
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow intramolecular coordination bonding (ICB) controls the homolysis of the C–ON bond in alkoxyamines†
Gérard Audrana,
Elena Bagryanskaya bc,
Irina Bagryanskayabc,
Mariya Edeleva*bc,
Jean-Patrick Jolya,
Sylvain R. A. Marque*ab,
Anna Iurchenkovac,
Polina Kaletinabc,
Sergey Cherkasovbc,
Tung To Haia,
Evgeny Tretyakovbc and
Svetlana Zhivetyevab
bc,
Irina Bagryanskayabc,
Mariya Edeleva*bc,
Jean-Patrick Jolya,
Sylvain R. A. Marque*ab,
Anna Iurchenkovac,
Polina Kaletinabc,
Sergey Cherkasovbc,
Tung To Haia,
Evgeny Tretyakovbc and
Svetlana Zhivetyevab
aAix Marseille Univ, CNRS, ICR, UMR 7273, Case 551, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France. E-mail: Sylvain.marque@univ-amu.fr
bN. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry SB RAS, 9 Pr. Lavrentjeva, Novosibirsk 630090, Russia. E-mail: edeleva@nioch.nsc.ru
cNovosibirsk State University, 2 Pirogova Str., Novosibirsk 630090, Russia
First published on 16th August 2019
Abstract
Because the C–ON bond homolysis rate constant kd is an essential parameter of alkoxyamine reactivity, it is especially important to tune kd without a major alteration of the structure of the molecule. Recently, several approaches have become known, e.g., protonation of functional groups and formation of metal complexes. In this paper, coordination reactions of [Zn(hfac)2(H2O)2] with a series of new SG1-based alkoxyamines affording complexes with different structures are presented. The kd values of the complexed forms of the alkoxyamines were compared to those of free and protonated ones to reveal the contribution of the electron-withdrawing property and structure stabilization. Together with previously published data, this work provides clues to the design of alkoxyamines that can be effectively activated upon coordination with metal ions. Furthermore, our results provide insight into the mechanism underlying the influence of complexation on the reactivity of alkoxyamines. This led us to describe different types of coordination: intramolecular in nitroxyl fragment, intramolecular in alkyl fragment, intramolecular between alkyl and nitroxyl fragment, and intermolecular one. All of them exhibit different trends which are dramatically altered by changes in conformation.
1 Introduction
Alkoxyamines are compounds that find a wide variety of applications ranging from valuable synthons to initiators of nitroxide-mediated polymerization (NMP)1 as well as theranostic agents.2 The rate constant of C–ON bond homolysis kd is important for these applications.3 Recently, several methods for tuning kd without alteration of alkoxyamine structure have become known, e.g., protonation of functional groups4–6 and formation of complexes with metal ions Zn2+,7 Cu2+,8 or Tb3+.9 Indeed, complexation is a valuable approach to influence the reactivity of alkoxyamines because it paves the way to metal–polymer complexes and to the tuning of the reactivity of theranostic agents upon reaction with metal-containing proteins in vivo.10,11 Furthermore, formation of complexes of the Zn(hfac)2 electron acceptor matrix with alkoxyamines has a positive effect on NMP because it enhances initiation efficiency.12As we have demonstrated earlier,7–9 alkoxyamine–metal complexes undergo equilibrium dissociation in solution. One can shift this equilibrium by adding a complexation agent for zinc ions, e.g., pyridine (Py) or bi-pyridine. Considering homolysis rate constants, the values of kd for complexes differ from the ones for free alkoxyamines because complexation causes a redistribution of C–ON bond polarity owing to the electron-withdrawing effect. Depending on the structure of the complex, researchers can expect either an increase or decrease of kd. Namely, when coordination involves an alkyl part of an alkoxyamine, kd can increase and vice versa.13 Shifting of solution equilibrium leads to a gradual change of homolysis rate constant kd, thus allowing for smart tuning of its value for optimization. Furthermore, the formation of a complex leads to stabilization of alkoxyamine structure. Consequently, we can expect that coordination should have an influence equivalent to both the electronic effect and structure stabilization.
Here we present the synthesis of Zn(hfac)2 complexes with polyfunctional alkoxyamines based on the SG1 nitroxyl radical and its derivatives (Chart 1). We measured homolysis rate constants kd to evaluate the influence of the coordination. The effect of coordination was compared to that of protonation, intramolecular hydrogen bonds (IHBs) and previously reported complexes (see Chart 2). Together with previously published data, the present work gives some clues to the design of alkoxyamines that can be effectively activated upon coordination with metal ions. Furthermore, our findings provide insight into the mechanism underlying the influence of complexation on reactivity of alkoxyamines. It led us to propose several type of intramolecular coordination bonding (ICB) on the same model as proposed for intramolecular hydrogen-bonding (IHB).14
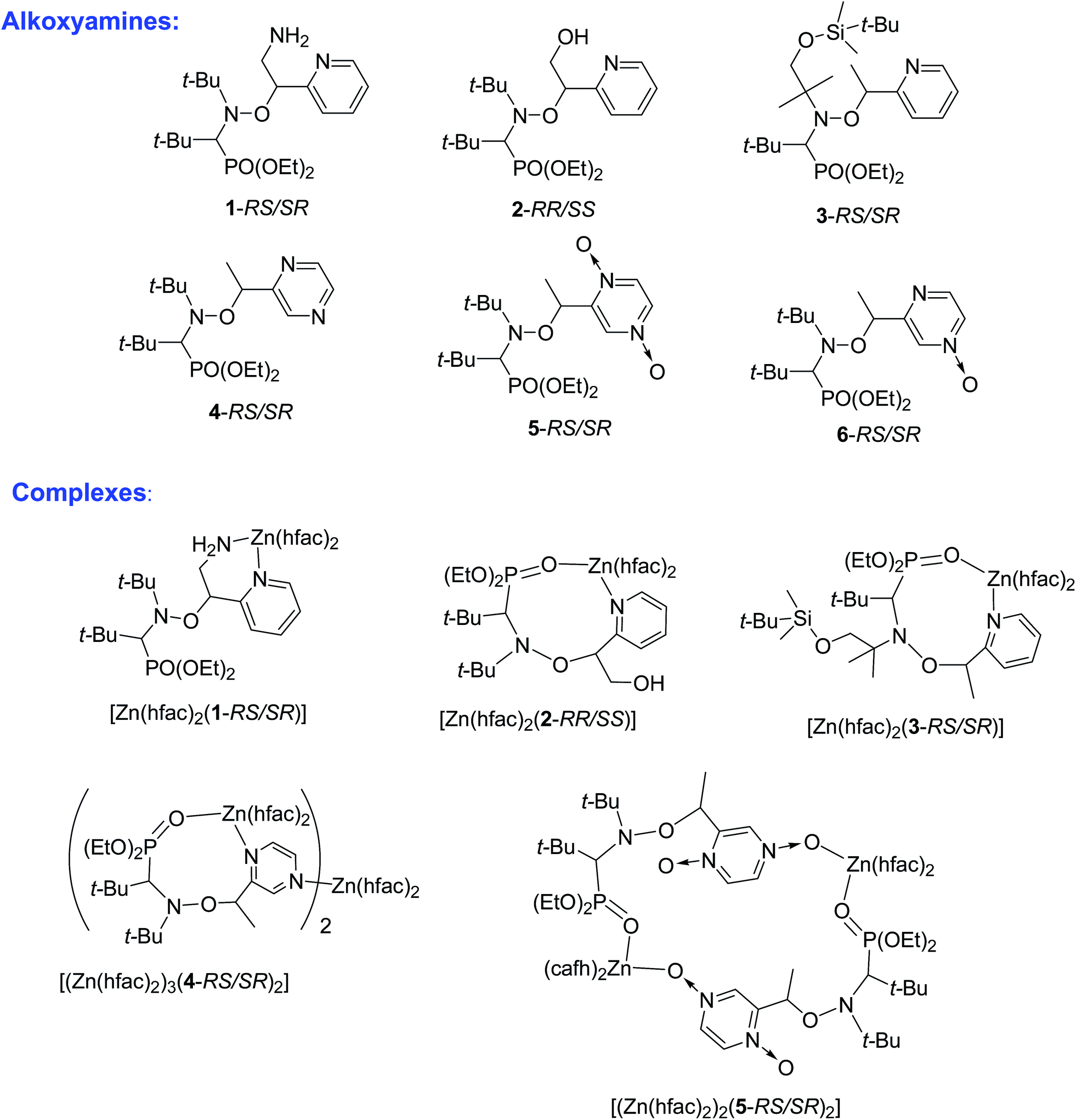 | ||
| Chart 1 The structures of alkoxyamines and their complexes with Zn(hfac)2 drawn on the basis of XRD data. | ||
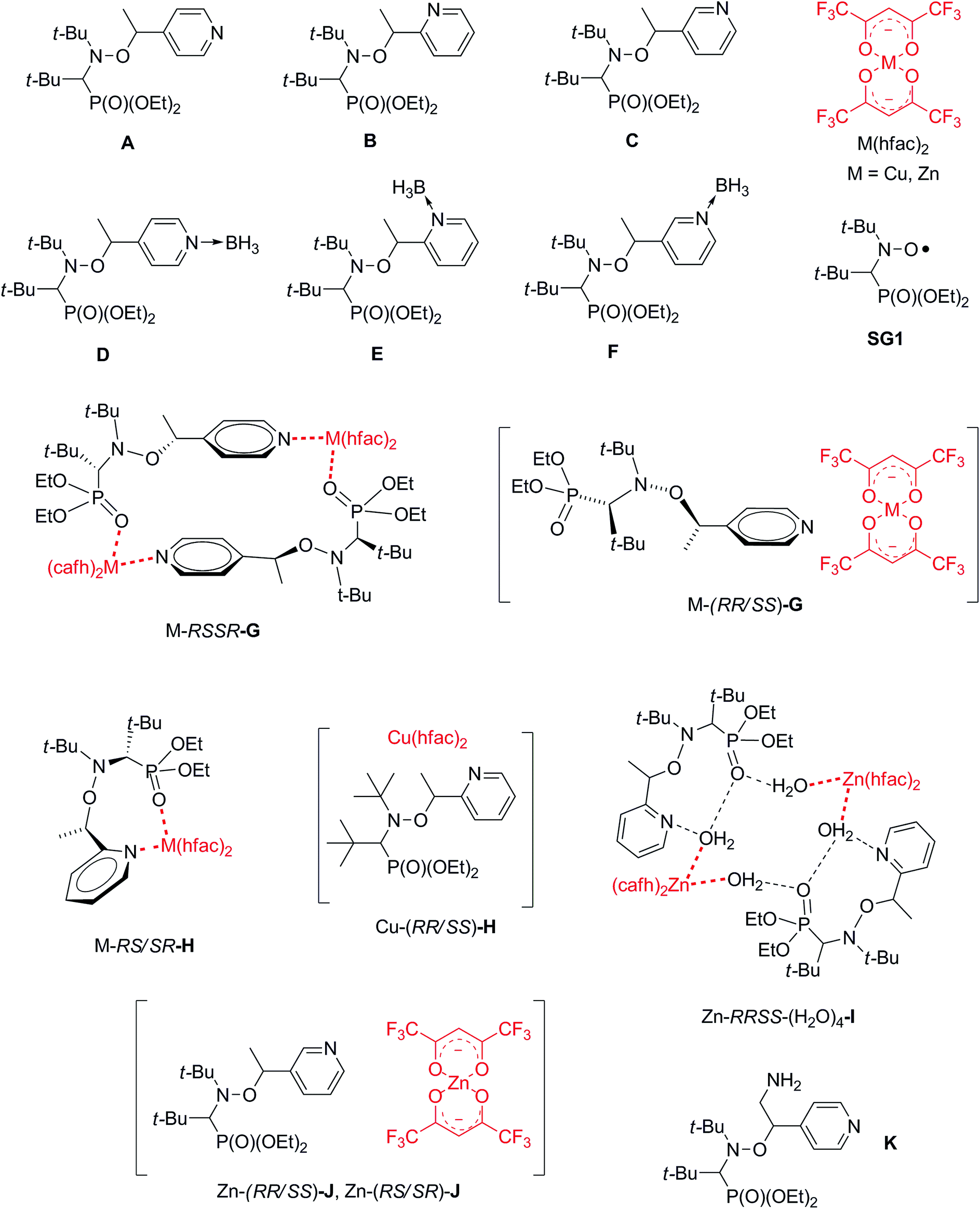 | ||
| Chart 2 Complexes and alkoxyamine models reported in literature. | ||
2 Results
Synthesis and structures of complexes
Syntheses of alkoxyamines 1–3 have been described in the literature.14,15 Alkoxyamines 4–6 are prepared as displayed in Scheme 1, using salen salt procedure and alkene as previously reported.16 Complexes were prepared according to previously reported procedures.7,12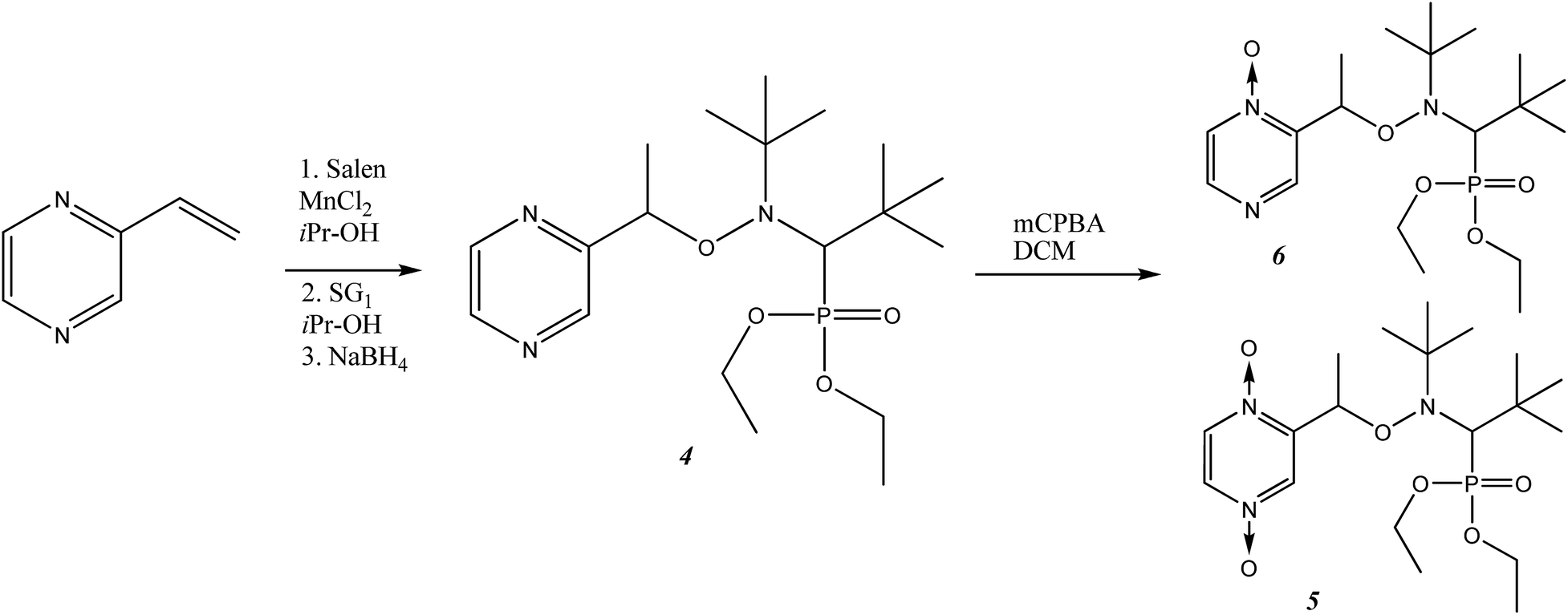 | ||
| Scheme 1 The synthetic scheme for the preparation of alkoxyamines 4–6. | ||
According to our previous observations, the use of the ratio [Zn(hfac)2(H2O)2]/Ln of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as well as the choice of an acetone–heptane mixture as the solvent appear to be suitable for obtaining high-quality crystals of complexes with alkoxyamines. Indeed, under these conditions, the reaction of [Zn(hfac)2(H2O)2] with a racemic mixture of diethyl 1-((2-amino-1-(pyridin-2-yl)ethoxy)(tert-butyl)amino)-2,2-dimethylpropylphosphonates (1-RS/SR) formed the cyclic complex [Zn(hfac)2(1-RS/SR)] with a high yield (89%). The complex was formed upon the coordination of alkoxyamine 1-RS/SR via the two N-atoms of a pyridyl moiety and of the NH2– group of the alkyl moiety (Fig. 1). According to X-ray analysis, the complex was isolated as a solvate with one molecule of acetone.
:1 as well as the choice of an acetone–heptane mixture as the solvent appear to be suitable for obtaining high-quality crystals of complexes with alkoxyamines. Indeed, under these conditions, the reaction of [Zn(hfac)2(H2O)2] with a racemic mixture of diethyl 1-((2-amino-1-(pyridin-2-yl)ethoxy)(tert-butyl)amino)-2,2-dimethylpropylphosphonates (1-RS/SR) formed the cyclic complex [Zn(hfac)2(1-RS/SR)] with a high yield (89%). The complex was formed upon the coordination of alkoxyamine 1-RS/SR via the two N-atoms of a pyridyl moiety and of the NH2– group of the alkyl moiety (Fig. 1). According to X-ray analysis, the complex was isolated as a solvate with one molecule of acetone.
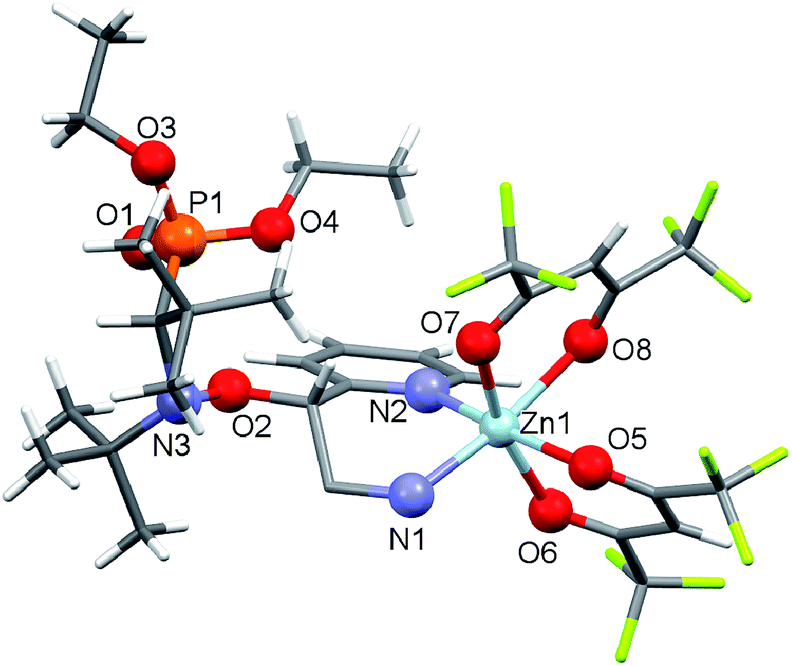 | ||
| Fig. 1 Molecular structure of [Zn(hfac)2(1-RS/SR)]. | ||
Under the same conditions, interaction of [Zn(hfac)2(H2O)2] with a racemic mixture of diethyl 1-(tert-butyl(2-hydroxy-1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropylphosphonates (2-RR/SS) or diethyl 1-((1-(tert-butyldimethylsilyloxy)-2-methylpropan-2-yl)(1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropylphosphonates (3-RS/SR) afforded cyclic complexes [Zn(hfac)2(2-RR/SS)] and [Zn(hfac)2(3-RS/SR)] containing bidentate alkoxyamines coordinated via the N-atom of pyridyl moiety and O-atom of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety (Fig. 2). Both complexes were isolated in the form of colorless crystals with high yields (>95%).
O moiety (Fig. 2). Both complexes were isolated in the form of colorless crystals with high yields (>95%).
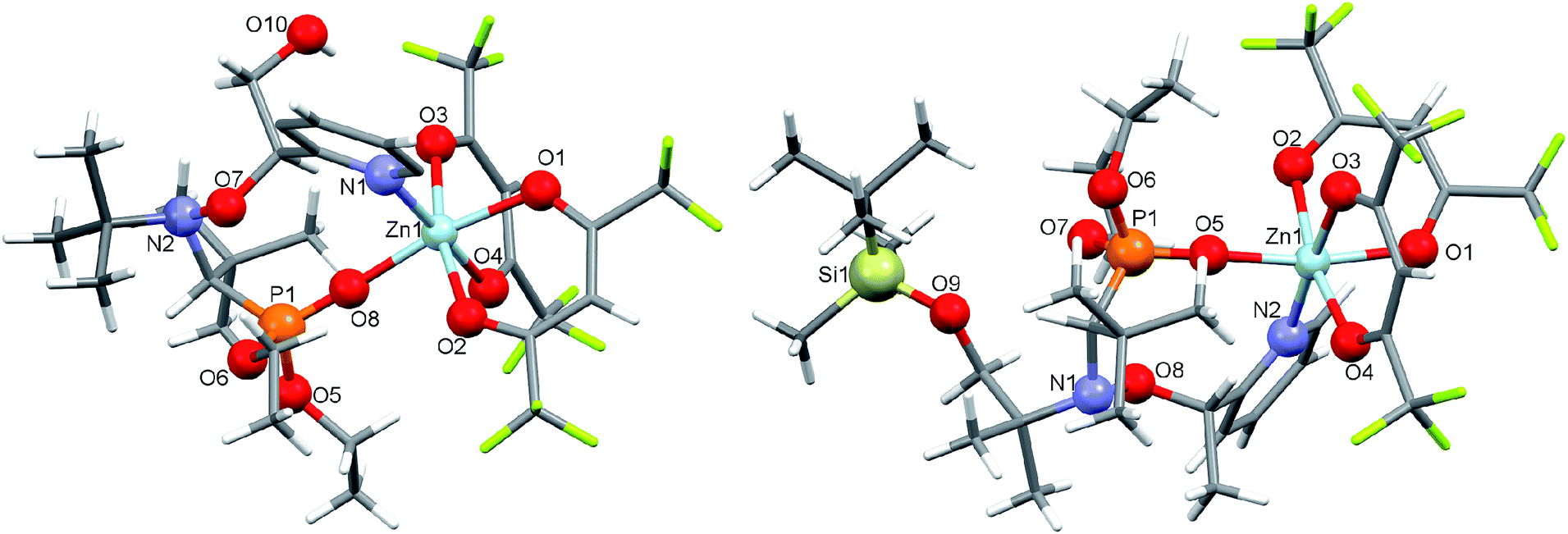 | ||
| Fig. 2 Molecular structures of [Zn(hfac)2(2-RR/SS)] and [Zn(hfac)2(3-RS/SR)]. | ||
Our experiments revealed that the reaction of [Zn(hfac)2(H2O)2] with the racemic mixture of diethyl 1-(tert-butyl(1-(pyrazin-2-yl)ethoxy)amino)-2,2-dimethylpropylphosphonates (4-RS/SR) in the molar ratio 1:1 led only to decomposition of initial alkoxyamine 4-RS/SR. The use of an appropriate excess of [Zn(hfac)2(H2O)2] in an acetone–heptane mixture resulted in trinuclear complex [(Zn(hfac)2)3(4-RS/SR)2] in a quantitative yield. In the [(Zn(hfac)2)3(4-RS/SR)2] complex, two cyclic parts Zn(hfac)2(4-RS/SR) containing bidentate coordinated ligand 4-RS/SR are bound together by the Zn(hfac)2 matrix (Fig. 3).
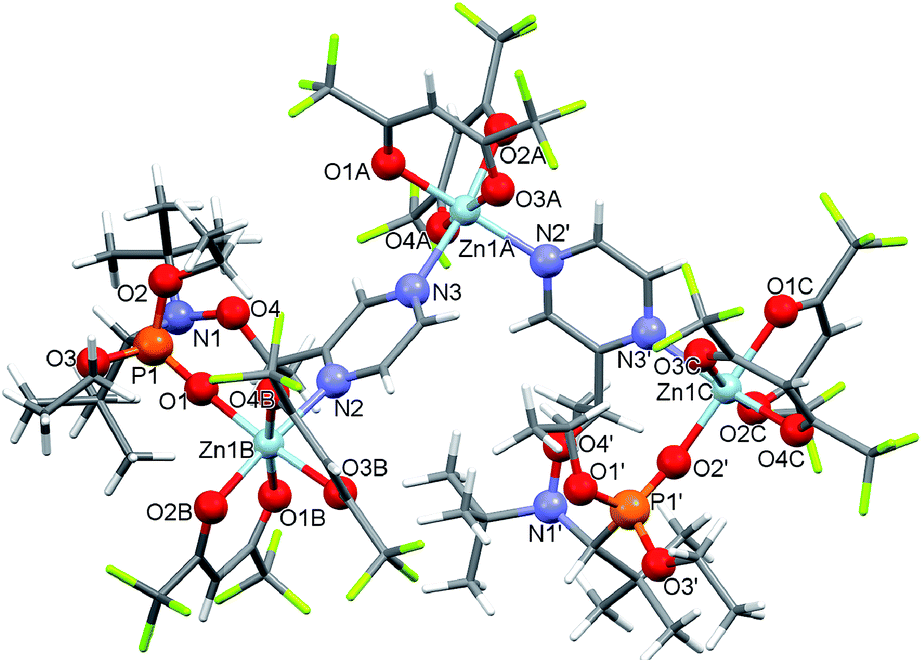 | ||
| Fig. 3 Molecular structure of [(Zn(hfac)2)3(4-RS/SR)2]. | ||
Under the same conditions, the interaction of [Zn(hfac)2(H2O)2] with the racemic mixture of the bis-N-oxide derivatives (5-RS/SR) in the molar ratio 1:1 afforded a centrosymmetric cyclic complex: [(Zn(hfac)2)2(5-RS/SR)2] (Fig. 4). The [(Zn(hfac)2)2(5-RS/SR)2] complex was isolated as pale pink crystals with a high yield (98%).
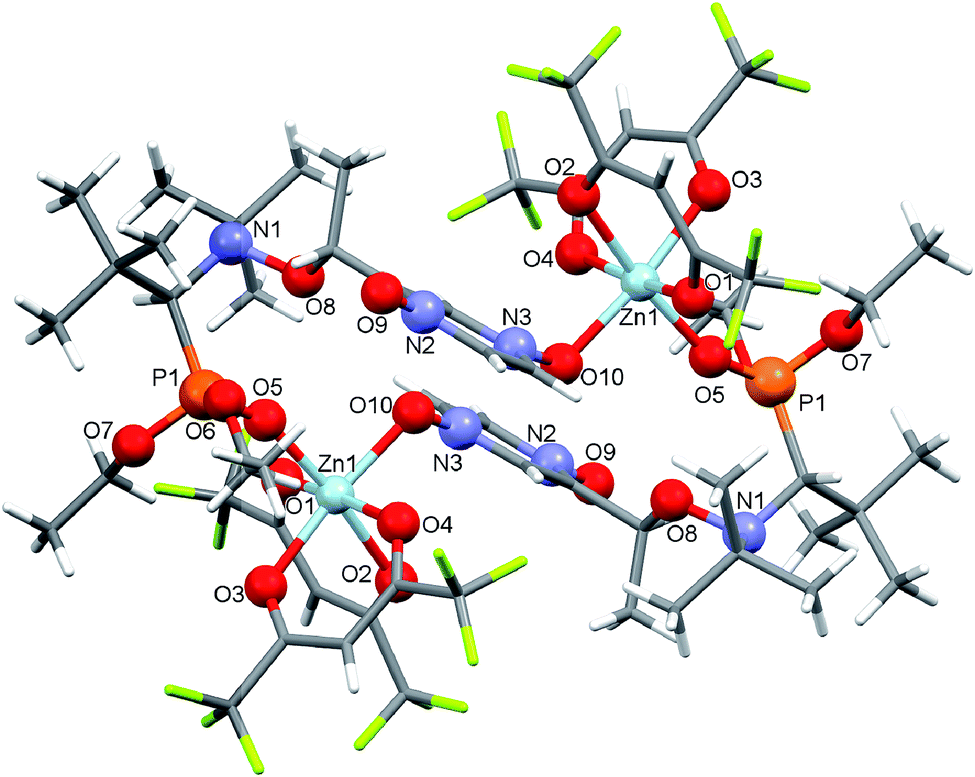 | ||
| Fig. 4 Molecular structure of cyclic complex [(Zn(hfac)2)2(5-RS/SR)2]. | ||
Thus, the interaction of the Zn(hfac)2 matrix with polyfunctional alkoxyamines 1–5 led to the formation of different complexes as the least soluble species: mononuclear and binuclear cyclic complexes and a trinuclear zinc complex as well. The molecular and crystal structures of all the complexes were solved by monocrystal X-ray diffractometry.
XRD structure of 6 was determined and show all geometrical features already reported for free alkoxyamines based on SG1-nitroxyl fragment and does not deserved more comments.
NMR analysis
The structure of alkoxyamines and complexes in solution was studied by 1H and 31P NMR analyses. The influence of the complexation was compared to that of protonation. In the solution of [Zn(hfac)2(1-RS/SR)], we observed single resonance at 23.6 ppm in a 31P NMR spectrum (Fig. 6a). The signal was broad, i.e., the width was 40 Hz due to slow chemical exchange between free and complexed forms of the alkoxyamine. Even though X-ray diffraction (XRD) data indicate that the diethylphosphono group is not involved in the complex formation, we observed a significant impact of the complexation on electronic structure of the alkoxyamine PO group because the 31P resonance of the alkoxyamine ligand differs significantly from that of the free alkoxyamine. Upon gradual addition of pyridine as a competitive ligand, we observed a downfield shift of the signal's chemical shift of the P atom. When 2 equiv. of pyridine were added, the line broadened, meaning an intermediate chemical exchange between different types of complexes present in the solution. Further addition of pyridine resulted in a gradual decrease in the effective concentration of the complex because we observed a constant increase in the 31P chemical shift with a final value of 24.2 ppm. Even in the presence of 100 equiv. of pyridine, we did not observe the complete decomposition of the complex because the value of the chemical shift was different from the free alkoxyamine. It should be noted that protonation of 1 upon addition of 1 equiv. of TFA causes a significant downfield shift of phosphorus signals to 25.0 ppm. In this case, the influence on the δ of the diethylphosphono group is caused by the electron-withdrawing effect of the protonated alkyl moiety of the alkoxyamine.
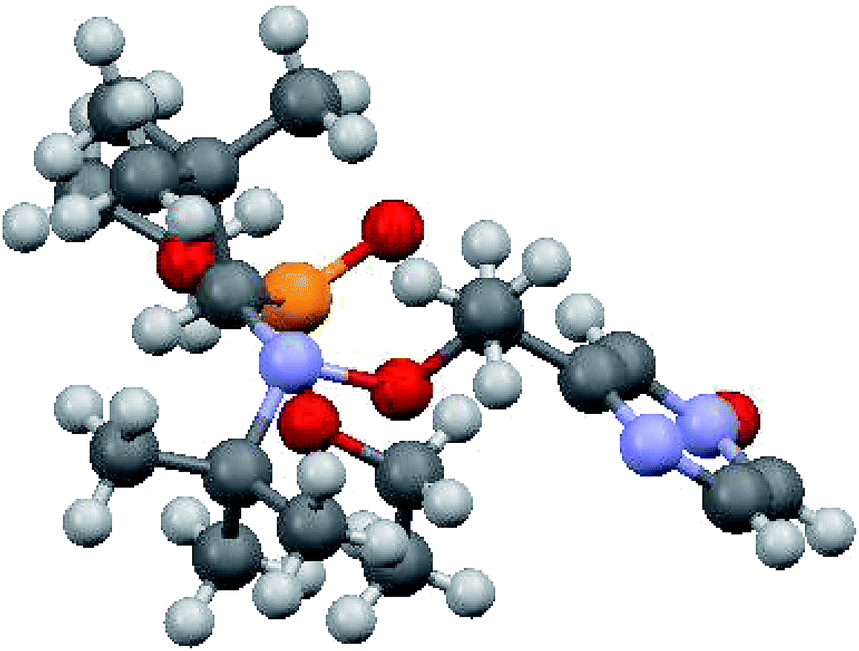 | ||
| Fig. 5 Molecular structure of 6. | ||
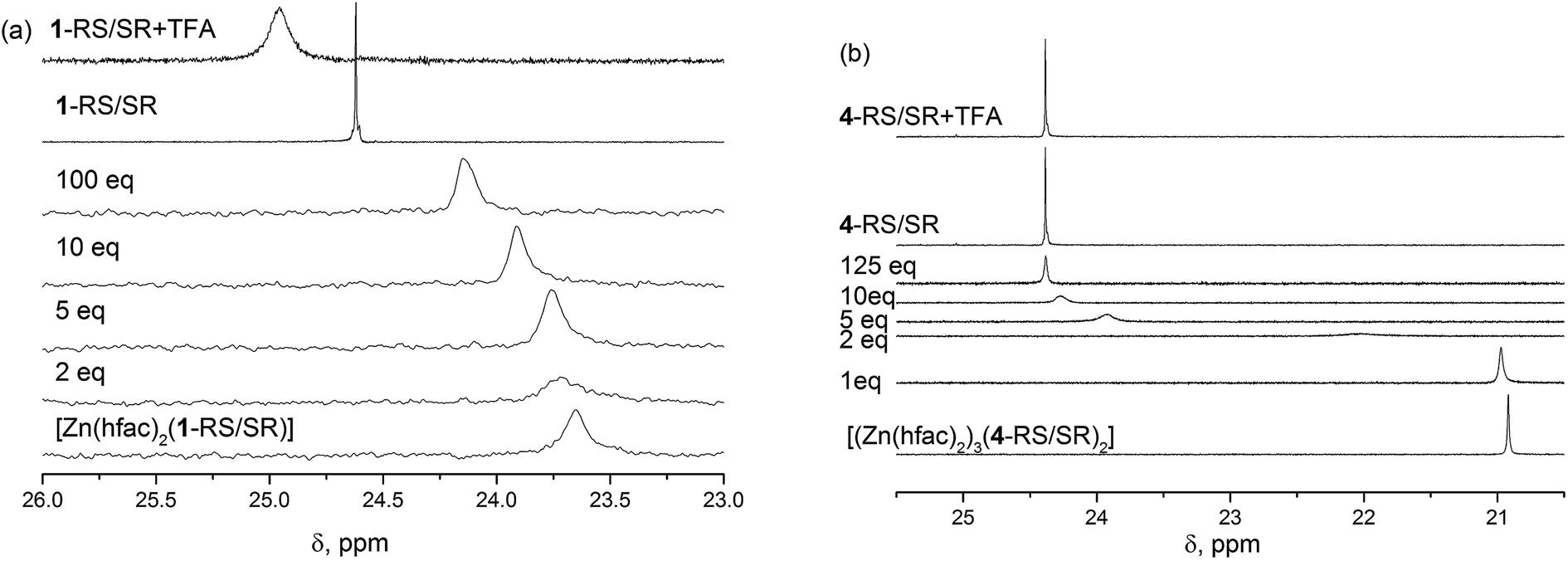 | ||
| Fig. 6 31P NMR spectroscopy of [Zn(hfac)2(1-RS/SR)] (a) and of [(Zn(hfac)2)3(4-RS/SR)2] (b) complex in C6D6 (0.02 M solution) with different amounts of pyridine as a competitor along with a free alkoxyamine and alkoxyamine with 0.02 M (1 equiv.) of trifluoroacetic acid (TFA). | ||
Complexes [Zn(hfac)2(2-RR/SS)] and [Zn(hfac)2(3-RS/SR)] show similar behavior. The corresponding 1H and 31P NMR spectra are presented as ESI.†
Similarly, the addition of pyridine to the solution of [(Zn(hfac)2)3(4-RS/SR)2] results in downshifting of the 31P signal. When 2 to 10 equiv. of pyridine were added, we observed broadening of the signal, implying intermediate chemical exchange. Although no changes are observed in 31P NMR upon addition of 1 equiv. of TFA (Fig. 6b), significant shifts are observed in the aromatic zone upon addition of both 1 and 2 equiv of TFA meaning that the two N-atom of the pyrazyl moiety are successively protonated (Fig. 7) despite pKa values estimated lower than 1 (see Scheme ESI†).
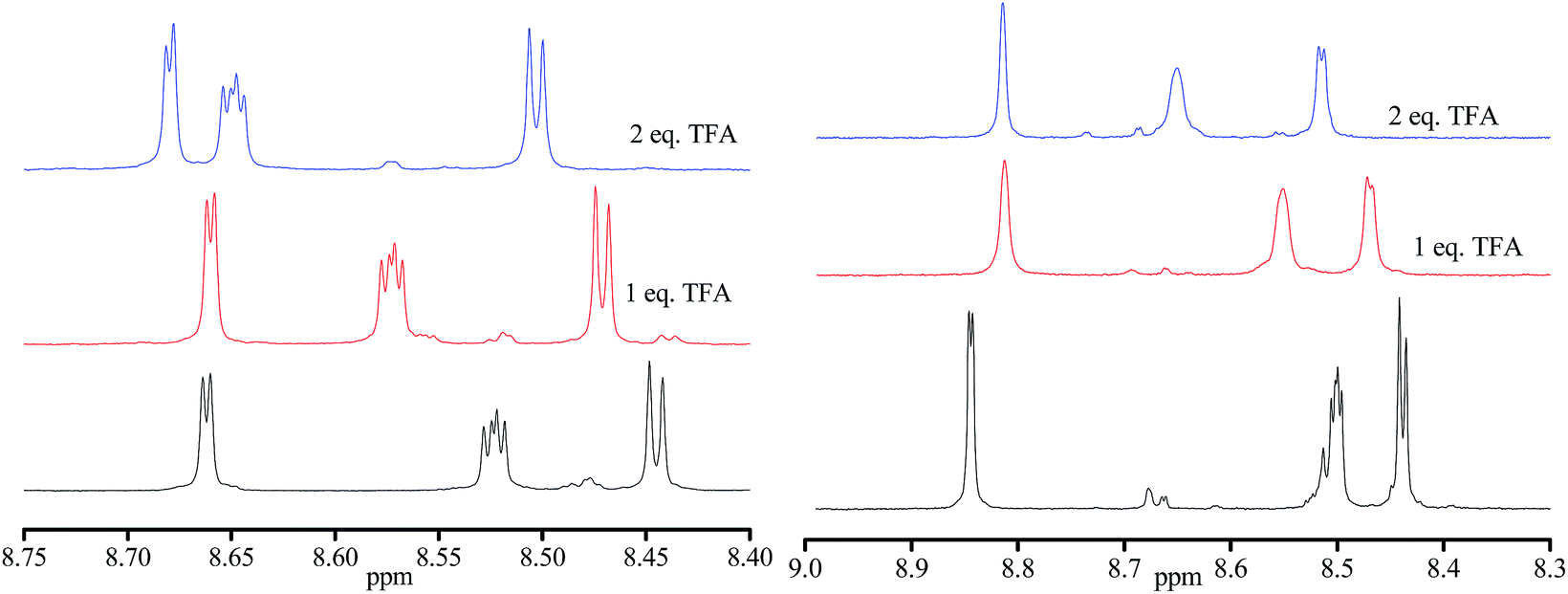 | ||
| Fig. 7 1H NMR of aromatic zone in CDCl3 for 0.02 M of RR/SS-4 (left) and RS/SR-4 (right): non-protonated 4 (bottom), 4 + one equivalent TFA (middle) and 4 + 2 equivalents TFA (top). | ||
Differences reported in the aromatic zone and in the zone of nitroxyl fragment in 31P and 1H NMR signals between [(Zn(hfac)2)2(5-RS/SR)2] and alkoxyamine 5 denote likely an equilibrium between different isomer of unimeric form as already reported and discussed for Zn-RSSR-G.7
Determination of homolysis rate constants kd
The results on homolysis rate constant kd measurements for coordinated, protonated, and pure forms of alkoxyamines are presented in Fig. 8 and in Table 1. For all the species, we observed monoexponential growth of a nitroxide concentration upon heating as displayed in Fig. 8a. In this case, to determine kd, we fitted the experimental data points to eqn (1):| I = I∞(1 − e−kdt) | (1) |
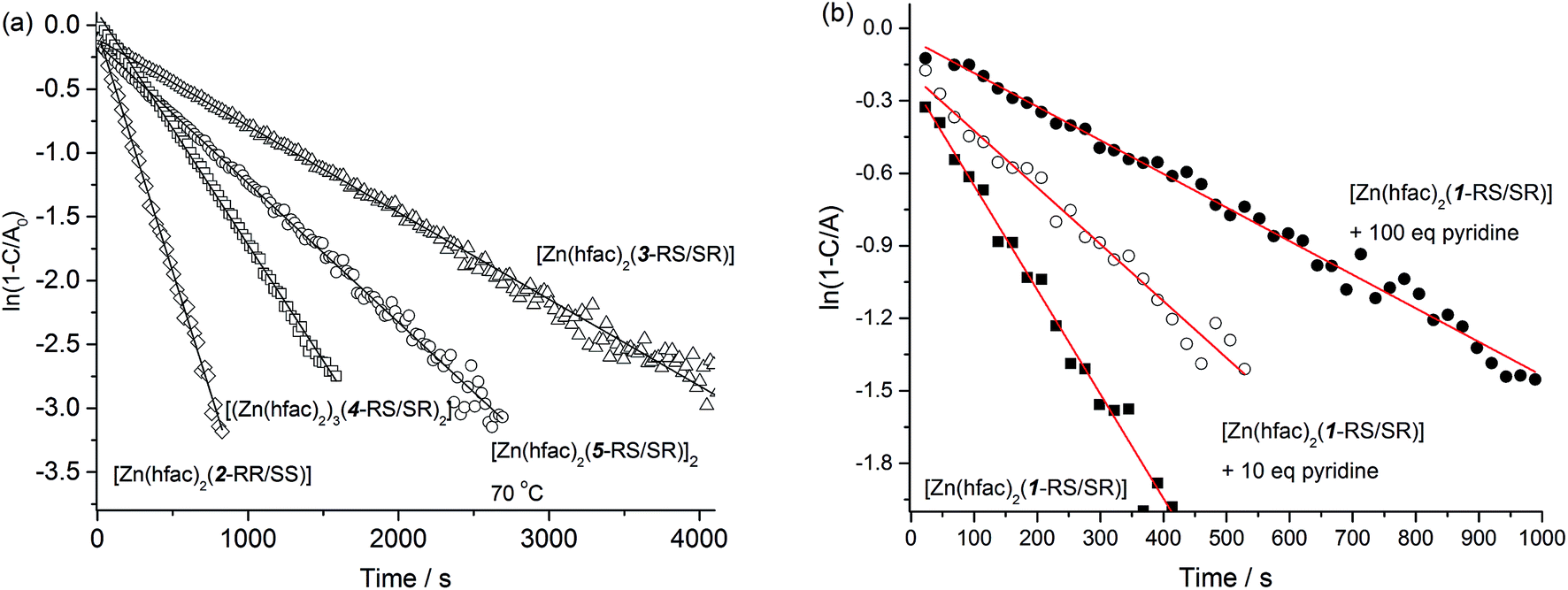 | ||
| Fig. 8 (a) Experimental kinetics (at 80 °C unless specified otherwise) of homolysis of a complex (in semi-logarithmic coordinates) and their subsequent fit to eqn (1). (b) Experimental kinetics of [Zn(hfac)2(1-RS/SR)] homolysis in the presence of various amount of Py at 80 °C. | ||
| Compound | Pyridine (equiv.) | T (°C) | kda (10−3 s−1) | Eab (kJ mol−1) | Ref. |
|---|---|---|---|---|---|
| a Error 5%.b Error at 1 kJ mol−1.c Not concerned.d At 100 °C, kd = 9.7 × 10−3 s−1, Ea = 120.5 kJ mol−1.e For RR/SS diastereoisomer, T = 80 °C, kd = 4.5 × 10−4 s−1, Ea = 119.8 kJ mol−1.f For RR/SS diastereoisomer, T = 55 °C, kd = 5.5 × 10−4 s−1, Ea = 110.8 kJ mol−1.g For RR/SS diastereoisomer, T = 70 °C, kd = 1.5 × 10−4 s−1, Ea = 119.5 kJ mol−1.h For RR/SS diastereoisomer, T = 84 °C, kd = 7.8 × 10−4 s−1, Ea = 119.5 kJ mol−1.i For RR/SS diastereoisomer, T = 86 °C, kd = 11.5 × 10−4 s−1, Ea = 119.8 kJ mol−1.j For RS/SR, T = 80 °C, kd = 2.4 × 10−4 s−1, 121.8 kJ mol−1, see ref. 17.k For RS/SR-1 + 1 equiv. TFA, T = 61 °C, kd = 7.2 × 10−4 s−1, Ea = 112.1 kJ mol−1. For RS/SR-1 + 2 equiv. of TFA, T = 61 °C, kd = 7.6 = 10−4 s−1, Ea = 111.9 kJ mol−1, see ref. 17.l Averaged value of duplicate experiments: T = 80 °C, kd = 4.5 × 10−4 s−1, Ea = 119.8 kJ mol−1 and T = 80 °C, kd = 7.0 × 10−4 s−1, Ea = 118.0 kJ mol−1.m Average value of duplicate experiments: T = 70 °C, kd = 2.5 × 10−4 s−1, Ea = 118.0 kJ mol−1 and T = 86 °C, kd = 13.0 × 10−4 s−1, Ea = 118.7 kJ mol−1. | |||||
| [Zn(hfac)2(1-RS/SR)] | 0 | 80 | 4.2 | 113.0 | This work |
| 1 | 80 | 3.2 | 114.0 | This work | |
| 10 | 80 | 2.4 | 115.0 | This work | |
| 100 | 80 | 1.4 | 116.5 | This work | |
| 1j-RS/SR | —c | 100 | 2.5 | 121.0 | This work |
| 1-RS/SR + 1 equiv. TFAk | —c | 80 | 2.6 | 114.5 | This work |
| [Zn(hfac)2(2-RR/SS)] | 0d | 90 | 3.8 | 120.5 | This work |
| 2-RR/SS | —c | — | — | 121.5 | 14 |
| 2-RR/SS + 1 equiv. TFA | —c | — | — | 118.0 | 15 |
| [Zn(hfac)2(3-RS/SR)] | 0 | 100 | 6.8 | 125.0 | This work |
| 3-RS/SR | —c | — | — | 122.0 | 14 |
| 3-RS/SR + 1 equiv. TFA | —c | — | — | 114.0 | 14 |
| [(Zn(hfac)2)3(4-RS/SR)2] | 0 | 100 | 1.8 | 122.0 | This work |
| 4e-RS/SR | —c | — | —l | 118.9l | This work |
| 4-RS/SR + 1 equiv. TFAh | —c | —m | —m | 118.3m | This work |
| 4-RS/SR + 2 equiv. TFAi | —c | 84 | 1.2 | 118.4 | This work |
| [(Zn(hfac)2)(5-RS/SR)]2 | 0 | 70 | 1.1 | 124.0 | This work |
| 5f-RS/SR | —c | 65 | 1.6 | 111.1 | This work |
| 5-RS/SR + 1 equiv. TFA | —c | 70 | 2.9 | 111.0 | This work |
| 6g-RS/SR | —c | 70 | 0.2 | 118.2 | This work |
Activation energies were estimated via preexponential factor A0 = 2.4 × 1014 s−1.
We also performed measurement of kd for [Zn(hfac)2(1-RS/SR)] in the presence of different amounts of pyridine to investigate the behavior of kd after a gradual decrease in the concentration of the complex. The kinetic curves still had the monoexponential profile, in this case meaning that the equilibrium between the free and complexed forms of an alkoxyamine is reached fast.
3 Discussion
General comments
During the 20 last years, effects ruling the C–ON bond homolysis have been thoroughly investigated.18,19 General trends have been observed:18,19 increasing kd values with increasing electronwithdrawing properties and bulkiness of the groups carried by the alkyl fragment,20,21 with stabilization of the released alkyl and nitroxyl radicals, and, on the other hand, anti-correlated trends22–24 were reported for the nitroxyl fragments. It led us to define some geometrical requirements and orbital interactions which have to be met at TS (Fig. 9),18,19,25 that is, dihedral angle θ 〈OCCX〉 close to 90°, dihedral angle 〈CONlone pair nN〉 close to 180°, and flattening at the N-atom of the C–ON moiety, and donation of the lone pair into the antibonding orbital of the C–O bond nN → σ*C–O, and then donation of the bonding orbital of the C–O bond into the antibonding orbital of the unsaturated moiety σC–O → π*CX.
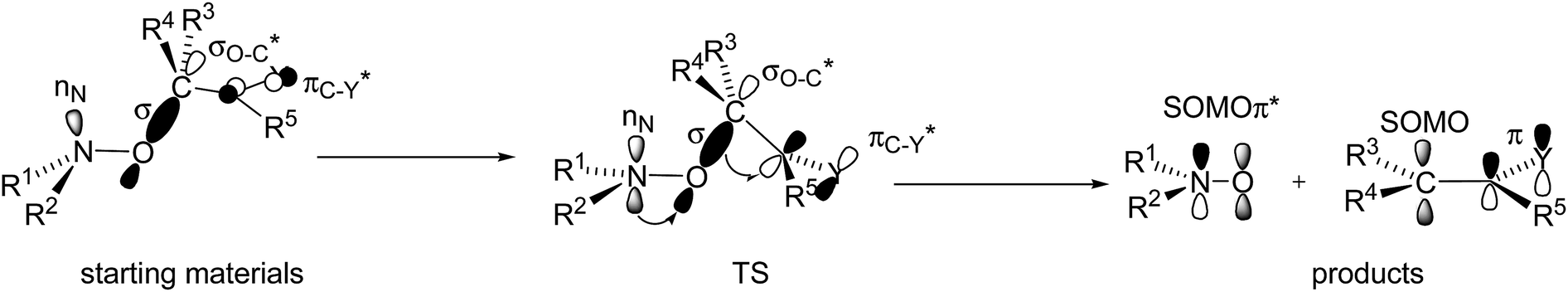 | ||
| Fig. 9 Orbital interactions and geometries in starting materials (left), at TS (middle), and in products (right). | ||
Based on a large set of data, several empirical or semi-empirical equations accounting for these effects have been proposed.20,22–24,26,27 However, in most cases, for the chemical activation of the alkoxyamine, these correlations failed to describe and to predict accurate values of kd although trends are still good. These trends are often modified by changes in conformation caused by large steric repelling interactions or IHB. Recently, we showed that different types of IHB – intraN, interN, intraR and interR (Fig. 10) – are possible and modify the basic trends in very different ways.14 Thus, we proposed to describe the different types of coordination in a similar way (Fig. 10): coordination by bidentate nitroxyl fragment (intraN), coordination by bidentate alkyl fragment (intraR), coordination by alkyl and nitroxyl fragments (interF), and intermolecular coordination bonding, i.e., metal cation coordinated at least by two alkoxyamines. For cases (b–d) and (g) in Fig. 10, the occurrence of IHB or ICB induce the formation of cyclic compounds and, hence, homolysis requires to cleave two bonds: the covalent C–O and the weaker IHB or ICB bond which increases activation energy. However, the occurrence of IHB or ICB implies often changes in conformation which may balance the effect of the second bond, or may strengthened its effect.
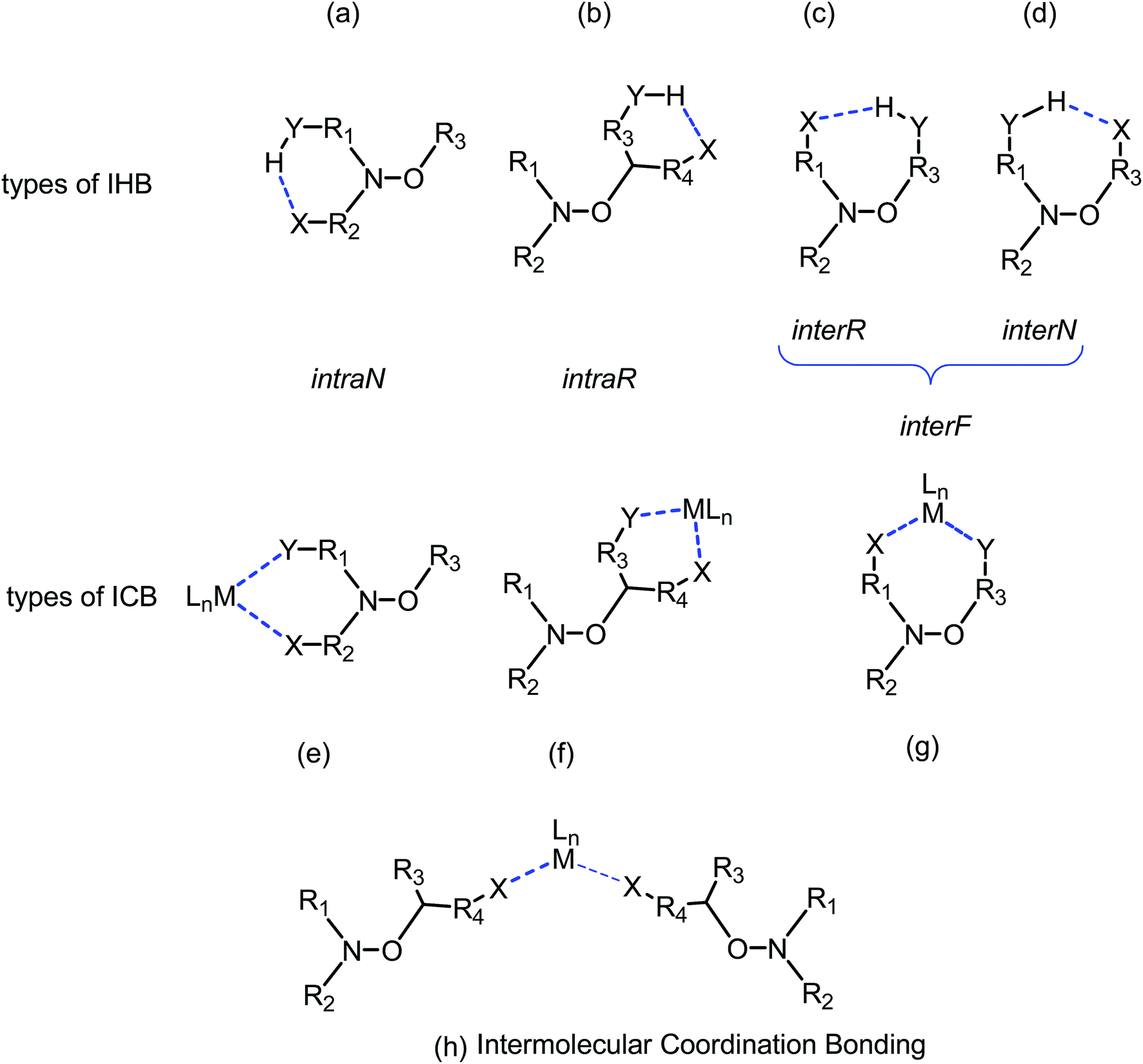 | ||
| Fig. 10 Various types of IHBs and ICB with metals (dashed blue lines): (a) IHB within the nitroxyl part (intraN), (b) IHB within the alkyl part (intraR), (c) IHB from an alkyl part to nitroxyl part (interR), and (d) IHB from a nitroxyl part to alkyl part (interN), (e) ICB within the nitroxyl part (intraN), (f) ICB within the alkyl part (intraR), (g) ICB between nitroxyl and alkyl parts (interF), and intermolecular coordination bonding (h). | ||
Kinetics for RS/SR-3, kinetics and IHB for RR/SS-2 have been previously reported and do not deserve more comments.14 Due to the silylation of the hydroxyl group, no IHB occurs in 3.
Surprisingly, Ea of 4 is 6 kJ mol−1 lower than regioisomers ortho B and meta C; which are pyridyl models. As stabilization of released radical from 4 is not expected larger than for radical released from B and C, and as electron withdrawing property of pyrazyl moiety is very close to those of pyridyl moieties, i.e., σI = 0.25 vs. σI = 0.33 and σI = 0.27,28 respectively, this difference in Ea is likely due to change in conformation (vide infra).
Alkoxyamines carrying regioisomer ortho (B); meta (C), and para (A) of pyridyl moiety have been oxidized into their corresponding N-oxide. Oxides of A29 and B30 exhibit a decrease of Ea by 10 kJ mol−1 according to the non-oxidized regioisomer (Ea ≈ 124 kJ mol−1) whereas only a decrease by 3 kJ mol−1 (ref. 31) is reported for the regioisomer meta. The activation is mainly ascribed to the extra stabilization of the released radical in oxidized form (caused by nitroxide mesomeric form) in comparison the non-oxidized alkyl radical.29–31 Thus, the increase in kd for the oxidized regio-isomer meta is ascribed to a small increase of the polarity. Then, the very close values of Ea for 4 to 6 agree with the non-activating oxidation at the position meta. On the other hand, upon oxidation of 4 into 5, and 5 into 6, the increase in Ea by 10 kJ mol−1 is attributed to effect of extra stabilization of the released alkyl radical as observed for oxidized pyridine ortho and para derivatives.
Thus, to compared the effect of mono-coordination with Zn2+ (vide infra), 1 equiv. of TFA is added to free alkoxyamine 5, for which no significant difference is observed, as expected due to the very low basicity of N-oxide function. Although pKa of 4 are very low, mono and diprotonation are observed by 1H NMR (Fig. 7). Nevertheless, no significant effect on kd (Table 1) is observed for the first protonation likely due to the protonation at the position meta.31 Disappointingly, no effect (Table 1) is observed for the second protonation as expected for protonation at the position ortho.30 This lack of effect of the protonation might be tentatively ascribed to 3 possibilities: (i) parabolic polar effect;32 (ii) occurrence of intimate ion pair33 and its decomposition at high temperature;29 (iii) conformational effect.§
IntraR model. At the difference of RS/SR-2 for which interR IHB is reported,14 not such an IHB is observed for RS/SR-1 meaning that replacing OH group by NH2 group, both being H-bond donor groups, changes strikingly the structure and the subsequent reactivity. Moreover, the mono protonation of RS/SR-1 shows a significant increase in kd and suggests a strong effect of the coordination of Zn(hfac)2 by 1. Hence, XRD structure shows a coordination of type intraR (Fig. 1 and 10), supported by 1H and 31P NMR analysis (vide supra) in sharp contrast to the interF model of coordination observed for Cu-RS/SR-B8 and Zn-RS/SR-C (vide infra).7 Consequently, a 10-fold increasing in kd is observed upon the coordination of metal cation Zn(II) by 1 in sharp contrast to intraR IHB model RR/SS-2 for which no significant differences where observed.14 Moreover, the change in electronegativity at N-atoms due to intraR-type ICB overbalances by 8 kJ mol−1 in Ea the entropic cost due to the strongly disfavoured conformation in starting materials accordingly to the geometrical requirements at TS, i.e. θ = 0° vs. θ = 90°, respectively (Fig. 10) (Fig. 11).18,19,25
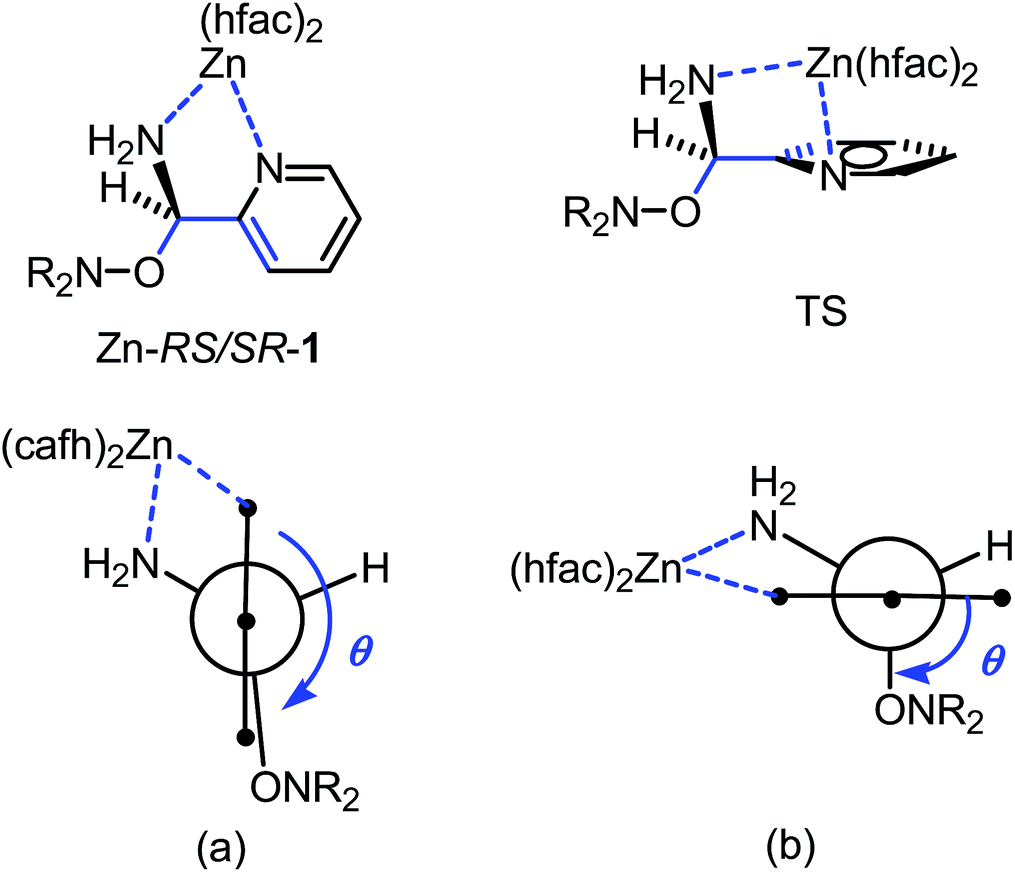 | ||
| Fig. 11 Newman projections around CO–Caryl bond in Zn-RS/SR-1 in XRD structure (a) and at TS (b). Blue arrow for dihedral angle θ 〈OCCC〉. | ||
InterF ICB. ICB of interF type was first reported for Cu-RS/SR-H and Zn-RS/SR-H. For diastereoisomer Cu-RR/SS-H, no crystals were available8 and for Zn-RR/SS-H dimer was formed involving water molecules bridging alkoxyamine and metal cation Zn2+.7 Different models were proposed to account for the kinetic results. That is, an equilibrium between Cu-RS/SR-H and Cu(II) coordinated with the alkyl fragment of B favouring an activation of the C–ON bond homolysis, i.e. a 4-fold increase in kd was observed,8 and an equilibrium in favour of Zn-RS/SR-H leading to a small decrease in kd because of matching between polar effect due to the coordination of the alkyl fragment and the formation of a bridge between alkyl and nitroxyl fragments, i.e. a formation of second bond to cleave.7 For RR/SS diastereoisomers, coordination of the alkyl fragment is favoured although affording weak activation. The formation of interF ICB for both Zn-RR/SS-2 and Zn-RS/SR-3 denotes that such ICB does not depend straight forwardly on the configuration but more on the conformations allowed by the steric repelling interactions which, in turn, are configuration dependent. As reported for Zn-RS/SR-H, a small 3-fold decrease in kd is observed for Zn-RR/SS-3 and no effect is observed for Zn-RS/SR-2. These differences are likely due to the difference in strength for the coordination bond between the metal cation and the phosphoryl group which is controlled by the repelling interactions between alkyl and nitroxyl fragments. InterF ICB involving one molecule of water bridging the metal cation Tb3+ and the phosphoryl group in B was also reported.9 In solution, such a bridge is expected to collapse and to afford only coordination bond between phosphoryl group and the oxophil metal cation Tb3+ affording less than a 3-fold decrease in kd.9 It means that the electronwithdrawing effect of the coordination of the phosphoryl group with metal cation is weak, i.e., weak destabilization of the nitroxyde and small increase in electronegativity at the O-atom of the C–ON moiety.
Intermolecular coordination bonding. Due to the pyrazine moiety in the alkyl fragment, alkoxyamine Zn-RS/SR-4 exhibits two sites of coordination: at the ortho position providing interF coordination between phosphoryl group in the nitroxyl fragment and pyrazine moiety in the alkyl fragment, and at the position meta providing, in that case, an intermolecular coordination bonding, i.e., metal cation is coordinated by two alkoxyamines through the meta position of the pyrazine moiety.
InterF coordination is expected to decrease kd as mentioned above and does not deserve more comments. On the other hand, activation at the position meta is in general reported as weak, i.e., not more than 4-fold increase in kd, whatever the mode of activation and the diastereoisomer excepted for Zn-RR/SS-J for which a 30-fold increase in kd is reported.
As dihedral angle θ 〈OCCaryl〉 is in the range of 70°,¶ the 3-fold decrease in kd is then mainly ascribed to the formation of ICB of interF type.
Miscellaneous coordination bonding. In this section are gathered all XRD structures which do not display intraN, intraR, and interF ICBs, and intermolecular coordination bonding, that is, dimer as Cu-RSSR-G and Zn-RSSR-G (para regioisomer A), polymer as Cu-RR/SS-G (para regioisomer A), bridge structure as Zn-RRSS-I (ortho regioisomer B), and unknown structures as Zn-RR/SS-G (para regioisomer A), Cu-RR/SS-H (ortho regioisomer B), Zn-RR/SS-J and Zn-RS/SR-J (meta regioisomer C, Chart 2).
Up to now, monocoordination of alkoxyamines has only been investigated with alkyl fragments carrying one pyridyl moiety. It was reported that the influence of coordination depended on the regioisomer, i.e., para isomers show an increase of kd, meta isomers show either an increase in kd (Zn-RR/SS-J) or a decrease in kd (Zn-RS/SR-J). Ortho isomers display, in general, bidentate coordination as discussed above. Interestingly, alkoxyamine Zn-RSSR-5 exhibits a dimer structure very similar to those reported for Zn-RSSR-G and Cu-RSSR-G. For both Zn-RSSR-G and Cu-RSSR-G, it was assumed that dimer structures were decomposed into unimeric species. We expect that these comments hold also for Zn-RSSR-5 and that the differences in 1H NMR observed between free alkoxyamine RS/SR-5 and Zn-RSSR-5 are due to the equilibrium between several conformers as already described.26 The 50-fold decrease in kd is the largest de-activation effect of the coordination of alkyl fragment reported up to now and in very sharp contrast with the moderate de-activation effect reported for interF and the weak activation effect reported for Zn-RS/SR-J and the strong activation effect for Zn-RR/SS-J. The coordination of the O-atom at the position meta should not change the stabilization of the released alkyl radical (same number of mesomer forms than the non-coordinated radical), should slightly increase the polarity of the pyrazinyl ring and should increase the primary steric effect, thus an increase in kd is expected in very sharp contrast with the 50-fold decrease observed. Thus, this decrease is better ascribed to a change in conformation due to strong repelling interactions between alkyl and nitroxyl fragments. Indeed, the increase in kd observed for Zn-RR/SS-J for the coordination at the position meta of C is ascribed to the change of conformation of the aromatic ring affording a conformation with angle θ close to 90° as required for TS (Fig. 12). Hence, entropic cost is lower in Zn-RR/SS-J than in “normal” alkoxyamines, with a value around 60–70° as in C, for which to open the angle at the required value has an entropic cost, and a lower entropic cost affords a higher value of kd. Thus, XDR of 6 (Fig. 5) shows θ = 73° and it is assumed the same value in 5. Hence, θ = 30° in Zn-RSSR-5 (Fig. 4) means a high entropic cost to reach the angle required at TS and, consequently, a decrease in kd from free alkoxyamine 5 to Zn-RSSR-5.
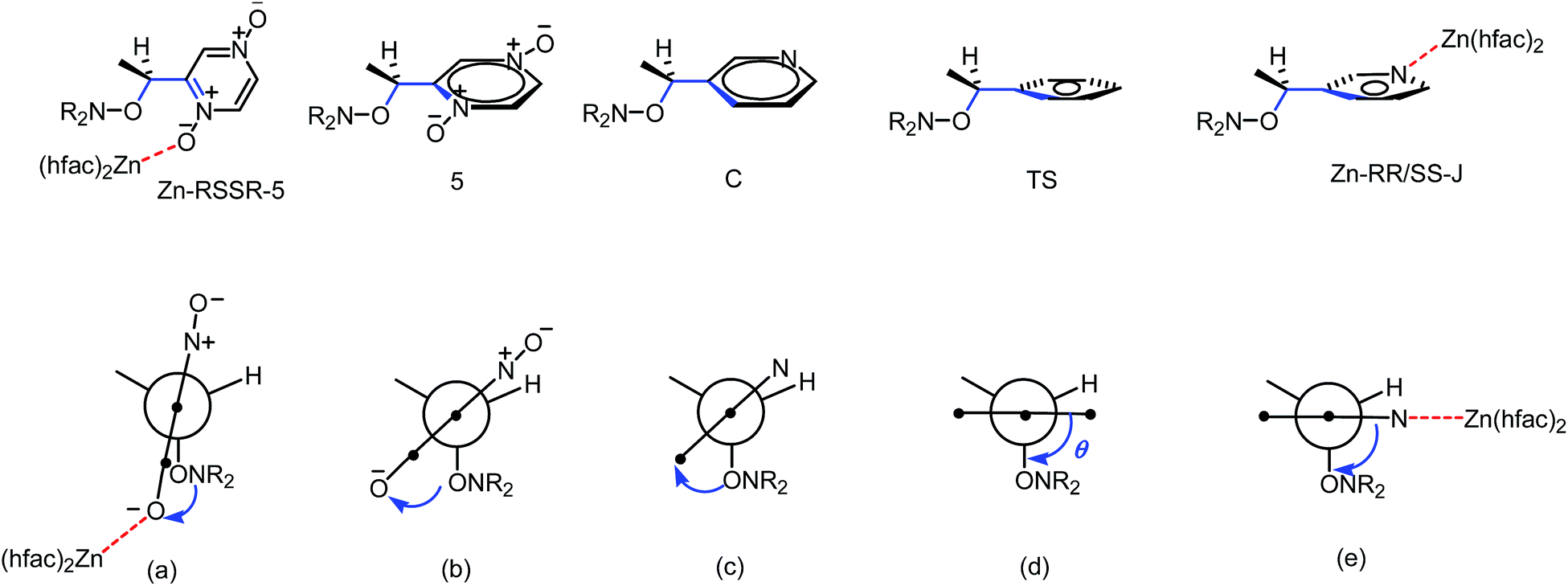 | ||
| Fig. 12 Newman projections along CO–Caryl bond for Zn-RSSR-5 (a), 5 (b), C (c), expected TS (d), and Zn-RR/SS-J (e). Blue bonds and blue arrows for dihedral angle θ 〈OCCaryl〉 and dotted red lines for coordination bonds. | ||
4 Conclusion
In this paper, we present the synthesis of five new complexes of alkoxyamines (based on the SG1 nitroxide) with [Zn(hfac)2(H2O)2]. Depending on the structure of the alkoxyamine ligand, different types of complexes were formed: a 1-to-1 complex with the alkyl part of an alkoxyamine acting like ligand, a 1-to-1 complex with both parts of the alkoxyamine involved in the complexation, a 1-to-2 complex, and ring-type complexes. With these examples, we highlighted several type of ICB as for IHB and we proposed a similar ranking but with strikingly different influence: intraN ICB affording a striking decrease in kd in sharp contrast to intraR IHB, intraR ICB affording a moderate increase in kd in contrast to intraR IHB for which no effect is observed, intraF ICB affording a moderate decrease in kd as observed for both interN and interR IHB. Details on intraN ICB are reported in a forthcoming article. In contrast to IHB, intermolecular coordination bond is also possible, and the effect of the example reported for Zn-RS/SR-4 is mainly controlled by the occurrence of interF ICB.ICB provides a new tool to control the homolysis of the C–ON bond in alkoxyamine in a different way of IHB but applying the same rules. Indeed, the high lability of alkoxyamines may rise several issues in storage, handling and shipping of alkoxyamines as for example 5 has t1/2 = 23 hours at 25 °C involving issues whereas Zn-RSSR-5 has t1/2 = 182 days making it easier to use.
5 Experimental
Infrared (IR) spectra were recorded on a Bruker Vector 22 spectrometer (KBr). The elemental analyses were performed on a Carlo Erba 1106 CHN elemental analyzer and Euro EA-3000 CHNS elemental analyzers. Solvents and reagents were of reagent quality. Dihydrate of bis(hexafluoroacetylacetonato)zinc(II) was prepared as previously reported.34Preparation of alkoxyamines
:2 mixture of diastereoisomers (31P-NMR ratio). The diastereomers were separated by automatic flash column chromatography (gradient of Et2O in petroleum ether: 100% EP to 100% Et2O) to obtain 4-RR/SS (0.54 g, 22%) and 4-RS/SR (1.41 g, 56%). 4-RR/SS; colorless oil; 1H NMR (300 MHz, CDCl3) δ: 8.61 (d, J = 1.5 Hz, 1H), 8.47 (dd, J = 2.6, 1.5 Hz, 1H), 8.39 (d, J = 2.6 Hz, 1H), 5.18 (q, J = 6.8 Hz, 1H), 4.59–4.23 (m, 1H), 4.21–3.78 (m, 3H), 3.31 (d, JH−P = 26.3 Hz, 1H), 1.61 (d, J = 6.8 Hz, 3H), 1.28 (m, 6H), 1.20 (s, 9H), 0.74 (s, 9H). 13C NMR (75 MHz, CDCl3) δ: 158.9 (C ar), 144.6 (CH ar), 143.8 (CH ar), 143.2 (CH ar), 82.8 (CH), 69.8 (d, J = 139.4 Hz, CH), 61.7 (d, J = 6.4 Hz, CH2), 61.4 (C), 59.1 (d, J = 7.5 Hz, CH2), 35.6 (d, J = 5.2 Hz, C), 30.4 (d, J = 5.9 Hz, CH3), 28.4 (CH3), 21.5 (CH3), 16.7 (d, J = 5.8 Hz, CH3), 16.3 (d, J = 6.6 Hz, CH3). 31P NMR (121 MHz, CDCl3) δ: 25.27. HRMS m/z (ESI) calcd for C19H37N3O4P [M + H]+ 402.2516, found: 402.2514. 4-RS/SR; colorless crystal; 1H NMR (300 MHz, CDCl3) δ: 8.77 (d, J = 1.5 Hz, 1H), 8.42 (dd, J = 2.6, 1.5 Hz, 1H), 8.36 (d, J = 2.5 Hz, 1H), 5.30 (q, J = 6.6 Hz, 1H), 4.21–3.78 (m, 2H), 3.66–3.45 (m, 2H), 3.37 (d, JH−P = 26.7 Hz, 1H), 1.55 (d, J = 6.6 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H), 1.15 (s, 9H), 1.11 (s, 9H), 0.93 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 157.6 (C ar), 144.6 (CH ar), 143.5 (CH ar), 143.2 (CH ar), 77.7 (CH), 69.7 (d, J = 138.9 Hz, CH), 61.5 (C), 61.1 (d, J = 6.9 Hz, CH2), 59.4 (d, J = 7.5 Hz CH2), 35.3 (d, J = 4.8 Hz, C), 30.6 (d, J = 6.0 Hz, CH3), 28.2 (CH3), 19.7 (CH3), 16.2 (d, J = 5.8 Hz, CH3), 16.1 (d, J = 5.8 Hz, CH3). 31P NMR (121 MHz, CDCl3) δ: 24.64. HRMS m/z (ESI) calcd for C19H37N3O4P [M + H]+ 402.2516, found: 402.2516.:1; 2 mL) was kept in a refrigerator at 5 °C for 72 h to prepare colorless crystals of the solvate complex.:2; 2 mL) was kept in a refrigerator at 5 °C for 7 days to obtain colorless crystals suitable for XRD.:1; 3 mL) was kept in a refrigerator at 5 °C for 5 days. Colorless crystals were separated and air dried.:2) at 5 °C.:3) to obtain 0.041 g of the title compound (98%). Anal. calc. for C58H76Zn2F24N6O20P2: C, 38.15; H, 4.20; N, 4.60; found: C, 38.32; H, 4.10; N, 4.50. IR (neat): 3423 vw, 3144 vw, 3124 vw, 3101 vw, 3001 w, 2983 w, 2937 w, 2883 vw, 1659 s, 1649 s, 1616 w, 1591 w, 1554 m, 1527 m, 1495 s, 1421 s, 1398 w, 1371 w, 1344 w, 1309 w, 1282 m, 1257 vs, 1201 vs, 1149 vs, 1093 m, 1053 m, 1026 m, 980 w, 968 w, 935 vw, 881 vw, 845 w, 833 w, 823 w, 795 m, 775 w, 766 w, 752 w, 742 w, 665 m, 621 w, 582 m, 534 w, 503 vw, 415 vw cm−1. Crystal growth: a solution of the complex in a CH2Cl2/heptane mixture (1:3; 4 mL) was kept in a refrigerator at 5 °C for 16 days. Pale pink crystals were separated and air dried.XRD analysis
X-ray crystallographic analyses of the crystals were carried out on a Bruker Kappa Apex II CCD diffractometer using φ,ω-scans of narrow (0.5°) frames with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. The structure was solved by direct methods and refined via a full-matrix least-squares method using the SHELX-97 software suite.35 The positions of hydrogen atoms were calculated via the riding model. Absorption corrections were applied by the empirical multiscan method in the SADABS software.36 The structure of [(Zn(hfac)2)2(5-RS/SR)2] is formed by crystallographically independent 1/2 part of the molecule. In all structures, the same part of CF3– groups is disordered by two positions. Notably, such disordering of CF3– groups is quite typical. Compounds [Zn(hfac)2(1-RS/SR)], [Zn(hfac)2(2-RR/SS)], and [(Zn(hfac)2)2(5-RS/SR)2] crystallized in monoclinic space groups P21/n, P21/c, P21/n, and Cc accordingly, whereas [Zn(hfac)2(3-RS/SR)] and [(Zn(hfac)2)3(4-RS/SR)2] crystallized in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Their crystallographic data are listed in Table 2. Molecular structures of [Zn(hfac)2(1-RS/SR)], [Zn(hfac)2(2-RR/SS)], [Zn(hfac)2(3-RS/SR)], [(Zn(hfac)2)3(4-RS/SR)2], and [(Zn(hfac)2)2(5-RS/SR)2] are shown in Fig. 1–5 with 30% thermal ellipsoid. The obtained crystal structures were analyzed for short contacts between nonbonded atoms in PLATON37,38 and MERCURY software.39 CCDC 1878897 ([Zn(hfac)2(1-RS/SR)]), 1878898 ([Zn(hfac)2(2-RR/SS)]), 1878899 ([Zn(hfac)2(3-RS/SR)]), 1878900 ([(Zn(hfac)2)3(4-RS/SR)2]), 1878902 ([(Zn(hfac)2)2(5-RS/SR)2]), 1904966 6-RS/SR contain the supplementary crystallographic data for this paper.
. Their crystallographic data are listed in Table 2. Molecular structures of [Zn(hfac)2(1-RS/SR)], [Zn(hfac)2(2-RR/SS)], [Zn(hfac)2(3-RS/SR)], [(Zn(hfac)2)3(4-RS/SR)2], and [(Zn(hfac)2)2(5-RS/SR)2] are shown in Fig. 1–5 with 30% thermal ellipsoid. The obtained crystal structures were analyzed for short contacts between nonbonded atoms in PLATON37,38 and MERCURY software.39 CCDC 1878897 ([Zn(hfac)2(1-RS/SR)]), 1878898 ([Zn(hfac)2(2-RR/SS)]), 1878899 ([Zn(hfac)2(3-RS/SR)]), 1878900 ([(Zn(hfac)2)3(4-RS/SR)2]), 1878902 ([(Zn(hfac)2)2(5-RS/SR)2]), 1904966 6-RS/SR contain the supplementary crystallographic data for this paper.
| Compound | [Zn(hfac)2(1-RS/SR)] | [Zn(hfac)2(2-RR/SS)] | [(Zn(hfac)2)(5-RS/SR)]2 |
| Empirical formula | C33H42F12N3O9PZn | C30H37F12N2O9PZn | C58H76F24N6O20P2Zn2 |
| Formula weight | 949.04 | 893.96 | 1825.93 |
| Temperature, K | 296(2) | 296(2) | 296(2) |
| Wavelength, Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c | P21/n |
| Unit cell dimensions a, Å | 12.4608(7) | 13.121(1) | 15.7902(6) |
| b, Å | 16.0495(7) | 12.1695(9) | 11.3758(4) |
| c, Å | 22.7935(11) | 25.591(2) | 23.2214(11) |
| α, ° | 90 | 90 | 90 |
| β, ° | 99.656(2) | 98.367(4) | 105.424(2) |
| γ, ° | 90 | 90 | 90 |
| Volume, Å3 | 4493.9(4) | 4042.7(6) | 748.9(1) |
| Z | 4 | 4 | 2 |
| Density (calcd), Mg m−3 | 1.403 | 1.469 | 1.508 |
| Abs. coefficient, mm−1 | 0.680 | 0.751 | 0.759 |
| F(000) | 1944 | 1824 | 1864 |
| Crystal size, mm3 | 0.15 × 0.60 × 0.90 | 0.04 × 0.20 × 0.60 | 0.15 × 0.25 × 0.40 |
| Θ range for data collection, ° | 3.1–27.5 | 3.1–25.0 | 3.1–26.0 |
| Index ranges | −16 ≤ h ≤ 16, −20 ≤ k ≤ 20, −29 ≤ l ≤ 29 | −15 ≤ h ≤ 15, −14 ≤ k ≤ 14, −30 ≤ l ≤ 30 | −19 ≤ h ≤ 19, −14 ≤ k ≤ 14, −25 ≤ l ≤ 28 |
| Reflections collected | 75372 |
56525 |
41980 |
| Independent reflections | 10286 R(int) = 0.048 |
7134 R(int) = 0.054 | 1480 R(int) = 0.045 |
| Completeness to θ, % | 99.8 | 99.7 | 99.8 |
| Data/restraints/parameters | 10286/0/529 |
7134/0/506 | 7893/13/594 |
| Goodness-of-fit on F2 | 1.07 | 1.02 | 1.06 |
| Final R indices I > 2σ(I) | R1 = 0.0529, wR2 = 0.1440 | R1 = 0.0702, wR2 = 0.1899 | R1 = 0.0453, wR2 = 0.1217 |
| Final R indices (all data) | R1 = 0.0805, wR2 = 0.1840 | R1 = 0.1027, wR2 = 0.2488 | R1 = 0.0612, wR2 = 0.1366 |
| Largest diff. peak/hole, e Å−3 | 0.81/−0.52 | 0.89/−0.85 | 0.68/−0.30 |
| Compound | [Zn(hfac)3(3-RS/SR)] | [(Zn(hfac)2)3(4-RS/SR)2] | 6-RS/SR |
| Empirical formula | C36H53F12N2O9PSiZn | C68H78F36N6O20P2Zn3 | C19H36N3O5P |
| Formula weight | 1010.23 | 2241.41 | 417.48 |
| Temperature, K | 296(2) | 200(2) | 293 |
| Wavelength, Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group | P |
P |
C2/c |
| Unit cell dimensions a, Å | 11.3993(6) | 15.4251(6) | 16.2198(9) |
| b, Å | 13.6801(8) | 17.7197(7) | 9.6226(6) |
| c, Å | 16.1856(9) | 19.8153(7) | 29.4018(18) |
| α, ° | 96.668(2) | 86.523(2) | 90 |
| β, ° | 94.471(2) | 70.3620(10) | 93.873(5) |
| γ, ° | 100.497(2) | 67.3500(10) | 90 |
| Volume, Å3 | 2452.3(2) | 4692.9(3) | 4578.5(5) |
| Z | 2 | 2 | 8 |
| Density (calcd), Mg m−3 | 1.368 | 1.586 | 1.211 |
| Abs. coefficient, mm−1 | 0.651 | 0.931 | 0.152 |
| F(000) | 1044 | 2264 | 1808.0 |
| Crystal size, mm3 | 0.20 × 0.30 × 0.35 | 0.03 × 0.15 × 0.40 | 0.36 × 0.28 × 0.06 |
| Θ range for data collection, ° | 3.1–27.5 | 3.0–26.0 | 5.584–49.426 |
| Index ranges | −14 ≤ h ≤ 14, −17 ≤ k ≤ 17, −21 ≤ l ≤ 21 | −19 ≤ h ≤ 19, −21 ≤ k ≤ 21, −24 ≤ l ≤ 24 | −19 ≤ h ≤ 19, −11 ≤ k ≤ 11, −22 ≤ l ≤ 34 |
| Reflections collected | 75434 |
83455 |
13148 |
| Independent reflections | 11207 R(int) = 0.040 |
18469 R(int) = 0.034 |
3887 R(int) = 0.0634 |
| Completeness to θ, % | 99.8 | 99.8 | 99.8 |
| Data/restraints/parameters | 11207/9/676 |
18469/26/1216 |
3887/0/262 |
| Goodness-of-fit on F2 | 1.05 | 1.04 | 1.078 |
| Final R indices I > 2σ(I) | R1 = 0.0448, wR2 = 0.1255 | R1 = 0.0573, wR2 = 0.1530 | R1 = 0.0735, wR2 = 0.2152 |
| Final R indices (all data) | R1 = 0.0686, wR2 = 0.1784 | R1 = 0.0740, wR2 = 0.1701 | R1 = 0.1172, wR2 = 0.3084 |
| Largest diff. peak/hole, e Å−3 | 0.60, −0.61 | 2.28, −1.10 | 0.56, −0.67 |
31P NMR analyses
31P NMR spectra were acquired for 0.01 M solutions of compounds [Zn(hfac)2(1-RS/SR)] (with respect to phosphorus) in deuterated benzene on a conventional NMR spectrometer operating at the resonance frequency of protons 500 MHz. For 31P spectroscopy, the signal of H3PO4 served as an external reference. A total of 1024 scans were collected to achieve a good signal-to-noise ratio.EPR-based determination of homolysis rate constants kd
EPR experiments were performed on a SpinScan EPR machine (Adani). The values of kd were measured by recording ESR spectra upon heating of 10−4 M toluene solutions of complexes and alkoxyamines in the presence of 3 equiv. of 2,2,6,6-tetramethylpiperidine-N-oxyl radical (TEMPO) as an alkyl radical scavenger. To generate protonated forms, TFA was used. The solutions were degassed by three cycles of freeze–pump–thaw and sealed in an argon atmosphere prior to measurements to ensure narrow ESR signals of nitroxide SG1 generated in the course of heating. Profiles of the relative concentration were obtained by integration of the low-field EPR line of SG1. Depending on the form of kinetics, experimental data points were fitted to monoexponential eqn (1) (see main text).Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge the Ministry of Education and Science of the Russian Federation (state contract No. 2017-220-06-7355) and RSF (grant No. 17-73-10101) for financial support. The authors are also grateful to the Multi-Access Chemical Research Center SB RAS for spectral and analytical measurements. SRAM, GA, THT and JPJ thank Aix-Marseille University and CNRS for support, and ANR for funding (ANR ANR-14-CE16-0023-01). The English language was corrected and certified by http://shevchuk-editing.com.References
- D. Gigmes, Nitroxide mediated polymerization: from fundamentals to applications in materials science, Royal Society of Chemistry, 2016 Search PubMed.
- G. Audran, P. Brémond, J.-M. Franconi, S. R. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Org. Biomol. Chem., 2014, 12, 719–723 RSC.
- F. Chauvin, P.-E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- P. Brémond and S. R. Marque, Chem. Commun., 2011, 47, 4291–4293 RSC.
- M. V. Edeleva, I. A. Kirilyuk, I. F. Zhurko, D. A. Parkhomenko, Y. P. Tsentalovich and E. G. Bagryanskaya, J. Org. Chem., 2011, 76, 5558–5573 CrossRef CAS PubMed.
- E. Bagryanskaya, P. Brémond, M. Edeleva, S. R. Marque, D. Parkhomenko, V. Roubaud and D. Siri, Macromol. Rapid Commun., 2012, 33, 152–157 CrossRef CAS PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, M. Edeleva, S. R. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, ChemistrySelect, 2017, 2, 3584–3593 CrossRef CAS.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, P. Brémond, M. Edeleva, S. R. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, Inorg. Chem. Front., 2016, 3, 1464–1472 RSC.
- G. Audran, E. G. Bagryanskaya, I. Y. Bagryanskaya, M. Edeleva, P. Kaletina, S. R. Marque, D. Parkhomenko, E. V. Tretyakov and S. I. Zhivetyeva, Inorg. Chem. Commun., 2018, 91, 5–7 CrossRef CAS.
- N. Popova, G. Sysoeva, V. Nikolin, V. Kaledin, E. Tretyakov, M. Edeeva, S. Balakhnin, E. Lushnikova, G. Audran and S. Mark, Bull. Exp. Biol. Med., 2017, 164, 49–53 CrossRef CAS PubMed.
- D. Moncelet, P. Voisin, N. Koonjoo, V. r. Bouchaud, P. Massot, E. Parzy, G. r. Audran, J.-M. Franconi, E. Thiaudière and S. R. Marque, Mol. Pharmaceutics, 2014, 11, 2412–2419 CrossRef CAS PubMed.
- G. Audran, E. Bagryanskaya, M. Edeleva, S. R. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, Aust. J. Chem., 2018, 71, 334–340 CrossRef CAS.
- M. V. Edeleva, S. R. Marque and E. G. e. Bagryanskaya, Russ. Chem. Rev., 2018, 87, 328 CrossRef CAS.
- G. Audran, R. Bikanga, P. Brémond, M. Edeleva, J.-P. Joly, S. R. Marque, P. Nkolo and V. Roubaud, Org. Biomol. Chem., 2017, 15, 8425–8439 RSC.
- P. Nkolo, Synthèse et étude physico-chimique de nouvelles alcoxyamines activables pour la lutte contre le paludisme, Aix-Marseille University, 2017 Search PubMed.
- J. P. Blinco, S. E. Bottle, K. E. Fairfull-Smith, E. Simpson and K. Thomas, in Nitroxide Mediated Polymerization; From Fundamentals to Applications in Materials Science, ed. D. Gigmes, 2015, pp. 114–152 Search PubMed.
- S. R. Marque, in preparation.
- D. Gigmes and S. R. A. Marque, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, 2012, DOI:10.1002/9781119953678.rad059.
- E. G. Bagryanskaya and S. R. A. Marque, in Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, ed. D. Gigmes, The Royal Society of Chemistry, 2016, pp. 45–113, 10.1039/9781782622635-00045.
- D. Bertin, D. Gigmes, S. R. Marque and P. Tordo, Macromolecules, 2005, 38, 2638–2650 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. Marque, S. Milardo, J. Peri and P. Tordo, Collect. Czech. Chem. Commun., 2004, 69, 2223–2238 CrossRef CAS.
- S. Marque, J. Sobek, H. Fischer, A. Kramer, P. Nesvadba and W. Wunderlich, Macromolecules, 2003, 36, 3440–3442 CrossRef CAS.
- S. Marque, J. Org. Chem., 2003, 68, 7582–7590 CrossRef CAS PubMed.
- H. Fischer, A. Kramer, S. R. Marque and P. Nesvadba, Macromolecules, 2005, 38, 9974–9984 CrossRef CAS.
- E. G. Bagryanskaya and S. R. Marque, Chem. Rev., 2014, 114, 5011–5056 CrossRef CAS PubMed.
- C. Y. Lin, S. R. Marque, K. Matyjaszewski and M. L. Coote, Macromolecules, 2011, 44, 7568–7583 CrossRef CAS.
- J. L. Hodgson, C. Yeh Lin, M. L. Coote, S. R. Marque and K. Matyjaszewski, Macromolecules, 2010, 43, 3728–3743 CrossRef CAS.
- C. Hansch, A. Leo and R. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- P. Brémond, A. Koïta, S. R. Marque, V. Pesce, V. Roubaud and D. Siri, Org. Lett., 2011, 14, 358–361 CrossRef PubMed.
- G. r. Audran, L. Bosco, P. Brémond, S. R. Marque, V. r. Roubaud and D. Siri, J. Org. Chem., 2013, 78, 9914–9920 CrossRef CAS PubMed.
- G. Audran, P. Brémond, M. B. B. Ibanou, S. R. Marque, V. Roubaud and D. Siri, Org. Biomol. Chem., 2013, 11, 7738–7750 RSC.
- P. Nkolo, G. Audran, R. Bikanga, P. Brémond, S. R. Marque and V. Roubaud, Org. Biomol. Chem., 2017, 15, 6167–6176 RSC.
- G. r. Audran, P. Brémond, S. R. Marque and G. Obame, J. Org. Chem., 2013, 78, 7754–7757 CrossRef CAS PubMed.
- A. Caneschi, D. Gatteschi, R. Sessoli, C. I. Cabello, P. Rey, A. L. Barra and L. C. Brunel, Inorg. Chem., 1991, 30, 1882–1886 CrossRef CAS.
- G. Sheldrick, SHELX97 Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, 1997 Search PubMed.
- Saint and SADABS, v. 2008-1, Bruker AXS Inc., Madison, WI, USA, 2008 Search PubMed.
- A. Spek, PLATON, a multipurpose crystallographic tool (Version 10M), Utrecht University, Utrecht, The Netherlands, 2003 Search PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. v. d. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1878897–1878900, 1878902, 1904966. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05334d |
| ‡ Article in preparation. |
| § The article considering this effect is in preparation. |
| ¶ For alkoxyamines described by multiparameter correlations, values of θ from XRD or DFT calculations are in the range 60–70°. |
| This journal is © The Royal Society of Chemistry 2019 |