
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
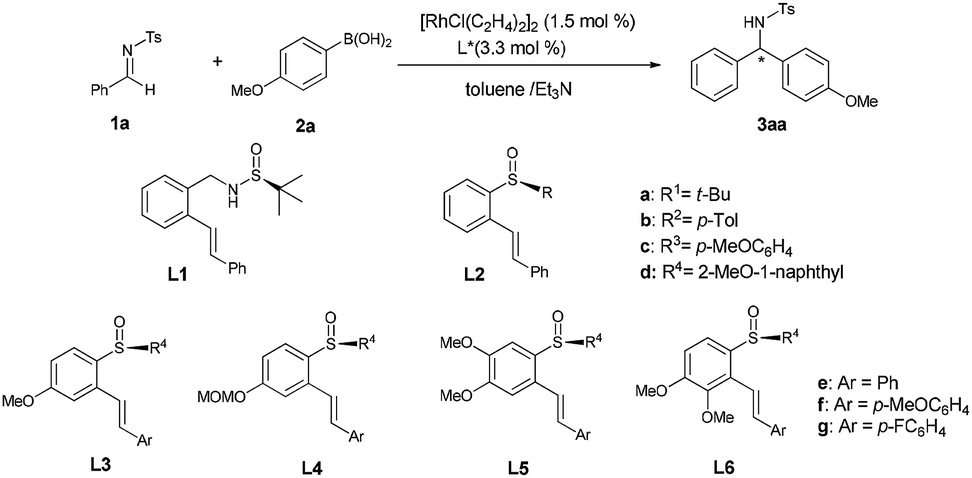
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral benzene backbone-based sulfoxide-olefin ligands for highly enantioselective Rh-catalyzed addition of arylboronic acids to N-tosylarylimines†
Feng Xue *a,
Qibin Liu*b,
Yong Zhua,
Yunfei Qinga and
Boshun Wan*c
*a,
Qibin Liu*b,
Yong Zhua,
Yunfei Qinga and
Boshun Wan*c
aKey Laboratory of Functional Organic Molecules of Xinxiang City Henan Province, College of Chemistry and Chemical Engineering, Henan Institute of Science and Technology, Xinxiang, Henan 453002, China. E-mail: fxuehist@sina.com
bDalian Allychem Co., Ltd, 5 Jinbin Road, Dalian 116620, China. E-mail: qliu@allychem.com
cDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, P. R. China. E-mail: bswan@dicp.ac.cn
First published on 14th August 2019
Abstract
An efficient Rh-catalyzed addition of arylboronic acids to N-tosylarylimines has been developed with chiral benzene backbone-based sulfoxide-olefin ligands, where 2-methoxy-1-naphthyl sulfinyl functionalized olefin ligands have shown to be more effective. The versatile method tolerates a wide range of functional groups and shows broad scope without regard to electronic or steric substitution pattern, allowing access to a broad range of chiral diarylmethylamines in high yields (up to 99%) with excellent enantioselectivities (up to 99% ee).
Introduction
Optically active diarylmethylamines are present in a variety of biologically significant structures.1 Due to their pharmaceutical importance, great attention has been drawn to the development of their synthesis.2 Rhodium-catalyzed asymmetric addition of organoboron reagents to aldimines has become a powerful strategy for the straightforward synthesis of the skeleton since the first report by Tomioka using L-valine-connected amidomonophosphane as chiral ligand in 2004.3 From then on, considerable efforts have been made and great progress has been achieved in the catalytic enantioselective arylation of N-tosyl or nosyl activated/protected imines.4 Among them, Hayashi and co-workers5 reported excellent enantioselectivities for the addition of aryl boroxines to N-tosylarylimines by employing chiral bicyclo[2.2.2]octadiene ligand. In 2006, Zhou and co-workers6 also reported an efficient asymmetric arylation of N-tosylarylimines using monodentate spiro phosphite (S)-ShiP. In 2007 and 2010, Lin and co-workers7 successively reported other examples in the highly efficient arylation of N-tosylarylimines with chiral bicyclo-[3.3.0]octadiene and dicyclopentadienes ligands. In 2014, Wu and co-workers8 adopted another diene ligand in enantioselective Rh-catalyzed arylation of N-tosyl and N-nosyl aldimines in methanol. Later, Dorta and co-workers9 employed chiral disulfoxide ligand for the efficient rhodium-catalyzed 1,2-addition of arylboroxines to N-tosylarylimines in 2016. Recently, Lin and co-workers applied chiral spiro monophosphite-olefin ligands in asymmetric addition of organoboronic acids to aldimines.10 Despite the remarkable advances with these ligands (Scheme 1a), the substrate generality is likely influenced by the electronic and steric substitution pattern of both reaction partners to show relatively narrow compatibility of reaction substrates. For example, these methods mainly focused on less sterically hindered para- as well as electron-rich substitution arylboron reagents. Moreover, limited research has been conducted on substrates with electron-poor groups and ortho-substituent groups. Thus, it is required to broaden the substrate scope of this Rh-catalyzed asymmetric arylation reaction with greater substitution tolerance. | ||
| Scheme 1 Rh-catalyzed asymmetric arylation of N-tosylarylimines. | ||
With o-phenylene as the linkage of olefin and sulfoxide, we11 have successfully developed a series of simple and easily tunable chiral benzene backbone-based olefin–sulfoxide ligands bearing different olefin and sulfinyl moieties for the Rh-catalyzed enantioselective addition reaction, where 2-methoxy-1-naphthyl sulfinyl functionalized olefin ligands11b,c have high efficiency for the conjugate addition of arylboronic acids to nitroalkenes and unsaturated esters. Meanwhile, other chiral sulfinyl-based olefin ligands (SOLs)12 have also been explored and applied in a range of Rh-catalyzed asymmetric transformations, which have obvious advantage over some conventional chiral ligands in terms of activity and selectivity. Recently, Xu and co-workers12j reported highly enantioselective addition of aryl boroxines to N,N-dimethylsulfamoyl-protected aldimines by employing chiral branched tert-butyl sulfonamide-based olefins ligands, in which limited examples were tentatively conducted on the reaction of N-tosylarylimines with arylboroxines without systematically examined the reaction (Scheme 1a). Despite the significant progress, the sulfinyl moieties of the reported sulfinyl-based olefin ligands were mostly limited to tert-butyl substitution, and the incorporation of alkenes with other sulfinyl groups remains underappreciated and far less explored. In view of the pharmaceutical importance of chiral diarylmethylamines in organic transformations, it is still highly desirable to develop effective catalytic systems that would successfully lead to a broad range of desired diarylmethylamines with excellent enantioselectivities. To this end, we explored the asymmetric addition of aryl boronic acids to N-tosylarylimines by using chiral benzene backbone-based sulfoxide-olefin ligands based on 2-methoxy-1-naphthyl sulfinyl moiety, affording a broad range of chiral diarylmethylamines in high yields (up to 99%) with excellent enantioselectivities (up to 99% ee) (Scheme 1b).
Results and discussion
We started with Rh-catalyzed conjugate addition of N-tosylphenylimine 1a with p-anisylboronic acid 2a in the presence of ligands L1–L6 (Table 1). Initially, the reaction proceeded in the presence of 1.5 mol% [RhCl(C2H4)2]2 and 3.3 mol% L1 or L2a bearing tert-butylsulfinyl moiety in Et3N/toluene, giving a trace amount of desired product (entries 1–2). To improve the activity, ligands L2b–L2d bearing different sulfinyl moieties were screened. It was found that ligand L2b with p-tolsulfiny moiety gave the expected product 3aa in 75% yield with 40% ee (entry 3), whereas ligand L2c bearing p-methoxybenzene sulfinyl moieties gave 60% ee (entry 4) and L2d with 2-methoxy-1-naphthyl sulfinyl moiety afforded higher yield (85%) and enantioselectivity (76% ee) (entry 5). With 2-methoxy-1-naphthyl moiety was established, ligands L3–L6 bearing different alkene moieties containing substituents with different steric and electronic natures were further examined, meanwhile, the substitution effects on the central benzene ring moiety were also explored (entries 6–11). It was speculated that the electronic property of the substitutes at the olefin moiety would have a significant effect on the enantioselectivity. Ligand L3f possessing an electron-donating para-methoxy group on the terminal benzene ring afforded the product with higher enantioselectivity (entry 7 vs. entries 6 and 8). Moreover, the electronic effect on the benzene backbone was also examined. Ligands L4f bearing a MOMO substituent on the benzene backbone showed similar reactivity as that of L3f (entry 9 vs. entry 7). However, when an additional methoxy group was introduced to the ortho-position of MeO on the benzene backbone of L3f, ligand L6f afforded the product with increased catalytic reactivity and enantioselectivity (92% yield, 95% ee, entry 11) while ligand L5f showed inferior result (80% ee, entry 10).
|
|||
|---|---|---|---|
| Entry | Ligand | Yieldb (%) | eec (%) |
| a The reaction was carried out with N-tosylphenylimine 1a (0.30 mmol), p-anisylboronic acid 2a (0.45 mmol), [RhCl(C2H4)2]2 (0.0045 mmol), ligand (0.0099 mmol, 1.1 equiv. to Rh), and 0.75 M Et3N (0.20 mL) in toluene (2.0 mL) at 50 °C for 5–6 h.b Yield based on N-tosylphenylimine.c Determined by HPLC analysis.d Not determined. | |||
| 1 | L1 | Trace | n.d.d |
| 2 | L2a | Trace | n.d.d |
| 3 | L2b | 75 | 40 |
| 4 | L2c | 80 | 60 |
| 5 | L2d | 85 | 76 |
| 6 | L3e | 86 | 79 |
| 7 | L3f | 83 | 91 |
| 8 | L3g | 87 | 85 |
| 9 | L4f | 91 | 90 |
| 10 | L5f | 90 | 80 |
| 11 | L6f | 92 | 95 |
Next, the influence of solvents and bases was further explored (Table 2). Other inorganic bases such as KOH, K2CO3, and K3PO4 did not improve the enantioselectivity (entries 1–3). When the reaction was conducted in aqueous KHF2/toluene solution, in which potassium aryltrifluoroborate can be generated in situ,13 no increase of reactivity and enantioselectivity was also observed (entry 4). Gratifyingly, KF afforded highest enantioselectivity (97% ee, entry 5). Furthermore, solvent screening showed that dioxane, THF, DCE and CH2Cl2 failed to improve the enantioselectivity (entries 6–9). In addition, other organoboron reagents and aldimines such as p-methoxy phenylboroxine and N-nosylphenylimine exhibited comparable results as those of p-anisylboronic acid 2a and N-tosylphenylimine 1a (entries 10–11).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Yieldb (%) | eec (%) |
| a The reaction was carried out with N-tosylphenylimine 1a (0.30 mmol), p-anisylboronic acid 2a (0.45 mmol), [RhCl(C2H4)2]2 (0.0045 mmol), ligand L6f (0.0099 mmol, 1.1 equiv. to Rh), and 1.5 M aq base (0.20 mL) in solvent (2.0 mL) at 50 °C for 5–6 h.b Yield based on N-tosylphenylimine.c Determined by HPLC analysis.d p-Methoxy phenylboroxine instead of p-anisylboronic acid.e N-Nosylphenylimine instead of N-tosylphenylimine. | ||||
| 1 | Toluene | KOH (0.75 M) | 95 | 91 |
| 2 | Toluene | K2CO3 (1.5 M) | 90 | 92 |
| 3 | Toluene | K3PO4 (1.5 M) | 90 | 92 |
| 4 | Toluene | KHF2 (1.5 M) | 90 | 93 |
| 5 | Toluene | KF (1.5 M) | 96 | 97 |
| 6 | Dioxane | KF (1.5 M) | 91 | 91 |
| 7 | THF | KF (1.5 M) | 93 | 91 |
| 8 | DCE | KF (1.5 M) | 92 | 89 |
| 9 | CH2Cl2 | KF (1.5 M) | 91 | 96 |
| 10d | Toluene | KF (1.5 M) | 94 | 97 |
| 11e | Toluene | KF (1.5 M) | 93 | 95 |
Having established the optimal reaction conditions, we then investigated the substrate scope of this Rh-catalyzed asymmetric arylation (Table 3). We were pleased to find that a variety of arylboronic acids bearing substituents with diverse electronic and steric properties all smoothly reacted with N-tosylarylimines to provide the desired product in high yields (90–99%) with excellent enantioselectivities (95–99%). In general, the electronic properties of the substituent did not significantly affect the reaction stereoselectivity. Extremely high enantiomeric excesses (98–99% ee) were attained with electron-poor or sterically encumbered arylboronic acids (entries 7–13 and 26–28). Furthermore, a broad range of electronically and sterically different aryl imines were tested. Regardless of the substitution pattern on the phenyl ring, all these imines reacted well with arylboronic acids to provide the corresponding products in high yields with excellent enantioselectivity (95–99% ee). Intriguingly, apart from almost same enantiocontrol happened in electron-poor imines 1f and arylboronic acids 2i (entry 9 vs. 20), significant enantiocontrol was observed with electron-rich imines; this trend is opposite to what is observed with arylboronic acids. Thus, in some cases such as for 1c and 1d, by simply reversing the corresponding Ar1 and Ar2 groups of the two substrates, the ee of the product can be readily enhanced (entry 1 vs. 18, entry 2 vs. 17, and also entry 32 vs. 24). These results indicate that the desired highly enantioenriched product could easily be furnished by switching the aryl acceptor/donor. Another interesting feature is that the reaction exhibits a truly remarkable ortho-substitution tolerance. In all cases with sterically encumbered substrates, as exemplified by 2d (entry 4), 2g (entry 7), 2m (entry 13), 1b (entry 16), 1c (entry 17), 1g (entry 21), nearly perfect enantiomeric excesses (97–99% ee) could be achieved. In addition, it is worth mentioning that when it comes to arylboronic acids with m-OMe and p-Me groups on the phenyl ring, higher yield and enantioselecitivity were obtained than those in previous work12j (entry 3, 95% yield, 97% ee vs. 67% yield, 94% ee and entry 6, 98% yield, 98% ee vs. 94% yield, 91% ee).
|
||||
|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Yieldb (%) | eec (%) |
| a The reaction was carried out with N-tosylarylimines (0.30 mmol), arylboronic acids (0.45 mmol), [RhCl(C2H4)2]2 (0.0045 mmol), ligand L6f (0.0099 mmol, 1.1 equiv. to Rh), and 1.5 M aq KF (0.20 mL) in toluene (2.0 mL) at 50 °C for 5–6 h.b Yield based on N-tosylarylimines.c Determined by HPLC analysis. | ||||
| 1 | Ph(1a) | 4-MeOC6H4(2a) | 96(3aa) | 97 |
| 2 | Ph(1a) | 2-MeOC6H4(2b) | 93(3ab) | 95 |
| 3 | Ph(1a) | 3-MeOC6H4(2c) | 95(3ac) | 97 |
| 4 | Ph(1a) | 2-MeC6H4(2d) | 94(3ad) | 97 |
| 5 | Ph(1a) | 3-MeC6H4(2e) | 96(3ae) | 98 |
| 6 | Ph(1a) | 4-MeC6H4(2f) | 98(3af) | 98 |
| 7 | Ph(1a) | 2-FC6H4(2g) | 99(3ag) | 99 |
| 8 | Ph(1a) | 4-ClC6H4(2h) | 99(3ah) | 98 |
| 9 | Ph(1a) | 4-BrC6H4(2i) | 98(3ai) | 98 |
| 10 | Ph(1a) | 3-CF3C6H4(2j) | 95(3aj) | 99 |
| 11 | Ph(1a) | 4-CF3C6H4(2k) | 96(3ak) | 98 |
| 12 | Ph(1a) | 4-t-BuC6H4(2l) | 94(3al) | 98 |
| 13 | Ph(1a) | 1-Naphthyl(2m) | 90(3am) | 98 |
| 14 | Ph(1a) | 3,4-diMeC6H3(2n) | 91(3an) | 96 |
| 15 | Ph(1a) | 3,4-diMeOC6H3(2o) | 92(3ao) | 96 |
| 16 | 1-Naphthyl(1b) | Ph(2p) | 97(3bp) | 99 |
| 17 | 2-MeOC6H4(1c) | Ph(2p) | 95(3cp) | 98 |
| 18 | 4-MeOC6H4(1d) | Ph(2p) | 96(3dp) | 99 |
| 19 | 4-MeC6H4(1e) | Ph(2p) | 95(3ep) | 97 |
| 20 | 4-BrC6H4(1f) | Ph(2p) | 97(3fp) | 98 |
| 21 | 2-ClC6H4(1g) | Ph(2p) | 95(3gp) | 97 |
| 22 | 2-MeOC6H4(1c) | 4-MeOC6H4(2a) | 93(3ca) | 95 |
| 23 | 4-MeOC6H4(1d) | 3-MeOC6H4(2c) | 95(3dc) | 98 |
| 24 | 4-MeOC6H4(1d) | 4-MeC6H4(2f) | 96(3df) | 99 |
| 25 | 4-MeOC6H4(1d) | 3-MeC6H4(2e) | 95(3de) | 96 |
| 26 | 4-MeOC6H4(1d) | 2-MeC6H4(2d) | 93(3dd) | 98 |
| 27 | 4-MeOC6H4(1d) | 4-CF3C6H4(2k) | 98(3dk) | 99 |
| 28 | 4-MeOC6H4(1d) | 4-ClC6H4(2h) | 99(3dh) | 98 |
| 29 | 4-MeOC6H4(1d) | 1-Naphthyl(2m) | 90(3dm) | 97 |
| 30 | 4-MeOC6H4(1d) | 2-Naphthyl(2n) | 93(3dn) | 98 |
| 31 | 4-MeC6H4(1e) | 2-MeOC6H4(2b) | 94(3eb) | 95 |
| 32 | 4-MeC6H4(1e) | 4-MeOC6H4(2a) | 97(3ea) | 95 |
| 33 | 4-MeC6H4(1e) | 4-ClC6H4(2h) | 96(3eh) | 98 |
| 34 | 4-MeC6H4(1e) | 1-Naphthyl(2m) | 90(3em) | 97 |
| 35 | 4-MeC6H4(1e) | 2-Naphthyl(2n) | 93(3en) | 97 |

On the basis of the reaction stereochemical outcome, an empirical transition state model14 is proposed (Fig. 1). We assume that there tends to be a π–π stacking between the phenyl ring of the ligand terminus and the metalated phenyl ring of phenylboronic acid in the reaction transition state. The Rh-complex recognizes the alkene moiety of N-tosylarylimine as a result of the steric repulsion between the 2-methoxy-1-naphthyl group of the ligand and tosyl group of the N-tosylarylimine. To minimize steric congestion, coordination of N-tosylarylimine from the less steric hindered side is more favorable, which leads to the S isomer and is consistent with the observed stereochemistry.
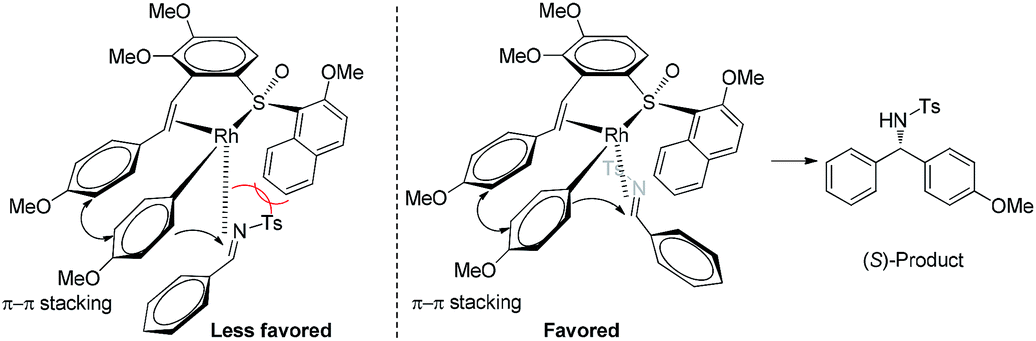 | ||
| Fig. 1 Proposed stereochemical pathway for the asymmetric arylation. | ||
Conclusion
In conclusion, an efficient Rh-catalyzed addition of arylboronic acids to N-tosylarylimines has been developed with chiral benzene backbone-based sulfoxide-olefin ligands, where 2-methoxy-1-naphthyl sulfinyl functionalized olefin ligands have shown to be more effective than the tert-butyl sulfinyl-based ones, allowing access to a broad range of chiral diarylmethylamines in high yields (up to 99%) with excellent enantioselectivities (up to 99% ee). Compared with the methods in the literature, both the electron effect and steric effect of the substituent group of the reaction substrate have no obvious influence on the reaction results, especially when the substrates with strong electron-withdrawing group or ortho-hindrance were involved in the reaction. Excellent yield and enantioselectivity could be also achieved, indicating that the catalytic system has a wide range of reaction substrate tolerance. This study sets the stage for further exploration of these recently developed ligands in other asymmetric transformations and the development of other kinds of unique olefin ligands. Further studies are underway and will be reported in due course.Experimental
General
All reactions were carried out under an atmosphere of nitrogen using the standard Schlenk techniques, unless otherwise noted. Solvents were dried and distilled by standard procedures. 1H NMR and 13C NMR spectra were recorded at room temperature in CDCl3 on 400 MHz and 600 MHz instrument with tetramethylsilane (TMS) as internal standard. Enantiomeric excess was determined by HPLC analysis, using chiral column described below in detail. Optical rotations were measured by polarimeter. Flash column chromatography was performed on silica gel (200–300 mesh). All reactions were monitored by TLC analysis.General procedure for the enantioselective Rh-catalyzed addition of arylboronic acids to N-tosylarylimines
Under nitrogen atmosphere, a mixture of [RhCl(C2H4)2]2 (1.8 mg, 0.0045 mmol) and ligand L6f (4.7 mg, 0.0099 mmol) in 1 mL toluene was stirred at room temperature for 1 h. At which time arylboronic acid (0.45 mmol) was added, followed by N-tosylarylimines (0.30 mmol), aqueous KF (1.5 M in H2O, 0.20 mL, 0.30 mmol) and toluene (1 mL). The reaction was stirred at 50 °C for 5–6 h. When the reaction was over, the reaction mixture was concentrated in vacuo and purified by silica gel flash column chromatography (petroleum ether/ethyl acetate as eluent) to afford the product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21172218) is gratefully acknowledged.Notes and references
- For representative examples, see: (a) C. M. Spencer, D. Foulds and D. H. Peters, Drugs, 1993, 46, 1055 CrossRef CAS; (b) S. N. Calderon, R. B. Rothman, F. Porreca, J. L. Flippen-Anderson, R. W. McNutt, H. Xu, L. E. Smith, E. J. Bilsky, P. Davis and K. C. Rice, J. Med. Chem., 1994, 37, 2125 CrossRef CAS PubMed; (c) M. J. Bishop and R. W. McNutt, Bioorg. Med. Chem. Lett., 1995, 5, 1311 CrossRef CAS; (d) S. Sakurai, N. Ogawa, T. Suzuki, K. Kato, T. Ohashi, S. Yasuda, H. Kato and Y. Ito, Chem. Pharm. Bull., 1996, 44, 765 CrossRef CAS PubMed.
- For representative examples, see: (a) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS PubMed; (b) C. Bolm, J. P. Hildebrand, K. Muñiz and N. Hermanns, Angew. Chem., Int. Ed., 2001, 40, 3284 CrossRef CAS; (c) F. Schmidt, R. T. Stemmler, J. Rudolph and C. Bolm, Chem. Soc. Rev., 2006, 35, 454 CAS; (d) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed.
- M. Kuriyama, T. Soeta, X. Hao, Q. Chen and K. Tomioka, J. Am. Chem. Soc., 2004, 126, 8128 CrossRef CAS PubMed.
- For representative examples, see: (a) T. Hayashi, M. Kawai and N. Tokunaga, Angew. Chem., Int. Ed., 2004, 43, 6125 CrossRef CAS PubMed; (b) Y. Otomaru, N. Tokunaga, R. Shintani and T. Hayashi, Org. Lett., 2005, 7, 307 CrossRef CAS PubMed; (c) D. J. Weix, Y. L. Shi and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 1092 CrossRef CAS; (d) Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) M. Trincado and J. A. Ellman, Angew. Chem., Int. Ed., 2008, 47, 5623 CrossRef CAS PubMed; (f) Z. Cui, H. J. Yu, R. F. Yang, W. Y. Gao, C. G. Feng and G. Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394 CrossRef CAS PubMed; (g) X. Gao, B. Wu, Z. Yan and Y. G. Zhou, Org. Biomol. Chem., 2016, 14, 55 RSC.
- N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584 CrossRef CAS PubMed.
- H. F. Duan, Y. X. Jia, L. X. Wang and Q. L. Zhou, Org. Lett., 2006, 8, 2567 CrossRef CAS.
- (a) Z. Q. Wang, C. G. Feng, M. H. Xu and G. Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336 CrossRef CAS; (b) C. Shao, H. J. Yu, N. Y. Wu, C. G. Feng and G. Q. Lin, Org. Lett., 2010, 12, 3820 CrossRef CAS PubMed.
- C. C. Chen, B. Gopula, J. F. Syu, J. H. Pan, T. S. Kuo, P. Y. Wu, J. P. Henschke and H. L. Wu, J. Org. Chem., 2014, 79, 8077 CrossRef CAS PubMed.
- G. Z. Zhao, G. Sipos, A. Salvador, A. Ou, P. C. Gao, B. W. Skelton and R. Dorta, Adv. Synth. Catal., 2016, 358, 1759 CrossRef CAS.
- H. Y. Shan, Q. X. Zhou, J. L. Yu, S. Q. Zhang, X. Hong and X. F. Lin, J. Org. Chem., 2018, 83, 11873 CrossRef CAS.
- (a) F. Xue, X. C. Li and B. S. Wan, J. Org. Chem., 2011, 76, 7256 CrossRef CAS; (b) F. Xue, X. C. Li and B. S. Wan, J. Org. Chem., 2012, 77, 3081 Search PubMed; (c) F. Xue, D. P. Wang, X. C. Li and B. S. Wan, Org. Biomol. Chem., 2013, 11, 7893 RSC.
- For representative examples, see: (a) T. Thaler, L. N. Guo, A. K. Steib, M. Raducan, K. Karaghiosoff, P. Mayer and P. Knochel, Org. Lett., 2011, 13, 3182 CrossRef CAS PubMed; (b) G. H. Chen, J. Y. Gui, L. C. Li and J. Liao, Angew. Chem., Int. Ed., 2011, 50, 7681 CrossRef CAS PubMed; (c) Z. Q. Liu, X. Q. Feng and H. F. Du, Org. Lett., 2012, 14, 3154 CrossRef CAS PubMed; (d) X. Feng and H. Du, Asian J. Org. Chem., 2012, 1, 204 CrossRef CAS; (e) H. Wang, T. Jiang and M. H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (f) F. Xue, C. G. Li, J. Chen and B. S. Wan, Chin. J. Org. Chem., 2014, 34, 267 CrossRef CAS; (g) Y. Li and M. H. Xu, Chem. Commun., 2014, 50, 3771 RSC; (h) Y. Li, Y. N. Yu and M. H. Xu, ACS Catal., 2016, 6, 661 CrossRef CAS; (i) Y. F. Zhang, D. Chen, W. W. Chen and M. H. Xu, Org. Lett., 2016, 18, 2726 CrossRef CAS PubMed; (j) T. Jiang, W. W. Chen and M. H. Xu, Org. Lett., 2017, 19, 2138 CrossRef CAS PubMed; (k) C. Y. Wu, Y. F. Zhang and M. H. Xu, Org. Lett., 2018, 20, 1789 CrossRef CAS PubMed; (l) Y. Li, B. Liu and M. H. Xu, Org. Lett., 2018, 20, 2306 CrossRef CAS.
- Z. G. Wang, C. G. Feng, S. S. Zhang, M. H. Xu and G. Q. Lin, Angew. Chem., Int. Ed., 2010, 49, 5780 CrossRef CAS.
- (a) T. Hayashi, M. Takahashi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052 CrossRef CAS PubMed; (b) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04836g |
| This journal is © The Royal Society of Chemistry 2019 |