
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance poly(urethane-co-amide) from CO2-based dicarbamate: an alternative to long chain polyamide†
Jiaxiang Qin‡
ab,
Junqiao Jiang‡a,
Shuxian Yea,
Shuanjin Wang*a,
Min Xiaoa,
Youji Taob,
Ganxin Jieb and
Yuezhong Meng *a
*a
aThe Key Laboratory of Low-carbon Chemistry & Energy Conservation of Guangdong Province, State Key Laboratory of Optoelectronic Materials and Technologies, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, PR China. E-mail: mengyzh@mail.sysu.edu.cn; wangshj@mail.sysu.edu.cn
bState Key Laboratory of Environmental Adaptability for Industrial Products, China National Electric Apparatus Research Institute Co., Ltd, Guangzhou, 510663, P. R. China
First published on 20th August 2019
Abstract
Due to its high strength, toughness, corrosion resistance and wear resistance, long chain polyamide (LCPA) has attracted broad interest. Nevertheless, its wide application in industrial fields is still being restricted because the starting material acquisition step involving diacid and diamine remains a major obstacle. Herein, we circumvent this obstacle by developing a novel polymer with similar properties by a green and efficient copolymerization process of carbon dioxide (CO2)-based dicarbamate with diamide diol under vacuum conditions, named poly(urethane-co-amide) (PUA). The semi-crystalline PUAs with high number-weight-average molecular weights (Mn, up to 41.3 kDa) were readily obtained, and these new polymers show high thermal stability (above 300 °C). Thanks to its unique chain structure, the amide, urethane and urea groups can endow the polymer with a high density cross-linking network via hydrogen bonds and high crystallinity that can result in high strength, up to 54.0 MPa. The dynamic thermomechanical analysis (DMA) results suggest that the phase separation exists within the new polymers, endowing the PUAs with a toughness higher than that of long chain polyamides. Consequently, this work not only develops a useful new polymer like commercial polyamides with high performance as a long chain polyamide candidate, but also provides a new way of utilizating CO2.
Introduction
Aliphatic polyamides, normally known as nylons, are a family of prominent semi-crystalline polymers possessing superior physical properties, among which, the most common ones are PA6 and PA66, extensively used in industrial fields, such as in electronic appliances, automobile manufacturing, and some commodity markets.1 Another attractive member in the polyamide series is LCPA, a long alkane segment polyamide with more than ten hydrocarbon repeat units involved in diacid or diamine segments. Because of the small average number of amide groups in the main chain, the hydrogen bond cross-linking density is low, endowing LCPA with flexible chain nature and major advantages such as high toughness, corrosion resistance, wear resistance and lower water absorption.2–7 Moreover, with the increasing length of carbon-chain in diacid or diamine, LCPA exhibits better processability, resulting in their wide application in electric/electronic and automobile industries.8,9 However, the monomer of LCPA is prepared by complex procedures, resulting in high price, which limits its application in extensive industrial fields. Therefore, it is of great significance to develop a novel, low-cost, high-performance and processable polyamide copolymer. Polyamide/polyamide blending has been reported as an easy way to obtain desirable properties that combine the properties of the different polyamides.10–14 However, most of these blends are confirmed to be phase separated, even with strong hydrogen bonding attraction and very similar chemical structures.Considering above reasons, poly(ether-block-amide) (PEBA),15–18 poly(ether ester amide) (PEEA),19–21 poly(ester amide) (PEA)22–25 and other poly(urethane amide) (PUA)26–29 segmented copolymers consisting of soft polyether or polyester blocks and hard PA blocks have been developed. Altering the composition and length of the soft and hard blocks provides a high-level of variability in copolymer physical properties. Although, the strain of segmented copolymers is high enough, because of the long chain of soft block, the tensile strength fails to reach the level of LCPA. Accordingly, the size of soft blocks should be controlled to balance the strength and toughness.
As an environmentally benign compounds and unique intermediates of versatile chemical products, dicarbamates play great roles as linkers in organic chemistry and amino groups' protectors in peptide chemistry.30,31 As well known, dicarbamates are industrially important precursors when serving as non-toxic substitutes in sustainable chemistry of polyurethanes since dicarbamates can be easily generated polyurethanes by reaction with oligomeric diol. In our previous work, the direct synthesis of dicarbamate from CO2, diamine and methanol was successfully carried out over various catalysts, which could serve as short blocks for the polyamide segmented copolymers. Following this idea, the purpose of the present study is to synthesize poly(urethane-co-amide) from CO2-dicarbamate with high performance, comparable with that of LCPA. The synthesis of poly(urethane-co-amide) segmented copolymers from CO2-dicarbamate may promise as one of the most attractive and effective approaches as such a method of environmentally friendly, which is not only for developing sustainable high-performance polymer but also for the utilization of greenhouse gas of carbon dioxide.32–35
Experimental and methods
Materials
Methanol, 1,6-hexanediamine (HDA), 1,8-octanediamine (ODA), 1,10-decanediamine (DDA), tetrabutyl titanate (TBT) and ε-caprolactone (CL) were purchased from Aladdin Co., Ltd (Shanghai China), while 1,3-bis(3-aminopropyl)tetramethyldisiloxane (BATMS) was from Tokyo Chemical Industry Co., Ltd. All the chemicals were analytically pure and used without further purification. High purity CO2 (>99.9999%) was obtained from Guangqi Gas Co., Ltd (Guangzhou China).Synthesis of dicarbamate
1,6-Hexanedicarbamate (HDC), 1,8-octanedicarbamate (ODC), 1,10-decanedicarbamate (DDC) synthesized from CO2, methanol and diamine was prepared according to previous work.36 Typically, 2.0 g CeO2 catalyst, 5.0 mol methanol, 50 mmol HDA and 200 mL NMP were put into the autoclave. CO2 was purged into reactor and replace the air inside for several times to make sure the oxygen-free reaction condition. The initial pressure of CO2 was set as 5 MPa and then reacted at 150 °C for 12 h. The resultants were filtrated by poly(ether sulfone) membrane (pore size 0.45um). The obtained dicarbamate was purified by recrystallization with ethanol/DI water (50/50) mixture as solvent. The corresponding structure of some important compounds are summarized in Table S1.†Preparation of diamide diol
The diamide diol was synthesized according to the report from Marcos-Fernández's group.28 HDA(0.08 mmol) and ε-CL (0.04 mmol) were added to a three-necked flask (25 mL), which was connected to a manifold equipped with vacuum and N2 gas lines, and equipped with a mechanical stirrer. The flask was made moisture-free by a nitrogen purge followed by use of a vacuum before was immersed in a silicone oil bath (120 °C) and the reaction lasted for 180 min. After reaction, N,N′-hexane-1,6-diylbis[6-hydroxyhexanamide] (HDHHA), the diamide diol from HDA, was poured into hexane and precipitated to remove the residual reactant. The final product was collected and dried in vacuum oven for 24 h. Using the same procedure, N,N′-1,3-bis(3-propyl) tetramethyldisiloxane-1,6-diylbis[6-hydroxyhexanamide] (BPTMSHHA), the diol from BATMS and CL, was also prepared.Synthesis of PUA copolymers
HDC (30 mmol) and equimolar diamide diol were added in 50 mL autoclave, which was connected with gas inlet and outlet adapters. The mixture was heated and then reacted at 160 °C under nitrogen atmosphere for pre-polymerization. It is well known that the polycondensation consists of pre-polymerization and polymerization reactions; in the process of pre-polymerization, CH3OH (0.6 mol) was collected using a trap device and the reaction was completed for about 3 h. Then appropriate amount of catalyst tetrabutyl titanate was added to the system and the viscous liquid was heated to 220 °C to undergo polymerization at 2.0 Torr for 1 h with the removal of generated CH3OH. At the end of the first-stage polymerization, the pressure in the reaction vessel was reduced to 0.5 Torr, and then the second stage polymerization proceeded for 2–3 h to further increase the molecular weight of the resulting linear PUA. After the polymerization was completed, yellowish solid was obtained. The resulting polymers were dissolved in dimethyl sulfoxide (DMSO) and then precipitated by being poured into vigorously stirred hexane. The final product was filtered and dried by lyophilization for 24 h. Eight PUAs containing different kind of diamide diols and dicarbamates were synthesized and their denotation, composition and structure are shown in Tables 1 and S1.†| Polymer | Composition of diamide diol/% HDHHA/BPTMSHHA | Dicarbamate | Urethanea/% | Ureaa/% | Mnb × 104 | Mwb × 104 | PDIb | |
|---|---|---|---|---|---|---|---|---|
| Feed | Founda | |||||||
| a Determined by 1H-NMR spectroscopy.b Measured by GPC. | ||||||||
| PUA-HDHHA-HDC | 100/0 | 100/0 | HDC | 70.4 | 29.6 | 4.13 | 6.24 | 1.51 |
| PUA-BPTMSHHA20-HDC | 80/20 | 75/25 | HDC | 62.7 | 38.3 | 2.96 | 4.85 | 1.64 |
| PUA-BPTMSHHA40-HDC | 60/40 | 52/48 | HDC | 63.7 | 36.3 | 2.87 | 5.34 | 1.81 |
| PUA-BPTMSHHA60-HDC | 40/60 | 37/63 | HDC | 66.0 | 34.0 | 2.52 | 4.12 | 1.63 |
| PUA-BPTMSHHA80-HDC | 20/80 | 17/83 | HDC | 74.1 | 25.9 | 2.31 | 4.01 | 1.74 |
| PUA-BPTMSHHA100-HDC | 0/100 | 0/100 | HDC | 70.7 | 29.3 | 2.74 | 4.16 | 1.52 |
| PUA-HDHHA-ODC | 100/0 | 100/0 | ODC | 79.4 | 20.6 | 3.85 | 5.51 | 1.43 |
| PUA-HDHHA-DDC | 100/0 | 100/0 | DDC | 80.6 | 19.4 | 3.56 | 6.23 | 1.75 |
Characterization of hydrolytic stability of PUA copolymers
Hydrolytic stability of PUA copolymers was determined at 25 °C in 0.1 M standard phosphate buffer solution (PBS, Ph = 7.4) obtained by mixing 0.2 M solution of NaH2PO4 and 0.2 M solution of Na2HPO4 in proportion 19/81 (wt/wt). Nine specimens of each PUA were washed with detergent solution, rinsed with distilled water thoroughly, dried on the filter paper, weighed and immersed in the buffer solution. After 1, 2 and 3 months, the three specimens were taken out from the buffer solution, dried on the filter paper, weighed, checked for any changes of appearance, and left for further investigation.Measurement
1H-NMR, 13C-NMR and 1H detected heteronuclear multiple bond correlation (HMBC) analysis of the PUAs were recorded on a Bruker DRX-500 NMR spectrometer at room temperature. Deuterated DMSO (D-DMSO) was used as solvent, chemical shifts were expressed in ppm with respect to tetramethylsilane (TMS). Mn and polydispersity index (PDI) of the resultant polymer product were measured using a gel permeation chromatography (GPC) system (Waters 515 HPLC Pump, Waters 2414 detector) with a set of three columns (Waters Styragel 500, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and 100000 A) and chloroform (HPLC grade) as eluent after carboxylation with trifluoroacetic anhydride. The GPC system was calibrated by a series of poly-styrene standards with polydispersities of 1.02 standards. TGA measurements were performed in a PerkinElmer Pyris Diamond TG/DTA analyzer under a protective nitrogen atmosphere. The temperature ranged from 50 to 500 °C with a heating rate of 10 °C min−1. The melting point (Tm), crystallization temperature (Tc) and glass transition temperature (Tg) of PUA copolymers were measured by a DSC (Netzsch Model 204) and the measurements were carried out under nitrogen flow from −70 to 180 °C at a heating rate of 10 °C min−1. Tg of the samples was determined from the second run. Dynamic mechanical analysis (DMA) (Netzsch Model 242) was carried out with a tension mode at 1 Hz, 3 N of static force and 3 °C min−1 from −60 to 70 °C. The tensile tests were performed using a temperature-controlled tensile tester (New SANS, Shenzhen, China) at 25 °C with a crosshead speed of 50 mm min−1. Five specimens of each sample were tested, and the average results were recorded. The Graves tear strength of the films was measured according to ASTM D 1004 with the same crosshead speed. Impact strength measurement was conducted based on a ZBC-4B Digital IZOD Impact Tester according to GB/T1843-2008 at room temperature. The morphology was characterized by a scanning electron microscope (SEM, FEI Nova Nano-SEM Quanta 400, 30 kV) after immersion period in PBS.
000, and 100000 A) and chloroform (HPLC grade) as eluent after carboxylation with trifluoroacetic anhydride. The GPC system was calibrated by a series of poly-styrene standards with polydispersities of 1.02 standards. TGA measurements were performed in a PerkinElmer Pyris Diamond TG/DTA analyzer under a protective nitrogen atmosphere. The temperature ranged from 50 to 500 °C with a heating rate of 10 °C min−1. The melting point (Tm), crystallization temperature (Tc) and glass transition temperature (Tg) of PUA copolymers were measured by a DSC (Netzsch Model 204) and the measurements were carried out under nitrogen flow from −70 to 180 °C at a heating rate of 10 °C min−1. Tg of the samples was determined from the second run. Dynamic mechanical analysis (DMA) (Netzsch Model 242) was carried out with a tension mode at 1 Hz, 3 N of static force and 3 °C min−1 from −60 to 70 °C. The tensile tests were performed using a temperature-controlled tensile tester (New SANS, Shenzhen, China) at 25 °C with a crosshead speed of 50 mm min−1. Five specimens of each sample were tested, and the average results were recorded. The Graves tear strength of the films was measured according to ASTM D 1004 with the same crosshead speed. Impact strength measurement was conducted based on a ZBC-4B Digital IZOD Impact Tester according to GB/T1843-2008 at room temperature. The morphology was characterized by a scanning electron microscope (SEM, FEI Nova Nano-SEM Quanta 400, 30 kV) after immersion period in PBS.
Results and discussion
Chemical structure characterization of PUAs
It has been well documented that the disadvantage of LCPA is high cost due to the complicated synthesis procedures, which restrains its wide applications. To address this issue, we synthesized PUA with high strength and toughness analogous containing urethane and amide segment by copolymerization of dicarbamate and diamide diol as shown in Scheme 1.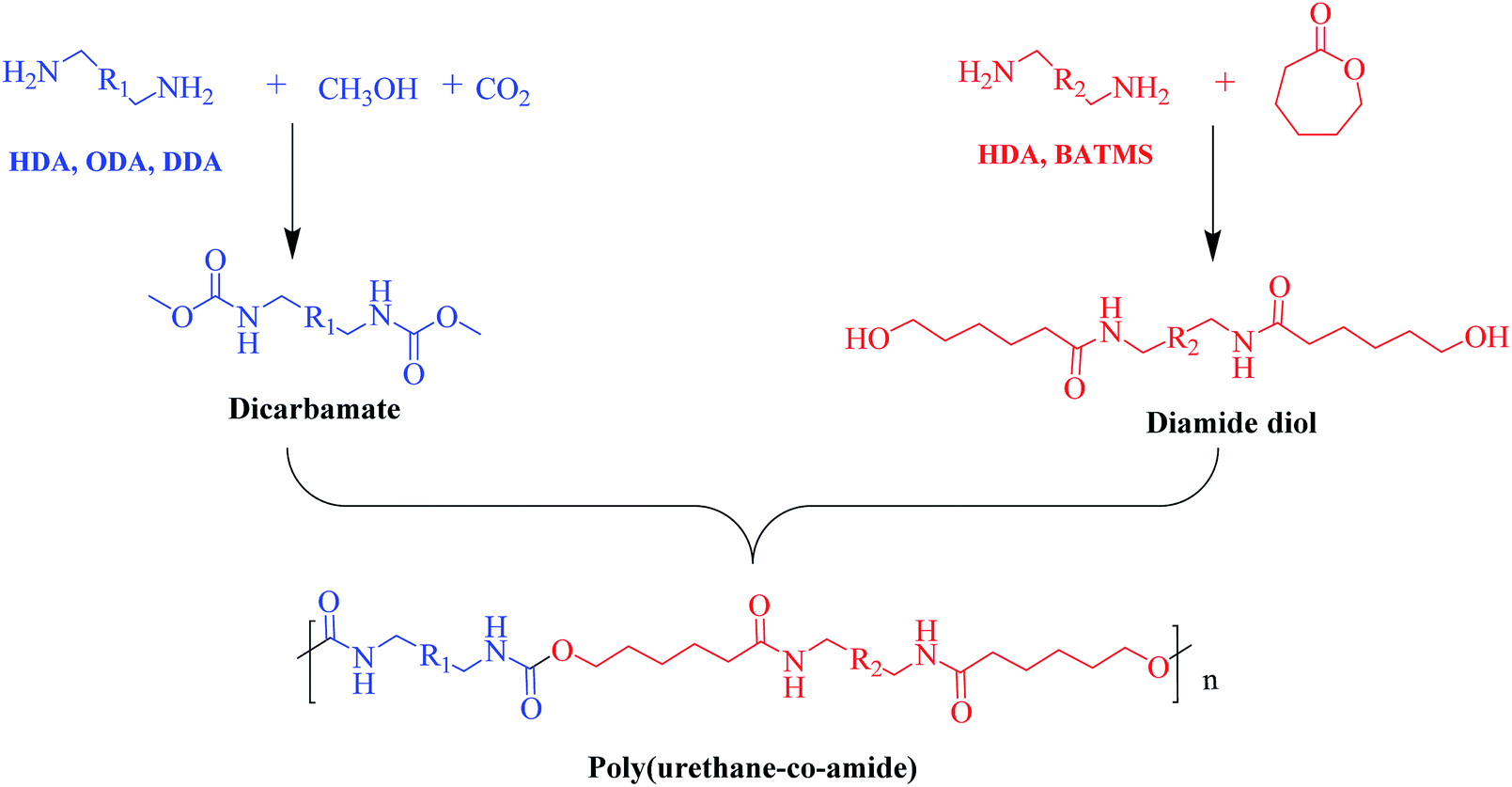 | ||
| Scheme 1 Schematic illustration of the synthesis of poly(amide-co-urethane) copolymer from CO2-based dicarbamate. | ||
Firstly, various dicarbamates were obtained from diamine, CO2 and methanol using CeO2 as catalyst, while diamide diol was synthesized by the ring-opening reaction of CL with diamine. The 1H-NMR spectra of dicarbamates and diamide diols are shown in the ESI (Fig. S1–S3†) indicating the successful synthesis of target compounds.
A series of PUA copolymers with different structure (PUA-HDHHA-HDC, PUA-BPTMSHHA20-HDC, PUA-BPTMSHHA40-HDC, PUA-BPTMSHHA60-HDC, PUA-BPTMSHHA80-HDC, PUA-BPTMSHHA100-HDC, PUA-HDHHA-ODC, PUA-HDHHA-DDC; Table S1†) were then synthesized to investigate the effect of segment composition on thermal and mechanical properties. The prepared PUAs copolymers were characterized by both 1H-NMR and 13C-NMR spectroscopy in D-DMSO, as shown in Fig. 1 and 2. In Fig. 1, the characteristic signal of methyls (–CH3, δ 3.5) from dicarbamates and hydroxyl (–OH, δ 3.6) of diamine diol disappear upon copolymerization. The N–H of urethane [–NH–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–, δ 7.19, 7.02 and 6.71] functional groups shows an obvious characteristics signal, which is different from dicarbamate, demonstrating that diamide diol HDHHA could be introduced into the main chain of PUA. The detected urea groups indicate that there is a transurethane reaction during the copolymerization between carbamate–carbamate, carbamate–urethane segment, and urethane–urethane segment, as shown in Scheme S1.† The urethane group in dicarbamate and formed polyurethane can react each other to form urea units and carbonate. Due to low stability, the dimethyl carbonate can evaporate and the carbonate in copolymer can further decompose into carbon dioxide and other by-products. As a result, the peak of carbonate didn't appear in Fig. 2. Moreover, 13C-NMR spectrum in Fig. 2 shows the peaks of the carbonyl zone for three different functional groups: (a) amide [–(CO)–NH–, δ 172.3], (b) urethane [O–(CO)–NH–, δ 156.8], and (c) urea [–NH–(CO)–NH–, δ 158.6], while the characteristic signals of methyl [–CH3, δ 53.1] of dicarbamate disappear after copolymerization. These evidences further confirm the presence of the functional groups attributed to PUAs, implying that PUAs were synthesized successfully from dicarbamate and diamide diol via polycondensation.
O)–O–, δ 7.19, 7.02 and 6.71] functional groups shows an obvious characteristics signal, which is different from dicarbamate, demonstrating that diamide diol HDHHA could be introduced into the main chain of PUA. The detected urea groups indicate that there is a transurethane reaction during the copolymerization between carbamate–carbamate, carbamate–urethane segment, and urethane–urethane segment, as shown in Scheme S1.† The urethane group in dicarbamate and formed polyurethane can react each other to form urea units and carbonate. Due to low stability, the dimethyl carbonate can evaporate and the carbonate in copolymer can further decompose into carbon dioxide and other by-products. As a result, the peak of carbonate didn't appear in Fig. 2. Moreover, 13C-NMR spectrum in Fig. 2 shows the peaks of the carbonyl zone for three different functional groups: (a) amide [–(CO)–NH–, δ 172.3], (b) urethane [O–(CO)–NH–, δ 156.8], and (c) urea [–NH–(CO)–NH–, δ 158.6], while the characteristic signals of methyl [–CH3, δ 53.1] of dicarbamate disappear after copolymerization. These evidences further confirm the presence of the functional groups attributed to PUAs, implying that PUAs were synthesized successfully from dicarbamate and diamide diol via polycondensation.
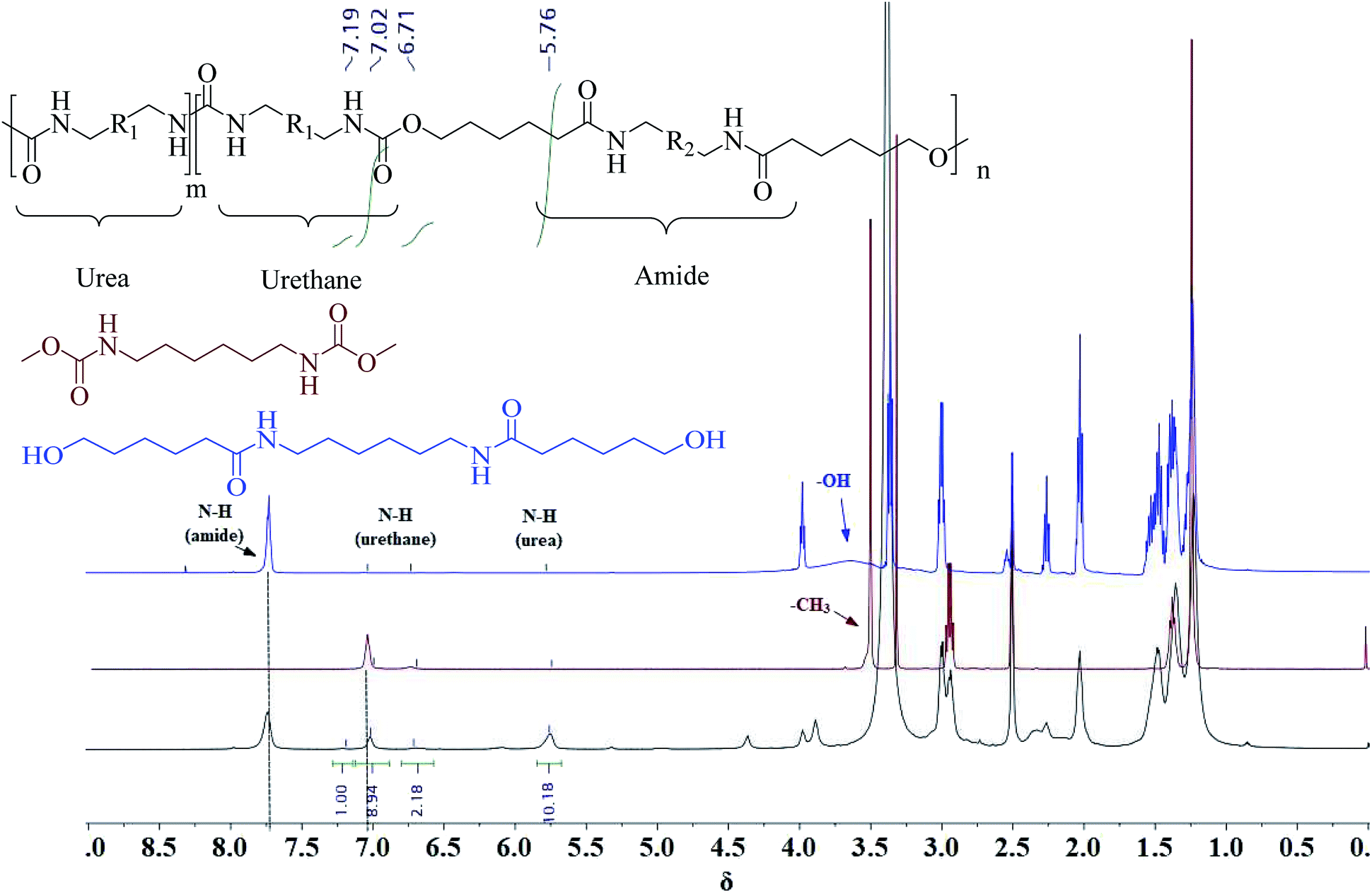 | ||
| Fig. 1 1H-NMR spectrum for HDC (auburn), HDHHA (blue) and resulted polymer structure (black). | ||
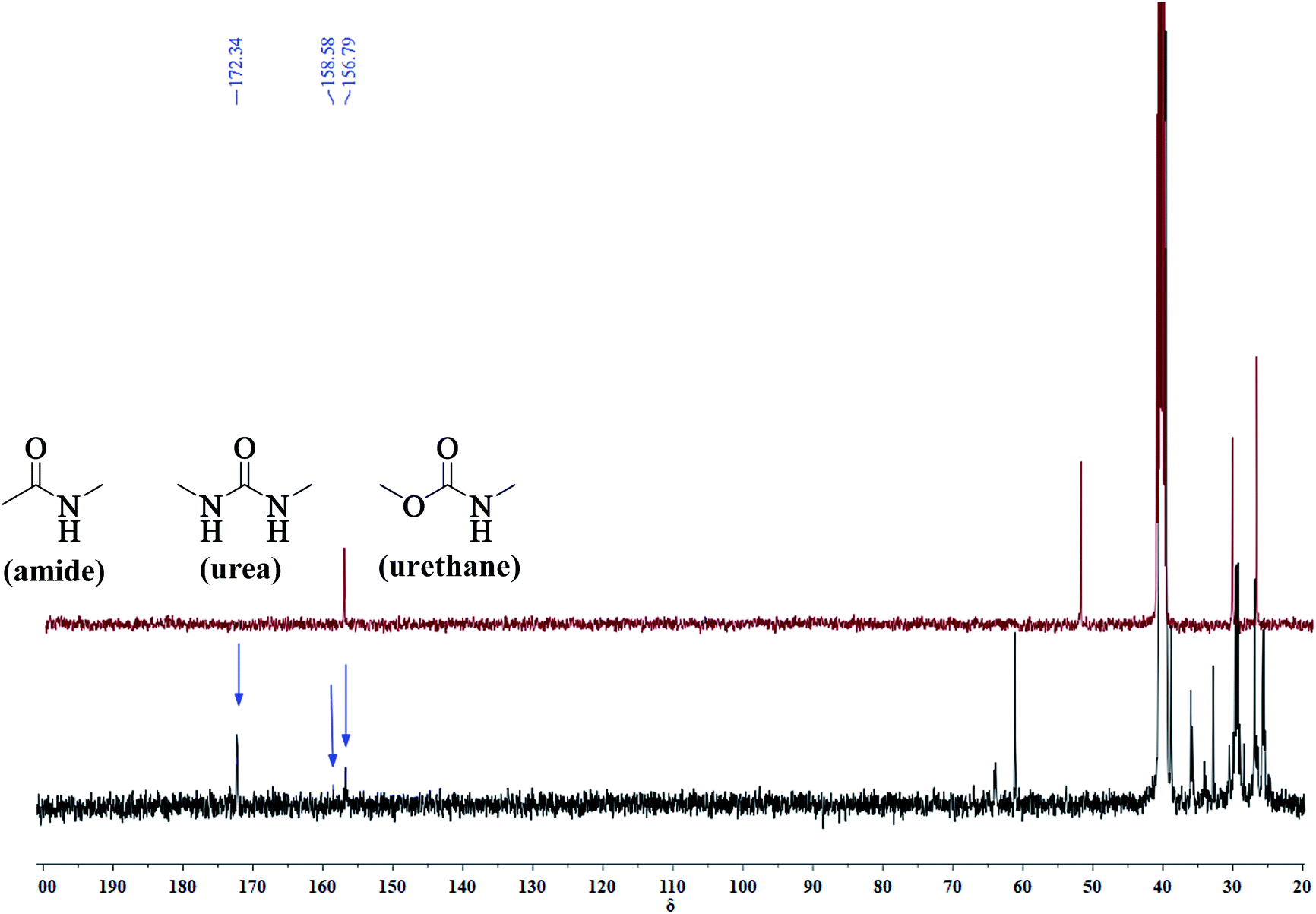 | ||
| Fig. 2 13C-NMR spectrum for HDC (auburn) and resulted polymer structure (blue). | ||
The unique chemical shifts observed in 1H-NMR spectroscopy not only provide insight into the successful copolymerization of dicarbamate and diamide diol, but also reveal the compositional structure of the resulted polymer chain. The proton resonances at 0.4–0.5 ppm correspond to –CH2– in BATMS sequence, next to Si atom, while the peaks at 7.7 ppm are attributed to the –NH– groups of amide. The molar ratio of BPTMSHHA segment can be calculated from the peak area of –CH2- in BATMS sequence and –NH– of amide groups from 1H-NMR spectroscopy (Fig. S4†), which is basically consistent with the feed ratio, as summarized in Table 1. Similarly, the degree of transurethane is also presented in Table 1 that was calculated from N–H signal area of urea and urethane. It suggests that amide, urethane and urea segments in backbone are random copolymerization structure.
The polymerization was further confirmed by 1H–13C detected heteronuclear multiple bond correlation (HMBC) NMR spectroscopy.37 HMBC NMR is a two dimensional NMR technique, which have been used for resonance assignments of biological and small organic molecules, because it can dramatically enhance the sensitivity of NMR for detecting the spectra of nuclei other than protons. In case of 13C, the sensitivity is theoretically enhanced 32-fold relative to standard direct 13C detection. This sensitivity enhancement is achieved using J couplings to promote coherence transfer from 13C nucleus to 1H followed by detection of much more sensitive 1H nucleus rather than 13C nucleus. Recently, these techniques of inverse detection have also been applied to structure determination of different polymers.38–41 The HMBC NMR of multiblock copolymer is realized in D-DMSO (Fig. 3). The HMBC spectrum illustrates that both 1H-NMR signal at H 3.0 ppm due to methylene proton in HA unit and H 2.2 ppm due to methylene proton in CL unit are related to 13C-NMR signals at C 172.3 ppm due to amide carbon, indicating that HA is bonded to CL covalently. 1H-NMR signal at H 3.9 ppm due to methylene proton in CL units is connected to 13C-NMR signals at C 156.8 ppm due to urethane carbon. This confirms the presence of both amide and urethane units within a polymer chain, i.e., the occurrence of the copolymerization between dicarbamate and diamide diol. Additionally, the proton at H 2.9 ppm is a direct correlation with carbon atom at C 158.6 ppm due to urea carbon, further suggesting the presence of transurethane reaction. The molecular weight and molecular weight distribution of the PUAs were measured using GPC as shown in Table 1. The results also suggest that the obtained copolymer is not a mixture of dicarbamate and diol but a copolymer. This fact is well consistent with above results of 1H-NMR, 13C-NMR and HMBC.
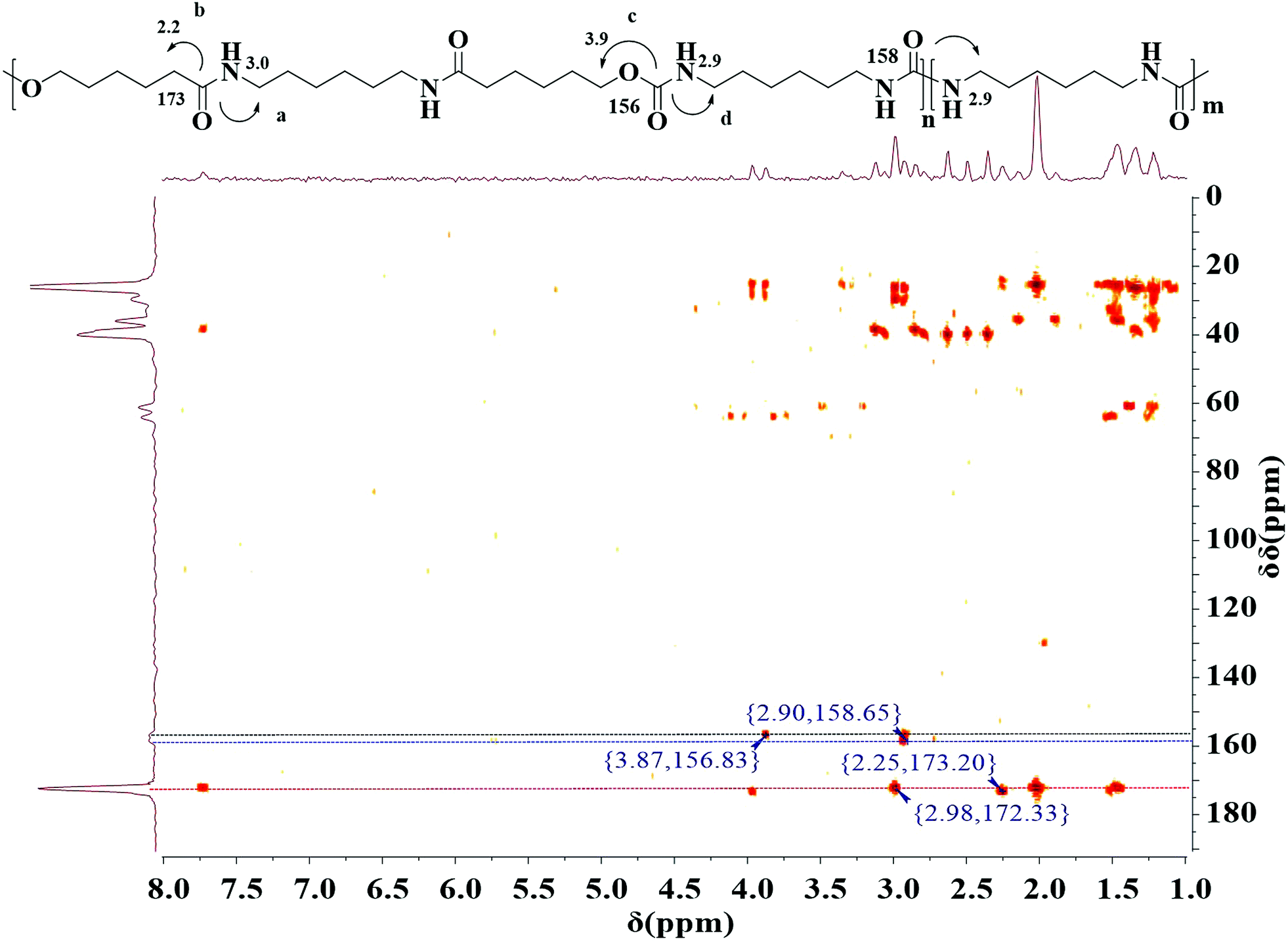 | ||
| Fig. 3 1H–13C HMBC spectrum of final polymer (entry 5, Table 1). | ||
Thermally oxidative stabilities
Table 2 shows the thermal properties of PEUAs obtained from Differential Scanning Calorimetry (DSC) measurements, and Fig. 4 displays typical curves of the PUAs derived from different dicarbamates. All PUA samples obtained from HDHHA and different dicarbamate are crystalline with a melting temperature (Tm) ranging from 150 to 168 °C. The greater the number of methylene in dicarbamate is, the lower the Tm of PUA is. Obviously, PUA-HDHHA-HDC shows the highest melting point of 168.5 °C. It is of importance to note that even at a highly cooling rate of 10 °C min−1, these PUAs show crystalline point. Moreover, due to the incomplete crystallization during cooling run, PUA-HDHHA-ODC derived from ODC and HDHHA shows an obvious cold crystallization around 50 °C.| Polymer | Diamide diol/% | Dicarbamate | Tm/°C | Tc/°C | T5%/°C | Tmax/°C | |
|---|---|---|---|---|---|---|---|
| BAD | BPTMSHHA | ||||||
| PUA-HDHHA-HDC | 1 | 0 | HDC | 168.5 | 122.8 | 310.9 | 459.4 |
| PUA-BPTMSHHA20-HDC | 0.8 | 0.2 | HDC | 137.2 | 60.8 | 310.7 | 447.7 |
| PUA-BPTMSHHA40-HDC | 0.6 | 0.4 | HDC | 107.7 | 60.7 | 309.4 | 433.7 |
| PUA-BPTMSHHA60-HDC | 0.4 | 0.6 | HDC | — | — | 301.9 | 446.2 |
| PUA-BPTMSHHA80-HDC | 0.2 | 0.8 | HDC | — | — | 307.6 | 433.7 |
| PUA-BPTMSHHA100-HDC | 0 | 1 | HDC | — | — | 301.3 | 446.5 |
| PUA-HDHHA-ODC | 1 | 0 | ODC | 156.3 | 72.8 | 308.0 | 458.2 |
| PUA-HDHHA-DDC | 1 | 0 | DDC | 150.0 | 85.6 | 300.5 | 466.0 |
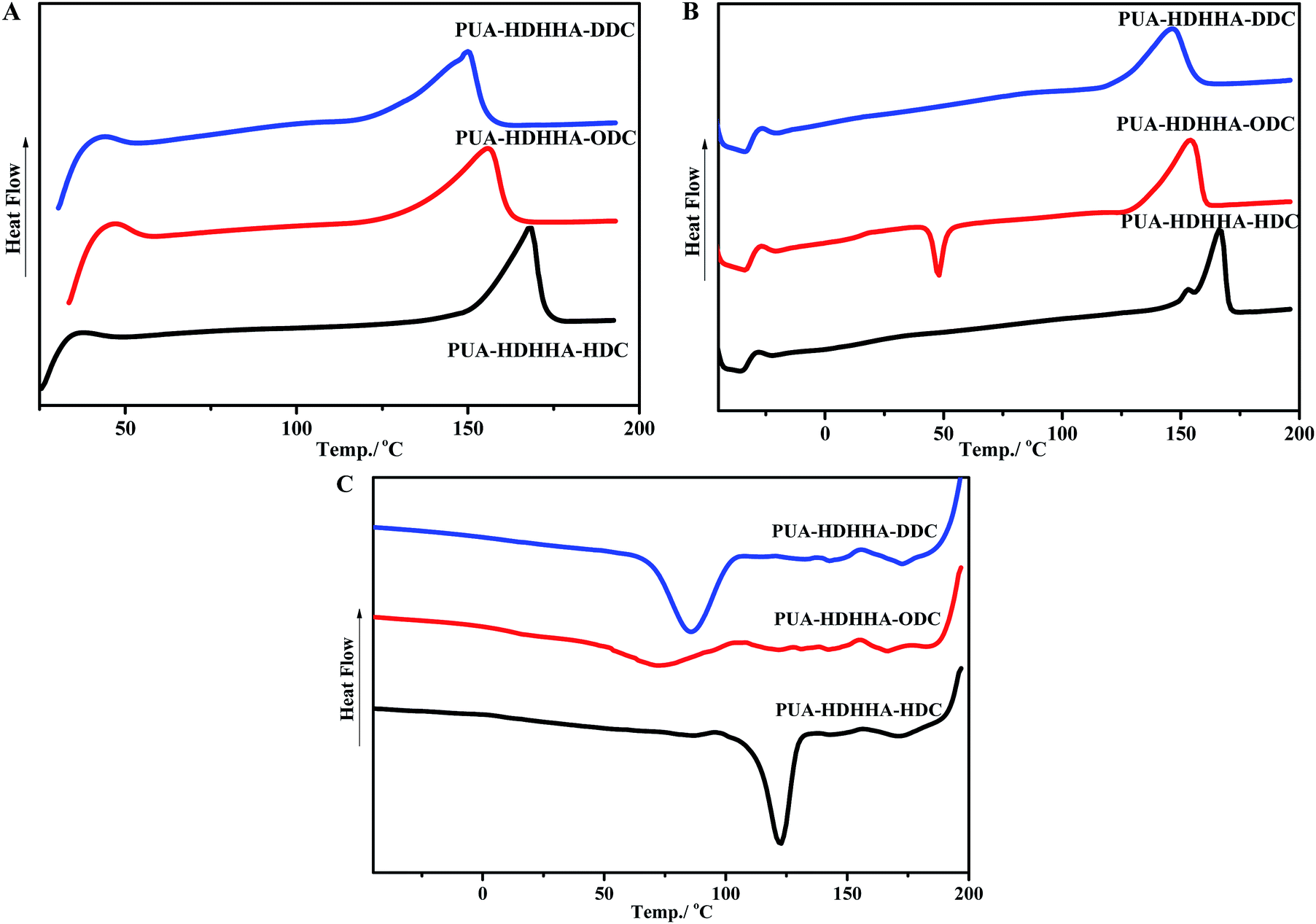 | ||
| Fig. 4 DSC testing of PUAs containing different size of dicarbamate with 10 °C min−1: (A) first heating run (B) second heating run (C) cooling run. | ||
As well known, dimethylsiloxane polymer has become the first choice for many flexible substrates, which shows availability, stable chemical properties, transparency and good thermal stability. In order to improve the flexibility of PUA, soft segment BPTMSHHA was introduced into the backbone of PUA-HDHHA-HDC. Because of the incorporation of soft segment, the movement and crystallization behavior of the hard segments in polymer chain are restricted, leading to an unfavorable crystallization dynamics and decrease in the degree of crystallinity. Consequently, cold crystallization happens in the second heating run for PUA-BPTMSHHA20-HDC and PUA-BPTMSHHA40-HDC and the Tm decreases to 107.7 °C with increasing BPTMSHHA content to 40%, as shown in Table 2 and Fig. S5.† The Tm disappears when the content of BPTMSHHA is higher than 60%. Due to the sensitivity of restriction, the Tg values for PUAs is rather difficult to be detected from −60 to 200 °C based on DSC test in the second heating run.
The synthesized PUAs were then evaluated using thermogravimetric analysis (TGA). Generally, polyamide shows highly thermal stability, while polyurethane is thermally unstable at high temperature. However, in the thermogravimetric analysis (TGA) for PUAs copolymerized from dicarbamate and diamide diol exhibited highly thermal stability as summarized in Table 2. It can be seen that 5% weight loss temperature (T5%d) of PUAs is higher than 300 °C, higher than classical polyurethane from diisocyanate.42,43 As shown in Fig. 5 and Table 2, the T5%d of the PUAs derived from HDHHA and different dicarbamates ranges from 300.5 to 310.9 °C, which decreases with increasing the size of dicarbamate segment. It demonstrates that the greater flexibility of PUAs depends upon more methylene groups in PUAs structure, which can then lead to lower crystallinity and thermal stability. Three peaks appeared in DTG curve (Fig. 5B) indicate three steps of thermal decomposition. It has been reported that when urea segments are present, the thermal stability of urea moiety containing polymer is higher due to its higher hydrogen-bonding capacity compared with urethane.44,45 Hence, the first weight loss temperature of PUAs is attributed to the degradation of urethane segment, while the second weight loss temperature is associated with urea segment decomposition. Similar behavior has been found in segmented poly(urethane–urea)s.42,43 Therefore, the third weight loss temperature is assigned to the degradation of amide segment. Due to the presence of crystalline structure, the PUA with amide segment exhibits the highest thermal stability.
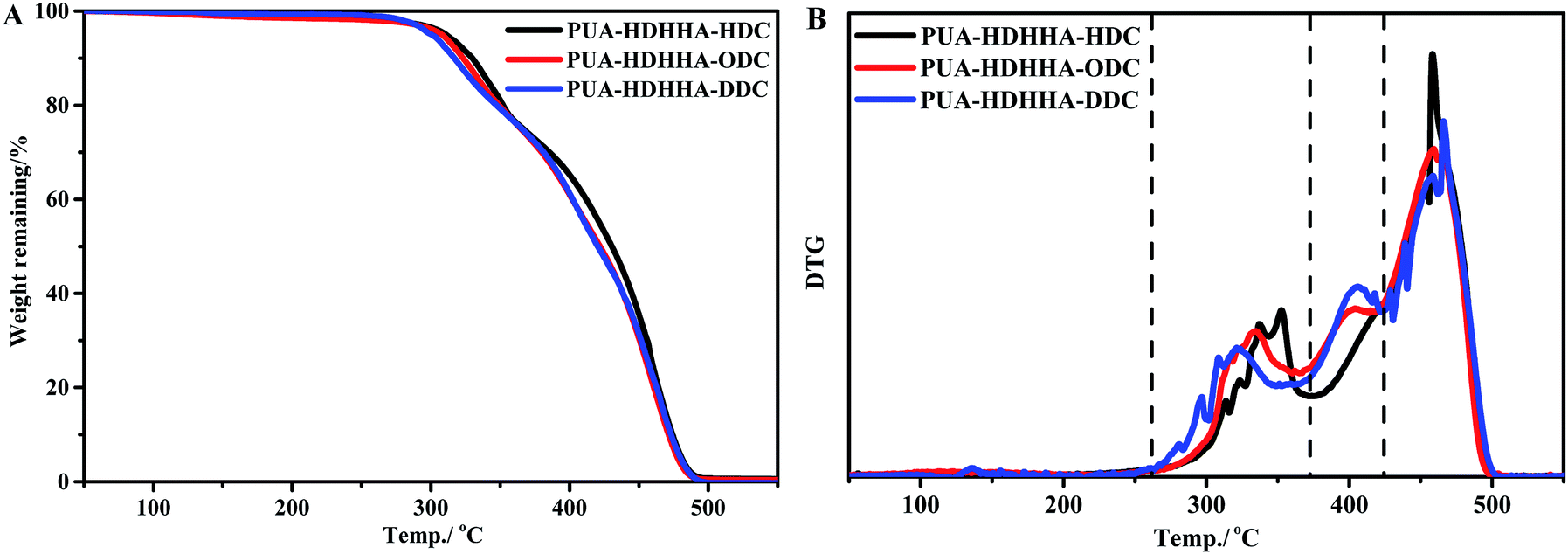 | ||
| Fig. 5 TGA (A) and DTG (B) curves of PUAs from HDHHA and various dicarbamate. | ||
The introduction of BPTMSHHA moieties into the PUA backbone can also lead to the decrease of T5%d and Tmax values as shown in Fig. S6† and Table 2. These can also be attributed to the decrease of crystallinity owing to the introduction of soft segment. From the data in Table 2, it can be deduced that for all prepared PUAs, the onset of degradation is higher than 140 °C that is above their melting points, endowing a wide melt processing temperature window.
Finally, the dynamic properties of PUAs were examined using dynamic thermomechanical analysis (DMA). The influences of the dicarbamates with different number of methylene and BPTMSHHA content on the viscoelastic properties of resulted PUA copolymers were investigated by dynamic thermomechanical analysis. The viscoelastic properties are presented as the storage modulus, E′ and loss factor, tanδ versus temperature at a frequency of 1 Hz. The E′ temperature dependence curves are given in Fig. 6A and S7A.† Due to the rigid natures of amide segment, PUA copolymers show high E′ value ranging from 2250 to 2900 MPa at low temperature. The E′ value of the copolymers increases with increasing the size of dicarbamate segment, following the trend PUA-HDHHA-DDC < PUA-HDHHA-ODC < PUA-HDHHA-HDC. Under ambient condition, the storage modulus of PUAs with different dicarbamate moieties still remain at a high level, up to 749.3 MPa, which is higher than conventional polyurethane.46 Moreover, the high density of hydrogen bond crosslinked network and crystallization endow the PUAs with considerable strength even at 50 and 100 °C (Table 3). The high mechanical strength over a wide range of temperature shows potential applications in the industrial fields. Because of the introduction of BPTMSHHA segment, the reduction of storage modulus of PUAs was observed, indicating that BPTMSHHA segment can increase the flexibility of copolymers gradually. The E′ value is lower than other copolymers without BPTMSHHA, such as PUA-HDHHA-DDC, PUA-HDHHA-ODC and PUA-HDHHA-HDC, because of the decrease or absence of crystalline region as evidenced by DSC curves in Fig. 3.
| Polymer | Tgs/°C | Tgh/°C | ΔTg | Modulus/MPa | ||
|---|---|---|---|---|---|---|
| 25/°C | 50/°C | 100/°C | ||||
| PUA-HDHHA-HDC | 30.9 | 84.0 | 53.1 | 530.0 | 107.2 | 5.700 |
| PUA-BPTMSHHA20-HDC | 27.7 | 83.1 | 55.4 | 419.4 | 86.60 | 0.900 |
| PUA-BPTMSHHA40-HDC | 27.9 | 77.4 | 49.5 | 423.2 | 99.80 | 25.80 |
| PUA-BPTMSHHA60-HDC | 6.3 | 54.4 | 48.1 | 118.0 | 39.50 | 10.20 |
| PUA-BPTMSHHA80-HDC | 7.4 | — | — | 113.8 | 50.70 | 20.50 |
| PUA-BPTMSHHA100-HDC | 10.0 | — | — | 82.6 | 18.10 | 4.200 |
| PUA-HDHHA-ODC | 37.3 | 85.8 | 48.5 | 726.0 | 102.9 | 4.200 |
| PUA-HDHHA-DDC | 38.8 | 86.9 | 48.1 | 749.3 | 126.1 | 34.90 |
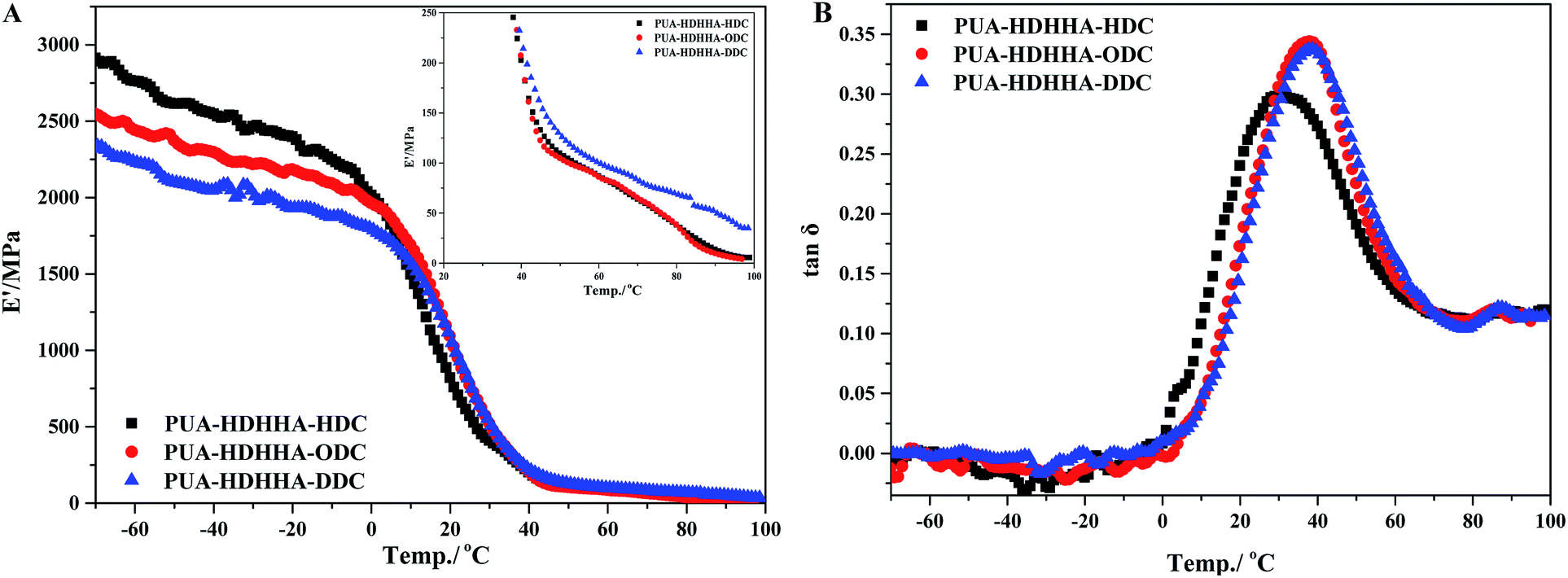 | ||
| Fig. 6 DMA single frequency temperature sweep experiment for PUAs containing HDHHA and various dicarbamate: (A) storage modulus (B) tanδ. | ||
Fig. 6B and S7B† show the loss factor (tanδ) curves of PUAs with different composition. Most samples present two tanδ peaks, and the peak at low temperature corresponds to the Tg of soft segments (Tgs), while the peak at high temperature is related to the Tg of hard segments (Tgh). It has been reported that the degree of microphase separation is reflected by the difference of Tgs and Tgh, namely ΔTg (Tgs − Tgh). The corresponding data of Tgs, Tgh, and ΔTg are summarized in Table 3. It is evident that the copolymerization of different dicarbamate and HDHHA diamide diol resulted in high Tg for both Tgs and Tgh (Fig. 6B). This is because the high hydrogen bonds cross-linking density of the PUA copolymers due to abundant amide, urethane and urea group in polymer chain. As listed in Table 3, the Tgs of PUA-HDHHA-DDC, PUA-HDHHA-ODC and PUA-HDHHA-HDC range from 30.9 to 38.8 °C, while the Tgh are above 84.0 °C. It was also found that the ΔTg of PUA samples containing different dicarbamate and diamide diol decrease in the order of PUA-HDHHA-DDC (48.1 °C) < PUA-HDHHA-ODC (48.5 °C) < PUA-HDHHA-HDC (53.1 °C). It is apparent that long dicarbamate can increase the flexibility of PUA and lead to low degree crystallinity and microphase separation.
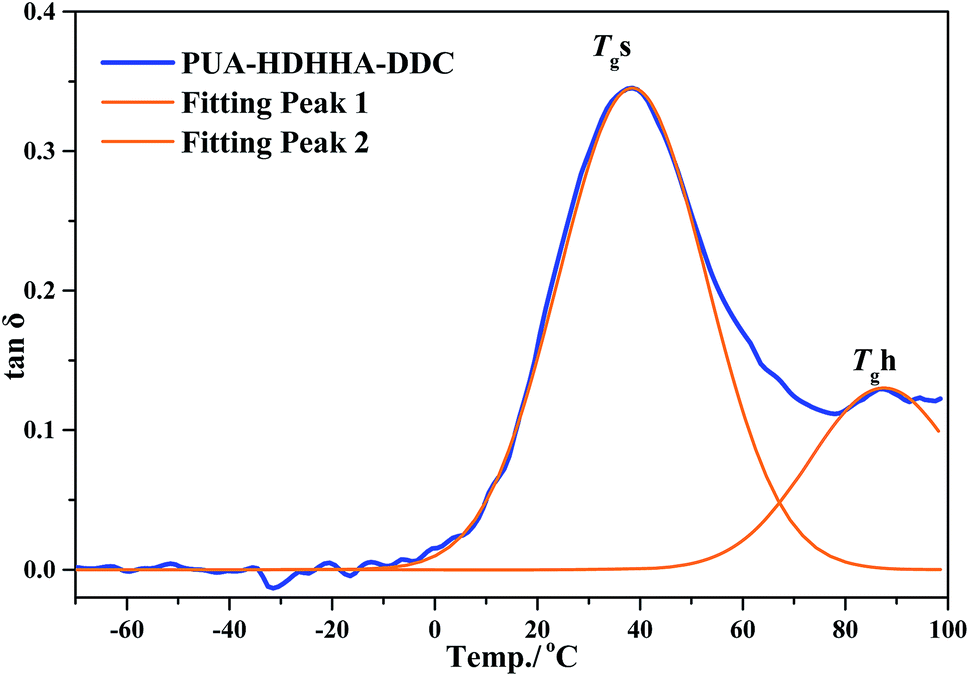 | ||
| Fig. 7 Tgs and Tgh of PUA-HDHHA-DDC from DMA experiment by fitting peak. | ||
Compared with PUA-HDHHA-DDC, PUA-HDHHA-ODC and PUA-HDHHA-HDC, PUAs containing BPTMSHHA unit show lower Tgs due to its lower crystallinity. The ΔTg decreases with increasing BPTMSHHA content, and only one Tg was observed when BPTMSHHA content is higher than 80%. This implies that the decrease of crystallinity results in a low degree microphase separation.
Mechanical properties of the synthesized PUAs
The mechanical properties of PUA copolymers were examined by tensile and impact test. It is well known that the backbone structure governs the mechanical properties of PUA, moreover, both crystallinity and phase separation in the polymer matrix can affect the mechanical strength. Fig. 8 shows the stress–strain curves, and the stress at break and strain at break are represented in Table 4. All polymers behave as tough materials. As shown in Fig. 8, it is clear that PUA-HDHHA-HDC exhibits the maximum stress at break, up to 54 MPa, while strain at break is 33.4%. The polymer shows a slightly higher strength and lower strain at break compared with long chain nylon 1010 and nylon 1212 (Table 4). Tensile strength decreases while strain at break increases with increasing the size of dicarbamate. There are two main reasons for this phenomenon. Firstly, all amide, urethane and urea can yield hydrogen-bond, leading to the formation of cross-linking network. With increasing the size of dicarbamate, the density of hydrogen bond cross-linking decreases, resulting in lower mechanical strength. Secondly, the long methylene segment in dicarbamate can endow highly flexible PUA and decrease the crystallinity of PUA, finally decreasing their tensile strength. Consequently, the tensile strength decreases and elongation at break increases gradually with increasing BPTMSHHA content in the polymer chain. The tensile strength was measured being 6.1 MPa when BPTMSHHA content was 100%.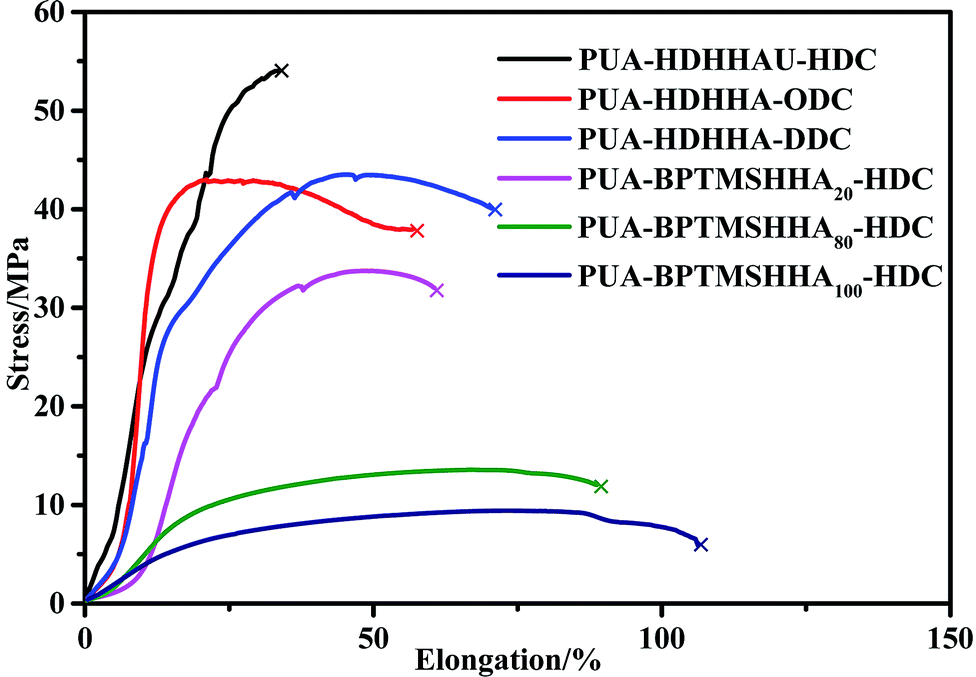 | ||
| Fig. 8 Stress–strain curves of PUAs based on CO2-based dicarbamate evaluated by tensile testing. | ||
| Polymer | Stress/MPa | Elongation/% | Impact strength/kJ m−2 |
|---|---|---|---|
| PUA-HDHHA-HDC | 54.0 | 33.4 | 6.93 |
| PUA-HDHHA-ODC | 43.0 | 57.0 | 8.14 |
| PUA-HDHHA-DDC | 43.5 | 70.5 | 8.98 |
| PUA-BPTMSHHA20-HDC | 31.8 | 60.6 | — |
| PUA-BPTMSHHA80-HDC | 11.9 | 89.0 | — |
| PUA-BPTMSHHA100-HDC | 6.10 | 106.5 | — |
| Nylon 1010 | 49.98 (ref. 47) | 48.59 (ref. 47) | 4.5 (ref. 48) |
| Nylon 1212 | 51.58 (ref. 49) | 289.3 (ref. 49) | 5.48 (ref. 49) |
For clearly comparison, Table 4 lists the impact strength of PUAs and long chain nylon. The PUAs containing BPTMSHHA segment are very flexible and show relatively low tensile strength. It is obvious that the impact strength of PUA containing different dicarbamates and HDHHA ranges from 6.93 to 8.98 kJ m−2, which is higher than long chain nylon. Therefore, the copolymerization of CO2-based dicarbamate and diamide diol exhibited significant enhancement in not only tensile strength, but also impact strength. The results are believed to be attributed to their phase separation structure and the hydrogen bond crosslinking network. Apparently, the copolymerization of dicarbamate and diamide diol segments can fully combine the advantages of polyamide and polyurethane and endow the copolymers with excellent mechanical properties, making it more potential candidate as industrial engineering plastics.
Surface morphology observation
SEM investigations were performed in order to examine any changes of the microstructure of PUA samples containing different dicarbamates and HDHHA after immersion in the buffer solution. Considering potential industrial applications, PUA-HDHHA-DDC, PUA-HDHHA-ODC and PUA-HDHHA-HDC were selected for the hydrolytic stability with SEM investigations and the images before and after immersion are presented in Fig. 9. It can be seen that it is quite different for PUA surface after hydrolytic treatment with raw PUA surface (Fig. 9, top), moreover, the weight loss of samples increases slightly (Fig. S8†). In other words, PUA copolymers from CO2-based dicarbamate and HDHHA show highly hydrolytic stability to meet the requirements of industrial applications. | ||
| Fig. 9 SEM micrographs of the surfaces of raw PUA (top) and the hydrolytically treated sample (bottom) by immersion in buffer solution for 3 months. | ||
Conclusions
In summary, we successfully developed a novel high-performance polymer from CO2-based dicarbamate and diamide diol by polycondensation, which is a brand new route for carbon dioxide utilization. The poly(urethane-co-amide)s can be readily synthesized in autoclave in the presence of tetrabutyl titanate catalyst under vacuum condition, with high molecular weight (Mn up to 41.3 kDa). Because of the co-existence of amide, urethane and urea moieties, the crystallinity, morphologies and hydrogen bond cross-linked network of the as-synthesized polymers endow these new polymers with both high tensile strength (54 MPa) and impact strength (6.93 kJ m−2) when compared with conventional long chain nylon. Moreover, the obtained PUA copolymers also possess high thermal stability and hydrolytic stability. In this sense, this work not only establishes an attractive polymerization to indirectly convert CO2 into useful polymers, but also provides a new poly(urethane-co-amide)s with high performance that can match conventional long chain polyamides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank National Natural Science Foundation of China (Grant No. 21376276), the Special-funded Program on National Key Scientific Instruments and Equipment Development of China (Grant No. 2012YQ230043) for financial support of this work.Notes and references
- M. I. Kohan, Nylon plastics, Wiley, 1973 Search PubMed
.
- L. Wang, X. Dong, P. Zhu, X. Zhang, X. Liu and D. Wang, Eur. Polym. J., 2017, 90, 171–182 CrossRef CAS
- N. A. Jones, E. D. T. Atkins and M. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 1209–1221 CrossRef CAS
- X. Yang, S. Tan, G. Li and E. J. M. Zhou, Macromolecules, 2001, 34, 5936–5942 CrossRef CAS
- N. A. Jones, E. D. T. Atkins, M. J. Hill, S. J. Cooper and L. J. M. Franco, Macromolecules, 1997, 30, 3569–3578 CrossRef CAS
- L. Wang, X. Dong, Y. Gao, M. Huang, C. C. Han, S. Zhu and D. Wang, Polymer, 2015, 59, 16–25 CrossRef CAS
- L. L. Wang, X. Dong, X. R. Wang, G. Y. Zhu, H. Q. Li and D. J. Wang, Chin. J. Polym. Sci., 2016, 34, 991–1000 CrossRef CAS
- D. A. Ruehle, C. Perbix, M. Castañeda, J. R. Dorgan, V. Mittal, P. Halley and D. J. P. Martin, Polymer, 2013, 54, 6961–6970 CrossRef CAS
- H. Zeng, C. Gao, Y. Wang, P. C. P. Watts, H. Kong, X. Cui and D. Yan, Polymer, 2006, 47, 113–122 CrossRef CAS
- T. S. Ellis, Macromolecules, 1990, 23, 1494–1503 CrossRef CAS
- Y. Li and G. Yang, Macromol. Rapid Commun., 2010, 25, 1714–1718 CrossRef
- A. M. Aerdts, K. L. L. Eersels and G. Groeninckx, Macromolecules, 1996, 29, 1041–1045 CrossRef CAS
- K. L. L. Eersels, A. M. Aerdts and G. Groeninckx, Macromolecules, 1996, 29, 1046–1050 CrossRef CAS
- X. Zhang, T. Xie and G. Yang, Polymer, 2006, 47, 2116–2126 CrossRef CAS
- S. Arsenault, Annu. Tech. Conf. Soc. Plast. Eng., 2008, 775–788 CAS
- M. C. Choi, J. Y. Jung, H. S. Yeom and Y. W. Chang, Polym. Eng. Sci., 2013, 53, 982–991 CrossRef CAS
- J. H. Kim, S. Y. Ha and Y. M. Lee, J. Membr. Sci., 2001, 190, 179–193 CrossRef CAS
- A. Gugliuzza, Poly(ether-block-amide) Copolymers Synthesis, Properties and Applications, John Wiley & Sons, Inc., 2011 Search PubMed
- S. D'Angelo, P. Galletti, G. Maglio, M. Malinconico, P. Morelli, R. Palumbo and M. C. Vignola, Polymer, 2001, 42, 3383–3392 CrossRef
- A. A. Deschamps, D. W. Grijpma and J. Feijen, J. Biomater. Sci., Polym. Ed., 2002, 13, 1337–1352 CrossRef CAS PubMed
- F. Quaglia, M. C. Vignola, G. D. Rosa, M. I. L. Rotonda, G. Maglio and R. Palumbo, J. Controlled Release, 2002, 83, 263–271 CrossRef CAS PubMed
- P. A. M. Lips, R. Broos, M. J. M. V. Heeringen, P. J. Dijkstra and J. Feijen, Polymer, 2005, 46, 7823–7833 CrossRef CAS
- G. Deshayes, C. Delcourt, I. Verbruggen, L. Trouillet-Fonti, F. Touraud, E. Fleury, P. Degée, M. Destarac, R. Willem and P. Dubois, Macromol. Chem. Phys., 2010, 210, 1033–1043 CrossRef
- H. Harashina, T. Nakane and T. Itoh, J. Polym. Sci., Part A: Polym. Chem., 2010, 45, 2184–2193 CrossRef
- X. Pang, J. Wu, C. Reinhart-King and C.-C. Chu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3758–3766 CrossRef CAS
- K. G. Marra, T. M. Chapman and J. M. Orban, Macromolecules, 1996, 29, 7553–7558 CrossRef CAS
- M. V. D. Schuur, B. Noordover and R. Gaymans, Polymer, 2006, 47, 1091–1100 CrossRef
- J. E. Báez, D. Ramírez, J. L. Valentín and Á. MarcosFernández, Macromolecules, 2012, 45, 6966–6980 CrossRef
- S. Mallakpour, M. Khani and F. Rafiemanzelat, J. Appl. Polym. Sci., 2010, 108, 2975–2982 CrossRef
- A. J. Wills, A. Yamuna Krishnanghosh and S. Balasubramanian, J. Org. Chem., 2002, 67, 6646–6652 CrossRef CAS PubMed
- I. Vauthey, F. Valot, C. Gozzi, F. Fache and M. Lemaire, Tetrahedron Lett., 2000, 41, 6347–6350 CrossRef CAS
- M. Honda, S. Sonehara, H. Yasuda, Y. Nakagawa and K. Tomishige, Green Chem., 2011, 13, 3406–3413 RSC
- A. Ion, C. V. Doorslaer, V. Parvulescu, P. Jacobs and D. D. Vos, Green Chem., 2008, 10, 111–116 RSC
- J. Shang, S. Liu, X. Ma, L. Lu and Y. Deng, Green Chem., 2012, 14, 2899–2906 RSC
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS
- K. A. Alferov, Z. Fu, S. Ye, D. Han, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2019, 7, 10708–10715 CrossRef CAS
- A. Bax and M. F. Summers, J. Am. Chem. Soc., 1986, 108, 2093–2094 CrossRef
- M. Tokles, P. A. Keifer and P. L. Rinaldi, Macromolecules, 1995, 28, 3944–3952 CrossRef CAS
- S. J. Oh, D. R. Kinney, W. Wang and P. L. Rinaldi, Macromolecules, 2002, 35, 2602–2607 CrossRef CAS
- D. Peng, G. Du, P. Zhang, B. Yao, X. Li and S. Zhang, Macromol. Rapid Commun., 2016, 37, 987–992 CrossRef CAS PubMed
- R. Abdul-Karim, A. Hameed and M. I. Malik, Eur. Polym. J., 2018, 105, 95–106 CrossRef CAS
- A. Marcos-Fernández, G. A. Abraham, J. L. Valentín and J. S. Román, Polymer, 2006, 47, 785–798 CrossRef
- J. M. Cervantes-Uc, J. I. M. Espinosa, J. V. Cauich-Rodríguez, A. Ávila-Ortega, H. Vázquez-Torres, A. Marcos-Fernández and J. San Román, Polym. Degrad. Stab., 2009, 94, 1666–1677 CrossRef CAS
- F. M. B. Coutinho and M. C. Delpech, Polym. Degrad. Stab., 2000, 70, 49–57 CrossRef CAS
- F. M. B. Coutinho, M. C. Delpech, T. L. Alves and A. A. Ferreira, Polym. Degrad. Stab., 2003, 81, 19–27 CrossRef CAS
- A. K. Barick and D. K. Tripathy, Mater. Sci. Eng. A, 2010, 527, 812–823 CrossRef
- X. Fang, X. Li, L. Yu and Z. Zhang, J. Appl. Polym. Sci., 2010, 115, 3339–3347 CrossRef CAS
- H. Yu, Y. Zhang and W. Ren, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 877–887 CrossRef CAS
- Y. Wang, W. Wang, F. Peng, M. Liu, Q. Zhao and P.-F. Fu, Polym. Int., 2009, 58, 190–197 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04646a |
| ‡ Jiaxiang Qin and Junqiao Jiang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |