
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoth chemical and crystalline phase configuration influence the molecular features of humic acids in humic–calcium–phosphates fertilizers
Roberto Baigorri *ab,
Javier Errob,
Óscar Urrutiab,
José Manuel Martinezb,
Marcos Mandadoc,
Manuel Martín-Pastord and
José M. García-Minab
*ab,
Javier Errob,
Óscar Urrutiab,
José Manuel Martinezb,
Marcos Mandadoc,
Manuel Martín-Pastord and
José M. García-Minab
aInnovation & Development Department, Timac Agro España S.A., Roullier Group, Barrio Feculas s/n, 31508 Lodosa, Spain. E-mail: roberto.baigorri@timacagro.es
bBACh Research Group, Department of Environmental Biology, University of Navarra, Irunlarrea No. 1, 31008, Pamplona, Spain
cPhysical Chemistry Department, Faculty of Chemistry, University of Vigo, Campus Universitario, Vigo, 36310, Spain
dDepartamento de NMR C.A.C.T.U.S., University of Santiago de Compostela, Santiago de Compostela 15706, Spain
First published on 16th August 2019
Abstract
Phosphate–metal–humic complexes are very relevant in nature due to their crucial role in phosphate availability for plants and microorganisms. Synthetic phosphate–calcium–humic acid (HA) complexes have proven to be efficient sources of available phosphorus for crops. However, the current knowledge about their structure and molecular features is very poor. The structural implications of phosphate interaction with humic binding sites through calcium bridges, in both monocalcium phosphate and dicalcium phosphate is investigated by using molecular modeling, 31P-NMR, 1H-NMR and X-ray diffractometry. The conformational changes in the molecular configuration of the humic acid involved in the interaction resulting from the synthetic process is also studied by using HPSEC and synchronous fluorescence. The results obtained allow us to identify the phosphate type in the crystalline phase that is involved in the interaction of humic acid binding sites and the different forms of calcium phosphate. Synchronous fluorescence also shows that whereas the conformational configuration of the HA binding site is only partially affected in the monocalcium phosphate interaction, it changes in the case of dicalcium phosphate showing simpler molecular arrangements. These changes in the molecular conformation of the binding site in HA in solution may influence the biological activity of the humic acid. On the other hand, HPSEC studies show that the humic–calcium–phosphate interaction is accompanied by increases in the humic acid apparent size distribution. This effect is more intense in the case of monocalcium phosphate system probably due the influence of pH.
Introduction
Soil Organic Matter (SOM) plays a very relevant role in the sustainability of soil fertility over time.1 Thus, SOM affects many soil physico-chemical features such as water retention capacity, plant nutrient availability, soil structure and porosity, and soil microbial activity.2 In other words, without SOM the soil cannot fulfil its ecological function of supporting life. In some ways, SOM connects the living world (plants, microorganisms, fungi…) with the inert world (inorganic and organic compounds, minerals, nutrients…) working as a very active interface layer. Thus, the relationships between SOM and minerals or nutrients are of great importance and could predetermine soil fertility. Among these minerals, phosphates are one of the most important sources of phosphorous (P), being P one of the principal nutrients for plants. Inorganic phosphates (Pi) in soil are compounds of poor plant availability in both acidic or alkaline soils. In acidic soils Pi are blocked with iron(III) and/or aluminum(III),3 whereas in alkaline soils Pi are blocked with calcium. Different forms of calcium-phosphates may be involved in the equilibrium between the soil solution and soil mineral surface. Among them, the most relevant are monocalcium phosphate (MCP), dicalcium phosphate (DCP) and tricalcium phosphate (TCP). The three phosphate forms have different water solubility, with MCP being water-soluble, DCP being soluble in weak organic acids and TCT being practically insoluble in water and weak organic acids. Several studies have shown that phosphate solubility and availability is affected by its interaction with dissolve organic matter (DOM) and more specifically with humic acids (HA).4,5 Several studies have reported that this increase in phosphate water solubility is associated with the formation of water-soluble humic–metal–phosphate complexes.6 This fact has prompted the development of special P-based fertilizers including HA–calcium (or other metals)–phosphate complexes, in agricultural markets. It is noteworthy that, besides improvements in Pi availability, the presence of HA per se is also relevant due to its capacity to enhance plant growth.6Among other possible methodologies, these types of special P-based fertilizers can be produced following two main general synthetic pathways:
(1) The reaction of sulfuric acid or/and phosphoric acid with rock phosphate in the presence of HA (the use of sulfuric acid leads to the formation of HA-complexed simple superphosphate (HA–SSP),7 whereas the use of phosphoric acid results leads to the formation of HA-complexed triple superphosphate (HA–TSP)7). Both HA–TSP and HA–SSP contain MCP as the main phosphate form.8
(2) The reaction of phosphoric acid with calcium carbonate and/or calcium oxide in the presence of HA and, in some cases, the presence of micronutrients with metallic character.9 Depending on the reaction stoichiometry we obtain HA-complexed MCP (MCP–HA) or HA-complexed DCP (DCP–HA); or products containing MCP–HA and DCP–HA.8
In previous studies we have characterized HA–calcium–phosphates in P-based fertilizers obtained by pathway 1.6,7 In these studies, the local emplacement of phosphate anion in crystalline phases was studied in depth as well as the main characteristics of phosphate binding in Ca–HA.10
In the present work we investigate the phosphate complexation process in calcium–HA binding sites in the P-based fertilizers obtained by pathway 2 (DCP–HA and MCP–HA) in the presence of different concentrations of an HA with sedimentary origin extracted from leonardite. The hypothesis underlying the work is that the pH changes involved in the two reactions will affect the molecular features of the HA imbibed in the crystalline phase. This fact, in turn, will influence phosphate binding in Ca–HA as well as the potential biological activity of HA.
Results and discussion
Structural changes induced by HA–Ca–phosphate interaction in MCP
The X-ray diffraction analysis (XRD) showed only a crystallographic phase that Bruker evaluation software assigned to mono-calcium phosphate mono hydrate (Ca(H2PO4)2·H2O) (Fig. 1). This result indicates that the amount of water that is added with HA is evaporated during the formation reaction and further curing.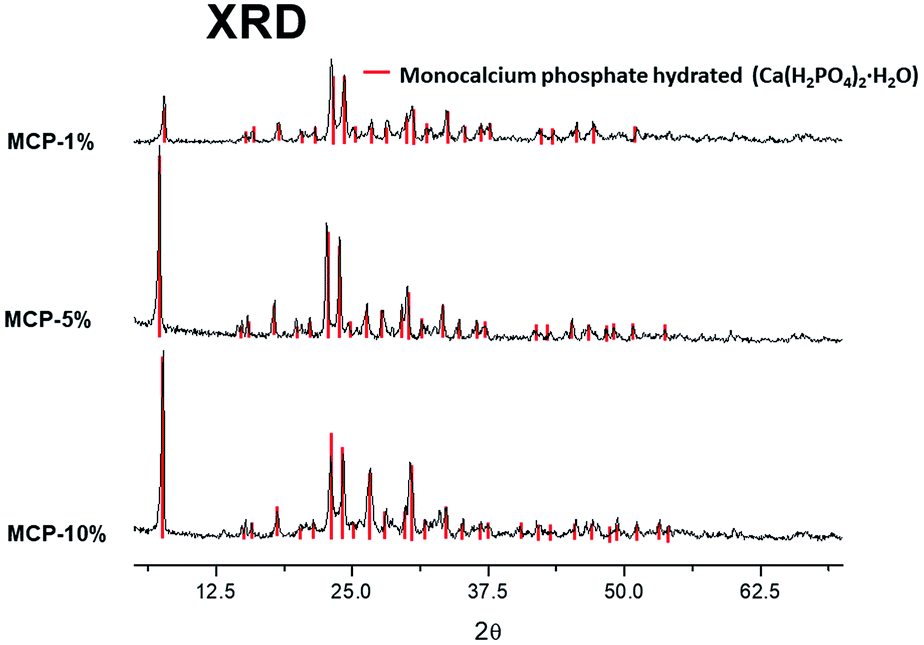 | ||
| Fig. 1 X-ray diffraction analysis (XRD) of crystallographic phase on MCP–HA (1,5 and 10%). | ||
On the other hand, phosphorus-31 nuclear magnetic resonance (31P-NMR) showed two peaks at 1.12 ppm and 5.64 ppm that are present in all samples and a peak at 4.38 ppm that only appears in those samples treated with HA (Fig. 2). Interestingly, as more HA is added the relative intensity of the new peak increases at the expenses of a reduction of the intensity of the peak at 5.64 ppm. This fact indicates that the peak at 4.38 ppm is probably reflecting the new electronic environment of phosphate that is linked to Ca–HA binding sites. This hypothesis is supported by the results obtained by molecular modelling (Fig. 3). Molecular modelling assisted the assignment of the 31P NMR peaks of MCP and MCP–HA. The peak at 1.12 ppm (Pm1) was assigned to the species coordinated to four calcium atoms and bound to two H+ ions. The peak at 5.64 ppm (Pm3) is coordinated to one calcium atom and bound to six H+ ions (Fig. 3). As mentioned above, the peak at 4.38 ppm (Pm2), which only appeared when HA is added to the reaction, has a chemical shift in-between Pm1 and Pm3, which suggests that its coordination with calcium and the binding with H+ ions in Pm2 is an intermediate situation between the two other species. The fact that Pm2 is negatively correlated with Pm1 (when Pm2 increase, the relative intensity of Pm1 decrease, Fig. 2) suggests that HA binding sites interacts with phosphate through Ca or H+ bounds involved in the Pm1 configuration.
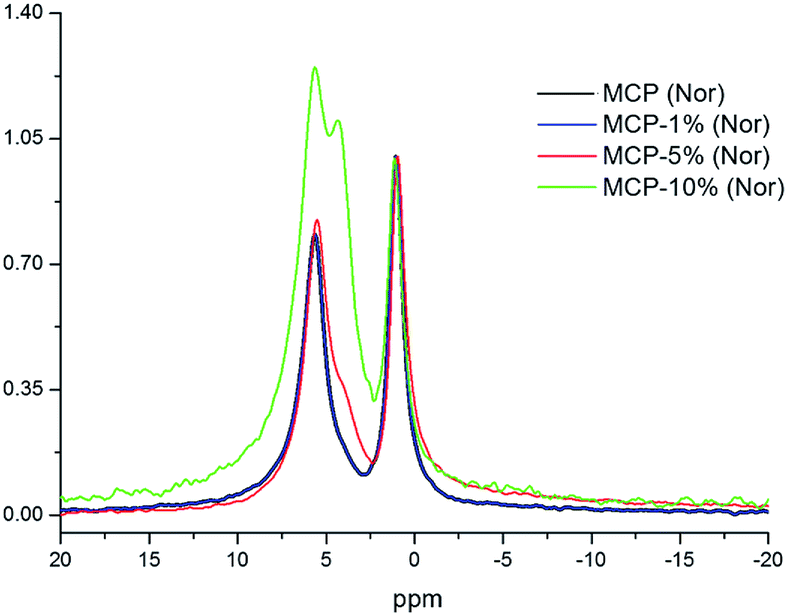 | ||
| Fig. 2 Superimposition of 1D 31P NMR spectra of MCP fertilizer samples prepared at different amounts of aqueous potassium humate (1, 5 and 10%). The intensities are normalized with respect to the signal at 1.12 ppm. | ||
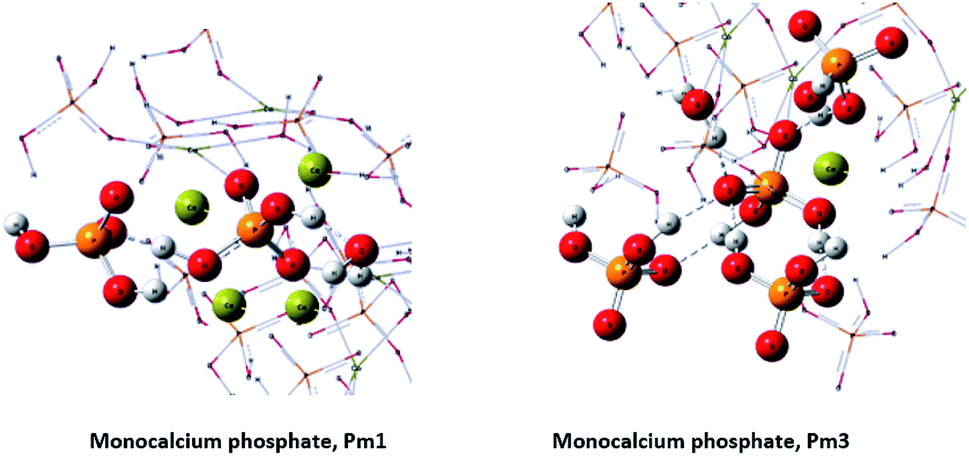 | ||
| Fig. 3 Crystallographic structures by molecular modelling of MCP. | ||
Two-dimensional 2D 1H–31P WISE NMR spectrum provided P–H correlations that are reflecting the chemical shift of those hydrogens that are close in space to a given P peak. The spectrum is useful to clarify how HA interacts with P phases (Fig. 4).
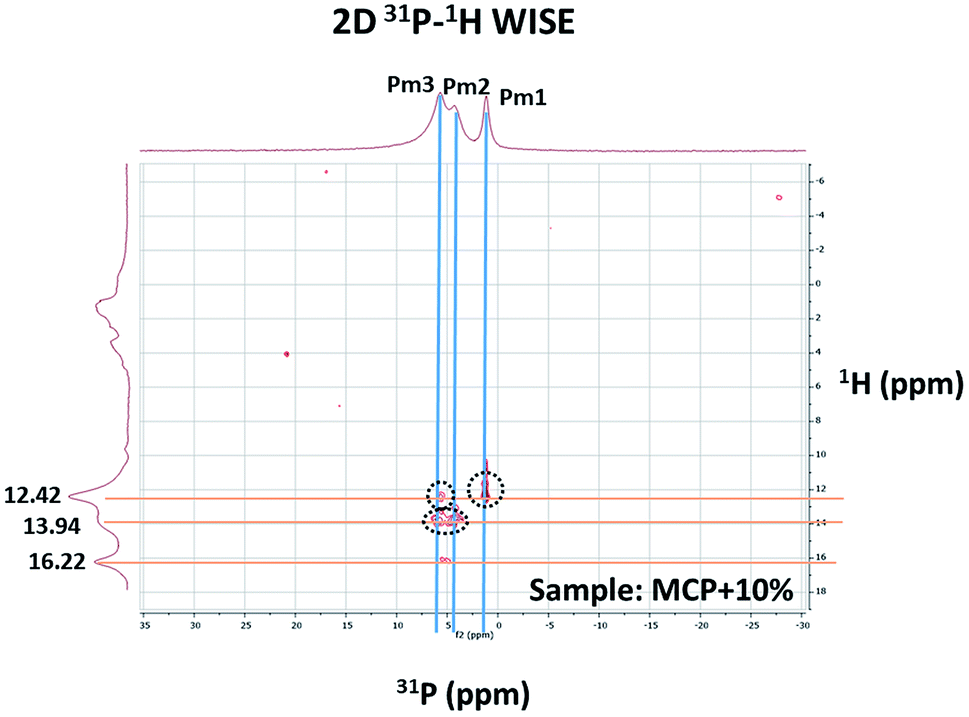 | ||
| Fig. 4 2D 1H–31P WISE spectrum of MCP sample prepared with 10% of aqueous potassium humate showing the assignment of the peaks. | ||
The 2D spectrum shows that the P peaks, Pm1, Pm2 and Pm3, are associated with H that resonates in the low field region, between 12–16.5 ppm, a region that is consistent with acidic structures of the type: H(Ca)PO4−.11,12 It is interesting to notice that although the amount of specie Pm2 detected by 31P-NMR signal increases as more HA in solution is added, this increase does not result from a hydration process since XRD shows not-hydrated phases (Fig. 1). This fact supports that Pm2 is reflecting phosphate interactions with HA binding sites through H+ bounds rather than with calcium bridges.
Focusing on in the effect of phosphate Ca–HA complexation on 1H signals, 2D 1H–31P WISE NMR spectra showed that the intensity of Pm2 and Pm3 correlations increases with the amount of HA used, which is also associated with a slight pH acidification (Fig. 4, Table 1). This behavior can be explained by considering that H+ ions from Hx(Ca)PO4 are released and converted into Hx−1(Ca)PO4− and free H+ causing the acidification of the medium (Table 1). On the other hand, phosphate–Ca complexation in HA did not affect the solubility patterns of MCP, either in water or neutral ammonium citrate (Table 1).
| Sample | % total P | % a.c.s. P | % w.s. P | pH |
|---|---|---|---|---|
| MCP-1% | 23.6 | 23.4 | 23.4 | 3.3 |
| MCP-5% | 23.5 | 23.0 | 22.9 | 3.2 |
| MCP-10% | 22.3 | 21.8 | 21.8 | 2.9 |
| DCP-1% | 22.3 | 21.2 | 0.0 | 7.3 |
| DCP-5% | 19.3 | 18.9 | 0.0 | 7.4 |
| DCP-10% | 14.1 | 10.7 | 0.0 | 7.6 |
Previous studies showed that phosphate complexation in SSP and TSP with HA through Ca bridges resulted in the variation of the P peak width.7,8 The study of this fact in MCP and MCP–HA samples is more complicated due to the appearance of a new peak associated (Pm2) with the disappearance of another P peak (Pm1). However, the deconvolution of 31P-NMR spectra of MCP–HA 10% showed that Pm2 is among Pm1 and Pm3 peaks (Fig. 5) and as 2D 1H–31P WISE NMR spectrum revealed no organic-attached H was related to that.11,12 This fact implies that MCP and HA interactions do not distort the P nucleus in the MCP crystallographic cells.
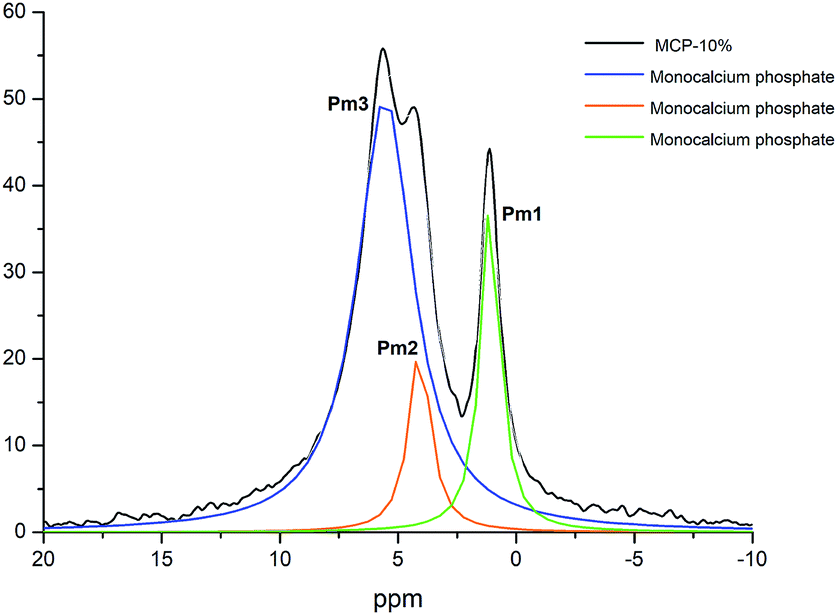 | ||
| Fig. 5 Deconvoluted peaks of 31P NMR spectrum of MCP sample prepared with 10% of aqueous potassium humate and crystallographic phase assignation. | ||
Structural changes induced by HA–Ca–phosphate interaction in DCP
The X-ray diffraction analysis (XRD) showed two different crystallographic phases for DCP and DCP–HA: monetite (anhydrous phase) and brushite (hydrated phase) (Fig. 6). Brushite appears indeed with the lower HA application (1%) and this phase increased as the aqueous HA amount applied was increased (Fig. 6).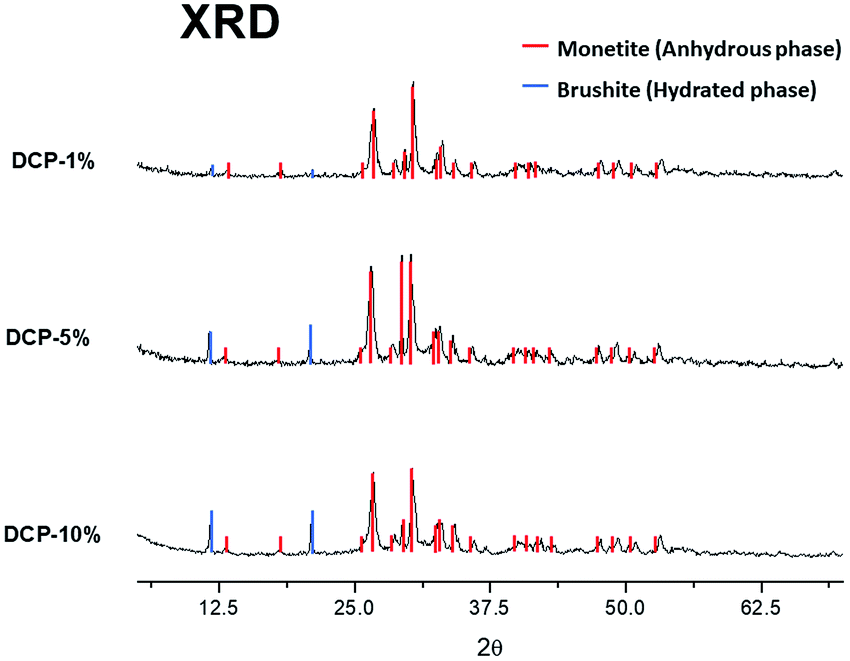 | ||
| Fig. 6 X-ray diffraction analysis (XRD) of crystallographic phases on DCP–HA (1, 5 and 10%) fertilizer samples. | ||
The 31P NMR spectrum of DCP showed three peaks at 4.20, 5.37 and 7.03 ppm (Fig. 7). In the case of DCP–HA (1%) the peaks at 7.03 and 5.37 ppm were hidden under the broad peak at 4.20 ppm. When HA was increased at 5% and 10% the peak at 7.03 ppm is clearer and increase as HA increase (Fig. 7). However, the peak at 5.37 ppm continued being hidden. Interestingly, a shoulder may be observed at approximately 8.40 ppm in DCP–HA that might be assigned to organic P species.
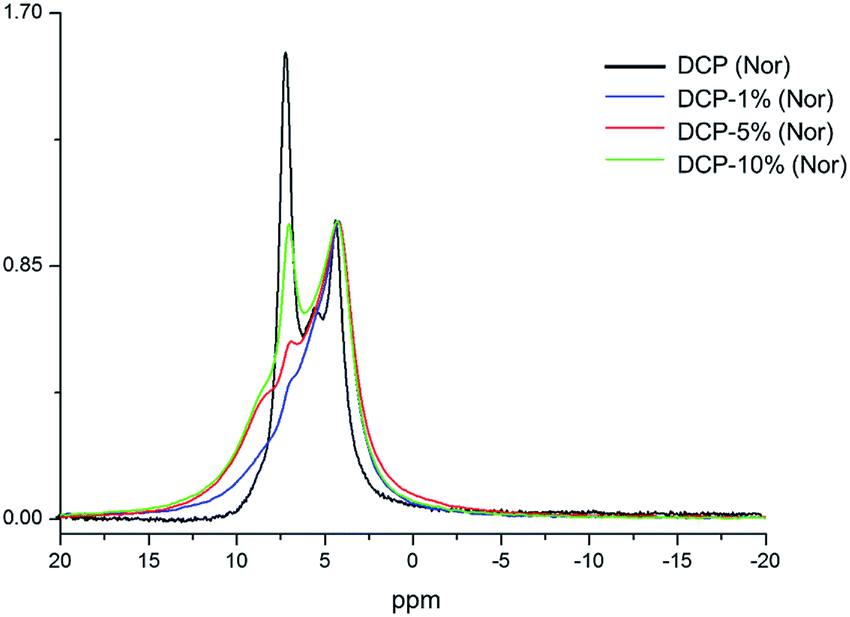 | ||
| Fig. 7 Superimposition of 31P NMR spectra of DCP fertilizer samples prepared at different amounts of aqueous potassium humate (1, 5 and 10%). The intensities are normalized with respect to the signal at 4.20 ppm. | ||
Molecular modelling assisted the assignment of the 31P-NMR peaks (Fig. 8). The peak at 4.20 ppm (Pd1) is assigned to the species coordinated to 6 calcium atoms. The peak at 7.03 ppm (Pd3) is assigned to the species coordinated to 4 calcium atoms. And again, we assigned the intermediate peak that appeared at 5.37 ppm (Pd2) to species coordinated to 7 calcium atoms.
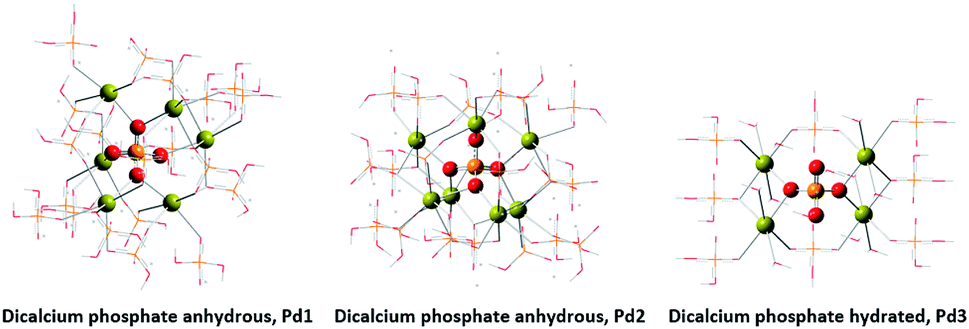 | ||
| Fig. 8 Crystallographic structures by molecular modelling of DCP. | ||
The 2D 1H–31P WISE spectrum (Fig. 9) indicated that two types of H signals are in proximity to the P peak of the species Pd1 assigned to H(Ca)PO42−. The P signal of Pd3 correlates with a proton a 10.65 ppm which is associated to hydrated form, Brushite.12,13 Correlations of the P peak of Pd2 with protons could not be identified in this spectrum due to their overlap with the broad peaks of Pd1. A fourth signal appears in the spectrum as a shoulder at ca. 8 ppm in the P dimension that correlates with protons at 0.49 ppm. This peak was assigned to a P-organic species (OrPd).13 Interestingly, the deconvolution of 31P-NMR spectra of DCP and DCP–HA showed that the intensity of OrPd peak increases as the amount of HA also increases (Fig. 10).
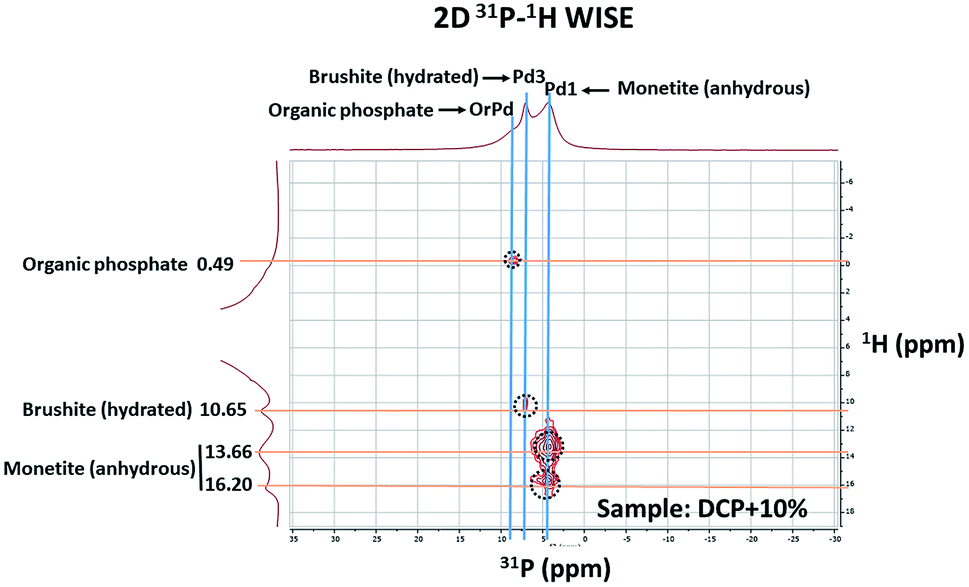 | ||
| Fig. 9 2D 1H–31P WISE spectrum of DCP prepared with 10% of aqueous potassium humate showing the assignment of the peaks. | ||
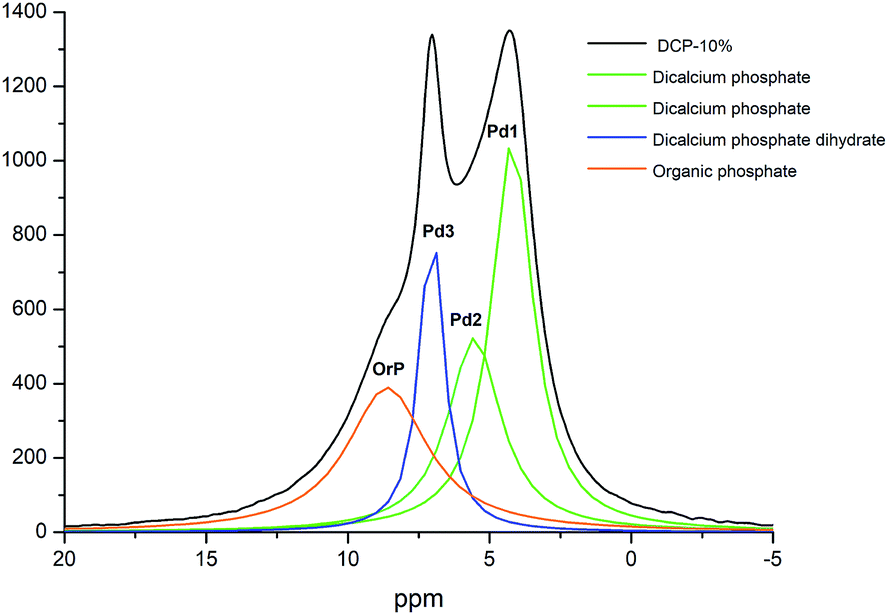 | ||
| Fig. 10 Deconvoluted peaks of 31P NMR spectrum of DCP sample prepared with 10% of aqueous potassium humate and crystallographic phase assignation. | ||
HA conformational changes induced by HA–Ca–phosphate interaction
To investigate the conformational and molecular changes experienced by HA because of phosphate–Ca–HA interaction, the organic fractions present in the samples of MCP–HA and DCP–HA were extracted with water. It is assumed that the HA fractions in water solution corresponds to the HA that is inserted in phosphate crystalline phases. This is because the high concentration of Ca in the medium will cause free HA to precipitate as Ca–HA complexes.The first remarkable observation is the pH of de water desorption solutions: acidic and around pH 3 for MCP and neutral and around pH 7.5 for DCP. Obviously, pH will affect HA molecular conformation in solution. In the case of MCP–HA, an acidic pH would be associated with the protonation of acidic functional groups favoring inter- and intra-molecular interactions through H bond.14 This fact would lead to increases in molecular size. In the case of DCP–HA, slightly basic pH will reduce inter- and intra-molecular assembling observed in MCP due to the repulsion between ionized acidic groups. Therefore, the molecular conformation in solution of MCP phosphate–Ca–HA and DCP phosphate–Ca–HA complexes would be different from each other. MCP phosphate–Ca–HA would present higher apparent molecular sizes than DCP phosphate–Ca–HA complexes.
This reasoning is supported by the results obtained in HPSEC study. MCP–HA (1%) complexes in solution present three main size populations, one around 5.5 ml of Volume of retention (Vr) that coincides with the size population of free HA, and two more peaks corresponding to 3.5 ml and 2 ml associated with higher molecular sizes. As the HA amount in complexes increases the 5.5 ml and 3.5 ml peaks disappeared and a new peak around 4.2 ml appeared. However, in the case of DCP–HA we observe the peak around 2 ml but the peaks around 5.5 ml remains. This result indicates that whereas in MCP–HA complexes HA molecular size increased very significantly, in DCP–HA there exists an increase in HA molecular size but remains a noticeable population of HA molecules with lower molecular size (Fig. 11).
 | ||
| Fig. 11 HPSEC of extracts from MCP- and DCP-fertilizer samples with different quantities of aqueous potassium humate (1, 5 and 10%). Dotted line in both figures is original humate spectra. | ||
As for the molecular features of HA involved in MCP–HA and DCP–HA complexes the information obtained from synchronous fluorescence spectra is very meaningful. In the case of MCP–HA the shape of the spectra was qualitatively similar to that of HA, but the intensity of some peaks varied. The peak at 270 nm, probably corresponding to aromatic amino acids in protein residues,15 increases as HA concentration in complexes also increases (Fig. 12). This fact suggests that phosphate–Ca–HA complexation may cause changes in HA molecular conformation favoring the release of fluorophores present in these residues that are quenched in free HA. As for the main peaks, the results suggest some changes in the populations corresponding to more condensed molecular conformations (480 nm) and simpler molecular conformations (380–425 nm).15 The lowest concentration of HA (1%) is associated with some molecular disaggregation reflected in the higher intensity of 380 nm peak comparing with the 480 nm peak. However, as HA concentration increases more complex, aggregated, conformations (480 nm peak) reappeared (Fig. 12). In the case of DCP–HA complexes, a major effect of molecular disaggregation from more complex-aggregated conformation to simpler conformations is associated with phosphate–Ca–HA interaction (Fig. 12). Whereas the peak at 480 nm tends to disappear with HA complexation, the intensity of peaks around 380 and 270 increased very significantly (Fig. 12). This fact may result from the acidification-realkalinization process associated with DCP–HA synthesis, since the first step of the reaction involves the presence of a fraction of free phosphoric acid that is neutralized as the reaction is completed. In this case, charge repulsion at basic pH will constrain the re-aggregation of HA molecular moieties in DCP–HA complexes. This phenomenon has been reported previously by different studies.14
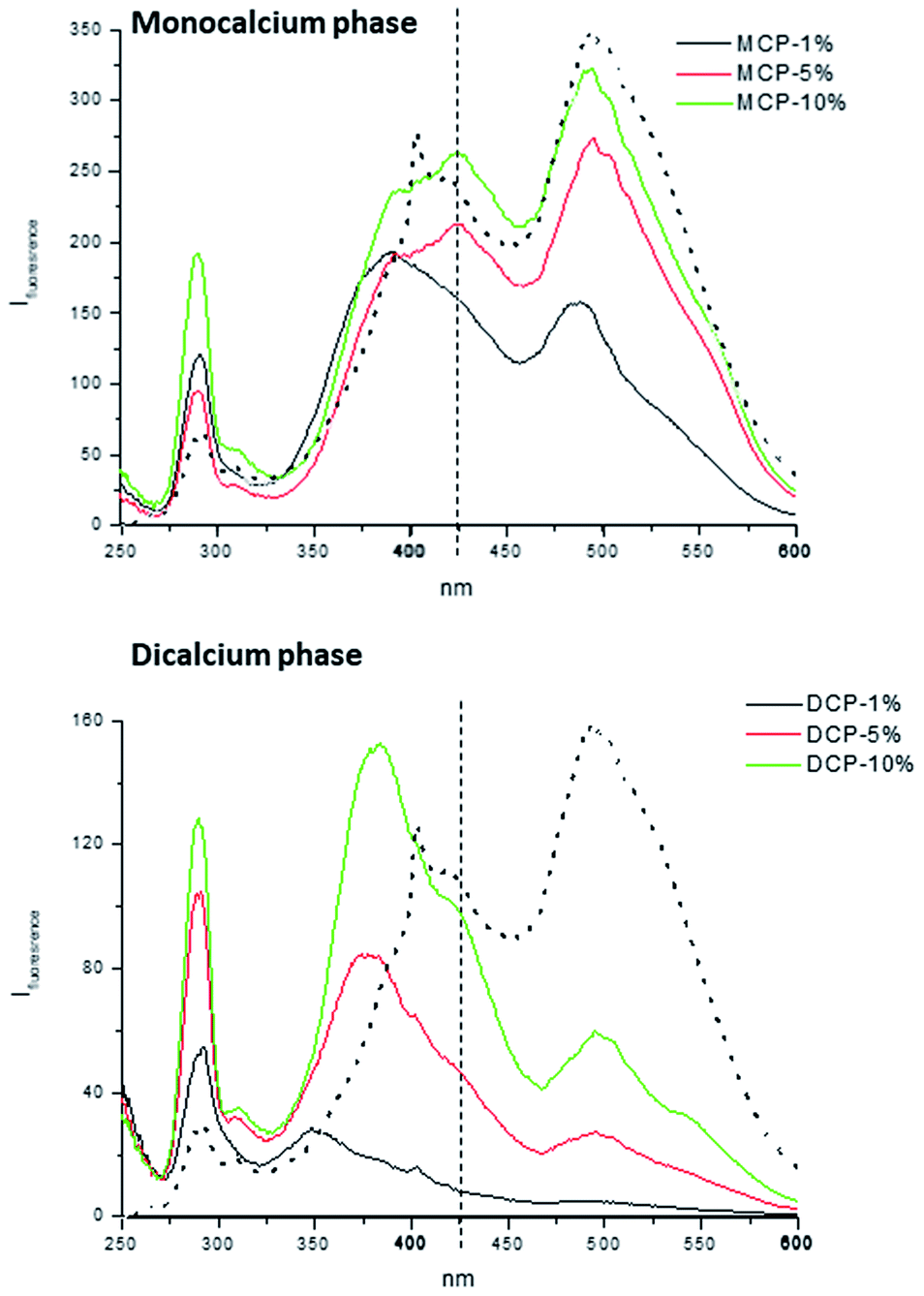 | ||
| Fig. 12 Synchronous fluorescence spectra of extracts from MCP- and DCP-fertilizer samples with different quantities of aqueous potassium humate (1, 5 and 10%). Dotted line in both figures is original humate spectra. | ||
It is noteworthy that the results obtained from HPSEC and those from synchronous fluorescence appear to be contradictory. Whereas HPSEC show a clear increase in molecular size with the complexation process, synchronous fluorescence suggests that complexation is associated with molecular disaggregation and structural simplification. This fact may be explained if the increase in molecular size involves non-fluorescent HA moieties such as aliphatic blocks including lipid-related and sugar-related structures. To some extent, synchronous fluorescence provides useful information about the binding site in HA that is involved in the interaction with calcium phosphate, whereas the information obtained from HPSEC is related to the bulk molecular system. In this sense, it is noteworthy that the results presented here support the relevant role of acidic groups (phenol, carboxylic) in aromatic regions on the interaction of HA with phosphate through Ca bridges. On the other hand, the different behavior of HA in MCP–HA and DCP–HA might be not only related to pH but also with the specific characteristics of MCP- and DCP-crystalline phases.
Conclusions
In consequence, the molecular features of HA in MCP–HA and DCP–HA are different from each other, and this fact might have important consequences in HA biological activity. Several studies have shown that the biological activity of HA varies depending on the origin and type (HA or FA) of HS.16 In general HA fractions with lower apparent molecular size and higher functionality present more mobility in soil solution and higher biological activity.16,17 Applying this concept to our results it would be possible that the HA in DCP is more active as biostimulant of biological processes than that in MCP. Further studies are needed to validate this hypothesis.Experimental
In this work, HA in different quantities (1%, 5% and 10%) were applied to obtain P-based fertilizers containing mono- and di-calcium phosphate as described in García-Mina.9 The P-based fertilizers were obtained by reaction of phosphoric acid with specific amounts of calcium carbonate and calcium oxide in the presence of HA as described in García-Mina.9Then, we finally obtained samples of monocalcium phosphate: MCP, MCP+1%, MCP+5% and MCP+10%, and from dicalcium phosphate: DCP, DCP+1%, DCP+5% and DCP+10%. HA was applied as potassium salt in water at a final concentration of 15% of HA.
Total P content, ammonium citrate soluble P content and water-soluble P content were determined by dissolving:
• Total P: 2.5 g of sample in a mixture of 20 ml concentrated nitric and 30 ml concentrated sulfuric acids and 30 min boiling. Afterwards 150 ml of water was added and 15 min of boiling.
• Ammonium citrate soluble P: 1 gr of sample in 100 ml of 1 N ammonium citrate after 1 hour of stirring at 65 °C.
• Water-soluble P: 5 gr of sample in 450 ml of deionized water after 30 min of stirring.
Solutions were diluted up to 500 ml and all P contents were determined by OES-ICP (iCAP 6500 by Thermo Fisher Scientific).
HA used was extracted from leonardite and afterwards was isolated following the International Humic Substances Society method. The elemental content yielded: 47.9% C, 3.97% H, 1.18% N and 46.9% O (by difference). Functional groups analysis by 13C-NMR showed: 14.4% alkyl groups, 10.4% O-alkyl groups, 47.8% aromatic groups, 11.2% phenolic groups, 14.2% carboxylic groups and 2.0% carbonylic groups. Aromaticity/aliphaticity relation was 2.38.
XRD diffractometry
Diffractograms in powder samples were obtained in a D2 Phaser by Bruker and interpreted under comparison with Bruker crystallographic data base.31P, 1H and 2D 1H–31P WISE NMR spectroscopy
31P-NMR spectra were obtained at room temperature using a Bruker XRD-500 Spectrometer, with a static field of 11.7 T. A 1H-BBI probe with Z gradient was used. Chemical shifts were calibrated with liquid H3PO4 as an external standard. The experiments were performed without sample rotation using a 31P pulse at 90° under H1 decoupling on Waltz16 scheme with a field of 3.13 kHz conditions. The relaxation delay was 3 s, the number of scans was 256. FID was acquired with 32 k complex points. All 31P spectra were treated with MestreNova software.Molecular modeling
Theoretical simulations were performed using quantum chemical methods with the electronic structure program package Gaussian09.18The crystal structures for monocalcium phosphate monohydrate, dicalcium phosphate anhydrous and dicalcium phosphate dihydrated were taken from references (Maclennan, G., Beevers, C. A. (1956): The crystal structure of monocalcium phosphate monohydrate, Ca(H2PO4)2·H2O, Acta Cryst., 9, 187–190), (Catti, M., Ferraris, G., Filhol, A. (1977): Hydrogen bonding in the crystalline state. CaHPO4 (monetite), P-1 or P1? A novel neutron diffraction study Locality: Synthetic. Acta Cryst., Section B 33, 1223–1229) and (Beevers, C.A. (1958): The crystal structure of dicalcium phosphate dihydrate, CaHPO4·2H2O, Acta Cryst., 11, 273–277), respectively.
The electronic densities were obtained using the Density Functional Theory (DFT) with the B3LYP exchange-correlation functional, which is a hybrid functional that combines the three-parameter exchange functional of Becke (Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A., 38, 3098–3100.) with the correlation functional of Lee, Yang and Parr (Lee, C., Yang, W., Parr, R.G. (1988): Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B., 37, 785–789.). To simulate the 31P NMR signals we have followed three steps:
(1) Identification of the phosphates containing a different chemical environment within the periodic crystal structure (non-symmetric phosphates).
(2) Generation of a reduced structure from the crystal structure for each non-symmetric phosphate. This reduced structure contains the central phosphate, the calcium atoms coordinated to this phosphate and the remaining phosphates coordinated to the calcium atoms. In order to impose the charge neutrality, the external phosphates were saturated with hydrogen atoms.
(3) Gauge including atomic orbitals (GIAO) method was applied to calculate the magnetic shielding of the central phosphorous. The DFT functional mentioned above was employed for the calculations together with the 6-31G(d,p) basis set.
Synchronous fluorescence spectroscopy
Synchronous fluorescence spectra were obtained in a Jasco FP-8200 Spectrofluorometer in the same conditions described in Peuravuori et al.15 after extraction of 1 g of sample in 100 ml of deionized water.HPSEC chromatography
Chromatography was carried out in an Alliance 2795 Waters HPLC followed by 2796 PDA Waters detector. For separation was used a column XBrigde Protein BEH SEC, 200 Å, 3.5 μm from waters. Spectra were obtained at 280 nm. Conditions were performed as follow, mobile phase: water type I ultrapure grade (pH 6.7), gradient: isocratic, flow: 1 ml min−1, column temperature: room temperature, sample temperature: room temperature, injection volume: 10 μl. Samples injected were the same that extracted for synchronous fluorescence.Conflicts of interest
There are no conflicts to declare.References
- F. Magdoff and R. Weil, Soil organic matter in sustainable agriculture, CRC Press LLC, Boca Raton, 2004 Search PubMed.
- F. J. Stevenson, Humus Chemistry: Genesis, Composition, Reactions, Wiley, New York, 1994 Search PubMed.
- J. Gerke, Commun. Soil Sci. Plant Anal., 1992, 23, 601, DOI:10.1080/00103629209368612.
- J. Gerke, Soil Sci., 2010, 175, 417, DOI:10.1097/SS.0b013e3181f1b4dd.
- I. Guardado, O. Urrutia and J. M. García-Mina, J. Agric. Food Chem., 2010, 53, 8673, DOI:10.1021/jf052031p.
- O. Urrutia, J. Erro, I. Guardado, S. San Francisco, M. Mandado, R. Baigorri, J. C. Yvin and J. M. Garcia-Mina, J. Plant Nutr. Soil Sci., 2014, 177, 128, DOI:10.1002/jpln.201200651.
- R. Baigorri, O. Urrutia, J. Erro, M. Mandado, I. Perez-Juste and J. M. Garcia-Mina, ChemSusChem, 2013, 6, 1245, DOI:10.1002/cssc.201300024.
- R. Alvarez, L. A. Evans, P. J. Milham and M. A. Wilson, Geoderma, 2004, 118, 245, DOI:10.1016/S0016-7061(03)00207-6.
- J. M. García-Mina, EU Pat., Fertilising composition EP1612200B1D, Munich, 2013.
- J. Riggle and R. von Wandruszka, Talanta, 2005, 66, 372, DOI:10.1016/j.talanta.2004.11.003.
- J. Riggle and R. von Wandruszka, Talanta, 2007, 73, 953, DOI:10.1016/j.talanta.2007.05.018.
- Z. R. Hinedi, l. S. Goldberg, A. C. Chang and J. P. Yesinowski, J. Colloid Interface Sci., 1992, 152, 141, DOI:10.1016/0021-9797(92)90015-E.
- J. P. Yesinowski and H. Eckert, J. Am. Chem. Soc., 1987, 109, 6274, DOI:10.1021/ja00255a009.
- A. Piccolo, Adv. Agron., 2002, 75, 57, DOI:10.1016/S0065-2113(02)75003-7.
- J. R. K. Peuravuori, R. Koivikko and K. Pihlaja, Water Res., 2002, 36, 4552, DOI:10.1016/S0043-1354(02)00172-0.
- A. Muscolo, M. Sidari, E. Attinà, O. Francioso, V. Tugnoli and S. Nardi, Soil Sci. Soc. Am. J., 2007, 71, 75, DOI:10.2136/sssaj2006.0055.
- W. Schmidt, S. Santi, R. Pinton and Z. Varanini, Plant Soil, 2007, 300, 259, DOI:10.1007/s11104-007-9411-5.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; J. M. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski; D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |