-NP by NaBH4 over PHAuNPs. C0 corresponds to the initial intensity of the absorption at 400 nm, and Ct is the absorption peak at different reaction time (t). (c) −ln(Ct/C0) versus reaction time for the reduction of 4-NP for PHAuNPs (red line), PSAuNPs (bule line) and commercial solid gold nanoparticles (black line), (d) Kapp values under different catalytic cycles in the reduction of 4-NP to 4-AP in the presence of PHAuNPs. | ||
In order to prove the enhancement in the catalytic activity of PHAuNPs, PSAuNPs and the commercial solid gold nanoparticles with the same size were used as catalysts for a comparison. The catalytic reduction of 4-NP to 4-AP was also used to discuss the catalytic activity of the PSAuNPs and commercial solid gold nanoparticles (Fig. S7 and S8†). Compared to PHAuNPs, the catalytic rates of the PSAuNPs and commercial solid gold nanoparticles were lower, with the Kapp being 4.6 × 10−3 s−1and 2.3 × 10−3 s−1, respectively (Fig. 4c). In other words, the as-prepared PHAuNPs show an accelerated reaction rate when they were employed as the catalysts, which was nearly 2.6 and 6 times that of PSAuNPs and commercial gold catalysts, respectively. The excellent catalytic performance of PHAuNPs was attributed to the porous and hollow structure. First, the porous cavity composed of about 5 nm gold nanoparticles endows the PHAuNPs with a larger specific surface area, which in turn provides more reactive sites for the same size. Second, the hollow structure was more favorable for the exchange of the substrate on the catalyst than the porous and solid structure. Furthermore, the surface of the samples was not covered by the surfactant. The PHAuNPs can thus provide more effective reactive sites. As we all know, recyclability is also very important for nanocatalysts in practical applications.29 The catalytic reduction product of 4-AP will adsorb on the surface of catalysts, which can lead to catalyst poisoning.30 We thus also investigated the recyclability of the PHAuNPs catalysts. Fig. 4d displays the values of Kapp for the successive reactions. It was found that the catalytic activity of the PHAuNPs slightly increased after the first run. Presumably, the catalyst was activated by NaBH4. The Kapp after ten cyclic measurements for the reduction of 4-NP could still reach 11.7 × 10−3 s−1, and exhibits almost no change compared with the initial cycles.
4. Conclusions
In summary, we have synthesized PHAuNPs using a surfactant-free method. The obtained Au nanocrystals show a novel porous and hollow structure, which provides high active sites for the catalytic reaction. Moreover, the surface of the PHAuNPs was clean, which is also beneficial for improving the catalytic activity. The catalytic performance of the as-prepared porous PHAuNPs toward the reduction of 4-NP to 4-AP revealed a 6-fold higher Kapp than that of the commercial gold nanoparticles. The stability was also found to be highly improved through cycling tests. Therefore, this study provides a new way to construct clean noble metal nanoparticles with special structures and properties in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Science Foundation of China (21301008, 21175002), Anhui Science and Technology Major Project (17030801021), Open Foundation of CAS Key Laboratory of Analytical Chemistry for Living Biosytems, ACL, Open Foundation of The Key Laboratory of Functional Molecular Solids, Ministry of Education (14007) and the members of Institute of Synthesis and Application of Medical Materials.References
- W. He, X. Wu, J. Liu, X. Hu, K. Zhang, S. Hou, W. Zhou and S. Xie, Chem. Mater., 2010, 22, 2988–2994 CrossRef CAS.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, J. Mater. Chem. B, 2014, 2, 4204–4220 RSC.
- H. Cai and P. Yao, Nanoscale, 2013, 5, 2892–2900 RSC.
- Y. Niidome, A. T. Haine and T. Niidome, Chem. Lett., 2016, 45, 488–498 CrossRef CAS.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed. Engl., 2012, 51, 602–613 CrossRef CAS PubMed.
- W. Si, J. Li, H. Li, S. Li, J. Yin, H. Xu, X. Guo, T. Zhang and Y. Song, Nano Res., 2013, 6, 720–725 CrossRef CAS.
- W. Yang, Q. Zhang and C. Peng, RSC Adv., 2019, 9, 931–939 RSC.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- H. Chen, F. Yuan, S. Wang, J. Xu, Y. Zhang and L. Wang, Biosens. Bioelectron., 2013, 48, 19–25 CrossRef CAS PubMed.
- J. Chen, C. Glaus, R. Laforest, Q. Zhang, M. Yang, M. Gidding, M. J. Welch and Y. Xia, Small, 2010, 6, 811–817 CrossRef CAS PubMed.
- H. Yuan, A. M. Fales and T. Vo-Dinh, J. Am. Chem. Soc., 2012, 134, 11358–11361 CrossRef CAS PubMed.
- G. W. Nyce, J. R. Hayes, A. V. Hamza, H. Joe and J. H. Satcher Jr, Chem. Mater., 2007, 19, 344–346 CrossRef CAS.
- Y. Qin, Y. Lu, W. Pan, D. Yu and J. Zhou, RSC Adv., 2019, 9, 10314–10319 RSC.
- Z. Qi, U. Vainio, A. Kornowski, M. Ritter, H. Weller, H. Jin and J. Weissmüller, Adv. Funct. Mater., 2015, 25, 2530–2536 CrossRef CAS.
- H. Wu, P. Wang, H. He and Y. Jin, Nano Res., 2012, 5, 135–144 CrossRef CAS.
- S. Pedireddy, H. K. Lee, W. W. Tjiu, I. Y. Phang, H. R. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 4947 CrossRef CAS PubMed.
- R. Kumar, A. N. Maitra, P. K. Patanjali and P. Sharma, Biomaterials, 2005, 26, 6743–6753 CrossRef CAS PubMed.
- F. Wang, C. Li, L. D. Sun, C. H. Xu, J. Wang, J. C. Yu and C. H. Yan, Angew. Chem., Int. Ed. Engl., 2012, 51, 4872–4876 CrossRef CAS PubMed.
- M. Chen, B. Wu, J. Yang and N. Zheng, Adv. Mater., 2012, 24, 862–879 CrossRef CAS PubMed.
- S. Wang, L. Kuai, Y. Huang, X. Yu, Y. Liu, W. Li, L. Chen and B. Geng, Chem.–Eur. J., 2013, 19, 240–248 CrossRef CAS PubMed.
- Q. Cao, K. Yuan, J. Yu, J. J. Delaunay and R. Che, J. Colloid Interface Sci., 2017, 490, 23–28 CrossRef CAS PubMed.
- T. T. Guo, G. Yu, Y. Zhang, H. Y. Xiang, F. F. Chang and C. J. Zhong, J. Phys. Chem. C, 2017, 121, 3108–3116 CrossRef CAS.
- A. T. Miah, S. K. Bharadwaj and P. Saikia, Powder Technol., 2017, 315, 147–156 CrossRef CAS.
- M. A. Wall, S. Harmsen, S. Pal, L. Zhang, G. Arianna, J. R. Lombardi, C. M. Drain and M. F. Kircher, Adv. Mater., 2017, 29, 1605622 CrossRef PubMed.
- C. H. Lai, I. C. Wu, C. C. Kang, J. F. Lee, M. L. Ho and P. T. Chou, Chem. Commun., 2009, 15, 1996–1998 RSC.
- M. Z. Guo, J. He, Y. Li, S. Ma and X. H. Sun, J. Hazard. Mater., 2016, 310, 89–97 CrossRef CAS PubMed.
- W. Wen, J. Yao, Y. Luo, L. Lai and J. Wu, Ceram. Int., 2018, 44, 4381–4384 CrossRef CAS.
- K. Gu, X. Pan, W. Wang, J. Ma, Y. Sun, H. Yang, H. Shen, Z. Huang and H. Liu, Small, 2018, 14, 1801812 CrossRef PubMed.
- X. Lu, X. Bian, G. Nie, C. Zhang, C. Wang and Y. Wei, J. Mater. Chem., 2012, 22, 12723 RSC.
- T. Yu, J. Zeng, B. Lim and Y. Xia, Adv. Mater., 2010, 22, 5188–5192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04372a |
| This journal is © The Royal Society of Chemistry 2019 |