
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-decorated vanadium pentoxide for room temperature ammonia sensing
Shobha N. Birajdara,
Neha Y. Hebalkarb,
Satish K. Pardeshi c,
Sulabha K. Kulkarni*a and
Parag V. Adhyapak*a
c,
Sulabha K. Kulkarni*a and
Parag V. Adhyapak*a
aCentre for Materials for Electronics Technology (C-MET), Panchwati, Off Pashan Road, Pune-411008, India. E-mail: adhyapak@cmet.gov.in; Fax: +91-20-25898180; Tel: +91-20-25899273
bInternational Advanced Research Centre for Powder Metallurgy and New Materials, Hyderabad-500005, India
cSavitribai Phule Pune University, Pune-411007, India
First published on 12th September 2019
Abstract
Layer structured vanadium pentoxide (V2O5) microparticles were synthesized hydrothermally and successfully decorated by a facile wet chemical route, with ∼10–20 nm sized ruthenium nanoparticles. Both V2O5 and ruthenium nanoparticle decorated V2O5 (1%Ru@V2O5) were investigated for their suitability as resistive gas sensors. It was found that the 1%Ru@V2O5 sample showed very high selectivity and sensitivity towards ammonia vapors. The sensitivity measurements were carried out at 30 °C (room temperature), 50 °C and 100 °C. The best results were obtained at room temperature for 1%Ru@V2O5. Remarkably as short a response time as 0.52 s @ 130 ppm and as low as 9.39 s @ 10 ppm recovery time at room temperature along with high selectivity towards many gases and vapors have been noted in the 10 to 130 ppm ammonia concentration range. Short response and recovery time, high reproducibility, selectivity and room temperature operation are the main attributes of the 1%Ru@V2O5 sensor. Higher sensitivity of 1%Ru@V2O5 compared to V2O5 has been explained and is due to dissociation of atmospheric water molecules on 1%Ru@V2O5 as compared to bare V2O5 which makes hydrogen atoms available on Brønsted sites for ammonia adsorption and sensing. The presence of ruthenium with a thin layer of oxide is clear from X-ray photoelectron spectroscopy and that of water molecules from Fourier transform infrared spectroscopy.
1 Introduction
Vanadium oxides and their composites are of considerable research interest due to their selective catalytic and sensing activity, potential applications in electrochemical capacitors or supercapacitors, as well as potential for thermoelectric energy application.1–6 Catalytic and sensing activities mainly arise due to the interesting electronic structure of vanadium and its oxides. Vanadium has 3d34s2 electrons in its outer shell, which enables it to be in various oxidation states viz. V2+, V3+, V4+ and V5+. This makes it a complex as well as interesting material. Amongst the various vanadium oxides, vanadium pentoxide (V2O5) is widely used and is stable. It has been synthesized using various physico-chemical methods such as co-precipitation, vapor-solid method of thermal evaporation, hydrothermal, pulsed laser deposition etc.3,4,7,8 The literature also reports wire, belt, tube, needle, hollow nano-assemblies or flower like morphologies for V2O5 which depend upon the precursors used and synthesis procedures.8–17 However there is no report so far on small V2O5 particles catalyzed by ruthenium in detection of ammonia at room temperature which is an important task. Vanadium oxide is a well-known catalyst in reduction of NOx, NH3.18,19 Theoretical framework for ammonia adsorption on vanadium oxide also is available.20,21 Moreover there are also some reports which have indicated role of RuO2 in increasing the sensitivity of tin oxide towards ammonia.22 Ruthenium nanoparticles are likely to be covered with thin oxide layer. Therefore we have chosen to use ruthenium decorated V2O5 as a novel sensor for ammonia detection.Ever increasing ammonia level in atmosphere as well as water is of great concern. Ammonia level from few ppb to tens of ppb in air is permissible. Ammonia is released by various rural and urban human activities like agriculture (use of fertilizers), husbandry, chemical industry, cold storage or refrigeration plants of food storage steadily causing increase of ammonia in air. Small quantities of dissolved ammonia in water can become threat to marine life. Continuous inhaling up to 35 ppm for 15 minutes and maximum 8 h exposure at 25 ppm is just safe according to the US Agency for Toxic Substances and Disease Registry (ATDSR CAS # 7664-41-7). Major threat of ammonia can be from industrial places and large cold storages of food as is evident from major accident like West Texas Fertilizer (USA) plant in 2013 which not only completely destroyed the establishment but also killed many lives. A similar accident took place in 2017 at a cold storage in Kanpur, India killing at least five workers. Recently, in 2019 also ammonia leakage is reported in Lucknow, India. Thus it is very important to control leakage of ammonia in its early stage. Proper alarm systems must be equipped with appropriate sensors i.e. fast response time and recovery time, selectivity, reproducible as well as operating at low temperature, preferably room temperature. Room temperature operation eliminates the cost of electricity needed for heating the sensor as well as avoids catching fire in case of any accidental explosion in its neighborhood.
There are some reports on the use of V2O5 as an ammonia gas sensor.10,12,17 However, the sensitivity of V2O5 towards ammonia is usually low and response as well as recovery times are large. Surface sensitization is often made to increase the sensing of V2O5/VO2 by using noble metals or graphene.3,13,23,24 Some nanocomposites also have been reported.25–28 V2O5 has also been deposited by many groups in porous silicon structure.29–31 Novel assemblies of V2O5 have been reported by Wang et al. which improved the hydrogen gas sensing activity.2 The attempts using graphene functionalized V2O5 could improve the sensitivity towards NH3 gas but response and recovery times were still few tens of seconds.3
Here we report, a facile method using hydrothermally synthesized V2O5 decorated with ruthenium nanoparticles (1%Ru@V2O5), which serves as a highly sensitive and selective sensor with 0.52 s @ 130 ppm and 1.57 s @ 10 ppm, response time for ammonia. The recovery times are 15.13 s @ 130 ppm and 9.39 s @ 10 ppm. This novel ruthenium nanoparticles decorated vanadium oxide sensor is capable of operating at room temperature. The sensitivity of the sensor is ∼4% for 130 ppm of ammonia, which is quite good for V2O5 ammonia sensor.
Field Emission Scanning Electron Microscopy (FE-SEM) images revealed that V2O5 had poly-dispersed microparticles having layered structure. 1%Ru@V2O5 had a more interesting morphology. One could clearly see that some Ru nanoparticles were decorated on the layered V2O5 microparticles. Energy Dispersive Analysis of X-rays (EDAX) of both V2O5 and 1%Ru@V2O5 also was carried out for determining the composition of the sample along with the presence of any contamination in the samples. Transmission Electron Microscopy (TEM) images also clearly showed presence of ruthenium particles on the V2O5 surface of 1%Ru@V2O5 sample. Further analysis using High Angle Annular Dark Field (HAADF) and Scanning Transmission Electron Microscopy (STEM) also was carried out to record more morphological, structural as well as compositional details. V2O5 had orthorhombic and ruthenium particles had hexagonal structure, as found from X-ray Diffraction (XRD) analysis. Sensitivity analysis was carried out by measuring the changes in the resistance of the samples with or without the presence of ammonia. X-ray Photoelectron Spectroscopy (XPS) analysis was carried out to find out the elemental composition as well as oxidation states of vanadium, ruthenium and oxygen in the samples under investigation.
2 Materials and methods
2.1 Materials
All A. R. grade reagents were used for the synthesis of materials. Ammonium vanadate, NH4VO3, (May and Baker laboratory chemicals, 99.0%), maleic acid (HO2CCHCHCO2H, 99.0%), and polyethylene glycol (C2nH4n+2On+1), PEG-400, (S.D fine chemicals), absolute ethanol (99.9%), ruthenium chloride hydrate RuCl3·xH2O (Sigma Aldrich 98%), hydrazine hydrate, H6N2O (Loba chemie, 99.0%) and deionised water were used in the experiment (Scheme 1).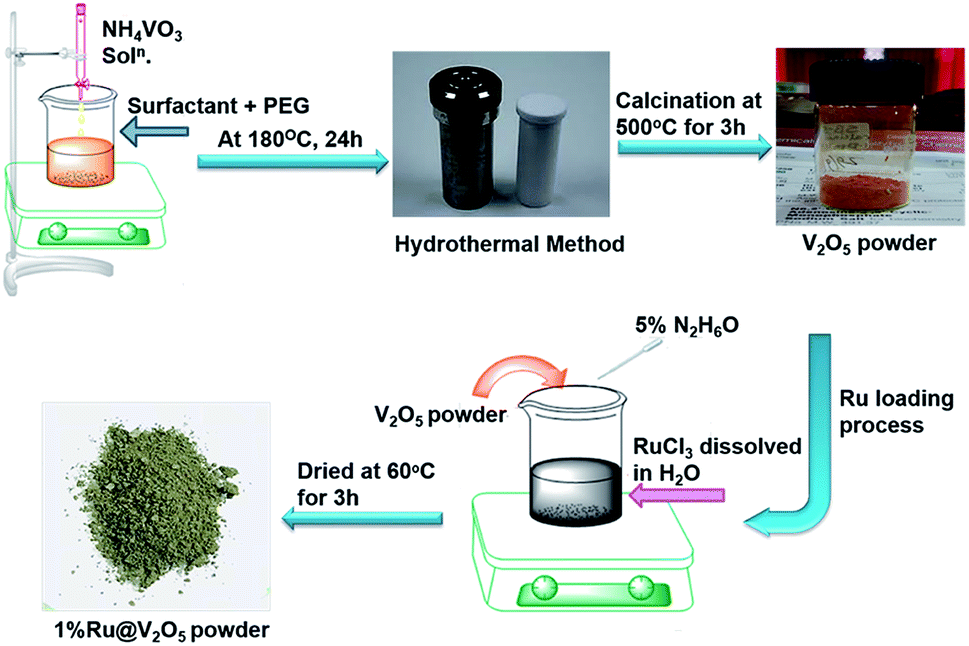 | ||
| Scheme 1 Schematic representation of V2O5 and 1%Ru@V2O5 synthesis. | ||
2.2 Synthesis of V2O5
Synthesis of V2O5 was performed by hydrothermal method as follows. 1 mole of maleic acid (surfactant) was dissolved in PEG-400-water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). Further, 0.5 mole of NH4VO3 was dissolved separately in 50 mL of deionized water. This solution was added dropwise to the maleic acid and PEG-water solution under constant stirring, which then turns from a colourless solution to orange solution. After complete addition, the solution was stirred for about 2 h at room temperature (∼30 °C). This reaction mixture was then transferred into 250 mL Teflon lined stainless steel autoclave, kept at 180 °C for 24 h. After this, autoclave was cooled to room temperature. Black precipitate was obtained when the solution was centrifuged at ∼5000 rpm for 15 min. The precipitate was then washed several times with deionized water and finally with ethanol. It was dried at 60 °C for 4–5 h. The dried product was calcined at 500 °C for 3 h. This resulted into a yellowish orange V2O5 powder.
:10). Further, 0.5 mole of NH4VO3 was dissolved separately in 50 mL of deionized water. This solution was added dropwise to the maleic acid and PEG-water solution under constant stirring, which then turns from a colourless solution to orange solution. After complete addition, the solution was stirred for about 2 h at room temperature (∼30 °C). This reaction mixture was then transferred into 250 mL Teflon lined stainless steel autoclave, kept at 180 °C for 24 h. After this, autoclave was cooled to room temperature. Black precipitate was obtained when the solution was centrifuged at ∼5000 rpm for 15 min. The precipitate was then washed several times with deionized water and finally with ethanol. It was dried at 60 °C for 4–5 h. The dried product was calcined at 500 °C for 3 h. This resulted into a yellowish orange V2O5 powder.
2.3 Synthesis of 1%Ru@V2O5
Precursor RuCl3 (0.0102 g) was taken in a beaker and 30 mL of deionised water was added in it with continuous stirring till it was completely dissolved giving a clear solution. V2O5 (0.4897 g) was then added to the above solution. In another beaker 1 mL of hydrazine hydrate was mixed in 19 mL deionized water and the solution was dropwise added to the precursor solution till it changed to olive green colour. The supernatant was removed and precipitate was dried at 60 °C for 3 h, which resulted into an olive green powder.2.4 Material characterization
The structural analysis of the V2O5 and 1%Ru@V2O5 was made using X-ray diffraction (XRD) technique. Rigaku Miniflex X-ray diffractometer equipped with copper target (CuKα, λ = 1.5406 Å) and nickel filter was used to record the diffraction patterns. Powder samples were held on glass substrates.Field Emission Scanning Electron Microscopy (FE-SEM) was performed with a JEOL-JSM Model 6700F having Energy Dispersive Analysis of X-rays (EDAX) attachment. For the analysis, silicon substrates were used on which a thin layer of powder samples was spread. Transmission Electron Microscopy (TEM) analysis of V2O5 and 1%Ru@V2O5 samples was carried using TEM 2200FS JEOL, equipped with High Angle Annular Dark Field (HAADF) and Scanning Transmission Microscopy (STEM) facilities, operated at 200 keV electron energy. The samples were prepared by putting a drop of solution containing samples on copper grids.
The reflectance spectra were recorded on Jasco-V-770 spectrophotometer. The samples were pressed in a quartz sample holder cavity and measurements were done in the spectral range 200 to 1200 nm.
Fourier transform infrared spectroscopy (FTIR) analysis of V2O5 and 1%Ru@V2O5 samples was carried out using a Bruker TENSOR37. Samples were kept in the stainless steel sample holder for measurements in the range from 400 to 4000 cm−1.
X-ray Photoelectron Spectroscopy (XPS) analysis was carried out using ESCA + model of Omicron, UK. AlKα (X-ray energy 1253.6 eV) was used as the source of X-rays. Photoelectron spectra were recorded using hemispherical analyzer held at 50 eV pass energy and CASA XPS software was used for the analysis of the spectra.
2.5 Fabrication and sensing measurements
Sensing measurements on V2O5 and 1%Ru@V2O5 samples were performed in a static system (shown in Scheme 2) as per the procedure reported earlier.32 Briefly it is made of an airtight glass dome with inlet and outlet for gases/vapors. The vapors can be injected using a syringe though a metallic tube connected to the steel base plate of the glass dome. A water suction pump is used to remove the gases from the chamber. There is a provision of heater along with thermocouple at the base of the dome. The alumina substrate (0.6 mm thick and 10 × 10 mm area) was used for the fabrication of interdigitated electrode (IDE) made up of silver that occupies an area of 8 × 8 mm. The width of the digits is 1 mm and the space between them is 0.5 mm. The IDE is kept on the heater and connected to a Keithley Digital Multimeter (model DMM 7510, 7½ Digital multimeter) equipped with computer. The change in resistance with time was recorded on computer with KickStart software.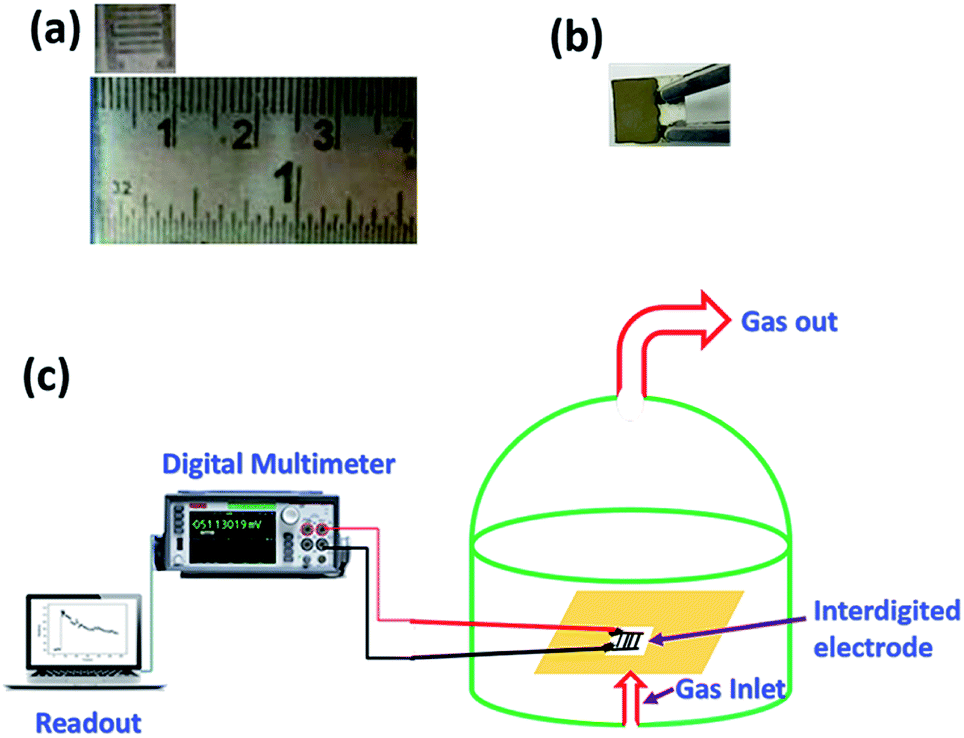 | ||
| Scheme 2 Photograph of (a) interdigitated electrode (IDE) (b) sample coated on the IDE (c) schematic representation of sensing set up. | ||
For sensing measurements, the sample is coated on IDE. The gases/vapors are injected through the syringe and corresponding change in resistance is measured. After stipulated time the resistance value starts dropping down due to desorption of gases. Then the desorbed gas is driven out of the chamber by using a water suction pump. Sensitivity of the sample can be then determined by finding out the change in the resistance from ambient and that occurs when the film is in contact with a gas. Thus, initial (in ambient air) resistance of the thin film if Ra and that in the contact with a test gas is Rg then the percentage sensitivity or the gas response S% can be calculated with the equation,
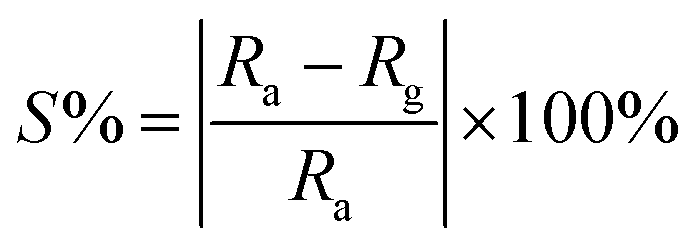 | (1) |
In these experiments various oxidizing gases such as SO2, NOx and reducing vapors like C2H6O (ethanol), CH4O (methanol), C3H6O (acetone), CH2O (formaldehyde), NH3 (ammonia), C6H15N (triethylamine), C3H9N (trimethylamine) were used.
3 Results and discussion
‘As received’ pristine V2O5 powder resulting from the hydrothermal synthesis was yellowish orange in color, characteristic of V2O5. The hydrothermal synthesis of V2O5 can be explained using following eqn (2)–(5).33| NH4VO3 → NH4+ + VO3− | (2) |
| 2VO3− + 8H+ → 2VO2+ + 4H2O | (3) |
| 2VO2+ + NH4+ + 2C4H4O4 → (NH4)2[2VO(C4H4O4)2] | (4) |
| (NH4)2[2VO(C4H4O4)2] + 8O2 → V2O5 + 2NH3 + 8CO2 + 5H2O | (5) |
There are three polymorphs of V2O5 viz. α-V2O5, β-V2O5 and γ-V2O5 having same yellowish orange color.34,35 The differences in the polymorphs arise due to their structures viz. their lattice parameters as well as the way in which the V2O5 pyramids are oriented or stacked. In α-V2O5, a = 11.516 Å, b = 3.5656 Å and c = 4.3727 Å [JCPDS card # 41-1426]. Whereas in β-V2O5, a = 7.1140 Å, b = 3.5718 Å and c = 6.2846 Å.20 β-V2O5 is metastable at room temperature and can be observed under high pressure. γ-V2O5 crystallizes with lattice parameters a = 9.9461 Å, b = 3.5852 Å and c = 10.0423 Å.20
As discussed in the introduction, the formation of vanadium oxides depends on the synthesis and processing procedure as well as precursors used. Therefore, we characterized the V2O5 powder using XRD. Lower curve in Fig. 1(a) illustrates the XRD pattern of the V2O5 sample synthesized here. It is in good agreement with the JCPDS card # 41-1426 for orthorhombic α-V2O5. This is also confirmed by the characteristic FTIR spectrum shown in Fig. 1(b). It was discussed by Pinna et al. that the absorption peak around 1009 cm−1 in FTIR, assigned to V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, splits into two components in γ-V2O5.36 Absence of such splitting in the peak at 1009 cm−1 confirms the assignment using XRD that the sample is indeed in α-V2O5 phase. We can also see that all the peaks of α-V2O5 appear in the sample without any peaks due to contaminants. Thus we have obtained a good quality α-V2O5 polycrystalline sample.
O stretching, splits into two components in γ-V2O5.36 Absence of such splitting in the peak at 1009 cm−1 confirms the assignment using XRD that the sample is indeed in α-V2O5 phase. We can also see that all the peaks of α-V2O5 appear in the sample without any peaks due to contaminants. Thus we have obtained a good quality α-V2O5 polycrystalline sample.
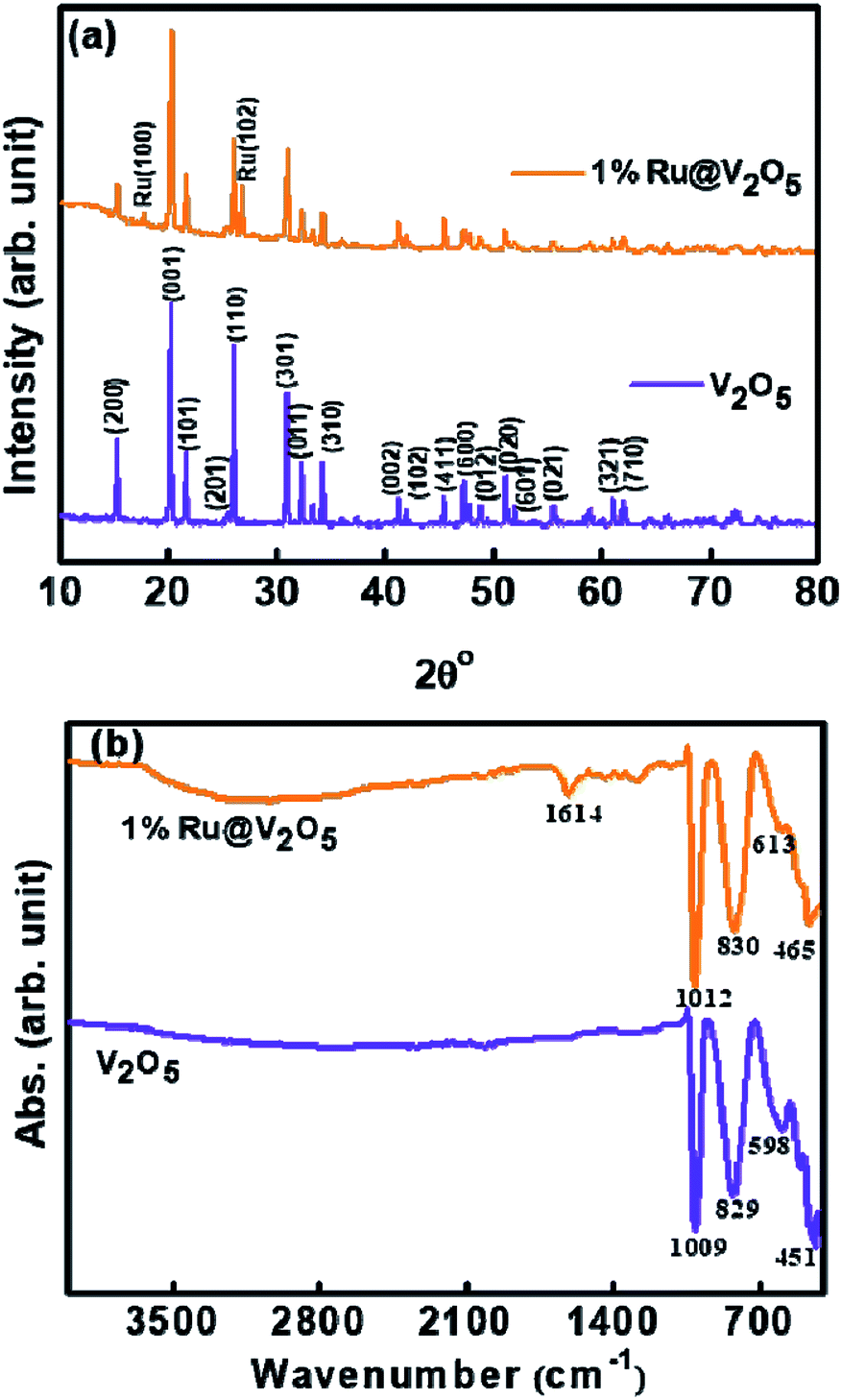 | ||
| Fig. 1 (a) X-ray diffraction and (b) FTIR of V2O5 and 1%Ru@V2O5 powders. | ||
XRD pattern (upper curve) of 1%Ru@V2O5 is also shown in Fig. 1(a). It is noticed that all the peaks of V2O5 are present along with two more peaks. These new peaks are due to (100) and (102) of hexagonal phase of ruthenium in agreement with (JCPDS card # 01-1256) for ruthenium. Careful analysis of XRD pattern suggests that these peaks belong to metallic ruthenium and there is no indication of any oxide formation. However, from XRD alone the possibility of any ruthenium oxide or sub-oxide on the surface as a thin layer cannot be confirmed.
Fig. 1(b) shows FTIR spectra of as-synthesized V2O5 (lower curve) and 1%Ru@V2O5 (upper curve) samples. In case of pristine V2O5 there are four peaks located at 451, 598, 829 and 1009 cm−1, which are characteristic peaks of orthorhombic V2O5.1
The peaks observed at 451 and 598 cm−1 correspond to symmetric and asymmetric vibrations of triply coordinated oxygen, whereas peaks located at 829 and 1009 cm−1 correspond to vibrations of bridge oxygen and stretching vibrations of V5+O respectively. It is observed that the peak at 829 cm−1 in FTIR of the pristine V2O5 is only slightly shifted to 830 cm−1 in case of 1%Ru@V2O5 sample. But the peaks at 451, 598 and 1009 cm−1 shift by larger amount to 461, 613 and 1012 cm−1 respectively in 1%Ru@V2O5 sample. The shift in the wavenumber to higher value here indicates that there are small bond length decreases in V2O5 due presence of Ru. Additionally, in case of 1%Ru@V2O5 two new peaks, one sharp at 1614 cm−1 and one very broad peak centered at ∼3000 cm−1 can be seen. The peak at 1614 cm−1 has been identified as hydrogen bonded surface water molecule.37 The peak centered around 3000 cm−1 can be attributed to adsorbed water molecules. The absence of peaks around 3000 cm−1 and 1614 cm−1 on pristine V2O5 surface are consistent with the theoretical calculations of Yin et al. which show that water adsorption is possible on V2O5 surface but water dissociation does not take place on V2O5.38
Fig. 2 depicts the FE-SEM patterns of V2O5 and 1%Ru@V2O5. The V2O5 sample in Fig. 2(a) and 1%Ru@V2O5 in Fig. 2(b) show formation of large, irregular flakes of couple of micrometer sizes with layered structure. EDAX (not shown here) analysis revealed that V is 8.82 at% and O is 91.82 at% in V2O5. No impurities were detected in EDAX. In case of 1%Ru@V2O5, although there is a presence of ruthenium, we do not clearly see the ruthenium particles in the FESEM images. Therefore, we performed TEM analysis of the V2O5 and 1%Ru@V2O5 samples.
 | ||
| Fig. 2 FE-SEM images of (a) V2O5 and (b) 1%Ru@V2O5. | ||
The TEM images of V2O5 and 1%Ru@V2O5 are shown in Fig. 3. In Fig. 3(a) one can see a number of particles of few hundreds on nm in agreement with FESEM of Fig. 2(a) for V2O5. The typical lattice fringes (Fig. 3(b)) show d spacing of 0.40 nm which is the lattice spacing in (001) set of planes for V2O5. Electron diffraction pattern of V2O5 as obtained from TEM is illustrated in Fig. 3(c). The intense diffractions spots without any rings indicate that there are single crystalline particles present in the V2O5 sample. The middle panel of Fig. 3 shows the TEM images of 1%Ru@V2O5. Fig. 3(d) shows the image of a typical particle in which particle surrounded by nanoparticles can be seen. For the sake of the guideline to the eye, the surface region with numerous particles is shown with the dotted lines. Using digital micrography software the ruthenium nanoparticle sizes were found to be 10–20 nm. The lattice fringes observed in Fig. 3(e) with lattice spacing ∼0.22 nm could be assigned to Ru (100) planes. A diffraction pattern in Fig. 3(f) shows well defined diffraction spots. Ru (100) spots could be identified in Fig. 3(f). Hexagonal structure of ruthenium also is clearly observed in this pattern. Thus the surface of V2O5 particles is decorated with crystalline ruthenium nanoparticles. The EDAX pattern in Fig. 3(g) also shows presence of ∼1.51 at% ruthenium in 1%Ru@V2O5 sample. Vanadium and oxygen at% is 76.35 and 22.14 respectively.
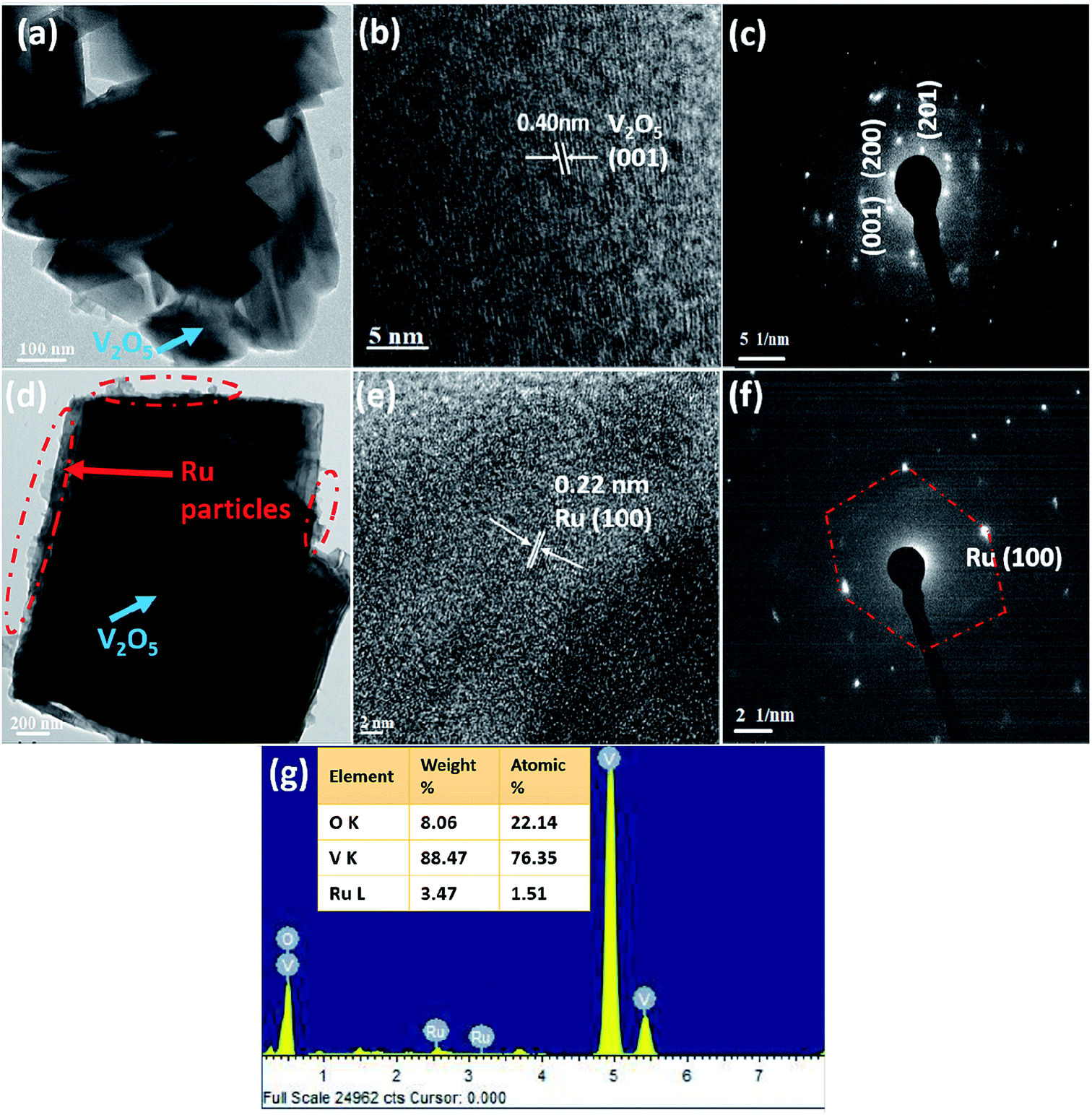 | ||
| Fig. 3 FE-TEM analysis (a) overview image of V2O5 particles (b) lattice resolved image of V2O5 (c) electron diffraction of V2O5 (d) a single particle of 1%Ru@V2O5 (e) lattice resolved image of 1%Ru@V2O5 (f) electron diffraction of 1%Ru@V2O5 (g) EDAX of 1%Ru@V2O5. | ||
Fig. 4(a) shows the representative HAADF image of an edge of a 1%Ru@V2O5 sample. Particles can be clearly seen but to know their composition corresponding elemental mapping was performed and shown in Fig. 4(b–d). The elemental mapping of 1%Ru@V2O5 reveals the distribution of O (green color) V (red color) and Ru (greenish blue). It is observed that at the edges of the flake, ruthenium particles are concentrated. Fig. 4(e) is the overlay of O, V and Ru in Fig. 4(b–d). It can be seen that except for the colors it is similar to Fig. 4(a), a TEM image of the same portion of the sample.
 | ||
| Fig. 4 (a) HAADF-STEM image of an edge portion of a 1%Ru@V2O5 particle (b) elemental mapping of oxygen (c) elemental mapping of vanadium (d) elemental mapping of ruthenium (e) overlay image of oxygen, vanadium and ruthenium. | ||
Presence of ruthenium in case of 1%Ru@V2O5 is accompanied by change in the resistance. At room temperature, the resistance of the V2O5 thin films used in the sensing measurements was found to be 67.09 kΩ but that of 1%Ru@V2O5 was 20.71 kΩ. This dramatic reduction of resistance of thin films of material with same dimensions indicates that metal deposition is responsible in reducing the resistance of 1%Ru@V2O5 by such a large value. The changes are also accompanied by band gap reduction of 1%Ru@V2O5 as compared to pristine V2O5 sample. This is clearly seen in Fig. 5(a) optical absorption spectra (b) reflectance spectra (c and d) corresponding Kubelka–Munk function plots of both the samples. Kubelka–Munk k/f function is calculated using formula,
| k = (intensity/100)2, f = intensity/100 | (6) |
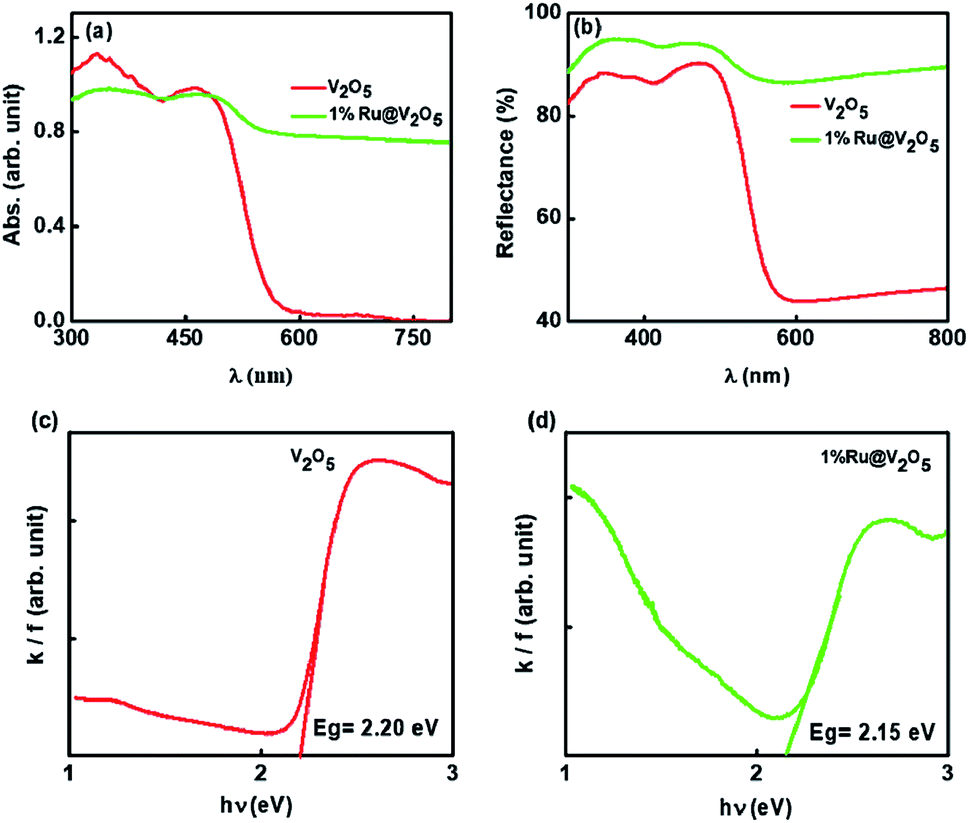 | ||
| Fig. 5 (a) Optical absorption spectra (b) reflectance spectra (c and d) corresponding k/f vs. hυ Kubelka–Munk plots of V2O5 and 1%Ru@V2O5. | ||
In Fig. 5 the extrapolation of the leading edges to x-axis gives the energy gap. The energy gap for the V2O5 sample using Kubelka–Munk function is 2.20 eV. This is in close agreement with the value reported for V2O5.39,40 The energy gap was found to be 2.15 eV for 1%Ru@V2O5. Thus band gap for 1%Ru@V2O5 has decreased by 0.05 eV. Moreover the band edge for 1%Ru@V2O5 is not as sharp as that for V2O5 and is tailing even beyond 2 eV, probably due to increase in diffuse interface (presence of particles at the surfaces in case of 1%Ru@V2O5). In any case, it can be deduced that ruthenium has substantially influenced the V2O5 sample and probably is responsible for the reduction in the resistance of the sample. As we will see below, such changes in the material are also responsible for the sensing change of 1%Ru@V2O5 sample compared to V2O5.
Many transition metal oxide sensors operate effectively at high temperature. This can be attributed to the higher reaction rates at high temperature as well as increased conductivity of metal oxides at high temperatures due to the formation of oxygen defects. We therefore first made the sensitivity measurements for both V2O5 and 1%Ru@V2O5 samples at three temperatures viz. room temperature (30 °C), 50 °C and 100 °C. After measuring the initial resistances of the samples at these temperatures in air (Ra, without any gas injection in the chamber) the changes in the resistances in presence of different gas ambient (Rg) at these temperatures for fixed quantity viz. 130 ppm, were measured. Literature on V2O5 semiconductor shows interesting resistivity behavior. It is p-type, n-type or even shows transition from n to p or vice a versa.9,14 An n-type semiconductor shows reduction in the resistance in presence of a reducing gas. Here we have used vapors of ethanol, methanol, acetone, formaldehyde, ammonia, triethylamine and trimethylamine as reducing gases. We also checked the sensing for the oxidizing gases SO2 and NOx.
Fig. 6(a) shows the sensitivity obtained using the sensitivity equation,1 at three temperatures viz. room temperature (30 °C), 50 °C and 100 °C for various gases at 130 ppm. Similarly sensing measurements were carried out for the sample 1%Ru@V2O5 and shown in Fig. 6(b). A dramatic difference is seen for the sensing of all the gases investigated here as seen in Fig. 6(b) when V2O5 is replaced with 1%Ru@V2O5. First of all there is an overall increase in the sensitivities at room temperature for all the gases. But more striking change occurs for NH3 vapor sensing and will be the focus of our discussion. It can be seen from Fig. 6(b) that the sensitivity of ammonia has increased to ∼4% at room temperature. This probably is the highest value obtained so far for any functionalized or nanocomposite or otherwise modified V2O5 sample, with exception of graphene composites. With increase of temperature to 50 °C, sensitivity of 1%Ru@V2O5 towards NH3 reduced to ∼3% and then further went to its lowest value of ∼0.5% at 100 °C. Corresponding bar graphs for V2O5 and 1%Ru@V2O5 are illustrated in Fig. 6(c) and (d) respectively. From Fig. 6(d) one can see that sample 1%Ru@V2O5 shows remarkable sensitivity at room temperature and it is very selective. For 1%Ru@V2O5 the response to other gases is relatively negligible (<0.5%).
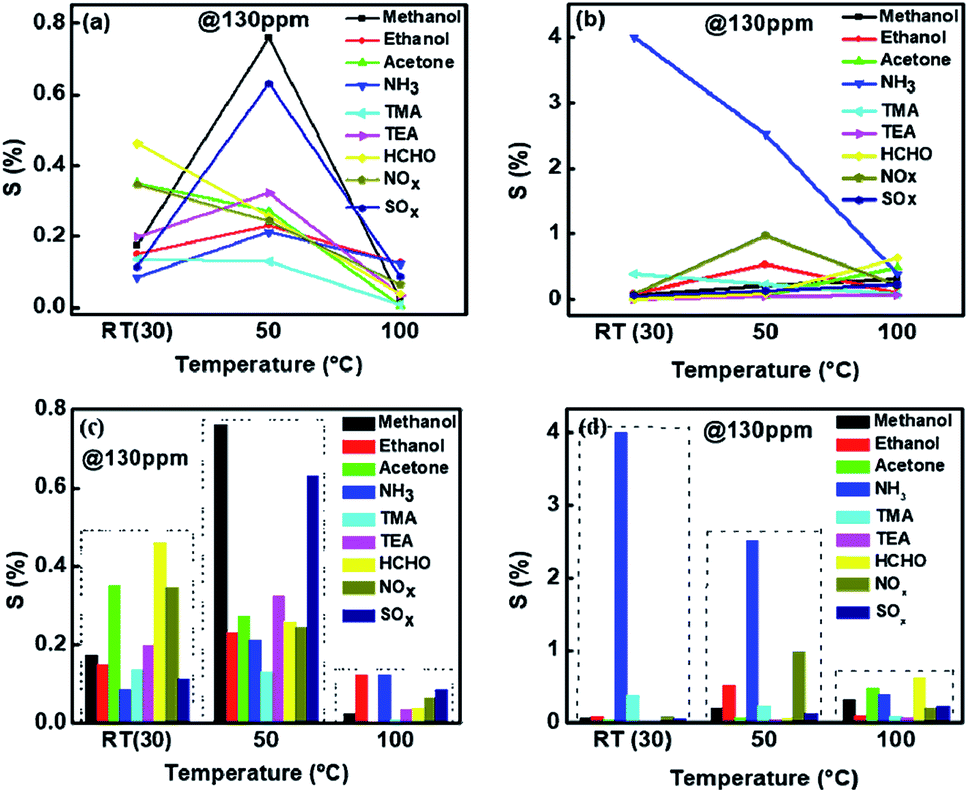 | ||
| Fig. 6 Comparison of (a) sensitivity of V2O5 and (b) 1%Ru/V2O5 towards different gases. Sensitivity shown as bar graph, at different temperatures, for (c) V2O5 and (d) 1%Ru/V2O5. | ||
This makes 1%Ru@V2O5 a very good ammonia sensor in terms of selectivity and sensitivity at room temperature. Obtaining room temperature operating sensor is very important as it can be used in places where the gas needs to be stored at high pressure and needs to be detected for any leaks. Particularly in case a gas cylinder containing ammonia gas explodes it can lead to a disaster, as it is a toxic gas. Operation at high temperature of sensors can not only be expensive but also dangerous as gases can further catch fire on exploding.
Before proceeding with further analysis of 1%Ru@V2O5, we measured the effect of humidity on 1%Ru@V2O5. This is particularly essential when a sensor is to be used at room temperature, as in many places there can be large humidity at room temperature. It can be seen from Fig. 7 that over the entire range, less than 0.1% contribution can come from humidity. Thus the effect of humidity on our measurements is negligible.
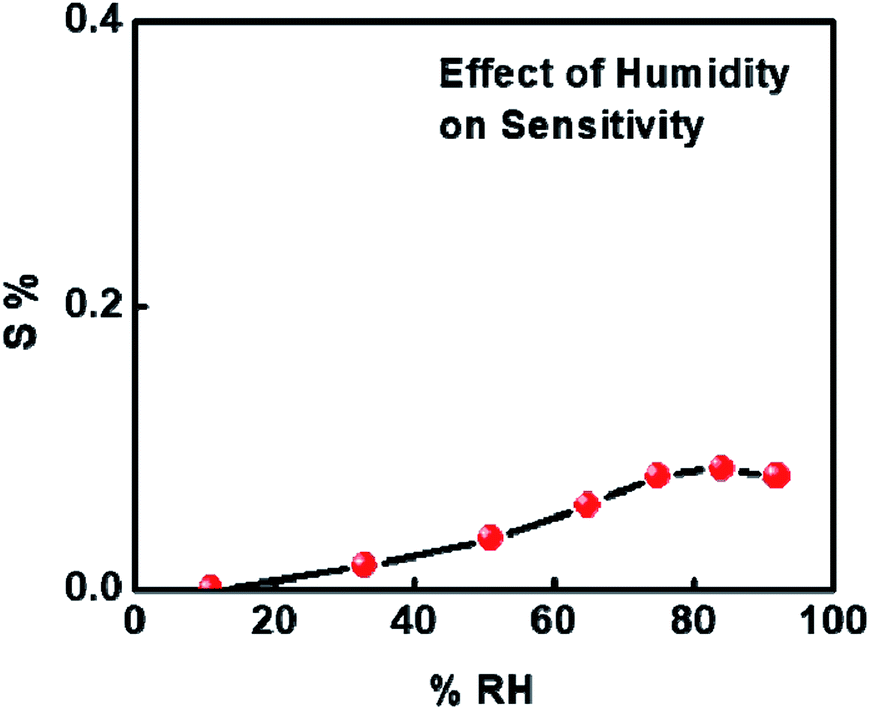 | ||
| Fig. 7 Humidity sensing of 1%Ru@V2O5. | ||
As the sensitivity for NH3 was found to be highest and sensor was also selective, further measurements were made on NH3 interaction with 1%Ru@V2O5. Fig. 8 shows the sensitivity plot for NH3 for the fixed concentration of NH3 on 1%Ru@V2O5. It can be seen that the repeatability of the material is quite good. Also the sensor shows instantaneous response and recovery without any saturation.41–43
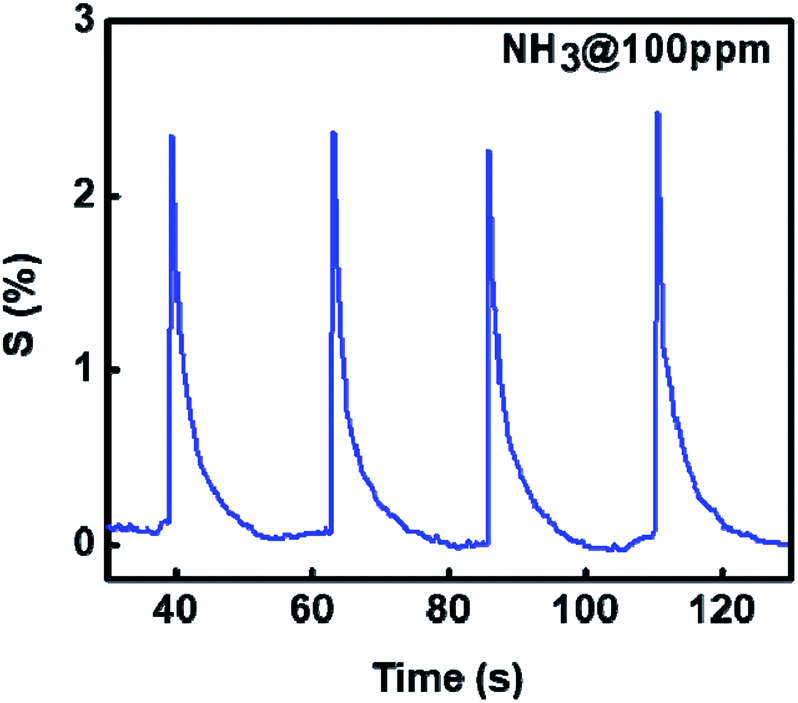 | ||
| Fig. 8 Sensitivity of Ru@V2O5 as a function of time at constant NH3 concentration (100 ppm), shown for 4 cycles. | ||
Further we show the response of the sensor for different concentrations of ammonia in Fig. 9(a). It can be seen that the sensitivity decreases with reduction in the gas concentration. The sensitivity with different NH3 concentrations can be seen from Fig. 9(b) is linear on the log–log scale over the entire range (10–130 ppm) investigated here. Use of log–log scale has been made earlier for plotting the sensitivity.44,45
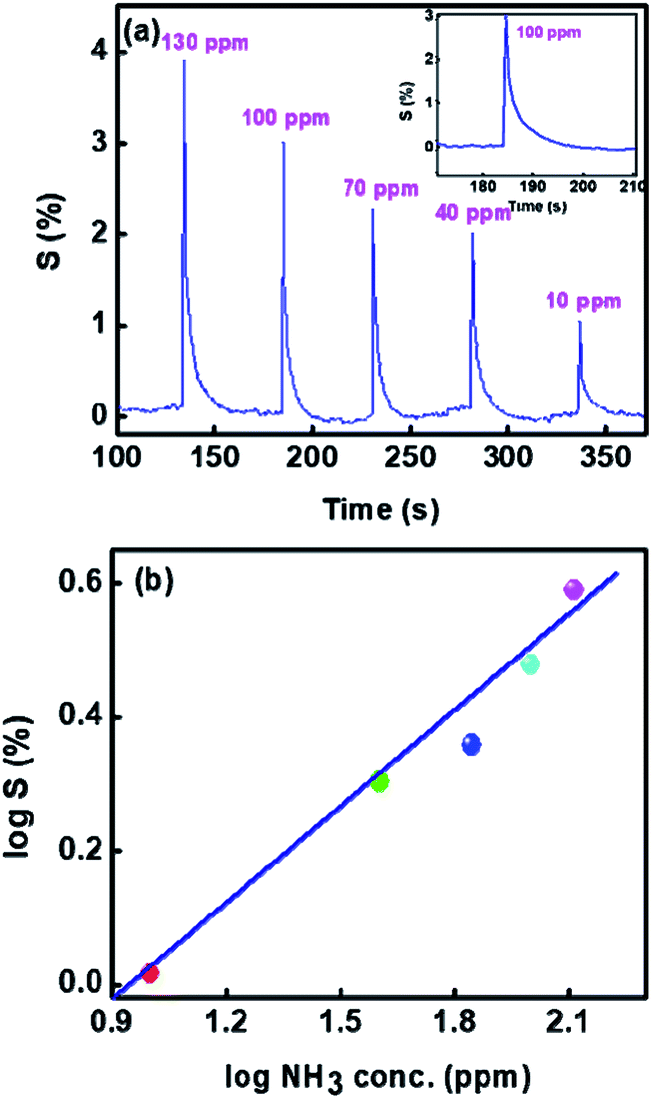 | ||
| Fig. 9 Sensitivity of 1%Ru@V2O5 (a) as a function of time for various concentrations of NH3 and (b) log–log plot of sensitivity vs. ammonia concentrations. | ||
We have also determined the response time and recovery times for each concentration from Fig. 9(a). It is also enlarged and shown for one concentration in the inset of Fig. 9(a). The response time is considered as the time required to reach the lowest resistance in the presence of gas and recovery time is taken as the time needed to attain the 90% of the original value of resistance viz. Ra.
Fig. 10(a) shows the dynamic response and recovery time for 100 ppm towards ammonia gas at room temperature. In Fig. 10(b) we have plotted response and recovery time for each concentration (obtained from Fig. 9(a)). As can be seen from Fig. 10(b), the maximum response time is less than 2 seconds and the recovery time is less than 12 seconds. These to our knowledge are the best response and recovery times for V2O5 sensor so far. For comparison see Table 1, where we have listed the ammonia sensing data of different V2O5 morphologies and also the temperatures or concentrations.
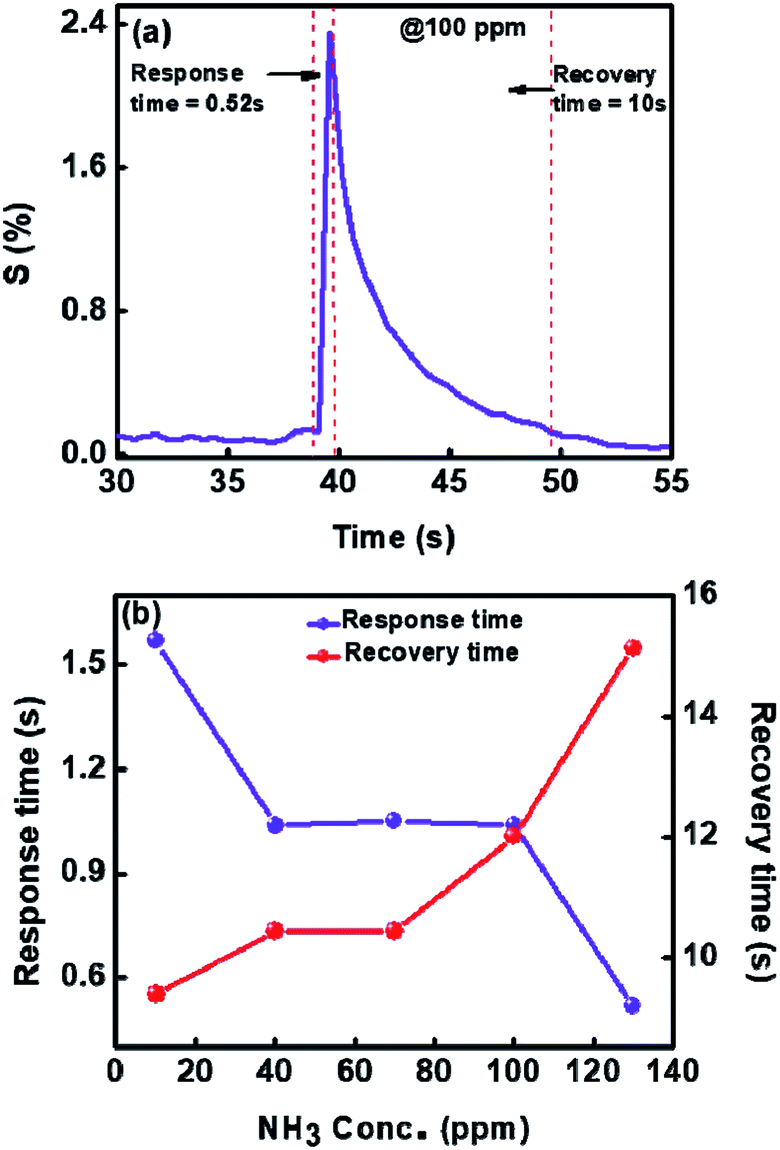 | ||
| Fig. 10 (a) Dynamic response to NH3 (b) response and recovery times of 1%Ru@V2O5 at 30 °C for different concentrations of NH3 gas. | ||
| Sr. no. | Metal oxides used | Gas detected | Sensitivity (%) | Operating temperature (°C) | Response/recovery time | Ref. |
|---|---|---|---|---|---|---|
| 1 | V2O5 nanorods | Ethanol (100 ppm) | 1.03 | RT | 2.5 s (res. time from graph) | 10 |
| Ammonia (100 ppm) | 1.018 | 5 s (res. time from graph) | ||||
| 2 | V2O5/PVAC-fibres | Ammonia (0.8–8.5 ppm) | 30 (from graph) | 200–250 | 50 s/350 s | 12 |
| 3 | V2O5 nanofibers | 1-Butylamine (10 ppm) | 42 | RT | Not mentioned | 17 |
| Ammonia (10 ppm) | 1.8 | |||||
| 4 | V2O5@TiO2 | Ammonia (5 ppm) | 60 | 365 | 9 s/6 s | 46 |
| 5 | 2 wt% Sn doped V2O5 nanoparticle | Ammonia (5–50 ppm) | 77.84 at 50 ppm | RT | Not mentioned | 47 |
| 6 | V2O5/PVP | Ammonia (0.1–0.8 ppm) | 6 (0.8 ppm) from graph | 260 | 25 s/60 s (from graph) | 48 |
| 7 | V2O5–WO3–TiO2 (potentiometric sensor) | Ammonia (10–320 ppm) | Not mentioned | 550 | 5–8 s/8–12 s | 49 |
| 8 | V2O5/PANI amperometric sensor | Ammonia (0–54 ppm) | 175 (from graph) | Not mentioned | 45 min/60 min (from graph) | 50 |
| 9 | pTSA doped V2O5@Pani nanofibers | Ammonia (0.1 M) | Not mentioned | RT | 1 min/1.5 min (from graph) | 18 |
| 10 | V2O5 + V7O16 | Ammonia (200 ppb) | Not mentioned | 350 | 60 m/60 min (from graph) | 19 |
| 11 | V2O5 nanopillars | Ammonia (2.5–20 ppm) | Not mentioned | 350 | 15 min/15 min (from graph) | 21 |
| 12 | V2O5 + V7O16 | Ammonia (1 ppm) | 32 (from graph) | 350 | 10 min/10 min (from graph) | 22 |
| 13 | V2O5 + V7O16 | Ammonia (40 ppb) | Not mentioned | 350 | 10 min/10 min | 51 |
| 14 | 1%Ru@V2O5 | Ammonia (10 ppm) | 4 | RT | 1.5 s/9.3 s | Present work |
3.1 Ammonia sensing mechanism of V2O5 and 1%Ru@V2O5
Vanadium pentoxide (V2O5) is a very common and stable oxide (melting point 690 °C). It is known to be a good catalyst, particularly for selective catalytic reduction of NOx along with NH3.52–54 V2O5 (010) surface of orthorhombic V2O5 is commonly studied theoretically, as it is found to be most important and exposed surface in most of the catalytic reactions.54 Interaction of NH3 with V2O5 (010) also has been studied theoretically.38,55 It was found that NH3 adsorbs preferentially on three Brønsted sites shown in Fig. 11 which is constructed based on the Fig. 1 from the work of Yin et al.38 Accordingly, three oxygen atoms have been identified viz., O1, O2, O3 and two types of vanadium V1, V2 on bare (010) surface.38 They find that adsorption probability on O1H > O2H > O3H sites. As H2O cannot dissociate on bare V2O5 they had assumed that already atomic hydrogen (H) was present on these sites so that NH4+ formation could take place.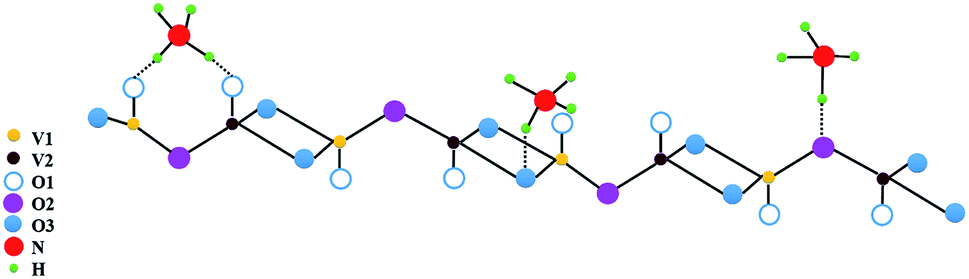 | ||
| Fig. 11 Adsorption sites for NH3 adsorption at Brønsted acid sites consisting of singly coordinated, di-coordinated and tri-coordinated oxygen sites on V2O5 surface atoms. | ||
On Lewis sites adsorption of NH3 is in any case less but it was necessary that presence of hydrogen atom on oxygen or bonding with oxygen was necessary for the formation of NH4+ species. Here we do not further discuss exact charge transfers etc. between NH3 and H or vanadium at different sites which are discussed in ref. 38 and 55 but only use their conclusions viz. hydrogen atom needs to be present at Brønsted site and adsorption of NH3 to form NH4+ is preferentially on O1 > O2 > O3 site with more charge transfer to surface at O1 > O2 > O3 sites.38 Assuming the formation of NH4+ one can write,
| NH3 + H+ ⇌ NH4+ + e− | (7) |
| 2NH3 + [4O(ads.)−] → 4N2O + 3H2O + 4e− | (8) |
| 2NH3 + [5O(ads.)−] → 2NO + 3H2O + 5e− | (9) |
For reaction pathways (8) and (9) presence of adsorbed oxygen is necessary. Which reaction pathway among (7)–(9) is chosen is difficult to decide here, as this would need in situ determination of released gas species in the reaction (which is not possible here).
It can be seen in Fig. 1(b) that V2O5 does not show any prominent OH related vibrational mode over a wide range from 4000 cm−1 to 450 cm−1. However, as was discussed with reference to Fig. 1(b), 1%Ru@V2O5 does have this possibility.
FTIR spectrum clearly shows a sharp vibrational mode at 1614 cm−1 and a broad band around 3000 cm−1 indicative of the presence of dissociated water molecules.35,37
Presence of ruthenium in 1%Ru@V2O5 is also evident from XPS. XPS is a versatile technique to know the oxidation states or charge transfers between the surface atoms. The XPS peak shift to higher binding energy when charge from the corresponding atom is removed and peak shifts to lower binding energy if it gains the more charge. This type of charge transfer takes place amongst the neighboring atom.
In Fig. 12(a) the survey scans for V2O5 and 1%Ru@V2O5 are shown. One can see that in V2O5 sample only vanadium, oxygen and carbon related peaks are present, suggesting that the sample is free of any impurities. The 1%Ru@V2O5 has slight indication of additional peak of N 1s at ∼400 eV. In Fig. 12(b) the plots are made of narrow region from 276 to 294 eV. The V2O5 spectrum in the carbon region is deconvoluted into three peaks positioned at 284.6 eV (C1), 286.7 eV (C2) and 288.5 (C3) eV, all due to C 1s. The peak at 284.6 eV is assigned to adventitious carbon and treated as the reference for the whole analysis. C2 and C3 can be identified as due to C–O and CO respectively. In the same spectral region for the 1%Ru@V2O5 sample we observe that there also C 1s peak has C1, C2 and C3 components present but there are two additional peaks, which can be fitted. These can be identified as spin–orbit split Ru 3d5/2 at 282.2 eV and Ru3/2 at 286.0 eV positions. The binding energy of Ru 3d5/2 and Ru3/2 as well as their spin–orbit splitting of 3.8 eV suggests that Ru is in the Ru3+ state.56,57 The Ru 3d5/2 and Ru 3d3/2 peaks are separated by 3.8 eV in close agreement with reference handbook, which showed the splitting to be 4.1 eV for Ru metal. This suggests that ruthenium is forming RuO2 layer. Ru is probably forming contact with the V2O5 (also evident from STEM in Fig. 4). Schematic diagram depicting this is given in Fig. 12 and discussed later. From TEM we know that ruthenium nanoparticles of ∼10–20 nm size are formed on V2O5. Penetration depth of XPS is ∼2–3 nm. Therefore these particles could have ruthenium metal inside with thin layer of ruthenium oxide as suggested from XPS here but also consistent with the Ru metal peaks observed in Fig. 1, in XRD of 1%Ru@V2O5. Considering that XPS signal appears from very shallow surface (<3 nm) it can be safely assumed that RuO2 is present along with the underneath ruthenium metal in the hexagonal phase.
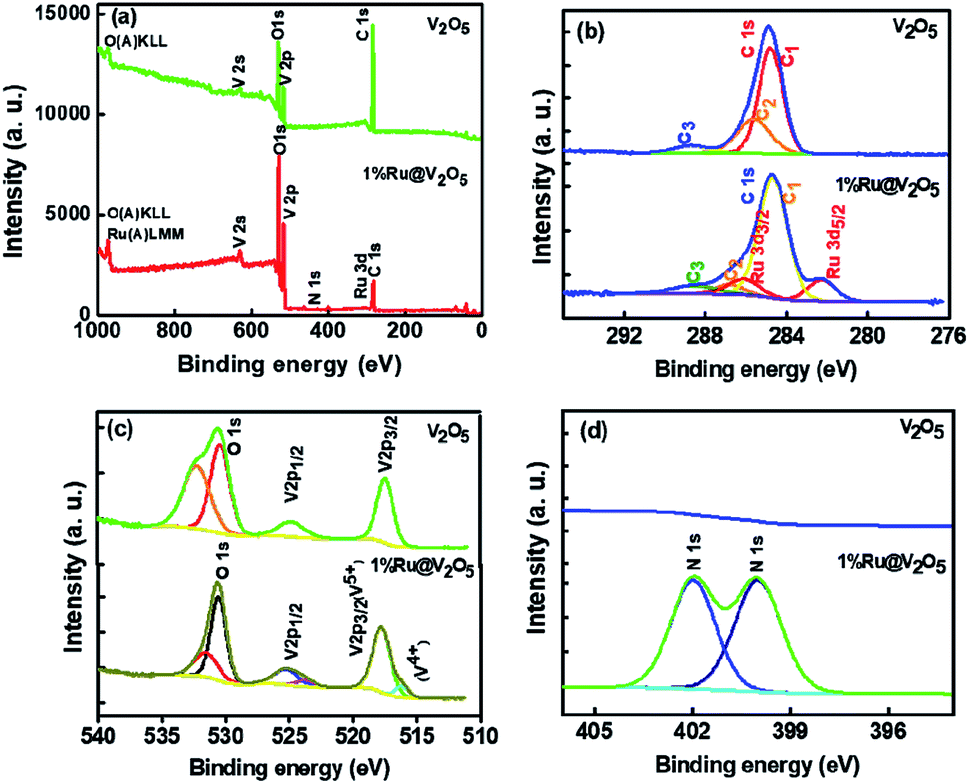 | ||
| Fig. 12 XPS spectra of V2O5 and 1%Ru@V2O5. | ||
In Fig. 12(c) vanadium and oxygen region is plotted from 510 to 540 eV. We can clearly see two peaks at 517.3 eV and 524.6 eV which can be assigned to 2p3/2 and 2p1/2 of V5+ state.56 The other two peaks at 530.3 eV and 532.1 eV, could be due to oxygen and molecularly adsorbed H2O respectively. For 1%Ru@V2O5 also two peaks can be fitted for 2p3/2 and 2p1/2 of V5+ at 517.7 and 525.2 eV, whereas O 1s peaks appear at 531.1 and 530.5 eV respectively. In addition we observed small nitrogen impurity peaks at 399.9 and 401.9 eV respectively, probably arising from hydrazine hydrate used in the synthesis of 1%Ru@V2O5.
The small nanoparticles (10–20 nm) formation of ruthenium covered with thin ruthenium oxide layer on V2O5, plays a very important role in the improved sensitivity of V2O5 towards ammonia. Small crystals would have in addition to surface oxide, surface defects due to kinks, steps, and corners etc. which are very surface active.58 The presence of RuO2 on tin oxide has shown high ammonia sensitivity.56 This is probably due to supply of necessary H atoms to vanadyl oxygen (O1). In fact RuO2 is considered to be a better catalyst in the synthesis of ammonia oxidation and decomposition.59
Further, in catalysis interaction of adsorbate molecule, structure of the molecule, structure of catalyst at adsorption site, subsequent bond adjustments or breaking of bonds and finally desorption are important factors to be considered. Sensing also depends upon local interactions. Therefore the process is similar to catalysis and was as discussed so far. However, sensing action is investigated often by measuring the electric resistance as in here. Therefore transport of charge carriers also needs to be taken into account i.e. locally liberated electrons as suggested earlier should be transferred to the valence band of the n-type V2O5 in this case. This can be seen schematically from Fig. 13.
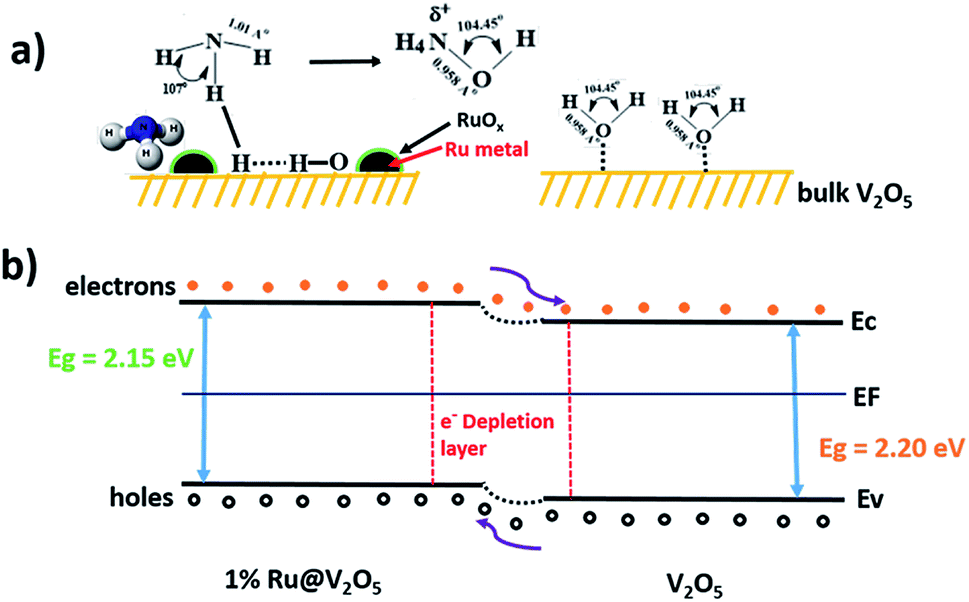 | ||
| Fig. 13 Schematic of (a) gas sensing mechanism and (b) energy band structure diagrams of V2O5 and 1%Ru@V2O5. | ||
Fig. 13(a) shows bare and 1%Ru@V2O5 surfaces. On V2O5 as no RuO2/Ru particles are present, there is no water adsorption and consequently no NH3 adsorption shown. This results into a situation as depicted in Fig. 13(b) as far as the bands are concerned. When there is no ruthenium oxide on the surface and bare V2O5 is exposed to the surface, less charge carriers are produced and resistance of V2O5 does not change much. When RuO2/Ru nanoparticles are present on the V2O5 surface they form a semiconductor–semiconductor contact, which in turn forms a depletion layer similar to that on V2O5.57 Now, unlike on V2O5, water molecules dissociate on ruthenium (or oxide) leading to liberation of electrons, which are easily transferred to V2O5 reducing the resistance. Thus higher sensitivity of 1%Ru@V2O5 compared to bare V2O5 is followed. It can be inferred from above discussion that Ru/RuO2 nanoclusters on the V2O5 microcrystallites assist the dissociation of water molecules. This is helpful for V2O5 to dissociate NH3, which in turn liberates free electrons.
Thus, it can be inferred from the discussion so far that,
(1) Water molecules do not dissociate on pristine V2O5 but dissociate on 1%Ru@V2O5.
(2) Ru/RuO2 nanoclusters on V2O5 directly participate in ammonia sensing.
(3) Ru/RuO2 forms a heterojunction with V2O5.
(4) Depletion layer formed between Ru/RuO2 and V2O5 reduces upon NH3 adsorption on 1%Ru@V2O5.
(5) Sensor 1%Ru@V2O5 is selective, sensitive, has low response and recovery time. Here oxygen defects are not probably playing important role, as such centers would be thermally activated. We obtain more sensitivity at room temperature than at higher temperatures.
4 Conclusions
We have successfully fabricated a resistive type ultrasensitive, 1%Ru@V2O5 ammonia sensor which is operated at room temperature, is selective and has short response and recovery time. Ru forms metallic nanoparticles having a thin surface oxide layer on V2O5. The high sensitivity obtained is due to small amount of dissociation of water molecules in presence of RuO2/Ru clusters, which participate in high catalytic sensing of ammonia molecules. However, the reduced response time and recovery time as well as optimum ruthenium concentration on the V2O5 surface need to be investigated further.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
PA thanks Director General, C-MET for permission to carry out this work. SK thanks INSA, Delhi, India for the financial assistance under INSA senior scientist program, INSA sanction no. SP/SS/216/1318. The authors also acknowledge the help of Dr J. Ambekar for recording FESEM and help of Dr M. Shinde of CMET, Pune for TEM analysis.References
- Y. Wu, G. Gao and G. Wu, J. Mater. Chem. A, 2015, 3, 1828 RSC.
- Y. T. Wang, W. T. Whang and C. H. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 8480 CrossRef CAS PubMed.
- M. Kodu, A. Berholts, T. Kahro, M. Kook, P. Ritslaid, H. Seemen, T. Avarmaa, H. Alles and R. Jaaniso, Beilstein J. Nanotechnol., 2017, 8, 571 CrossRef CAS PubMed.
- K.-Y. Pan and D.-H. Wei, Nanomaterials, 2016, 6, 140 CrossRef PubMed.
- C. Bianchi, L. M. Ferreira, J. Loureiro, A. Rodrigues, P. Duarte, A. C. Baptista and I. M. Ferreira, J. Electron. Mater., 2016, 45, 1987 CrossRef CAS.
- M. A. Centeno, I. Carrizosa and J. A. Odriozola, Appl. Catal., B, 2001, 29, 307 CrossRef CAS.
- J. Livage, Vanadium pentoxide gels, Chem. Mater., 1991, 3, 578 CrossRef CAS.
- J. Livage, Materials, 2010, 3, 4175 CrossRef CAS PubMed.
- M. Yu, X. Liu, Y. Wang, Y. Zheng, J. Zhang, M. Li, W. Lan and Q. Su, Appl. Surf. Sci., 2012, 258, 9554 CrossRef CAS.
- D. Raj, T. Pazhanivel, P. S. Kumar, D. Mangalaraj, D. Nataraj and N. Ponpandian, Curr. Appl. Phys., 2010, 10, 531 CrossRef.
- M. Wu, X. Zhang, S. Gao, X. Cheng, Z. Rong, Y. Xu, H. Zhao and L. Huo, CrystEngComm, 2013, 15, 10123 RSC.
- V. Modafferi, G. Panzera, A. Donato, P. L. Antonucci, C. Cannilla, N. Donato, D. Spadaro and G. Neri, Sens. Actuators, B, 2012, 163, 61 CrossRef CAS.
- W. Jin, S. Yan, L. An, W. Chen, S. Yang, C. Zhao and Y. Dai, Sens. Actuators, B, 2015, 206, 284 CrossRef CAS.
- Y. Qin, G. Fan, K. Liu and M. Hu, Sens. Actuators, B, 2014, 190, 141 CrossRef CAS.
- S. A. Hakim, Y. Liu, G. S. Zakharova and W. Chen, RSC Adv., 2015, 5, 23489 RSC.
- B. J. Liu, X. Wang and Q. Peng, Materials, 2005, 17, 764 Search PubMed.
- I. Raible, M. Burghard, U. Schlecht, A. Yasuda and T. Vossmeyer, Sens. Actuators, B, 2005, 106, 730 CrossRef CAS.
- M. Hasan, M. O. Ansari, M. H. Cho and M. Lee, J. Ind. Eng. Chem., 2015, 22, 147 CrossRef CAS.
- J. Huotari, J. Lappalainen, J. Eriksson, R. Bjorklund, E. Heinonen, I. Miinalainen, J. Puustinen and A. Lloyd Spetz, J. Alloys Compd., 2016, 675, 433 CrossRef CAS.
- J. M. Cocciantelli, P. Gravereau, J. P. Doumerc, M. Pouchard and P. Hagenmuller, J. Solid State Chem., 1991, 93, 497 CrossRef CAS.
- J. Huotari, V. Kekkonen, J. Puustinen, J. Liimateinen and J. Lappalainen, Procedia Eng., 2016, 168, 1066 CrossRef CAS.
- J. Huotari, R. Bjorklund, J. Lappalainen and A. Lloyd Spetz, Sens. Actuators, B, 2015, 217, 22 CrossRef CAS.
- W. Jin, S. Yan, W. Chen, S. Yang, C. Zhao and Y. Dai, Mater. Lett., 2014, 128, 362 CrossRef CAS.
- J. Liang, K. Zhu, R. Yang and M. Hu, Ceram. Int., 2018, 44, 2261 CrossRef CAS.
- P. Dutta and A. D. Adeyemo, 14th Int. Meet. Chem. Sensors., 2012, p. 152 Search PubMed.
- J. M. Patil, S. B. Patil, R. H. Bari and A. N. Sonar, Int. J. Chem. Concepts, 2016, 2, 12 CAS.
- R. Wang, S. Yang, R. Deng, W. Chen, Y. Liu, H. Zhang and G. S. Zakharova, RSC Adv., 2015, 5, 41050 RSC.
- F. Zhang, X. Wang, J. Dong, N. Qin and J. Xu, Sens. Actuators, B, 2013, 186, 126 CrossRef CAS.
- K. Chebout, A. Iratni, A. Bouremana, K. Mhammedi, H. Menari, A. Keffous and N. Gabouze, The Third Int. Conf. Sens. Devices & Tech. Appl., 2012, ISBN, 978-1-61208-208-042 Search PubMed.
- K. Chebout, R. Tala-Ighil, K. Mhammedi, S. Sam and N. Gabouze, Res. Rev.: J. Eng. Technol., 2018, 7, 24–29 Search PubMed , E-ISSN: 2319-9873.
- W. Yan, M. Hu, J. Liang, D. Wang, Y. Wei and Y. Qin, Nano, 2016, 7, 1650079 CrossRef.
- P. V. Adhyapak, A. D. Bang, P. More and N. R. Munirathnam, RSC Adv., 2018, 8, 34035 RSC.
- G. P. Nagabhushana and G. T. Chandrappa, J. Mater. Chem. A, 2013, 1, 11539 RSC.
- V. V. Porsev, A. V. Bandura and R. A. Evarestov, Acta Mater., 2014, 75, 246 CrossRef CAS.
- V. P. Filonenko, M. Sundberg, P. E. Werner and I. P. Zibrov, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 375 CrossRef CAS PubMed.
- N. Pinna, M. Willinger, K. Weiss, J. Urban and R. Schlo, Nano Lett., 2003, 3, 1131 CrossRef CAS.
- R. A. Elsalamony and S. A. Mahmoud, Arabian J. Chem., 2017, 10, 194 CrossRef CAS.
- X. Yin, H. Han, I. Gunji, A. Endou, S. S. Cheettu Ammal, M. Kubo and A. Miyamoto, J. Phys. Chem. B, 1999, 103, 4701 CrossRef CAS.
- A. A. Bahgat, F. A. Ibrahim and M. M. El-Desoky, Thin Solid Films, 2005, 489, 68 CrossRef CAS.
- P. Singh and D. Kaur, J. Appl. Phys., 2008, 4, 103 Search PubMed.
- J. Wu, K. Tao, J. M. Miao and L. Norford, ACS Appl. Mater. Interfaces, 2015, 7(49), 27502 CrossRef CAS PubMed.
- F. Hoshyargar, M. Shafiei, C. Piloto, N. Motta and P. A. O. Mullane, J. Mater. Chem. C, 2016, 4, 11173 RSC.
- S. Cui, J. Wang and X. Wang, RSC Adv., 2015, 5, 58211 RSC.
- V. Khambalkar, S. Birajdar, P. Adhyapak and S. Kulkarni, Nanotechnology, 2019, 30(10), 105501 CrossRef CAS PubMed.
- A. Marikutsa, A. Sukhanova, M. Rumyantseva and A. Gaskov, Sens. Actuators, B, 2018, 255(30), 3523 CrossRef CAS.
- H. Fu, X. Yang, X. An, W. Fan, X. Jiang and A. Yu, Sens. Actuators, B, 2017, 252, 103 CrossRef CAS.
- N. Singh, A. Umar, N. Singh, H. Fouad, O. Y. Alothman and F. Z. Haque, Mater. Res. Bull., 2018, 108, 266 CrossRef CAS.
- V. Modafferi, S. Trocino, A. Donato, G. Panzera and G. Neri, Thin Solid Films, 2013, 548, 689 CrossRef CAS.
- C. Wang, X. Li, Y. Yuan, B. Wang, J. Huang, F. Xia, H. Zhang and J. Xiao, Sens. Actuators, B, 2017, 241, 268 CrossRef CAS.
- M. C. Santos, O. H. C. Hamdan, S. A. Valverde, E. M. Guerra and R. F. Bianchi, Org. Electron., 2018, 65, 116 CrossRef.
- J. Huotari, R. Bojrklund, J. Lappalainen and A. Spetz, Procedia Eng., 2014, 87, 1035 CrossRef CAS.
- A. Bielański, J. Piwowarczyk and J. Poźniczek, J. Catal., 1988, 113, 334 CrossRef.
- D. Sun, Q. Liu, Z. Liu, G. Gui and Z. Huang, Appl. Catal., B, 2009, 92, 462 CrossRef CAS.
- M. Inomata, A. Mlyamoto and Y. Murakami, J. Phys. Chem., 1981, 85, 2372 CrossRef CAS.
- A. Chakrabarti, K. Hermann, R. Druzinic, M. Witko, F. Wagner and M. Petersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 10583 CrossRef CAS.
- A. Marikutsa, V. Krivetskiy, L. Yashina, M. Rumyantseva, E. Konstantinova, A. Ponzoni, E. Comini, A. Abakumov and A. Gaskov, Sens. Actuators, B, 2012, 175, 186 CrossRef CAS.
- J. F. Moulder, E. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy Search PubMed.
- V. Kumar, V. Patil, A. Apte, N. Harale, P. Patil and S. Kulkarni, Langmuir, 2015, 31(48), 13247 CrossRef CAS PubMed.
- Y. Wang, H. Shang, T. Chou and G. Cao, J. Phys. Chem. B, 2005, 109(22), 11361 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |