
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonomer sequence design at two solvent interface enables the synthesis of highly photoactive carbon nitride†
Susmita Dolai‡
,
Neeta Karjule‡,
Adi Azoulay and
Jesús Barrio *
*
Department of Chemistry, Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel. E-mail: barriohe@post.bgu.ac.il
First published on 20th August 2019
Abstract
Structural modifications in carbon nitrides and related carbon-based materials have been achieved in recent years by organizing their monomers into versatile supramolecular structures that serve as reactants for the high temperature solid-state reaction. To date, the organization is usually carried out in one solvent where the building blocks must be dispersed. Here, we show the utilization of a molecule with both hydrogen bond donor and acceptor sites for constructing hydrogen bonded frameworks in interfacial systems. The chemical and electronic properties of the carbon nitride materials after calcination are strongly altered showing enhanced photocatalytic performance in different model reactions. This work shows a new large-scale pathway for the synthesis of highly photoactive carbon nitride with tailored properties and morphology by employing novel supramolecular assemblies prepared in the interface between two solvents, and furthermore opens new opportunities in the rational design of different carbon–nitrogen based materials utilizing supramolecular structures.
Introduction
The tunable chemical and electronic properties as well as high stability and low cost of polymeric carbon nitride (CN)1,2 allow its exploitation in many fields including photo- and electrocatalysis,3–5 CO2 reduction,6,7 water treatment,8,9 sensing,10,11 LEDs,12 and more.13,14 Despite this wide research, tailoring its chemical and photophysical properties has represented a continual challenge due to its solid-state synthesis, which yields unordered materials with low photoactivity. To overcome these drawbacks, doping with heteroatoms,15,16 the use of hard and soft templates,17,18 and the formation of heterojunctions with different materials19–22 have been utilized in order to improve CN surface area,23 as well as to promote charge transfer and separation properties and overall catalytic activity.24 In the past six years, the use of ordered supramolecular structures comprised of C/N monomers has emerged as an alternative approach for synthesizing CN, nitrogen-doped carbon and carbon materials for various applications.25 A tailored monomer sequence permits the design of electronic structures with enhanced light harvesting properties26,27 or novel and ordered morphologies without the need of templating methods like hollow tubes,28,29 rods,30,31 spheres,32,33 or dots34,35 with enhanced number of active sites and overall enhanced photocatalytic activity.36 Our group and others have identified the importance of solvents in the supramolecular assembly as well as in the final CN formation and properties such as morphology, size and activity.37,38 Recently we demonstrated that a sequential treatment of supramolecular structures with different solvents can produce either a complete rearrangement or just surface modifications depending on the polarity of the solvent.39 Furthermore, we illustrated that the interfacial supramolecular organization on two non-miscible solvents40 leads to a significant modification of all CN parameters, e.g., morphology, surface area, and photo-physical properties.41 In this work we show the design of unique supramolecular assemblies in a water/chloroform system using melamine,42 cyanuric acid,43 and the amino acid creatine.44 The starting monomers will be preorganized in a water–chloroform interfacial system, where creatine selectively binds melamine units in the water phase acting as hydrogen bond acceptor, and also the cyanuric acid moieties in the chloroform phase, acting as hydrogen bond donor. 1H-NMR, XRD and FT-IR have proven the formation of hydrogen-bonded frameworks and the selective binding of creatine to both melamine and cyanuric acid units in the interface. At high temperatures creatine condensates to guanidine-like structural motifs that interlink the heptazine units and alter the photo-physical and morphological properties of the materials leading to an enhanced conductivity and photocatalytic activity in the hydrogen evolution reaction and the degradation of rhodamine B dye.Experimental section
Synthesis of CN materials
630 mg (5 mmol) of melamine were dispersed in 25 mL of deionized water and different amounts of creatine were added (32, 64 and 168 mg, 0.25, 0.5 and 1.25 mmol respectively) forming melamine–creatine complexes (M–C). Afterwards, a dispersion of cyanuric acid in chloroform (645 mg, 5 mmol in 25 mL) was added to the M–C suspensions, and the formation of modified cyanuric acid–melamine complexes (CM)45 happens at the interface between the two solvents. The solids were filtered, washed with distilled water, dried at 60 °C in vacuum overnight and labelled as CMCx (were x corresponds to the amount of creatine in molar ratio versus the cyanuric acid and melamine, namely 0.05, 0.1 and 0.25). The resulting powders were calcined at 550 °C for 4 h (heating rate of 2.3 °C min−1) under an inert nitrogen atmosphere in order to obtain modified carbon nitride materials (CMCx–CN). A reference material was synthesized by following the same procedure in the absence of creatine (CM–CN).Characterization
Scanning electron microscopy (SEM) images, energy dispersive X-ray (EDX) spectroscopy and elemental mapping were obtained using a JEOL JSM-7400F system operated at 3 kV equipped with Thermo Scientific NORAN System SIX. Transmission electron microscopy (TEM) images were recorded using a FEI Tecnai T-12 G2 TWIN transmission electron microscope operated at 120 kV. A JEOL JEM-2100F analytical TEM operated at 200 kV as used for high-resolution transmission electron microscopy (HRTEM) imaging. X-ray diffraction patterns (XRD) of the synthesized powders were obtained using an Empyrean powder diffractometer (Panalytical). The Fourier-transform infrared spectroscopy (FTIR) analysis was performed on a Thermo Scientific Nicolet iN 10Mx infrared microscope. 1H NMR spectra was recorded on a 400 MHz NMR spectrometer (Bruker DPX 400). Solid state 13C NMR spectra were recorded on a Bruker Biospin GmbH. Ultraviolet-visible spectroscopy (UV-vis) spectra were collected using a Cary 100 spectrophotometer. Fluorescence measurements were collected using an Edinburgh Instruments FLS920P spectrofluorimeter. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Fisher Scientific ESCALAB 250 using monochromated Al Kα X-rays (1486.6 eV). Elemental analysis (EA) results were collected using a Thermo Scientific Flash Smart elemental analyzer OEA 2000. The apparent surface area of the final CN products was calculated by applying the Brunauer–Emmett–Teller (BET) model to the isotherm data points of the adsorption branch recorded on a Quantachrome NOVAtouch LX3 surface analyzer (N2 at 77 K).Hydrogen generation test under visible light
Hydrogen evolution measurements were performed in a Schlenk flask thermally regulated with a cooling system and a white LED array as the irradiation source, with 3% Pt as a co-catalyst and triethanolamine (TEOA) as a hole scavenger in a previously reported setup.9 Hydrogen evolution experiments were conducted under Ar atmosphere and a constant temperature of 25 °C. In a H2 evolution measurement, 15 mg of the sample were placed inside a 50 mL Schlenk flask with a solvent mixture (19 mL) composed of water and TEOA in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio and 19.6 μL of a H2PtCl6 solution, (8% in water). After stirring for 30 min in the dark under constant Ar flow, the reaction was started by switching on a 50 W (the illumination intensity on the photocatalyst surface is 10000 μmol s−1 m−2) white LED array (Bridge lux BXRA-50C5300; λ > 410 nm). In order to confirm that the evolved gas is hydrogen, the headspace of the reactor was analyzed by Gas Chromatography (Agilent 7820 GC System). Quantum-yield measurements were performed in a sealed reactor connected to an argon line and an Agilent 7820 GC system.46 Argon gas was continuously flowing through the reactor in the dark to remove the existing hydrogen, nitrogen, oxygen and other gases and the purging process was monitored by automatically sampling every 11 min. After the purge, a Thorlabs LED (405 nm) was turned on, the hydrogen produced was recorded and the integrated area was used for the calculation of the average quantum yield (AQY%) which was performed as follows: AQY = Ne/Np × 100% = 2M/Np × 100%, where Ne is the amount of reaction electrons, Np is the incident photons, and M is the amount of H2 molecules.
:1 (v/v) ratio and 19.6 μL of a H2PtCl6 solution, (8% in water). After stirring for 30 min in the dark under constant Ar flow, the reaction was started by switching on a 50 W (the illumination intensity on the photocatalyst surface is 10000 μmol s−1 m−2) white LED array (Bridge lux BXRA-50C5300; λ > 410 nm). In order to confirm that the evolved gas is hydrogen, the headspace of the reactor was analyzed by Gas Chromatography (Agilent 7820 GC System). Quantum-yield measurements were performed in a sealed reactor connected to an argon line and an Agilent 7820 GC system.46 Argon gas was continuously flowing through the reactor in the dark to remove the existing hydrogen, nitrogen, oxygen and other gases and the purging process was monitored by automatically sampling every 11 min. After the purge, a Thorlabs LED (405 nm) was turned on, the hydrogen produced was recorded and the integrated area was used for the calculation of the average quantum yield (AQY%) which was performed as follows: AQY = Ne/Np × 100% = 2M/Np × 100%, where Ne is the amount of reaction electrons, Np is the incident photons, and M is the amount of H2 molecules.
Dye degradation under visible light
The photocatalytic activity was further evaluated by the degradation of rhodamine B (RhB). In a typical photocatalytic degradation experiment RhB (20 mL, 20 mg L−1) and carbon nitride (20 mg) were mixed in a glass vial in the dark under continuous stirring until the adsorption–desorption equilibrium between the dye and the catalyst was obtained. After turning on the light, aliquots were withdrawn from the suspension at a given time interval. The concentration of the remaining dye in solution was monitored by optical absorption measurements (measured at λmax = 554).Electrochemical measurements
Electrochemical measurements were recorded using a three-electrode system on an Autolab potentiostat (Metrohm, PGSTAT 101). A Pt-foil electrode and an Ag/AgCl (3 M KCl) electrode were used as the counter and reference electrodes, respectively. Photocurrent measurements and impedance spectroscopy were conducted with KOH (0.1 M) as electrolyte, saturated Ag/AgCl as reference electrode and Pt as counter electrode. Photocurrents were measured at 1.23 V vs. RHE under the illumination of a solar simulator (Newport 300 W Xe arc lamp, equipped with air mass AM 1.5G and water filters) at the power density of 100 mW cm−2. For photoelectrochemical measurements, the working electrode was prepared by dispersing 10 mg of CN materials into 4 mL of deionized water followed by sonication to get slurry. Then the slurry was drop casted onto cleaned FTO. After air-drying, the working electrode was further dried at 300 °C for 1 h to improve adhesion. Electrochemical impedance spectroscopy (EIS) was measured in dark and light (under illumination with a solar simulator with a power density of 100 mW cm−2). Nyquist plots of the prepared electrodes were measured in the frequency range from 100 kHz to 10 MHz at an applied voltage of 1.23 V vs. RHE.Results and discussion
Here, we took advantage of the tendency of melamine and cyanuric acid to preorganize in the interface between two solvents for tailoring new supramolecular assemblies. We envisioned that the bonding possibilities of creatine will allow its selective interaction with melamine and cyanuric acid populations in the interface between water and chloroform. Melamine (M) and creatine (C) were dissolved in water to form the MC complex. The interaction between both monomers was proven by 1H-NMR, FTIR and XRD (discussion in Fig. S1 and S2.† We note that we focused on 1:0.1 M:C ratio, 5 mmol melamine and 0.5 mmol creatine, due to the higher photocatalytic performance of the resulting material, as shown later in this paper). The formation of cyanuric acid–melamine derivatives was achieved by adding 25 mL of a cyanuric acid solution in chloroform to the MC solution in water. After certain time, the CMCx supramolecular assembly is formed in the interface between both solvents (Scheme 1), and the solids are collected and dried in vacuum overnight.
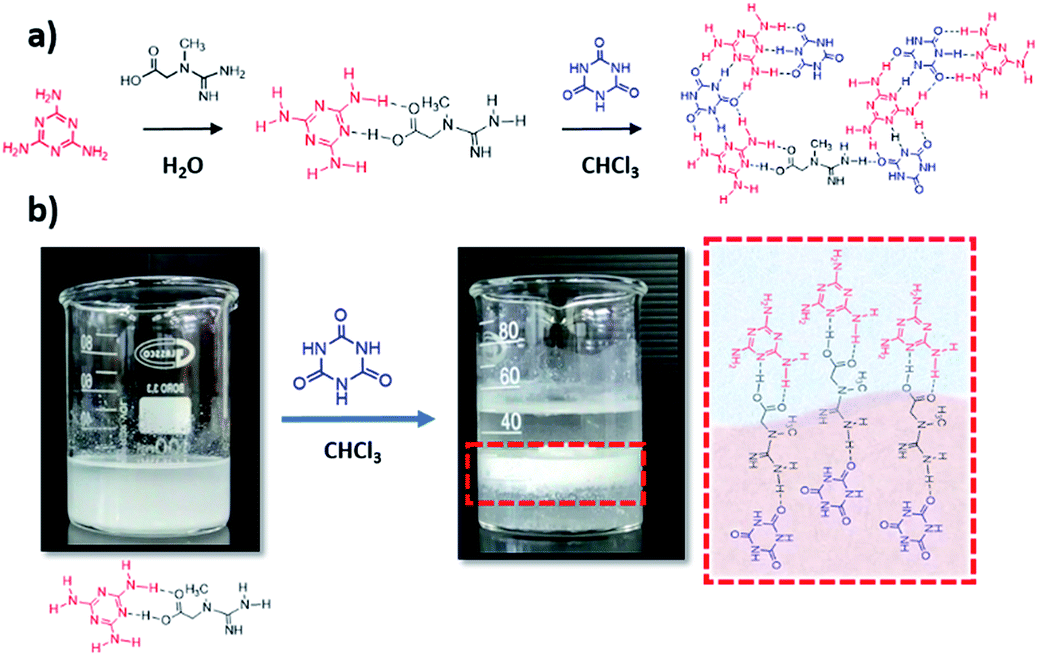 | ||
| Scheme 1 Synthetic pathway of the CMC assemblies, (a) and illustration of the hydrogen bond generation in the interface (b). | ||
The structural and chemical modifications induced by creatine in the cyanuric acid–melamine complex were studied by several means; FTIR (Fig. S3a†) proves the modification of the CM structure at high creatine concentrations, shown by the presence of typical vibrational modes of creatine, as the O–H, C–H stretching vibrations coming from the carboxylic acid and methyl group, at 3201, 3050 cm−1 respectively, C–O stretching mode at 1088 cm−1 or C–H bending vibration at 760 cm−1. The rearrangement in the crystal structure was confirmed by the XRD patterns (Fig. S3b†), the strong lamellar stacking peak in the CM unit at 27.9° disappears and three peaks at 17.6, 26.2 and 29.7 emerge, corresponding to the (110), (121), and (002) crystal planes. Elemental analysis (Table S1†) shows increasing C/N ratios with higher creatine amounts (0.6 in CMC0.25 vs. 0.51 in CM), confirming its insertion within the structure. XPS measurements (Fig. S4†) further prove the chemical modifications in the CM lattice (for this analysis just CM and CMC0.1 were analysed due to the high photocatalytic performance of the corresponding carbon nitride, as shown later in this paper); C1s spectra of the CM complex shows four different chemical states at 284.8, 286.0, 287.8 and 289.7 eV, which correspond to C–C and C–O adventitious bonds, C–(N)3 from melamine units and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–(N)2 from cyanuric acid, respectively. The formation of CMCx complexes increases dramatically the atomic weight percentage of the C–(N)3 due to a synergistic contribution of melamine and creatine groups and as well the C–O, coming from the carboxylic group in creatine. N1s spectrum of CM displays three different binding energies at 398.8, 400.0 and 401.1 eV, that correspond to CN–C of triazine rings, hydrogen bonded NH–O groups, and remaining amino groups, respectively, the N1s spectra doesn't show pronounced differences, due to the similarity in the N species in the CM lattice and creatine. The morphology of the CMCx assemblies, studied by SEM, shows that the insertion of creatine directs the growth of the supramolecular units towards one direction, resulting in micrometre-long aggregates, while the CM assembly in the absence of creatine results in a stone-like morphology (Fig. 1). Despite the previous report from our group proving that the utilization of an interfacial system for the synthesis of hydrogen-bonded frameworks strongly modifies their morphology,41 we wanted to make sure that in this work the preorganization in the interface between water and chloroform was providing a unique morphology. Hence, we synthesized the CMC0.1 complex just in water and analysed its morphology by SEM (Fig. S5†), confirming that its structural features strongly differ to the ones of CMC0.1 synthetized in an interfacial system. We observed that the size of the aggregates is considerably smaller in the absence of chloroform, probably due to the fact that the preorganization in between solvents allows a better rearrangement in plane, resulting in bigger stick-like aggregates (Fig. 1c).
C–(N)2 from cyanuric acid, respectively. The formation of CMCx complexes increases dramatically the atomic weight percentage of the C–(N)3 due to a synergistic contribution of melamine and creatine groups and as well the C–O, coming from the carboxylic group in creatine. N1s spectrum of CM displays three different binding energies at 398.8, 400.0 and 401.1 eV, that correspond to CN–C of triazine rings, hydrogen bonded NH–O groups, and remaining amino groups, respectively, the N1s spectra doesn't show pronounced differences, due to the similarity in the N species in the CM lattice and creatine. The morphology of the CMCx assemblies, studied by SEM, shows that the insertion of creatine directs the growth of the supramolecular units towards one direction, resulting in micrometre-long aggregates, while the CM assembly in the absence of creatine results in a stone-like morphology (Fig. 1). Despite the previous report from our group proving that the utilization of an interfacial system for the synthesis of hydrogen-bonded frameworks strongly modifies their morphology,41 we wanted to make sure that in this work the preorganization in the interface between water and chloroform was providing a unique morphology. Hence, we synthesized the CMC0.1 complex just in water and analysed its morphology by SEM (Fig. S5†), confirming that its structural features strongly differ to the ones of CMC0.1 synthetized in an interfacial system. We observed that the size of the aggregates is considerably smaller in the absence of chloroform, probably due to the fact that the preorganization in between solvents allows a better rearrangement in plane, resulting in bigger stick-like aggregates (Fig. 1c).
 | ||
| Fig. 1 SEM images of CMCx supramolecular assemblies. | ||
The formation of graphitic carbon nitride materials was achieved by the pyrolysis of the supramolecular assemblies at 550 °C for 4 h under N2 atmosphere. We expect, that during the pyrolysis, the creatine motifs interlinking the cyanuric acid–melamine lattices condensate to form a guanidine-like motifs, [HNC(NH2)2], which bind the heptazine units and modifies the properties of the material (Scheme S1†).47 The formation of CN-like structures was confirmed by several means, FTIR shows the typical stretching vibrational modes of C–N heterocycles between 1200–1600 cm−1 and the breathing modes of the triazine units at 810 cm−1 (Fig. S6†). Furthermore, XRD suggest the modification of the in-plane structure of the materials, as the peak at 13.1°, corresponding to the (100) plane, in CM–CN is shifted towards higher angles, probably due to the inclusion of molecular linkers within the heptazine units. Meanwhile the peak at 27.1° which belong to the (002) crystal plane remains unmodified with higher concentrations of creatine in the starting assembly. XPS measurements (discussion in Fig. S7†) further prove the formation of carbon nitride-like materials. The morphology of the CN materials, studied by SEM (Fig. 2), shows that upon calcination the CM assembly turns into hollow tubes, as previously reported by our group, and the insertion of guanidine like units in the supramolecular assembly modifies it towards more hollow layers. TEM imaging (Fig. S8†) confirms also the formation of hollow tubes and thin nanosheets upon addition of higher creatine amounts. The increasing porosity in CMCx is reflected also in the specific surface area as (from 54 m2 g−1 in CM–CN to 75 m2 g−1 in CMC0.1–CN, Fig. S9†).
 | ||
| Fig. 2 SEM images of CMCx–CN materials. The insets show digital images of the CMCx–CN powders. | ||
The chemical environment of C atoms in CM and CMC0.1–CN were examined by 13C CP-MAS NMR analysis (Fig. S10†). In both spectrum, two clear signals were observed around 164 and 157 ppm. The peak at ∼164 ppm is ascribed to C atoms bonded to the external –NH2 groups (N2C–NH2 unit), whereas peaks at ∼157 ppm are attributable to the sp2 species linked trigonally to three N atoms (CN3) which stands for the presence of typical tri-s-triazine or heptazine rings. The resolution of these peaks and the signal to noise ratio of 13C NMR spectra become gradually lower indicating a feasible disturbance of the polymeric matrix due to the linking units created by the condensation of creatine.
The photophysical properties of the as-prepared materials were studied by UV-vis absorbance and photoluminescence measurements. This study revealed that the modifications imprinted within the carbon nitride networks change the electron distribution and alter the fundamental electronic/optical properties of resultant CN. UV-vis absorbance (Fig. 3a) shows the shift of the band corresponding to the π–π* excitation from 370 (in CM–CN) to 395 nm (CMCx–CN) as well as the appearance of new peaks at 470 and 700 nm indicating that the resulting modified CN shows extended π-conjugation and enhanced light harvesting properties. Overall, with the increasing amount of creatine content, the C/N ratio has steadily increased in the final materials which is confirmed by elemental analysis (Table S2†). Photoluminescence measurements suggest a new, non-radiative path for photoexcited charges which result in the quenching in their fluorescence intensity (Fig. 3b). With all data in hand we can confirm that the monomer sequence alters the electronic structure of the final materials towards a more efficient charge separation properties, beneficial for photocatalysis.
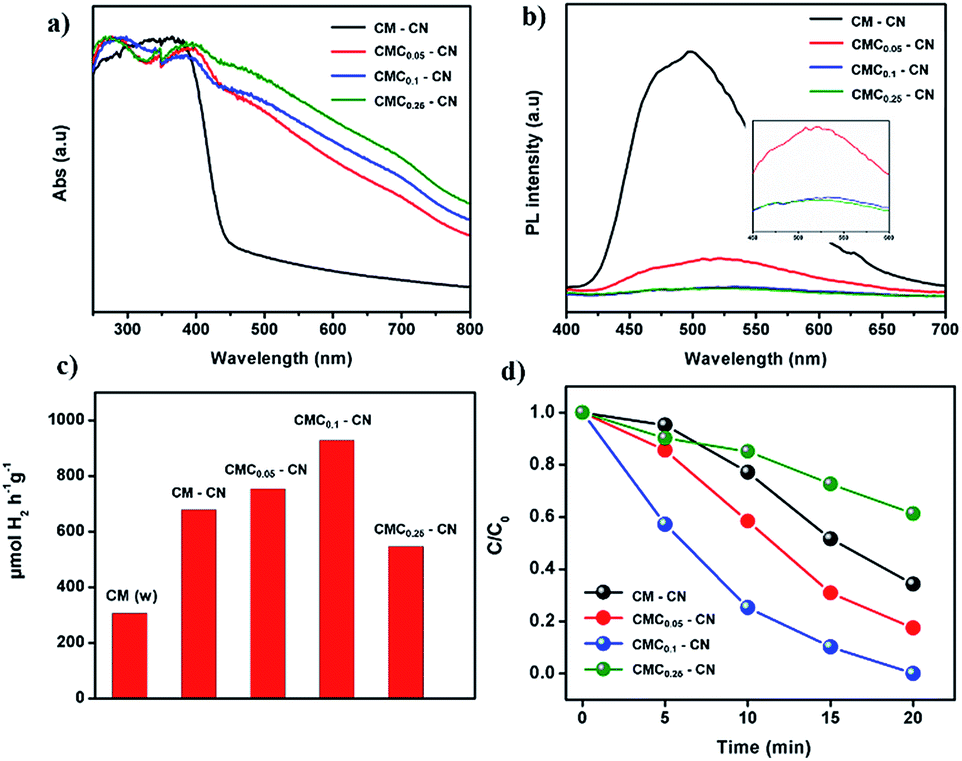 | ||
| Fig. 3 UV-vis absorbance spectra (a), fluorescence spectra (b), hydrogen evolution performances (c), and RhB degradation (d) performances of CM–CN and CMCx–CN materials. | ||
The photocatalytic activity of the materials was tested by performing the hydrogen evolution reaction in a water/triethanolamine solution with Pt as a co-catalyst (Fig. 3c), CMC0.1 showed the highest photocatalytic activity, resulting in 930 μmol H2 h−1 g−1 with a remarkable stability (92% of the initial performance after three consecutive cycles, Fig. S11†) and an average quantum yield (AQY) at 405 nm of 2.6%, this value is relatively high considering that no templating or ultrasonication techniques were employed. Additionally, the structure and morphology of the material remained intact after almost 15 h of photocatalytic hydrogen evolution. High resolution TEM images (HRTEM) of the material before and after the photocatalytic hydrogen evolution (Fig. S12†) show that the wrinkled nanosheet structure remains the same and just a slight decrease in the particle size is observed, and XPS measurements confirm as well that the chemical structure of the CN material remains undamaged (discussion in Fig. S13†). Further proof of the high photocatalytic activity was given by the degradation of the rhodamine B dye (RhB) under visible light (Fig. 3d), where CMC0.1–CN showed the fastest photodegradation performance (20 min).
In addition, the photocurrent responses of the CM–CN and CMC0.1–CN films were measured in 0.1 M KOH at 1.23 V vs. RHE under 1 sun illumination, where the CMC0.1–CN electrode exhibited the highest photocurrent response (Fig. 4a). The photocurrent results are further supported by electrochemical impedance spectra (EIS). The measurements are performed in dark and under one sun illumination, showing in both cases a lower charge transfer resistance for CMC0.1–CN (Fig. 4b).
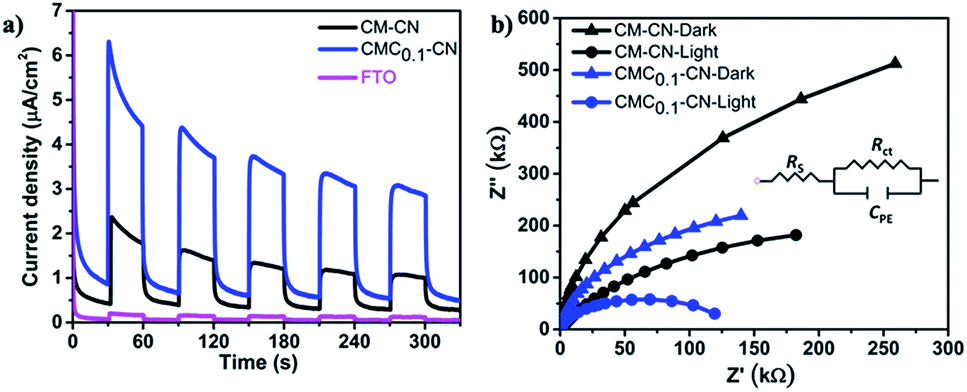 | ||
| Fig. 4 Photocurrent response under visible light irradiation in 0.1 M KOH at 1.23 (V) vs. RHE (a) and electrochemical impedance spectroscopy plots in the dark and under one sun illumination (b) (inset shows equivalent circuit for fitting the Nyquist plots). | ||
Conclusions
In summary, we showed the formation of modified CN materials derived of a rationally designed supramolecular assembly. The utilization of a molecule that can bind selectively both melamine and cyanuric acid in the interface between water and chloroform allows a robust grafting and interlink of cyanuric acid–melamine hydrogen bonded frameworks. This new pathway strongly modifies the chemical and electronic properties of the final CN materials toward an efficient photocatalytic performance. We believe that this work represents a step forward in understanding the rational design of carbon–nitrogen based materials properties utilizing supramolecular structures and in the further establishment of structure–activity relationships. Additionally, it opens the gate towards the utilization of interfacial systems for the preorganization of supramolecular frameworks that modulate the performance of a given material in different energy-related and environmental fields.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Dr Michael Volokh Dr Alexander Upcher, and Mr Jonathan Tzadikov for assistance with electronic microscopy measurements and Prof. Menny Shalom for fruitful discussion. This research was funded by the Israel Science Foundation (ISF), grant No. 1161/17, and supported by the Minerva Center No. 117873.Notes and references
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- J. Safaei, N. A. Mohamed, M. F. Mohamad Noh, M. F. Soh, N. A. Ludin, M. A. Ibrahim, W. N. Roslam Wan Isahak and M. A. Mat Teridi, J. Mater. Chem. A, 2018, 6, 22346–22380 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CrossRef CAS PubMed.
- J. Barrio and M. Shalom, ACS Appl. Mater. Interfaces, 2018, 10, 39688–39694 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298–2321 RSC.
- L. Chen and J. Song, Adv. Funct. Mater., 2017, 27, 1702695 CrossRef.
- J. Xu, M. Shalom, F. Piersimoni, M. Antonietti, D. Neher and T. J. K. Brenner, Adv. Opt. Mater., 2015, 3, 913–917 CrossRef CAS.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- J. Xu and M. Shalom, ChemPhotoChem, 2019, 3, 170–179 CrossRef CAS.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- X. Wu, F. Chen, X. Wang and H. Yu, Appl. Surf. Sci., 2018, 427, 645–653 CrossRef CAS.
- Z. Yang, Y. Zhang and Z. Schnepp, J. Mater. Chem. A, 2015, 3, 14081–14092 RSC.
- J. Liu, J. Yan, H. Ji, Y. Xu, L. Huang, Y. Li, Y. Song, Q. Zhang, H. Xu and H. Li, Mater. Sci. Semicond. Process., 2016, 46, 59–68 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- J. Barrio, C. Gibaja, J. Tzadikov, M. Shalom and F. Zamora, Adv. Sustainable Syst., 2019, 3, 1800138 CrossRef.
- J. Wang, J. Chen, P. Wang, J. Hou, C. Wang and Y. Ao, Appl. Catal., B, 2018, 239, 578–585 CrossRef CAS.
- T. Wu, P. Wang, J. Qian, Y. Ao, C. Wang and J. Hou, Dalton Trans., 2017, 46, 13793–13801 RSC.
- M. Shalom, S. Inal, D. Neher and M. Antonietti, Catal. Today, 2014, 225, 185–190 CrossRef CAS.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- J. Barrio and M. Shalom, ChemCatChem, 2018, 10, 5573–5586 CrossRef CAS.
- S. Wan, M. Ou, Q. Zhong, S. Zhang and W. Cai, Adv. Opt. Mater., 2017, 5, 1700536 CrossRef.
- M. Shalom, M. Guttentag, C. Fettkenhauer, S. Inal, D. Neher, A. Llobet and M. Antonietti, Chem. Mater., 2014, 26, 5812–5818 CrossRef CAS.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- T. Jordan, N. Fechler, J. Xu, T. J. K. Brenner, M. Antonietti and M. Shalom, ChemCatChem, 2015, 7, 2826–2830 CrossRef CAS.
- J. Barrio, L. Lin, P. Amo-Ochoa, J. Tzadikov, G. Peng, J. Sun, F. Zamora, X. Wang and M. Shalom, Small, 2018, 14, 1800633 CrossRef PubMed.
- J. Wang, H. Xu, X. Qian, Y. Dong, J. Gao, G. Qian and J. Yao, Chem.–Asian J., 2015, 10, 1276–1280 CrossRef CAS PubMed.
- Y. S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- Q. Li, J. Yang, D. Feng, Z. Wu, Q. Wu, S. S. Park, C.-S. Ha and D. Zhao, Nano Res., 2010, 3, 632–642 CrossRef CAS.
- J. Barrio, A. Grafmüller, J. Tzadikov and M. Shalom, Appl. Catal., B, 2018, 237, 681–688 CrossRef CAS.
- Q. Cui, J. Xu, X. Wang, L. Li, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2016, 55, 3672–3676 CrossRef CAS PubMed.
- X. Wu, D. Gao, H. Yu and J. Yu, Nanoscale, 2019, 11, 9608–9616 RSC.
- P. Li, H. D. Arman, H. Wang, L. Weng, K. Alfooty, R. F. Angawi and B. Chen, Cryst. Growth Des., 2015, 15, 1871–1875 CrossRef CAS.
- V. R. Pedireddi and D. Belhekar, Tetrahedron, 2002, 58, 2937–2941 CrossRef CAS.
- J. Sun, J. Xu, A. Grafmueller, X. Huang, C. Liedel, G. Algara-Siller, M. Willinger, C. Yang, Y. Fu, X. Wang and M. Shalom, Appl. Catal., B, 2017, 205, 1–10 CrossRef CAS.
- Q. Bo, Z. Shuai, S. Qiao, H. Zehuan, X. Jiang-Fei and Z. Xi, Angew. Chem., Int. Ed., 2017, 56, 7639–7643 CrossRef PubMed.
- S. Dolai, J. Barrio, G. Peng, A. Grafmüller and M. Shalom, Nanoscale, 2019, 11, 5564–5570 RSC.
- B. Roy, P. Bairi and A. K. Nandi, RSC Adv., 2014, 4, 1708–1734 RSC.
- C. Li, B. J. Cafferty, S. C. Karunakaran, G. B. Schuster and N. V. Hud, Phys. Chem. Chem. Phys., 2016, 18, 20091–20096 RSC.
- G. Li, J. Shi, G. Zhang, Y. Fang, M. Anpo and X. Wang, Res. Chem. Intermed., 2017, 43, 5137–5152 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- P. Kalisman, Y. Nakibli and L. Amirav, Nano Lett., 2016, 16, 1776–1781 CrossRef CAS PubMed.
- B. Wang, T. Dong, A. Myrlie, L. Gu, H. Zhu, W. Xiong, P. Maness, R. Zhou and J. Yu, Green Chem., 2019, 21, 2928–2937 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05264j |
| ‡ Susmita Dolai and Neeta Karjule contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |