
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium decorated on a new dendritic complex with nitrogen ligation grafted to graphene oxide: fabrication, characterization, and catalytic application†
Mohsen Golestanzadeh ab and
Hossein Naeimi*a
ab and
Hossein Naeimi*a
aDepartetment of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, 8731751167, Iran. E-mail: naeimi@kashanu.ac.ir; golestanzadeh@grad.kashanu.ac.ir; Fax: +98-31-55912397; Tel: +98-31-55912388
bChild Growth and Development Research Center, Research Institute for Primordial Prevention of Non-Communicable Disease, Isfahan University of Medical Sciences, Isfahan, 8174673461, Iran
First published on 2nd September 2019
Abstract
Immobilized Pd nanoparticles on a new ligand, namely, tris(pentaethylene-pentamine)triazine supported on graphene oxide (Pdnp-TPEPTA(L)-GO) was introduced as a novel and robust heterogeneous catalyst for use in C–C bond formation reaction. The Pdnp-TPEPTA(L)-GO catalyst was synthesized by complexation of Pd with TPEPTA as a ligand with high N-ligation sites that were supported on graphene oxide through 3-chloropropyltrimethoxysilane. The prepared catalyst was characterized using some microscopic and spectroscopic techniques. The TPEPTA(L)-GO substrate is a 2D heterogeneous catalyst with a high specific surface area and a large amount of N-ligation sites. The Pdnp-TPEPTA(L)-GO catalyst used in the C–C bond formation reaction between aryl or heteroaryl and phenylboronic acid derivatives was applied towards the synthesis of biaryl units in high isolated yields. Notably, a series of competing experiments were performed to establish the selectivity trends of the presented method. Also, this catalyst system was reusable at least six times without a significant decrease in its catalytic activity.
Introduction
In 1803, palladium was discovered by Wollaston in his investigation on refining platinum. Palladium has the ground state electronic configuration of [Kr] 4d10 and belongs to the fifth period and 10th group of the periodic table. Palladium has different oxidation states including 0, +1, +2 (the most stable for Pd), and +4. Also, the other unstable oxidation states of palladium are +3, +5, and +6.1–3 Most of the Pd(0) complexes are simply oxidized in air and therefore, the development of new Pd(0) complexes is still appealing. Importantly, the heterogeneous stabilized Pd(0) in less conventional ways should be cited. The ability of palladium(0) to construct new bonds between carbon–carbon and carbon–heteroatom is important in organic synthesis (Scheme 1).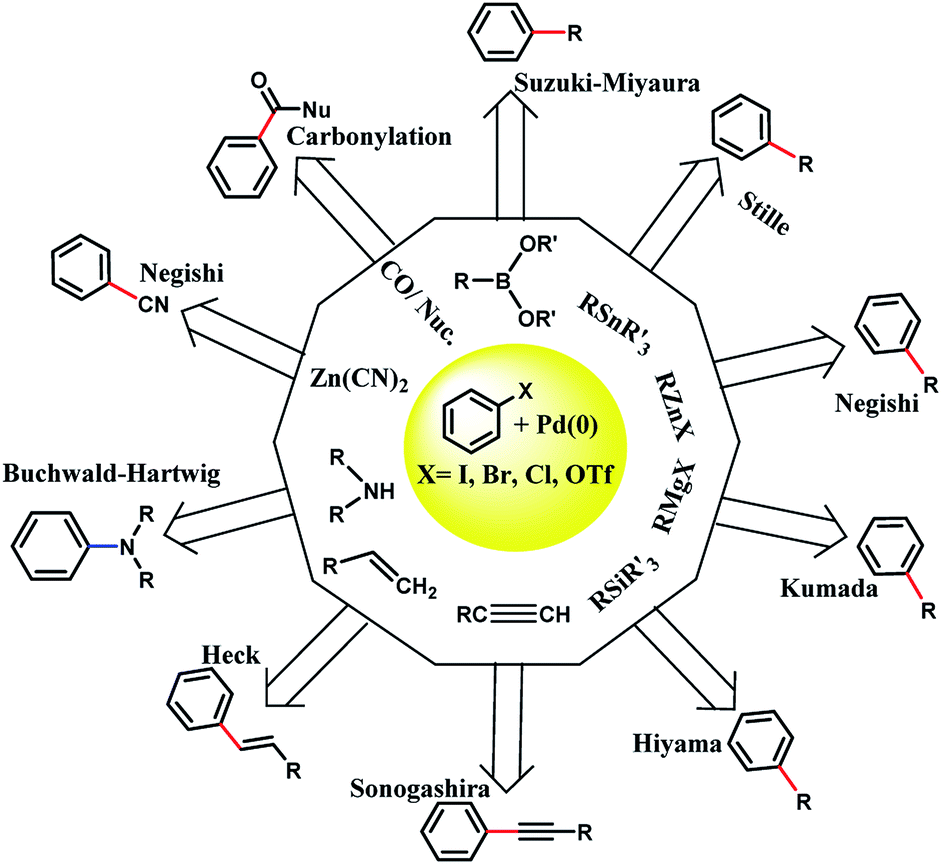 | ||
| Scheme 1 The cross-coupling organic reactions catalyzed by Pd(0). Palladium(0) catalysis has gained widespread use in industrial and academic synthetic chemistry laboratories as a powerful methodology for the formation of carbon–carbon and carbon–heteroatom bonds. R: usually sp2 hybridized carbon; the nature of R′ and M is dependent on the specific coupling being performed. The new carbon–carbon bond is shown in red color and the new carbon–nitrogen bond is shown in blue color. | ||
For example, the Suzuki–Miyaura cross coupling reaction is a highly efficient and robust organic reaction that includes cross coupling between a sp2-hybridized halide and sp2-hybridized boronic acid to form a Csp2–Csp2 bond. The biaryl or biheteroaryl motifs are ubiquitous among the wide range of industrially and pharmaceutically important compounds (Fig. 1).
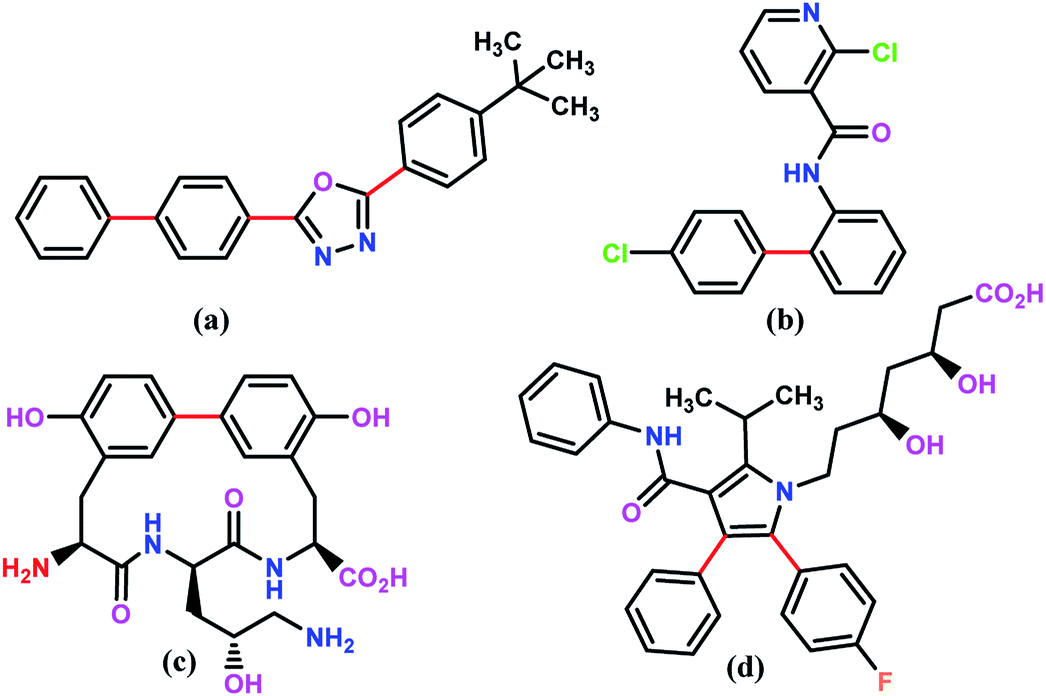 | ||
| Fig. 1 The chemical structure of selected industrially important compounds containing bi(hetero)aryl moiety where Suzuki–Miyaura coupling is used to form the key carbon–carbon bonds (shown in red); (a) is a component of OLEDs (organic light-emitting diodes) or electron conducting material (PBD). (b) is an agrochemical that is most commonly used as a fungicide (Boscalid). (c) is an antibiotic (Biphenomycin B). (d) is a pharmaceutical compound that lowers blood cholesterol (Lipitor). | ||
Generally, the homogeneous catalysts of Pd complexes catalyze the cross coupling Suzuki–Miyaura reaction.4–6 However, using homogeneous Pd complexes has several disadvantages including low efficiency in the separation of the catalyst and high cost of the Pd element at the industrial scale. Therefore, the immobilization of Pd complexes or Pd nanoparticles on different organic, inorganic, and hybrid supports has been made to provide heterogeneous Pd catalysts. Unfortunately, the supported Pd complex catalysts often suffer from low specific surface area (SSA), space confinement in mesoporous supports, less reactivity of support, reduced availability of the metal complex, and low thermal and chemical stability with respect to leaching of metals.7–9
Recently, two-dimensional supports such as graphene,10 graphene oxide (GO),11 and reduced graphene oxide (RGO)12 have attracted extensive attention in heterogeneous catalysis due to their high SSA,13 more surface active sites,14 excellent photocatalyst support,15 superior electron mobility,16 the availability of functional groups from all side of the sheets,17,18 and other excellent properties.19,20 As examples of the utilization of two-dimensional graphene in coupling reactions, we have considered previously reported papers. Very recently, Huang and co-workers reported a robust 3D ionic liquid supported on RGO/Pd nanocomposites for the Suzuki cross coupling reaction.21 Sengupta et al. reported an efficient aminobis(phosphine)-PdII complex on GO in different coupling reactions.22 Also, the effect of GO support on the catalytic performance of Pd–Fe3O4/GO, Pd–Co3O4/GO and Pd–Ni(OH)2/GO in the Suzuki cross coupling reaction was considered by Elazab and co-workers.23,24 In accordance with the various, constantly evolving fields, the development of efficient two-dimensional graphene/Pd nanocomposites is of enormous importance.25–43 In addition, Li et al. reported a new catalyst (palladium decorated on nitrogen-doped graphene nanoshells) for N-allylation reaction.44 Herein, it is notable that most of the reported catalysts applied in coupling reactions were unstable in air, less reusable or totally non-reusable, and had low loading of active sites. Undoubtedly, the design, preparation, and application of superior heterogeneous catalysts with high SSA and more reactive sites as well as highly coordinating donor atoms for the stabilization of Pd(0) in cross coupling reactions is still appealing. In continuation of our previous studies on two-dimensional heterogeneous catalysts,45–48 this research highlights the application of graphene oxide modified with a novel ligand with high N-ligation sites for the stabilization of palladium nanoparticles with oxidation state (0) in the Suzuki–Miyaura cross coupling reaction. The schematic representation of Pdnp-TPEPTA(L)-GO is shown in Fig. 2.
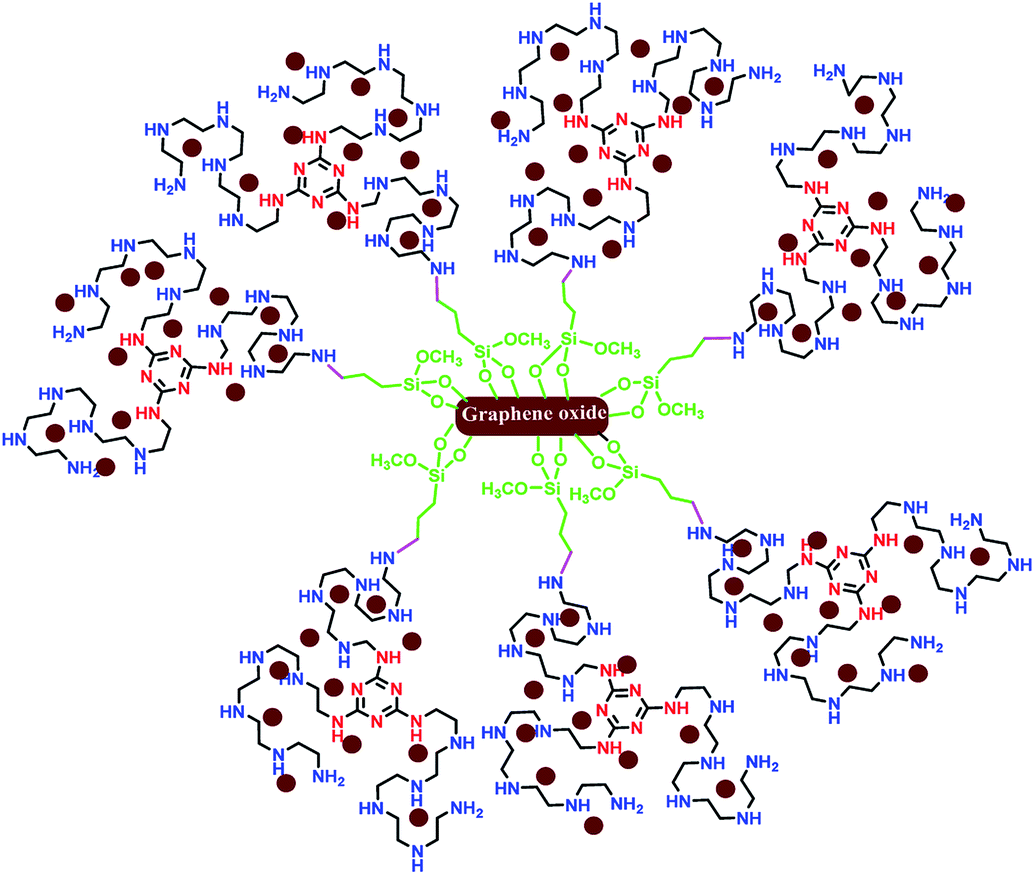 | ||
| Fig. 2 A representation of the chemical structure of Pdnp-TPEPTA(L)-GO. The nitrogen atoms of triazine are shown in red color and the nitrogen atoms in pentaethylenepentamine are shown in blue color. The new bond between the nitrogen atom of the ligand and the carbon atom of propyl trimethoxysilane supported on graphene oxide is shown in pink color. | ||
Result and discussion
Catalyst synthesis and its characterization
The supported palladium nanoparticles on graphene oxide including tris-pentaethylenepentaminetriazine as a novel ligand (Pdnp-TPEPTA(L)-GO) were prepared according to the following synthetic route, as shown in Scheme 2, through three steps. First, the preliminary ligand was prepared from the reaction of 2,4,6-trichloro-1,3,5-triazine (TCT) and pentaethylenehexamine (PEHA). We checked the reaction of TCT and PEHA under different conditions to obtain a high yield of tris(pentaethylene-pentamine)triazine (TPEPTA).49–53 This step has a vital role in the preparation of TPEPTA with high N-ligation sites because a decrease in the substitution of PEHA with chlorine atom in TCT and the total yield of TPEPTA have a significant effect on the highimmobilization of palladium nanoparticles. To obtain the optimized reaction conditions for this step, the reaction of TCT and PEHA was investigated under different circumstances and the obtained results are shown in Table 1S (see ESI†). In addition, TPEPTA was analyzed using a CHNS analyzer and the obtained results confirmed the successful substitution of three equivalents of PEHA on TCT substrate (see ESI†).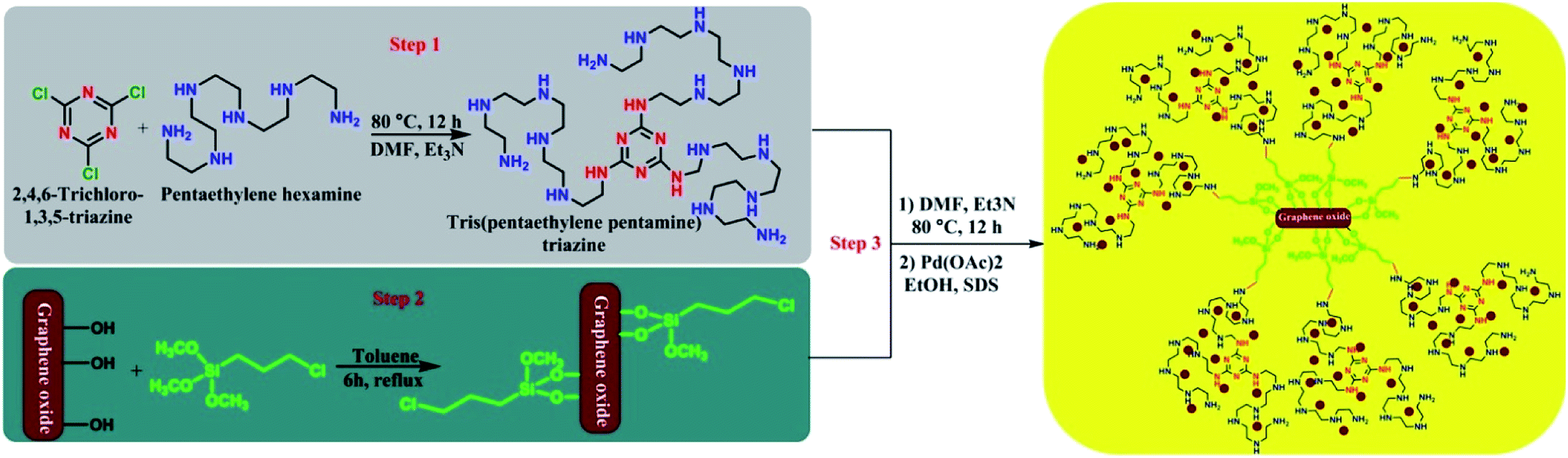 | ||
| Scheme 2 A synthetic pathway for the synthesis of Pdnp-TPEPTA(L)-GO. | ||
In the second step of the preparation of the Pdnp-TPEPTA(L)-GO catalyst, 3-chloropropyltrimethoxysilane was anchored on graphene oxide, according to the previous literature with some modification.54,55 Similar to the first step, the second step is also very important because a large amount of 3-chloropropyl on GO results in the stabilization of the highest amount of ligand on the surface of GO. We determined the amount of chloropropylsilyl groups on the surface of GO using Mohr's method (see ESI†).56,57 The total density of chloropropylsilyl groups on GO was estimated to be about 0.95 mmol g−1. The chloropropylsilyl groups not only cover the GO sheets but also make them ready for further surface functionalization.54 Finally, in the third step, through two sequential steps, the stabilization of TPEPTA on the surface of GO was performed at 80 °C for 12 h. Then, TPEPTA(L)-GO was reacted with Pd(OAc)2, leading to the immobilization of palladium nanoparticles in the presence of sodium dodecyl sulfate (SDS) as the surfactant for controlling the size of palladium particles58,59 in order to produce Pdnp-TPEPTA(L)-GO catalyst.
To confirm the functionalization of GO, the Pdnp-TPEPTA(L)-GO catalyst was characterized by the following techniques: Scanning Electron Microscopy (SEM), Energy Dispersive X-ray spectroscopy (EDX), Transmission Electron Microscopy (TEM), Fourier Transform Infrared Spectroscopy (FT-IR), Powder X-ray Diffraction (XRD), and Thermogravimetric Analysis (TGA).
In order to study the surface morphologies of GO and the Pdnp-TPEPTA(L)-GO catalyst, SEM images were recorded. As shown in Fig. 3a, the plate-like forms including the flaky layered texture of GO was observed. The EDX analysis of GO is shown in Fig. 3b. The results of EDX analysis show the presence of carbon, nitrogen, and oxygen. The absence of metal elements such as potassium and sodium clearly showed that the washing step was successful. The surface morphology of the Pdnp-TPEPTA(L)-GO catalyst was also studied by SEM (Fig. 3c). As shown in this figure, the sheets of GO were covered by the foreign matter Pdnp-TPEPTA(L). In this image, both the plate-like sheets of GO and Pd nanoparticles were observed. Moreover, the Pdnp-TPEPTA(L)-GO catalyst was investigated by EDX (Fig. 3d). This analysis clearly shows the presence of Pd and N in the concentration of 30.2 wt% and 46.3 wt%, respectively. According to the Pd wt%, the total mmol of Pd nanoparticles on TPEPTA(L)-GO was calculated to be 2.58 mmol g−1. Notably, the amount of Pd element in the Pdnp-TPEPTA(L)-GO catalyst measured using atomic absorption spectrometry (AAS) was 28 wt% (see ESI†). To the best of our knowledge, this amount is the highest amount of Pd immobilized on GO or 2D supports. In addition, the amount of TPEPTA ligand supported on the GO sheets was calculated using EDX analysis.60 Based on this method, the total amount of TPEPTA ligand detected from the nitrogen content was 1.43 mmol g−1 (see ESI†). In order to quantify the amount of Pd in the catalyst, ICP analysis was carried out after its treatment with HCl (37%) and HNO3 (65%). Based on the ICP results, the Pd quantity was measured to be 286 mg L−1 (286 ppm). This amount is equal to 28.6 wt% (∼2.57 mmol g−1). The quantitative analysis is in good agreement with the EDX data.
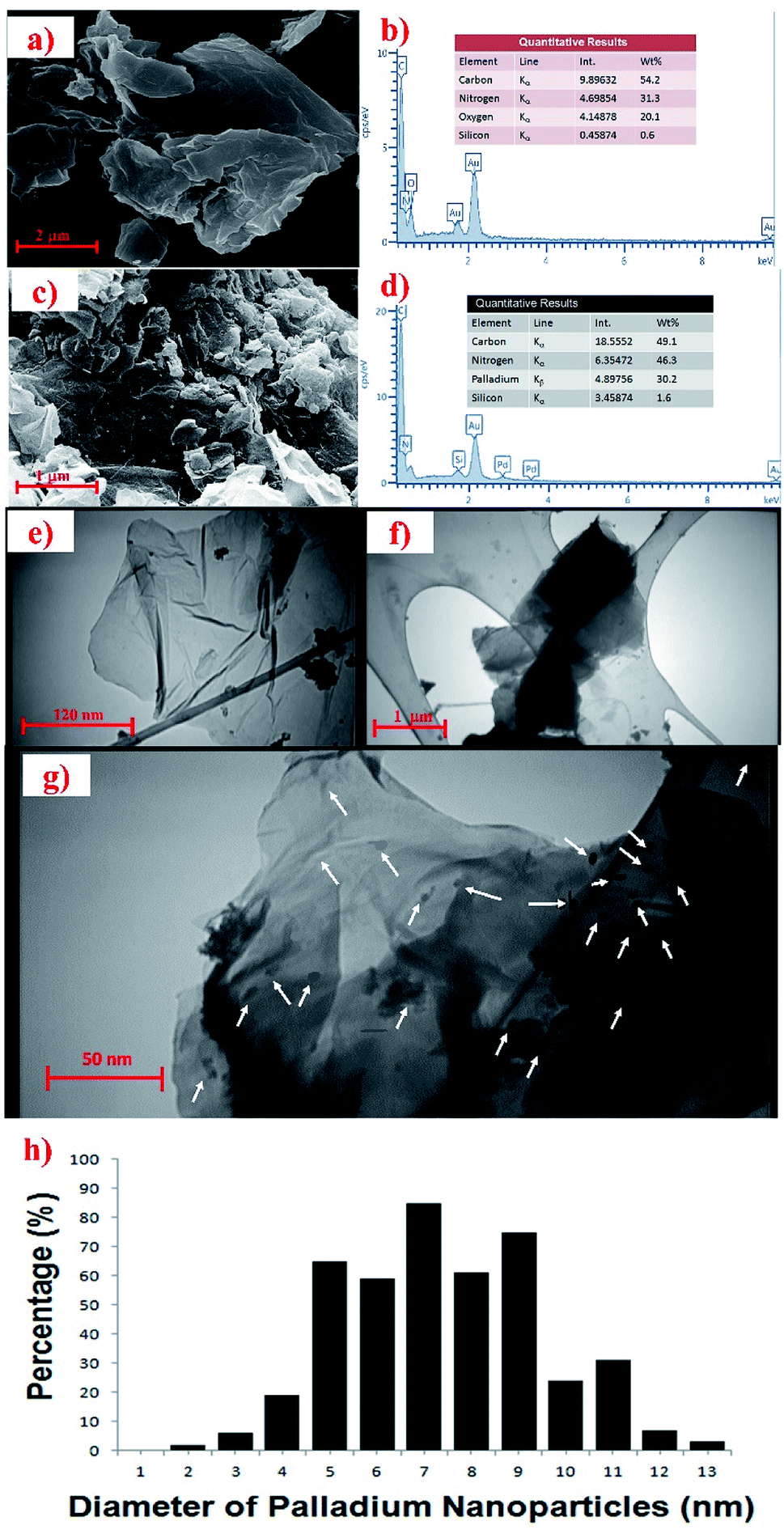 | ||
| Fig. 3 a) FE-SEM of GO [details of imaging: scale bar 2 μm, SEM HV: 15.00 kV, WD: 6.060 mm, view field: 8.668 μm, SEM magnification: 25.00k×]. (b) The EDX analysis of synthesized GO. (c) FE-SEM of Pdnp-TPEPTA(L)-GO [details of imaging: scale bar 1 μm, EHT: 15.00 kV, WD: 8.9 mm, signal A: SE2, SEM magnification: 10.000k×]. (d) The EDX analysis of Pdnp-TPEPTA(L)-GO catalyst. (e), (f), and (g) TEM images of the Pdnp-TPEPTA(L)-GO catalyst with different scale bars (1 μm, 120 nm, and 50 nm) and situations [details of imaging: 80 kV, limit of detection of 2 nm or less] (the white arrows on (g) show the palladium nanoparticles). (h) Particle size distribution results for the Pdnp-TPEPTA(L)-GO catalyst. | ||
Then, the Pdnp-TPEPTA(L)-GO catalyst was investigated by TEM in order to investigate the size of Pd nanoparticles and 2D layers of the GO support (Fig. 3e–g). From the TEM images in Fig. 3f and g, the layers of GO were found to be covered by functional groups such as the Pdnp-TPEPTA complexes. Also, the particle size distribution of the Pd nanoparticles was estimated using Image J software and the average size was about 7.63 ± 0.5 nm.61 This data indicates that the Pd nanoparticles did not aggregate on TPEPTA(L)-GO and the SDS surfactant assisted this event as well as delete the formation of smaller sized Pd particles.62
The FT-IR spectrum of the Pdnp-TPEPTA(L)-GO catalyst and its comparison with GO and TPEPTA ligand shows the peaks that confirm the successful synthesis of the abovementioned catalysts (Fig. 4). Investigating the FT-IR spectrum of GO, the bands appearing at 3394, 1719, 1573, and 1198 cm−1 may be attributed to the stretching vibrations of O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and C–O, respectively.63 According to the FT-IR spectrum of the TPEPTA ligand, the absorption peaks observed at 3412, 2925, 1575, and 1091 cm−1 are associated with the vibrations of N–H, C(sp3)–H, CN, and C–N, respectively.64,65 The presence of vibration bands of GO and the TPEPTA ligand in the FT-IR spectrum of Pdnp-TPEPTA(L)-GO confirms the successful synthesis of the target catalyst. The observation of a slight shift (∼4 cm−1) in the N–H bands and other peaks is due to the chemical environment of Pd–N coordination.66–68
O, CC, and C–O, respectively.63 According to the FT-IR spectrum of the TPEPTA ligand, the absorption peaks observed at 3412, 2925, 1575, and 1091 cm−1 are associated with the vibrations of N–H, C(sp3)–H, CN, and C–N, respectively.64,65 The presence of vibration bands of GO and the TPEPTA ligand in the FT-IR spectrum of Pdnp-TPEPTA(L)-GO confirms the successful synthesis of the target catalyst. The observation of a slight shift (∼4 cm−1) in the N–H bands and other peaks is due to the chemical environment of Pd–N coordination.66–68
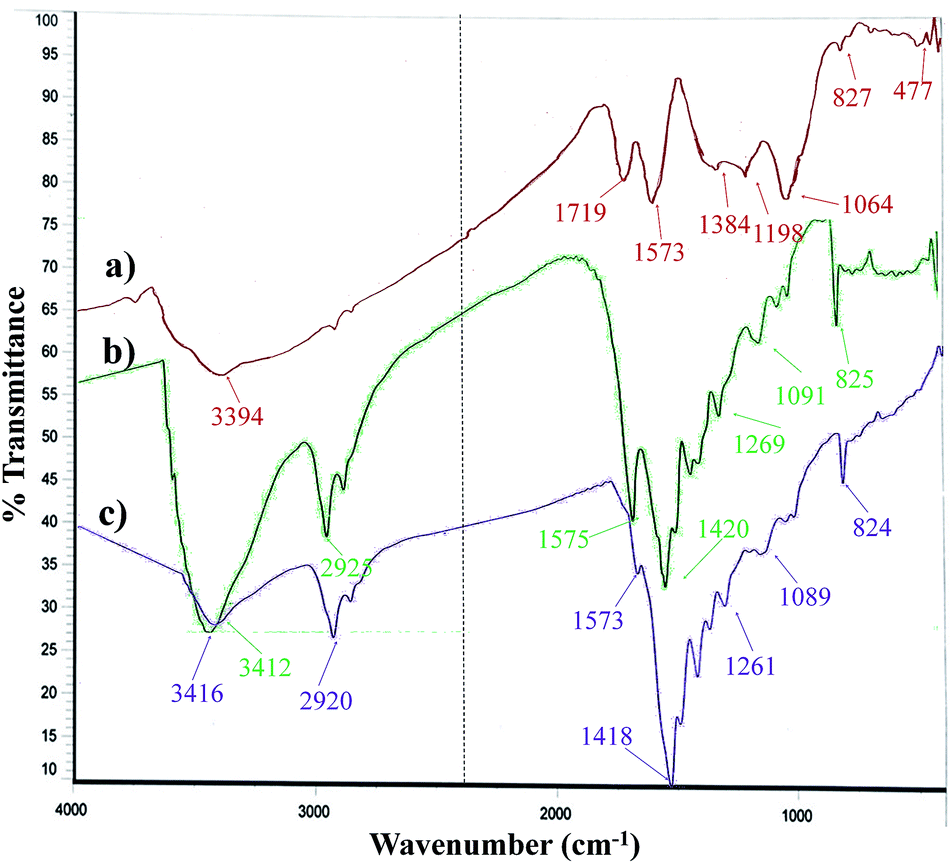 | ||
| Fig. 4 FT-IR spectra of (a) GO, (b) TPEPTA ligand, (c) Pdnp-TPEPTA(L)-GO catalyst. | ||
In addition, the crystalline structure of Pdnp-TPEPTA(L)-GO was studied by XRD analysis and the obtained data are displayed in Fig. 5. In the XRD pattern of GO, the peak at 26.4° (002),69 which is related to the crystalline structure of graphite, is moved to a lower Bragg angle of 12.1° in the crystalline structure of GO corresponding to the (001) plane with d-spacing of 0.92 nm.70
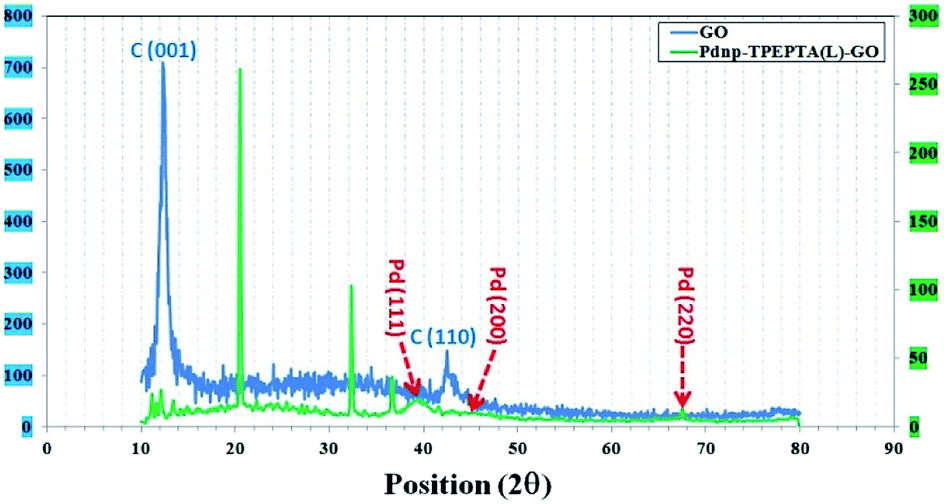 | ||
| Fig. 5 The XRD patterns of GO (blue) and the Pdnp-TPEPTA(L)-GO catalyst (green); the vertical axes at left and right are the intensity values of GO and the Pdnp-TPEPTA(L)-GO catalyst, respectively. | ||
The increase in d-spacing value is due to the presence of different functional groups including oxygen motif between the graphite layers during the oxidation process using KMnO4 and HNO3.69 However, in the XRD patterns of Pdnp-TPEPTA(L)-GO, the peaks are indexed as the (111) 38.4°, (200) 44.1°, and (220) 67.8° plane, which can be attributed to the face-centered-cubic (fcc) structure of the palladium nanoparticles on TPEPTA(L)-GO (JCPDS card 26-1081 and 01-0646).71,72 Also, a peak appeared at 21.2° (002), whereas the reflection corresponding to the (001) plane of GO at 12.1° disappeared. This observation confirms the functionalization of GO sheets with TPEPTA ligand.73 Furthermore, using Scherrer's equation, the size of the palladium nanoparticles 10.5 nm was calculated.74
TGA analysis was used to investigate the thermal stability of the Pdnp-TPEPTA(L)-GO catalyst (Fig. 6). The TGA of the catalyst shows two main weight losses. The first weight loss was obtained at 88–112 °C, which is attributed to the trapped water and organic solvents.75 The second weight loss of the catalyst occurred at 197–239 °C, which can be related to the TPEPTA ligand.76 Notably, the remaining non-decomposable sample was approximately 29 wt%, which released the amount of Pd immobilized on the structure of the catalyst. In addition, in accordance with the weight loss amount, the concentration of the TPEPTA ligand per C atoms in the graphene layers was measured, which was found to be one ligand per ninety six carbon atoms (see ESI†).
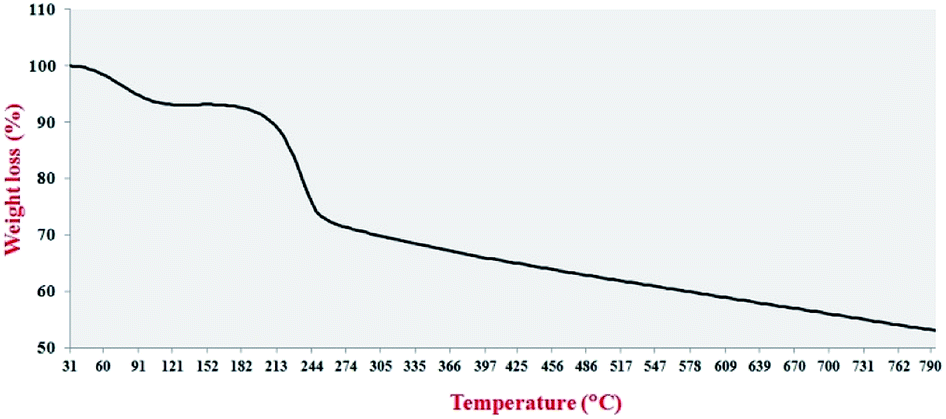 | ||
| Fig. 6 The thermogravimetric analysis curve of Pdnp-TPEPTA(L)-GO. Details of this experiment: sample: 3.0800 mg, range of temperature: 25–800 °C, rate of temperature change: 20 K min−1, flow rate of N2 gas: 50.0 mL min−1, synchronization enabled, sample holder: alumina 70 μL. | ||
Pdnp-TPEPTA(L)-GO-catalyzed Suzuki–Miyaura cross coupling reaction
First, the Suzuki–Miyaura cross coupling reaction of phenylboronic acid (1) and 4-iodoanisole (2) in the presence of Pdnp-TPEPTA(L)-GO catalyst was selected as the model reaction for the optimization of reaction parameters such as type of base, molar ratio of the reactant, loading of the catalyst, type of the solvent, and temperature of the reaction (Table 1). The reaction was performed in the presence of different bases including Et3N, NaOH, Na3PO4, Na2CO3, and K2CO3 (Table 1, entries 1–5). However, amongst them, K2CO3 was found to be the most efficient base.77 The main role of the base in the Suzuki reaction is to convert the phenylboronic acid to the more reactive organoborate (PhB(OH)3−), which facilitates the trans-metalation step with the Pd(II)-halide motif to afford Pd(II)-Ph intermediate.78 Then, the model reaction was performed in the presence of different organic solvents (Table 1, entries 6–11). Among the solvent experiments, DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (2:1) was selected as the best solvent for the reaction. In these experiments, the influence of solvent on the catalytic activity of Pdnp-TPEPTA(L)-GO catalyst has been highlighted including the solvent effects on the solubility of the reagents, mass transfer, interactions with the starting materials or products that activate or deactivate the reaction or selectivity in the reaction, solvent interaction with the catalyst, and transition state stabilization.79 In addition, DMF is an organic solvent with high dielectric constant with bulk properties similar to water and high ability to stabilize ionic species.80 Furthermore, the influence of catalyst amount and the molar ratio of phenylboronic acid and 4-iodoanisole were also considered (Table 1, entries 12, 13). The best results were obtained for the ratio of 1.2:1:2 for (1):(2):base. Also, the effect of catalyst amount (Table 1, entries 14, 15) and temperature (Table 1, entries 16, 17) were explored. Therefore, we obtained and concluded that the optimum reaction conditions were (1) (1.2 mmol), (2) (1 mmol), K2CO3 (2 mmol), DMF:H2O (2:1) (6 mL), T: 80 °C, and 30 mg of Pdnp-TPEPTA(L)-GO catalyst (Table 1, entry 5).
:H2O (2:1) was selected as the best solvent for the reaction. In these experiments, the influence of solvent on the catalytic activity of Pdnp-TPEPTA(L)-GO catalyst has been highlighted including the solvent effects on the solubility of the reagents, mass transfer, interactions with the starting materials or products that activate or deactivate the reaction or selectivity in the reaction, solvent interaction with the catalyst, and transition state stabilization.79 In addition, DMF is an organic solvent with high dielectric constant with bulk properties similar to water and high ability to stabilize ionic species.80 Furthermore, the influence of catalyst amount and the molar ratio of phenylboronic acid and 4-iodoanisole were also considered (Table 1, entries 12, 13). The best results were obtained for the ratio of 1.2:1:2 for (1):(2):base. Also, the effect of catalyst amount (Table 1, entries 14, 15) and temperature (Table 1, entries 16, 17) were explored. Therefore, we obtained and concluded that the optimum reaction conditions were (1) (1.2 mmol), (2) (1 mmol), K2CO3 (2 mmol), DMF:H2O (2:1) (6 mL), T: 80 °C, and 30 mg of Pdnp-TPEPTA(L)-GO catalyst (Table 1, entry 5).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Ratio (1:2:base) |
Catalyst (mg) | Solvent | T (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: phenylboronic acid (1) (x mmol), 4-iodoanisole (2) (y mmol), base (z mmol), solvent (6 mL).b Isolated yields.c N-Methylpyrrolidone.d The Pd-TPEPTA catalyst without graphene oxide as the support was used. | |||||||
| 1 | Et3N | 1.2:1:2 |
30 | DMF:H2O (2:1) |
80 | 60 | 20 |
| 2 | NaOH | 1.2:1:2 |
30 | DMF:H2O (2:1) |
80 | 40 | 45 |
| 3 | Na3PO4 | 1.2:1:2 |
30 | DMF:H2O (2:1) |
80 | 24 | 50 |
| 4 | Na2CO3 | 1.2:1:2 |
30 | DMF:H2O (2:1) |
80 | 15 | 75 |
| 5 | K2CO3 | 1.2:1:2 |
30 | DMF:H2O (2:1) |
80 | 10 | 95 |
| 6 | K2CO3 | 1.2:1:2 |
30 | DMF:H2O (1:1) |
80 | 15 | 79 |
| 7 | K2CO3 | 1.2:1:2 |
30 | H2O | Reflux | 20 | 75 |
| 8 | K2CO3 | 1.2:1:2 |
30 | DMF | 80 | 15 | 75 |
| 9 | K2CO3 | 1.2:1:2 |
30 | EtOH:H2O (1:1) |
Reflux | 25 | 65 |
| 10 | K2CO3 | 1.2:1:2 |
30 | NMPc:H2O (1:1) |
80 | 20 | 78 |
| 11 | K2CO3 | 1.2:1:2 |
30 | NMPc:H2O (2:1) |
80 | 15 | 87 |
| 12 | K2CO3 | 1.2:1:1.5 |
30 | DMF:H2O (2:1) |
80 | 20 | 55 |
| 13 | K2CO3 | 1:1:2 |
30 | DMF:H2O (2:1) |
80 | 20 | 70 |
| 14 | K2CO3 | 1.2:1:2 |
25 | DMF:H2O (2:1) |
80 | 15 | 72 |
| 15 | K2CO3 | 1.2:1:2 |
35 | DMF:H2O (2:1) |
80 | 10 | 93 |
| 16 | K2CO3 | 1.2:1:2 |
30 | DMF:H2O (2:1) |
100 | 10 | 95 |
| 17 | K2CO3 | 1.2:1:2 |
30 | DMF:H2O (2:1) |
60 | 15 | 60 |
| 18b | K2CO3 | 1.2:1:2 |
80 | DMF:H2O (2:1) |
80 | 25 | 85 |

With optimization results in hand, the generality and scope of this methodology was evaluated in the synthesis of diverse biaryl units using reactions of different substrates containing I, Br, and/or Cl with phenylboronic acid derivatives (Scheme 3). As shown in Scheme 3, the target products (P1-P21) were obtained in 85–97% and 82–96% yields from the reaction of aryl iodides and bromides involving electron-donating and electron-withdrawing substituents, and phenylboronic acid and 4-methoxyboronic acid as the starting materials, respectively. As we know from the previously reported literature, the presence of electron-withdrawing groups in aryl halide or electron-donating groups in aryl boronic acid enhances the reaction rate and yield of the Suzuki cross coupling reaction due to the facilitation of the rate limiting oxidative addition step.81–83 It is notable in this research that the catalyst can improve the Suzuki reaction irrespective of the nature of the substituent (electron-withdrawing or electron-donating) on the aryl halides and aryl boronic acids, and the type of halogen in the aryl halide, compared with the previously reported methods.21,37,40
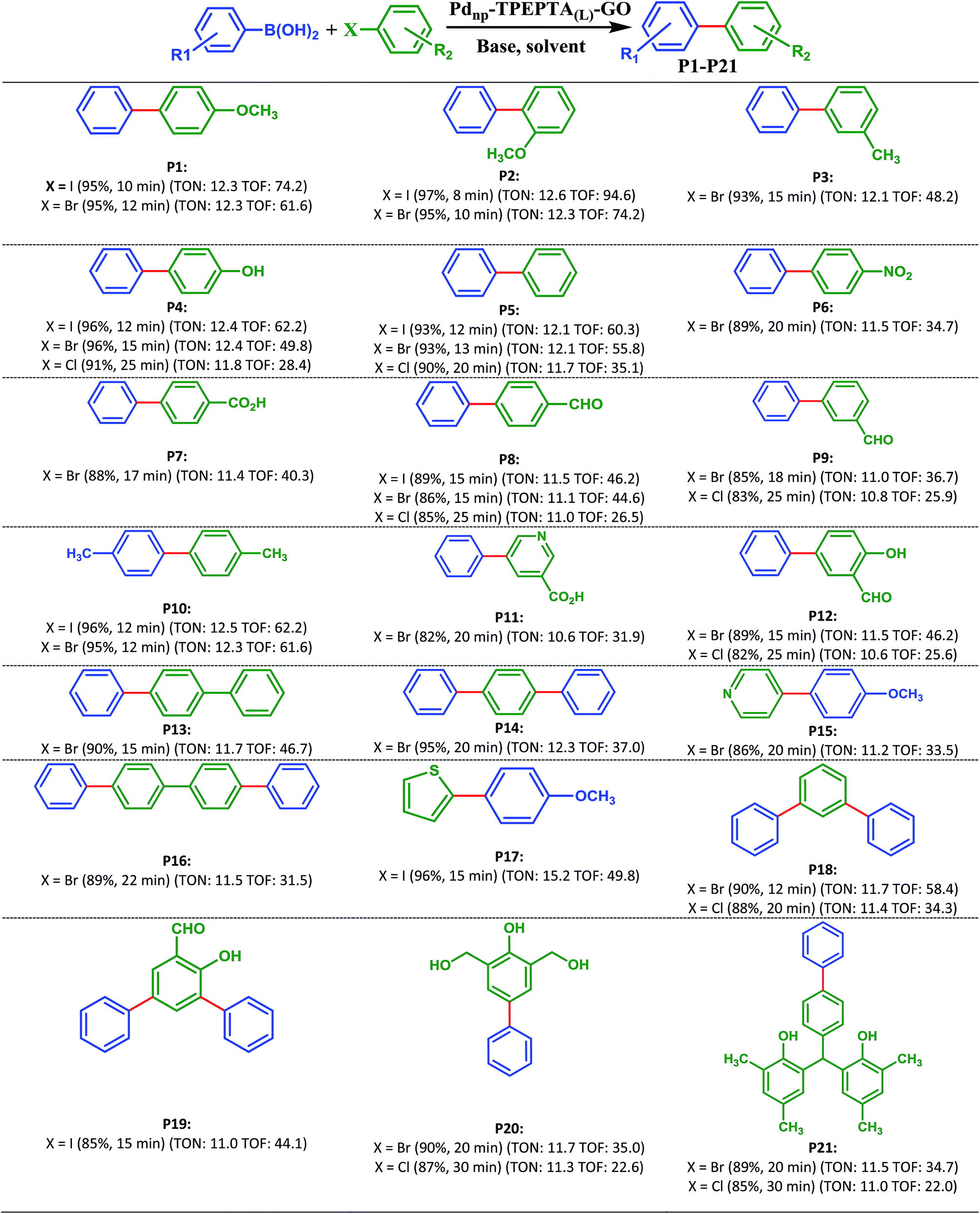 | ||
| Scheme 3 Products of Suzuki–Miyaura reaction catalyzed by Pdnp-TPEPTA(L)-GO. Reaction conditions: aryl(hetero)halide (1 mmol) (shown in green), phenylboronic acids (1.2 mmol) (shown in blue), K2CO3 (2 mmol), solvent: DMF:H2O (2:1) (6 mL), Pdnp-TPEPTA(L)-GO (30 mg), T: 80 °C. All yields refer to isolated products. New carbon–carbon bond is shown in red. When the aryl halide had two halogens, phenylboronic acids (2.4 mmol) and K2CO3 (4 mmol) were used (for products: P14, P16, P18, and P19). TON: turnover number (mmol of product/mmol of catalyst), TOF: turnover frequency ([mmol of product/mmol of catalyst]/time of the reaction) (unit: h−1). | ||
In addition, it is important to note that the aryl chlorides are less reactive than aryl iodides and bromides in the Suzuki cross coupling reaction due to the bond strength. However, many reported papers have focused on the utilization of aryl chlorides in the Suzuki cross coupling reaction.84–90 All of the papers report interesting results but research is still appealing in this field. Due to the less reactivity of aryl chlorides, some researchers used a higher amount of the catalyst.91–94 In order to study the applicability and efficiency of the Pdnp-TPEPTA(L)-GO catalyst, the Suzuki cross coupling reaction using aryl chlorides to produce products P4, P5, P8, P9, P12, P18, P20, and P21 was studied using the same amount of catalyst as that for the Suzuki reaction using aryl iodides and aryl bromides. The obtained results were very promising. The products were obtained in 82–91% isolated yields in 20–30 minutes. When the reactions were carried out at 120 °C, the isolated yields of the mentioned products increased slightly but the time of the reactions decreased similar to the time of the reactions for aryl iodides and bromides (see ESI-Table 2S†).
Interestingly, the preparation of compound P21 was studied directly from the reaction of 2,4-dimethylphenol (3) and 4-phenylbenzaldehyde (4) by a condensation reaction in the presence of RGO-SO3H as the catalyst under solvent free conditions, according to a previous work71 and the Suzuki cross coupling reaction through two synthetic pathways (Scheme 4). As can be seen, the approach (b) is more efficient and interesting when the researchers do not use 4-phenylbenzaldehyde. Also, the approach (a) is less unfavorable due to the formation of O-alkylated product during the condensation reaction95,96 and higher reaction time.
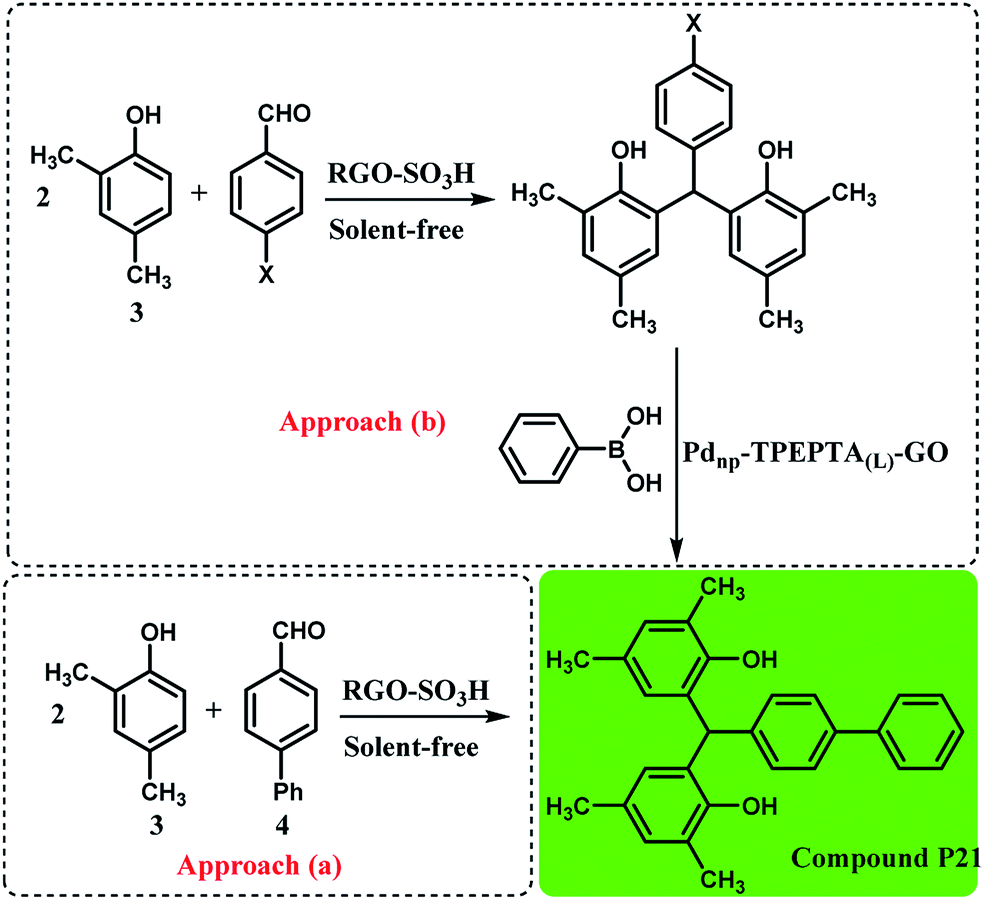 | ||
| Scheme 4 Synthesis of compound 21 using two synthetic pathways, reaction conditions for the approach (a): 2,4-dimethylphenol (3) (6 mmol), 4-phenylbenzaldehyde (4) (2 mmol), solvent-free, T: 100 °C, RGO-SO3H (sulfonated reduced graphene oxide): 40 mg, time: 2.5 h, isolated yield: 81%. Reaction conditions for approach (b): the condensation step was similar to the approach (a) and X: Br or Cl, the reaction conditions for the Suzuki reaction are the same as the footnote in Scheme 3. Time: 90 min for X: Br and 110 min for X: Cl, and for X: Br and X: Cl the final yields obtained were 91% and 86%, respectively (see ESI†). | ||
As shown in Scheme 5, it is possible to obtain the compound P22 (as an important chemical feedstock, CAS-RN: 118727-34-7 and Reaxys-RN: 7822565) using different approaches (Scheme 5, approaches: a–f). Despite much attempts for the synthesis of compound P22 using different starting materials, the successful synthesis with high yields and lower reaction time have been largely overlooked. For example, Schwab and co-workers reported the synthesis of compound P22 using [Pd(PPh3)4] catalyst in 62% isolated yield (Scheme 5, approach b).97 Also, for different approaches with two sequential steps, the total yields were lower than those for the one step approaches (Scheme 5, approaches c and e).98,99 Therefore, some experiments were conducted to obtain an efficient method for the preparation of the desired product (Scheme 5, approaches g–j). The cyclotrimerization of three equivalents of 4-aminoacetophenone was carried out using RGO-SO3H as the heterogeneous acid catalyst and the target product was produced in 62% yield within 16 h (approach g vs. approach d100,101 in Scheme 5). However, the presence of NH2 group on the starting material caused a higher amount of the used catalyst to be used and the amount of by-product produced was higher during the process. To overcome this problem, we protected the NH2 group using tert-butyloxycarbonyl ((Boc)2O), and then applied RGO-SO3H and de-protection of N-Boc to obtain the compound P22 (Scheme 5, approach h). Due to the three reaction steps, the total yield of the desired product was lower than that in our previous approach. These experiments show the significant role of Pdnp-TPEPTA(L)-GO catalyst in the preparation of compound P22 (Scheme 5, approaches i and j). The yield of the desired product increased to 89% isolated yield when the reaction conditions for approach (i) were used. Furthermore, using diverse starting materials, the desired product was obtained in good yield (80%) (Scheme 5, approach j).
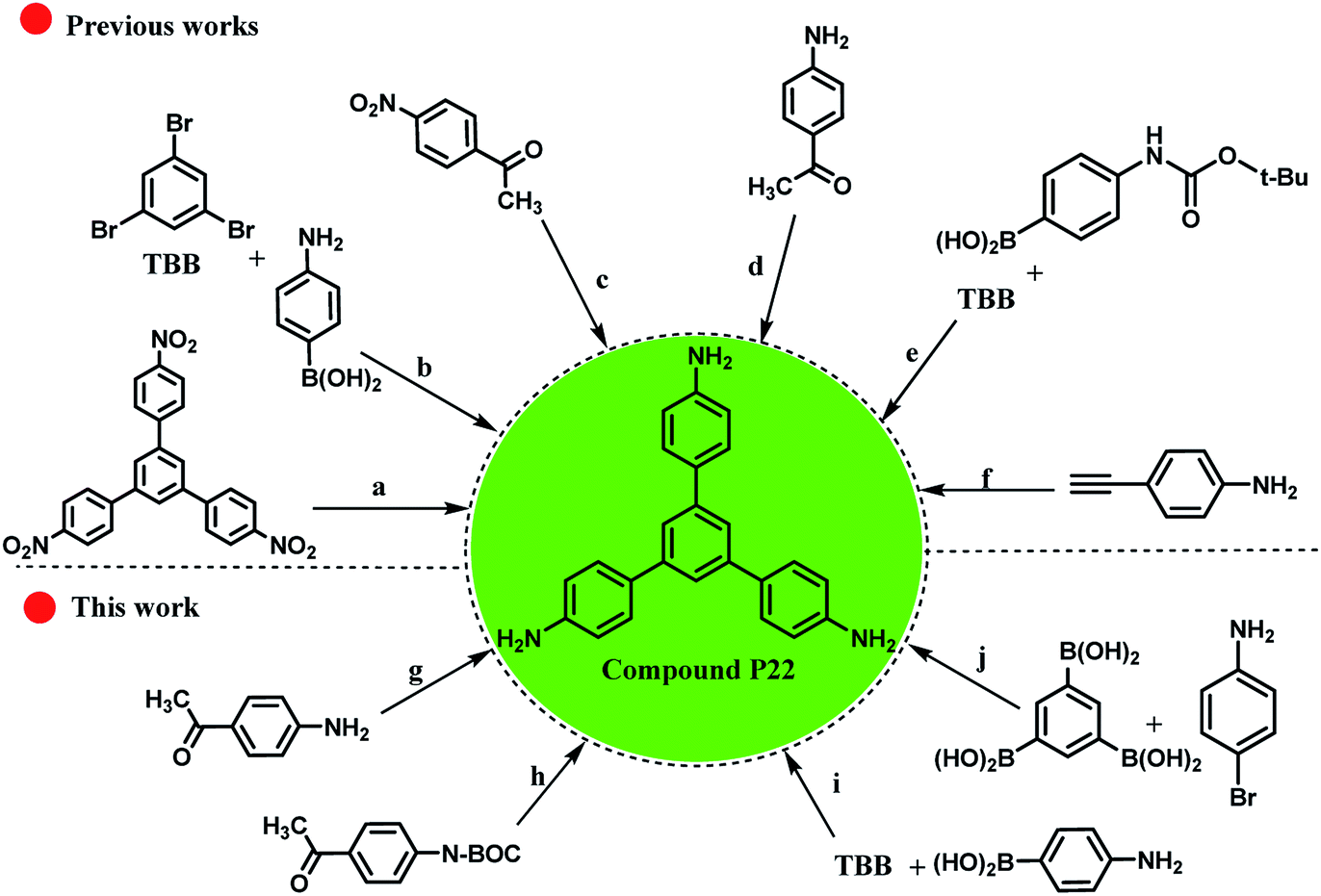 | ||
| Scheme 5 Different approaches for the synthesis of compound P22 from various starting materials (A comparison study between our work and previously reported works). Reaction conditions for (a): 10% palladium on activated charcoal, N2H4 in ethanol, time: 10.5 h, T: 70 °C, yield: 85%.102 (b) tetrakis(triphenylphosphine)palladium(0), Aliquat 336, K2CO3 in H2O, toluene, time: 24 h, T: 100 °C, inert atmosphere, yield: 62%, TBB is 1,3,5-tribromobenzene.97 (c) Two reaction steps, 1: CF3SO3H, toluene, heating, 2:32%, hydrazine hydrate, RANEY® nickel, THF, time: 6 h, T: 85 °C.98 (d) Toluene-4-sulfonic acid, H2O, neat (no solvent), time: 16 h, T: 142 °C, yield: 71% (ref. 100) and toluene-4-sulfonic acid, time: 16 h, T: 145 °C, yield: 25%.101 (e) Two reaction steps, 1: Na2CO3 in H2O, tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4], 1,2-dimethoxy-ethane, time: 18 h, heating, 2: 76%, CF3SO3H, CH2CL2, time: 2 h, T: 20 °C.99 (f) Arachno-[(η5-C5Me5RuCO)2B2H6] in toluene, time: 40 h, T: 75 °C, inert atmosphere.103 (g) Cyclotrimerization reaction, RGO-SO3H (80 mg), toluene, reflux, time: 16 h, yield: 62% (see ESI†) [this work]. (h) Three reaction steps, 1: protection of amino groups using (Boc)2O (tert-butyloxycarbonyl), 2: cyclotrimerization reaction, RGO-SO3H (80 mg), toluene, time: 12 h, reflux, 3: de-protection of N-Boc, total yield: 45%, (see ESI†) [this work]. (i) Pdnp-TPEPTA(L)-GO (30 mg), DMF:H2O (2:1) (6 mL), K2CO3, T: 80 °C, time: 1 h, yield: 89% (see ESI†) [this work]. (j): Pdnp-TPEPTA(L)-GO (60 mg), DMF:H2O (2:1) (6 mL), K2CO3, T: 100 °C, time: 1.5 h, yield: 80% (see ESI†) [this work]. | ||
For extended study, a series of competing experiments were also performed to establish the selectivity trends of the presented method (Scheme 6). First, the competing reactions were carried out between two boronic acids including phenylboronic acid (PBA) and 4-methoxyphenylboronic acid (4-OMe-PBA) with 1.2 mmol, 1.0 mmol of bromobenzene, and different amounts of K2CO3 as the base (Scheme 6, experiments 1–3). The typical procedure for each experiment is illustrated in ESI.† The researchers studying the Pd catalyst have found that a relationship exists between the Suzuki–Miyaura reaction catalytic cycle and acid–base chemistry.78 Notably, in the Suzuki cross coupling reaction, the main role of the base such as K2CO3 is to increase the reactivity of PBA towards the Ph-halide complex by changing it into the corresponding phenylborate (PBO). Hence, the pKa values of different PBA were reported in the literature.104,105 Therefore, a lower pKa value of PBA directly captures the greater tendency to obtain the OH− anion. Among PBA and 4-OMe-PBA, PBA has a lower pKa value.105 In the competing experiment 1 (CE-1), a limited amount of K2CO3 was used and good selectivity was observed for the two expected products 5a and 5b. When 1.0 mmol of K2CO3 was used, PBA with stronger acidity (lower pKa) reacts more extensively with the OH− anion and the compound 5a was obtained in 75% yield. For further investigation of the amount of base in the selectivity of the present method, we performed CE-2 and CE-3 with 2.0 and 4.0 mmol of K2CO3, respectively. As shown in these experiments, the selectivity decreases with increase in the stoichiometry of K2CO3. Additionally, we performed CE-4 and CE-5 for the study of para and meta isomers of arylbromides including electron-withdrawing groups (EWG) in the presence of PBA and 4-OMe-PBA in isolated experiments. In CE-4, 4-bromobenzaldehyde was more reactive relative to 3-bromobenzaldehyde in the presence of PBA. According to the reported literature,106 arylhalides with EWG accelerate the oxidative addition step in the mechanism of the Suzuki reaction. Also, this differentiation between 8a and 8b is related to the influence of inductive and resonance effects on the electronic charge stabilization in the Suzuki cross coupling reaction pathway. As shown in Scheme 6, CE-5, 4-OMe-PBA is less reactive towards the base (OH− anion) and a very good selectivity was observed for the two compounds 9a and 9b. Finally, in CE-6, the competition between aryl bromides including EWG and electron-donating groups (EDG) at the para positions were performed and we exclusively observed that aryl bromide including EDG cannot react with 4-OMe-PBO in the presence of aryl bromide including EWG. This observation refers to the mechanistic aspect of the Suzuki reaction progress, thus representing the existence of EDG and EWG on the aryl halides.106
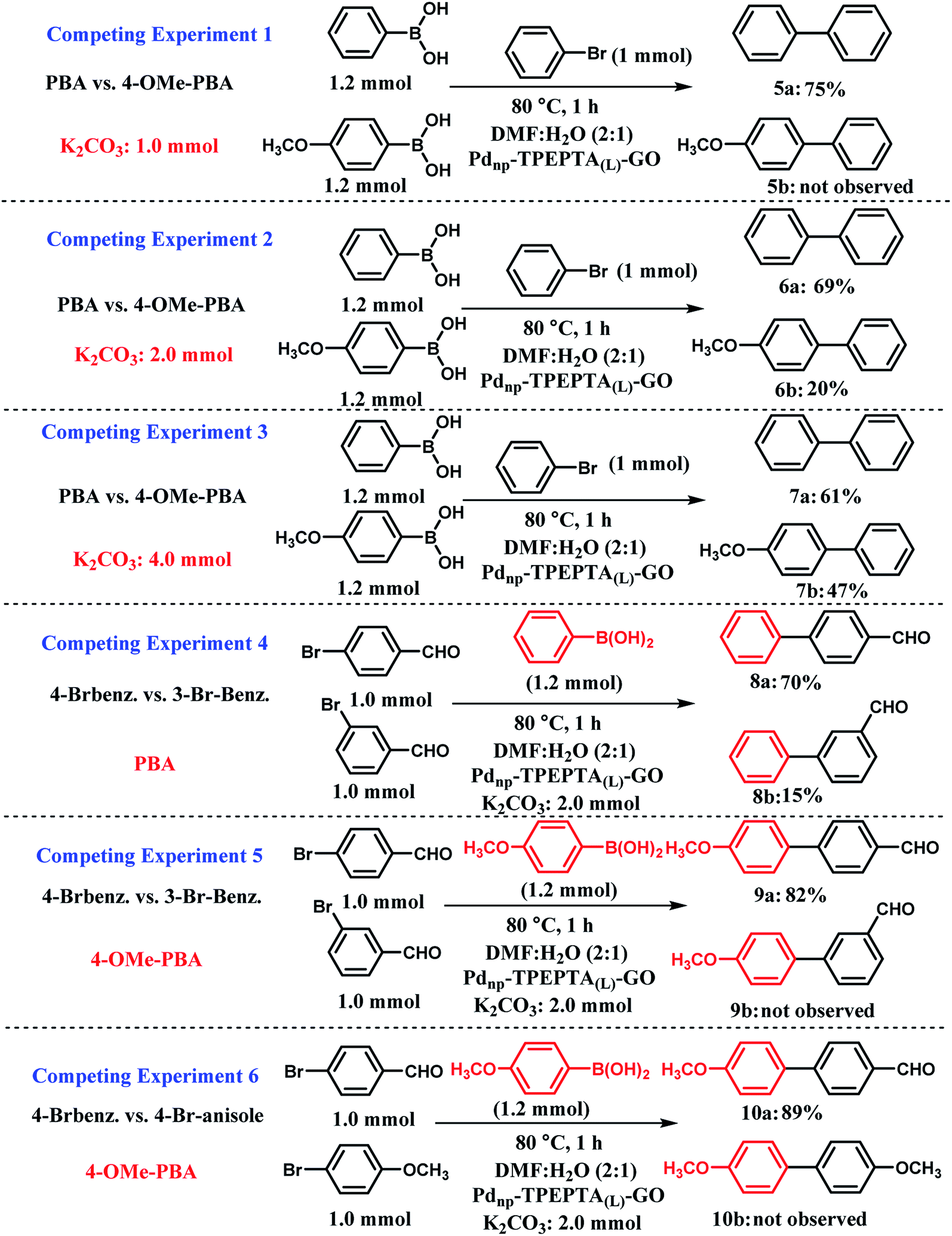 | ||
| Scheme 6 Competing experiments in the Suzuki–Miyaura cross coupling reaction; PBA is phenylboronic acid, 4-OMe-PBA is 4 methoxyphenylboronic acid. Experiments 1–3 for competition between PBA and PBA including electron-donating groups such as OMe at different mmol of K2CO3 as the base in the presence of the same arylbromide. Experiment 4 for studying the effect of electron-withdrawing group at meta or para positions in the presence of the same phenylboronic acid. Experiment 5 for the investigation of the effect of electron-withdrawing group at meta or para positions in the presence of the same phenylboronic acid including electron-donating groups such as OMe. Experiment 6 for studying the effect of the electron-donating and electron-withdrawing groups on the aryl halide in the Suzuki–Miyaura cross coupling reaction in the presence of the same phenylboronic acid with methoxy group at the para position. General reaction conditions for all the competing experiments: solvent (6 mL), Pdnp-TPEPTA(L)-GO catalyst (30 mg). All the yields refer to the isolated yield. For the typical procedures and separation of the desired compound from the reaction mixture, please see ESI.† | ||
Usually, there are two competing reactions with the main coupling reaction (Scheme 7, part A). In this study, all the possible products including the homocoupling, deboronation, and Suzuki–Miyaura reactions were monitored during the processes using gas chromatography (GC). These analyses revealed that in all the experiments, the conversion of PBA and aryl halides was 100% and the reaction occurred until completion. Also, in all the experiments, the main product of Suzuki reaction was obtained in large yields and two other byproducts from the homocoupling and deboronation reactions were detected at 0.1–0.5% and 2.3–3.0%, respectively. Interestingly, the present method is highly appealing and selective in limiting the production of by-products relative to the other methods.
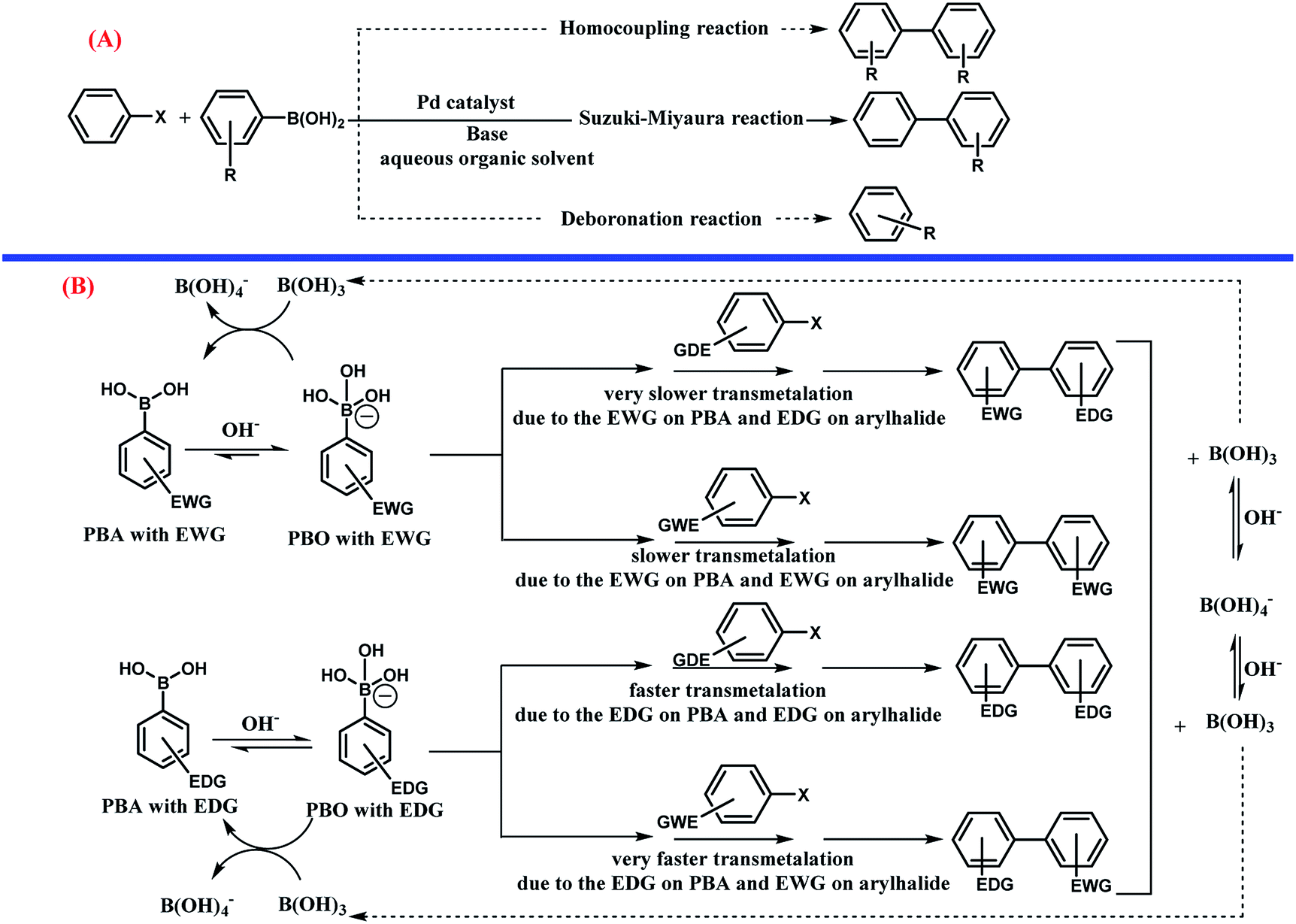 | ||
| Scheme 7 (A) The competing reactions (homocoupling and deboronation) with the Suzuki–Miyaura cross coupling reaction (----: competition reaction,—: main reaction). (B) The effect of electron-withdrawing group (EWG) and electron-donating group (EDG) on phenylboronic acid (PBA), phenylborate (PBO), and aryl halides on the rate of the Suzuki–Miyaura cross coupling reaction. | ||
It seems that the influence of the substituent plays a dual role in the mechanism of the Suzuki–Miyaura cross coupling reaction (Scheme 7 part B).78 As can be seen, PBA with EWG at different positions, especially the para position, increases the acidity of PBA due to delocalization of the negative charge on PBO as a conjugate base by the resonance effect. The EWG substitution on PBA decreased the nucleophilicity of PBO towards the electrophilic Pd complex, which can decrease the rate of the transmetalation step.107 The aryl halide with EDG substitution has a slower rate of transmetalation than that for the aryl halide with EWG. The presence of EDG on PBA has the opposite effects. However, according to some previous studies,78 a higher rate of transmetalation can be obtained when the EDG and EWG exist on PBA and the aryl halide, respectively.
We have proposed a plausible reaction mechanism for this protocol, according to the literature,108–110 as shown in Scheme 8. Depending on the reaction conditions such as the concentration and type of the base, the stability of Pd nanoparticles immobilized on the ligand and reduced reaction conditions, the reaction can be performed through different pathways. First, the free Pd(0) was released in the reaction medium near the high SSA of the Pdnp-TPEPTA(L)-GO catalyst. Then, the oxidative addition of Pd(0) to the aryl halide to produce the organopalladium complex intermediate (I) occurred. The two main pathways (a) and (b) can be proposed according to the concentration and power of the base in the reaction medium. When the base has a significant nucleophilicity to replace X as the leaving group from the complex (I), the pathway (a) was followed. However, in the opposite conditions, the base was attached to PBA and formed PBO (III) (pathway b). Subsequently, the migration of the Ph group to intermediates (I) or (II) took place in pathways (a) or (b) to form the intermediate (IV) through the transmetalation step. The Suzuki product was obtained through the reductive elimination of the Pd(II) complex (IV). At this point, the Pd(II) complex (V) was reduced and free Pd(0) can follow two main pathways including rebounding to the catalyst via pathway (c) or the initiation of the next catalytic cycle via pathway (d).
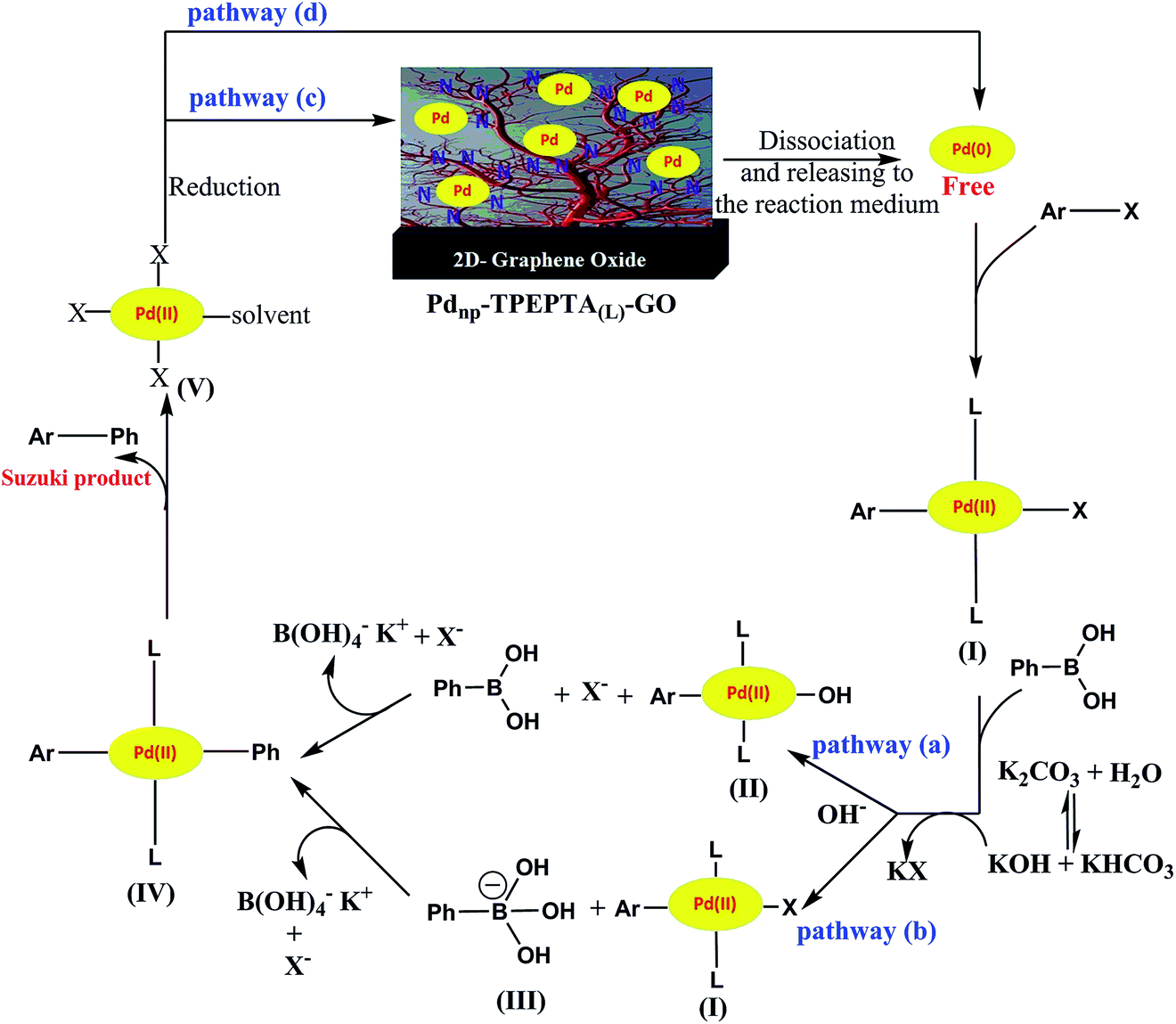 | ||
| Scheme 8 The proposed reaction pathway for Suzuki reaction using the Pdnp-TPEPTA(L)-GO catalyst. | ||
The greatest advantages of Pdnp-TPEPTA(L)-GO catalyst are the ease of separation and compatibility with the organic compounds as reactants due to the large organic ligand supported on GO as the inorganic support. Also, the reusability and recyclability of Pdnp-TPEPTA(L)-GO catalyst is very important from the economic, environmental, and industrial points of view. Therefore, the reusability of the Pdnp-TPEPTA(L)-GO catalyst was tested in the Suzuki–Miyaura cross coupling reaction for the preparation of compound P1 under the optimized conditions within 10 minutes. The Pdnp-TPEPTA(L)-GO catalyst showed a reusability of at least six runs without a remarkable decrease in its catalytic activity performance (Table 2). As shown in this table, the first catalytic run resulted in the production of 95% of the desired product within 10 minutes. In the 5th catalytic run, approximately 87% of the final product was obtained. According to the recovered catalyst values in each run, about 97% of the Pdnp-TPEPTA(L)-GO catalyst remained until the 6th run. However, the decreasing catalytic activity in the 6th run can be related to the lost catalyst during catalyst recovery and not to palladium leaching. During the reusability experiments, the Pd content of the catalyst decreased by only 6 ppm (about 2.3%) of the total Pd content immobilized on TPEPTA(L)-GO. Therefore, this observation showed that the Pdnp-TPEPTA(L)-GO catalyst is a robust and satisfactory heterogeneous catalyst. For the completion of our observation related to the Pd leaching, the hot filtration study for this catalyst was performed and the catalyst immediately following the end of the reaction was isolated. According to the ICP analysis obtained for the hot filtration test, the Pd detected in the 1st, 3rd, and 6th runs were 0.08, 0.1, and 0.06 ppm, respectively. After the isolation of the catalyst, the hot filtration reaction mixture was charged with iodobenzene and PBA. The GC yields for the 1st, 3rd, and 6th runs were only < 2%. This experiment strongly revealed that the leaching of Pd nanoparticles during the reaction progress is very low and the Pdnp-TPEPTA(L)-GO catalyst is a reusable and air-stable heterogeneous catalyst in practice. For further investigation of the used catalyst after the 6th run, the morphology of Pdnp-TPEPTA(L)-GO was studied and the catalyst remained mostly unchanged after use in the Suzuki–Miyaura cross coupling reaction, according to the FT-IR and TEM studies (see ESI†).
| Run | First | Second | Third | Fourth | Fifth | Sixth |
|---|---|---|---|---|---|---|
| a 4-Iodoanisole (1 mmol), phenylboronic acids (1.2 mmol), K2CO3 (2 mmol), solvent: DMF:H2O (2:1) (6 mL), Pdnp-TPEPTA(L)-GO (30 mg), T: 80 °C.b Isolated yields.c Not determined.d GC yield. |
||||||
| Time (min) | 10 | 10 | 10 | 10 | 10 | 10 |
| Yield (%)b | 95 | 95 | 93 | 92 | 90 | 87 |
| TON | 12.3 | 12.3 | 12.1 | 11.9 | 11.7 | 11.3 |
| TOF (h−1) | 74.1 | 74.1 | 72.9 | 71.7 | 70.5 | 68.1 |
| Recovered catalyst (%) | >99 | >99 | >98 | >98 | >97 | >97 |
| Pd amount (ppm) | 258 | 257 | ND | ND | 253 | 252 |
| Pd leached after hot filtration test (ppm) | 0.08 | NDc | 0.1 | ND | ND | 0.06 |
| Yield of hot filtration test (%)d | <2 | ND | <2 | ND | ND | <2 |
Conclusion
In this research, we have introduced an efficient synthetic methodology for the synthesis of biaryl units using aryl halide and phenylboronic acid derivatives. A heterogeneous Pd catalyst system was developed by immobilizing Pd on TPEPTA as a ligand with high N-ligation sites. The complex was supported on graphene oxide through 3-chloropropyltrimethoxysilane. The synthetic route for the preparation of Pdnp-TPEPTA(L)-GO catalyst is simple and the starting materials are available and cheap. The Pdnp-TPEPTA(L)-GO catalyst shows high catalytic activity in the Suzuki–Miyaura cross coupling reaction. A range of aryl or heteroaryl halides including electron-withdrawing and electron-donating substituents and different halogens were employed. The catalyst was efficient and reusable for three types of halogens involving Cl, Br, and I in the aryl halides. The Pdnp-TPEPTA(L)-GO catalyst was recyclable for six runs. Moreover, a series of competing experiments was performed to establish the selectivity trends of the presented method.Experimental
General
All the solvents, materials, and reagents were used without further purification. The melting points of the synthesized compounds were determined with a Thermo-scientific micro apparatus in capillary tubes and are uncorrected. Graphite powder and the inorganic materials were purchased from Merck chemical company. The NMR spectra of the synthesized compounds were recorded on a Brucker DRX-400 MHz spectrometer. δ (ppm) chemical shifts are given relative to CDCl3 and DMSO-d6 as the solvents: the reference peak for CDCl3 were 7.24 ppm (1H NMR) and 77.0 ppm (13C NMR), and for DMSO-d6 were 2.47 ppm (1H NMR) and 39.94 ppm (13C NMR). The FT-IR spectra were recorded on a PerkinElmer 781 spectrophotometer in the range of 400–4000 cm−1 using KBr pellets.General procedure for the Suzuki–Miyaura cross coupling reaction using Pdnp-TPEPTA(L)-GO catalyst
Aryl halide (1.0 mmol), phenyl boronic acid derivatives (1.2 mmol), Pdnp-TPEPTA(L)-GO (30 mg), K2CO3 (2.0 mmol), and DMF:H2O (2:1) (6.0 mL) were placed in a 25 mL round-bottom-flask equipped with a condenser and magnetic stirring bar, and heated at 80 °C. The progress of the reaction was monitored using TLC until 100% conversion of the aryl halide was confirmed. After the completion of the reaction, 5.0 mL of hot water and 5.0 mL of ethyl acetate were added to the reaction mixture. The Pdnp-TPEPTA(L)-GO catalyst was separated under reduced pressure using a vacuum pomp over sintered-glass grade-4. The organic solution was evaporated on a rotary evaporator and the crude product was obtained. For further purification of each of the synthesized compounds, different techniques were applied (see ESI†).
Conflicts of interest
The authors declare no conflict of interest.Abbreviation
| TPEPTA | Tris(pentaethylene-pentamine)triazine |
| GO | Graphene oxide |
| 1D | One-Dimensional |
| 2D | Two-Dimensional |
| 3D | Three-Dimensional |
| SSA | Specific Surface Area |
| TCT | 2,4,6-Trichloro-1,3,5-triazine |
| PEHA | Pentaethylenehexamine |
| SDS | Sodium dodecyl sulfate |
| AAS | Atomic absorption spectrometry |
| PBA | Phenylboronic acid |
| 4-OMe-PBA | 4-Methoxyphenylboronic acid |
| PBO | Phenylborate |
| EWG | Electron-withdrawing group |
| EDG | Electron-donating group |
Acknowledgements
The authors are grateful to University of Kashan for supporting this work by Grant Number 159148/129.Notes and references
- G. Anderson and F. Hartley, Chemistry of Platinum Group Metals, Recent Developments, Elsevier, 1st edn, 1991 Search PubMed.
- G. Wilkinson and R. D. Gillard, Comprehensive coordination chemistry: the synthesis, reactions, properties & applications of coordination compounds, Middle transition elements, Pergamon, 1987 Search PubMed.
- S. A. Cotton, in Chemistry of Precious Metals, Springer Netherlands, Dordrecht, 1997, pp. 173–272, DOI:10.1007/978-94-009-1463-6_3.
- P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435–4437, 10.1039/B505424A.
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3–18 CrossRef CAS.
- M. L. Toebes, J. A. van Dillen and K. P. de Jong, J. Mol. Catal. A: Chem., 2001, 173, 75–98 CrossRef CAS.
- E. B. Mubofu, J. H. Clark and D. J. Macquarrie, Green Chem., 2001, 3, 23–25 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro, M. Antonietti and H. García, Chem. Soc. Rev., 2017, 46, 4501–4529 RSC.
- Q. Lyu, H. Yan, L. Li, Z. Chen, H. Yao and Y. Nie, Polymers, 2017, 9, 447 CrossRef PubMed.
- B. Wang, T. Yan, T. Chang, J. Wei, Q. Zhou, S. Yang and T. Fang, Carbon, 2017, 122, 9–18 CrossRef CAS.
- C. Wang, L. Zhang, Z. Guo, J. Xu, H. Wang, K. Zhai and X. Zhuo, Microchim. Acta, 2010, 169, 1–6 CrossRef CAS.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- Q. Fu and X. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 RSC.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Y. Huang, Q. Wei, Y. Wang and L. Dai, Carbon, 2018, 136, 150–159 CrossRef CAS.
- D. Sengupta, M. K. Pandey, D. Mondal, L. Radhakrishna and M. S. Balakrishna, Eur. J. Inorg. Chem., 2018, 2018, 3374–3383 CrossRef CAS.
- H. A. Elazab, S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, Catal. Lett., 2017, 147, 1510–1522 CrossRef CAS.
- H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Appl. Catal., A, 2015, 491, 58–69 CrossRef CAS.
- C. Wang, L. Salmon, R. Ciganda, L. Yate, S. Moya, J. Ruiz and D. Astruc, Chem. Commun., 2017, 53, 644–646 RSC.
- X. Wang, W. Chen and L. Yan, Mater. Chem. Phys., 2014, 148, 103–109 CrossRef CAS.
- H.-q. Song, Q. Zhu, X.-j. Zheng and X.-g. Chen, J. Mater. Chem. A, 2015, 3, 10368–10377 RSC.
- V. V. Singh, U. Kumar, S. N. Tripathi and A. K. Singh, Dalton Trans., 2014, 43, 12555–12563 RSC.
- X. Shi and C. Cai, New J. Chem., 2018, 42, 2364–2367 RSC.
- V. Sharavath and S. Ghosh, RSC Adv., 2014, 4, 48322–48330 RSC.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed.
- M. Sarvestani and R. Azadi, Appl. Organomet. Chem., 2017, 31, e3667 CrossRef.
- S. Rana, S. Maddila, K. Yalagala and S. B. Jonnalagadda, Appl. Catal.,A, 2015, 505, 539–547 CrossRef CAS.
- K. Qu, L. Wu, J. Ren and X. Qu, ACS Appl. Mater. Interfaces, 2012, 4, 5001–5009 CrossRef CAS PubMed.
- R. Nie, J. Shi, W. Du and Z. Hou, Appl. Catal., A, 2014, 473, 1–6 CrossRef CAS.
- M. Moghadam, H. Salavati and Z. Pahlevanneshan, J. Iran. Chem. Soc., 2018, 15, 529–536 CrossRef CAS.
- H. Mahdavi and O. Rahmani, Catal. Lett., 2016, 146, 2292–2305 CrossRef CAS.
- H. Joshi, K. N. Sharma, A. K. Sharma and A. K. Singh, Nanoscale, 2014, 6, 4588–4597 RSC.
- M. Gómez-Martínez, E. Buxaderas, I. M. Pastor and D. A. Alonso, J. Mol. Catal. A: Chem., 2015, 404–405, 1–7 CrossRef.
- R. Fareghi-Alamdari, M. G. Haqiqi and N. Zekri, New J. Chem., 2016, 40, 1287–1296 RSC.
- R. Hashemi Fath and S. J. Hoseini, J. Organomet. Chem., 2017, 828, 16–23 CrossRef CAS.
- S. Jafar Hoseini, M. Dehghani and H. Nasrabadi, Catal. Sci. Technol., 2014, 4, 1078–1083 RSC.
- R. Fareghi-Alamdari, M. Golestanzadeh and O. Bagheri, RSC Adv., 2016, 6, 108755–108767 RSC.
- X. Li, Q. Zhao, X. Feng, L. Pan, Z. Wu, X. Wu, T. Ma, J. Liu, Y. Pan, Y. Song and M. Wu, ChemSusChem, 2019, 12, 858–865 CrossRef CAS PubMed.
- M. Golestanzadeh, H. Naeimi and Z. Zahraie, ChemistrySelect, 2016, 1, 6490–6498 CrossRef.
- H. Naeimi and M. Golestanzadeh, New J. Chem., 2015, 39, 2697–2710 RSC.
- M. Golestanzadeh, H. Naeimi and Z. Zahraie, Mater. Sci. Eng., C, 2017, 71, 709–717 CrossRef CAS PubMed.
- H. Naeimi, M. Golestanzadeh and Z. Zahraie, Int. J. Biol. Macromol., 2016, 83, 345–357 CrossRef CAS PubMed.
- A. Dainyte, D. Gudeika, G. Buika and J. V. Grazulevicius, Mol. Cryst. Liq. Cryst., 2014, 590, 73–79 CrossRef CAS.
- J. M. Lupton, L. R. Hemingway, I. D. W. Samuel and P. L. Burn, J. Mater. Chem., 2000, 10, 867–871 RSC.
- B. P. Bandgar, N. S. Joshi and V. T. Kamble, Tetrahedron Lett., 2006, 47, 4775–4777 CrossRef CAS.
- G. Blotny, Tetrahedron, 2006, 62, 9507–9522 CrossRef CAS.
- S. Samaritani, G. Signore, C. Malanga and R. Menicagli, Tetrahedron, 2005, 61, 4475–4483 CrossRef CAS.
- A. H. Gemeay, M. E. El-Halwagy, R. G. El-Sharkawy and A. B. Zaki, J. Environ. Chem. Eng., 2017, 5, 2761–2772 CrossRef CAS.
- S. Sobhani, F. Zarifi and J. Skibsted, New J. Chem., 2017, 41, 6219–6225 RSC.
- H. W. Doughty, J. Am. Chem. Soc., 1924, 46, 2707–2709 CrossRef CAS.
- F. Mohanazadeh and H. Amini, Bull. Korean Chem. Soc., 2010, 31, 3038–3040 CrossRef CAS.
- G. Baier, A. Baki, S. Tomcin, V. Mailänder, E. Alexandrino, F. Wurm and K. Landfester, Macromol. Symp., 2014, 337, 9–17 CrossRef CAS.
- F. Mafuné, J.-y. Kohno, Y. Takeda, T. Kondow and H. Sawabe, J. Phys. Chem. B, 2000, 104, 9111–9117 CrossRef.
- R. Nielsen, P. Kingshott, T. Uyar, J. Hacaloglu and K. L. Larsen, Surf. Interface Anal., 2011, 43, 884–892 CrossRef CAS.
- W. D. Pyrz and D. J. Buttrey, Langmuir, 2008, 24, 11350–11360 CrossRef CAS.
- S. Mortazavi-Derazkola, S. Zinatloo-Ajabshir and M. Salavati-Niasari, RSC Adv., 2015, 5, 56666–56676 RSC.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- L. Clougherty, J. Sousa and G. Wyman, J. Org. Chem., 1957, 22, 462 CrossRef CAS.
- C. N. R. Rao and R. Venkataraghavan, Spectrochim. Acta, 1962, 18, 541–547 CrossRef CAS.
- J. R. Durig, R. Layton, D. W. Sink and B. R. Mitchell, Spectrochim. Acta, 1965, 21, 1367–1378 CrossRef CAS.
- M. Mizuguchi, M. Nara, Y. Ke, K. Kawano, T. Hiraoki and K. Nitta, Eur. J. Biochem., 1997, 250, 72–76 CrossRef CAS.
- R. Wahl, H. Engelhardt, W. Pompe and M. Mertig, Chem. Mater., 2005, 17, 1887–1894 CrossRef CAS.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- S. Some, Y. Kim, Y. Yoon, H. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 CrossRef PubMed.
- H. Naeimi and M. Golestanzadeh, RSC Adv., 2014, 4, 56475–56488 RSC.
- A. Khalafi-Nezhad and F. Panahi, Green Chem., 2011, 13, 2408–2415 RSC.
- J. R. Anasdass, P. Kannaiyan, R. Raghavachary, S. C. B. Gopinath and Y. Chen, PLoS One, 2018, 13, e0193281 CrossRef PubMed.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2005, 21, 1931–1936 CrossRef CAS PubMed.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- H. Zhang, F. Y. Kwong, Y. Tian and K. S. Chan, J. Org. Chem., 1998, 63, 6886–6890 CrossRef CAS PubMed.
- C. F. R. A. C. Lima, A. S. M. C. Rodrigues, V. L. M. Silva, A. M. S. Silva and L. M. N. B. F. Santos, ChemCatChem, 2014, 6, 1291–1302 CrossRef CAS.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- E. V. Anslyn and D. A. Dougherty, Modern physical organic chemistry, University science books, 2006 Search PubMed.
- I. J. S. Fairlamb, A. R. Kapdi and A. F. Lee, Org. Lett., 2004, 6, 4435–4438 CrossRef CAS.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- Z. Guan, B. Li, G. Hai, X. Yang, T. Li and B. Tan, RSC Adv., 2014, 4, 36437–36443 RSC.
- R. K. Arvela and N. E. Leadbeater, Org. Lett., 2005, 7, 2101–2104 CrossRef CAS PubMed.
- C. R. LeBlond, A. T. Andrews, Y. Sun and J. R. Sowa, Org. Lett., 2001, 3, 1555–1557 CrossRef CAS.
- S.-D. Cho, H.-K. Kim, H.-s. Yim, M.-R. Kim, J.-K. Lee, J.-J. Kim and Y.-J. Yoon, Tetrahedron, 2007, 63, 1345–1352 CrossRef CAS.
- A. John, M. M. Shaikh and P. Ghosh, Inorg. Chim. Acta, 2010, 363, 3113–3121 CrossRef CAS.
- L.-Y. Xu, C.-Y. Liu, S.-Y. Liu, Z.-G. Ren, D. J. Young and J.-P. Lang, Tetrahedron, 2017, 73, 3125–3132 CrossRef CAS.
- L. Liu, W. Wang and C. Xiao, J. Organomet. Chem., 2014, 749, 83–87 CrossRef CAS.
- J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS.
- A. Corma, H. García and A. Leyva, J. Catal., 2006, 240, 87–99 CrossRef CAS.
- T. Mino, Y. Shirae, M. Sakamoto and T. Fujita, J. Org. Chem., 2005, 70, 2191–2194 CrossRef CAS PubMed.
- I. D. Kostas, F. J. Andreadaki, D. Kovala-Demertzi, P. Christos and M. A. Demertzis, Tetrahedron Lett., 2005, 46, 1967–1970 CrossRef CAS.
- C.-L. Deng, S.-M. Guo, Y.-X. Xie and J.-H. Li, Eur. J. Org. Chem., 2007, 2007, 1457–1462 CrossRef.
- R. Fareghi-Alamdari, M. Golestanzadeh, N. Zekri and Z. Mavedatpoor, J. Iran. Chem. Soc., 2015, 12, 537–549 CrossRef CAS.
- R. Fareghi-Alamdari, F. Mansouri, M. Golestanzadeh and N. Zekri, Curr. Org. Chem., 2018, 22, 1373–1419 CrossRef CAS.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- P. Yan, A. Chowdhury, M. W. Holman and D. M. Adams, J. Phys. Chem. B, 2005, 109, 724–730 CrossRef CAS PubMed.
- T. Ishi-i, R. Kuwahara, A. Takata, Y. Jeong, K. Sakurai and S. Mataka, Chem.–Eur. J., 2006, 12, 763–776 CrossRef CAS PubMed.
- Y. Zhao, J. Li, C. Li, K. Yin, D. Ye and X. Jia, Green Chem., 2010, 12, 1370–1372 RSC.
- G. Gattuso, G. Grasso, N. Marino, A. Notti, A. Pappalardo, S. Pappalardo and M. F. Parisi, Eur. J. Org. Chem., 2011, 2011, 5696–5703 CrossRef CAS.
- W.-Q. Xu, Y.-Z. Fan, H.-P. Wang, J. Teng, Y.-H. Li, C.-X. Chen, D. Fenske, J.-J. Jiang and C.-Y. Su, Chem.–Eur. J., 2017, 23, 3542–3547 CrossRef CAS PubMed.
- K. Geetharani, S. Tussupbayev, J. Borowka, M. C. Holthausen and S. Ghosh, Chem.–Eur. J., 2012, 18, 8482–8489 CrossRef CAS PubMed.
- D. Hall, Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley, VCH, 2011, vol. 1 and 2 Search PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS.
- D. Zim, A. L. Monteiro and J. r. Dupont, Tetrahedron Lett., 2000, 41, 8199–8202 CrossRef CAS.
- C. F. R. A. C. Lima, J. E. Rodriguez-Borges and L. M. N. B. F. Santos, Tetrahedron, 2011, 67, 689–697 CrossRef CAS.
- N. Miyaura, J. Organomet. Chem., 2002, 653, 54–57 CrossRef CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS.
- C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for the preparation of the catalyst, synthesized compounds, spectroscopic data, and hard copies of the NMR spectra. See DOI: 10.1039/c9ra04511b |
| This journal is © The Royal Society of Chemistry 2019 |