
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivating molecular oxygen with Au/CeO2 for the conversion of lignin model compounds and organosolv lignin†
Wu-Lin
Song
 a,
Qingmeng
Dong
a,
Liang
Hong
ab,
Zhou-Qi
Tian
a,
Li-Na
Tang
a,
Wenli
Hao
a and
Hongxi
Zhang
*a
a,
Qingmeng
Dong
a,
Liang
Hong
ab,
Zhou-Qi
Tian
a,
Li-Na
Tang
a,
Wenli
Hao
a and
Hongxi
Zhang
*a
aDepartment of Chemistry and Applied Chemistry, Changji University, Changji 831100, Xinjiang, China. E-mail: cjxyzhx@sina.cn
bProduct Quality Inspection Institute of Changji Hui Autonomous Prefecture, Changji 831100, Xinjiang, China
First published on 2nd October 2019
Abstract
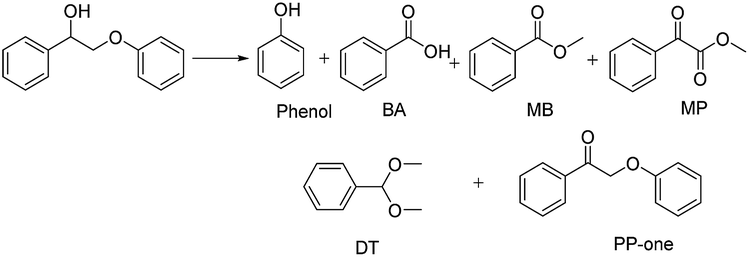
Au/CeO2 was demonstrated to be a high efficiency catalyst for the conversion of 2-phenoxyacetophenol (PP-ol) employing O2 as an oxidant and methyl alcohol as the solvent without using an erosive strong base or acid. Mechanistic investigations, including emission quenching experiments, electron spin-resonance (ESR) and intermediate verification experiments, were carried out. The results verified that the superoxide anion activated by Au/CeO2 from molecular oxygen plays a vital role in the oxidation of lignin model compounds, and the cleavage of both the β-O-4 and Cα–Cβ linkages was involved. Au/CeO2 also performed well in the oxidative conversion of organosolv lignin under mild conditions (453 K), producing vanillin (10.5 wt%), methyl vanillate (6.8 wt%), methylene syringate (3.4 wt%) and a ring-opened product. Based on the detailed characterization data and mechanistic results, Au/CeO2 was confirmed to be a promising catalytic system.
Introduction
Despite their excellent performance and broad applicability, fossil fuel reserves are dwindling and their use causes environmental pollution. Green and renewable resources are considered key to the continuous development of society. Lignocellulosic biomass can continuously and steadily provide chemicals and fuel with zero net carbon emissions. In recent years, there has been increasing interest in the industrial production of organic compounds and fuels from lignocellulosic biomass.1 As a primary constituent of lignocellulosic biomass (15–30% by weight and 40% by energy),2 lignin consists mainly of three monolignols: coniferyl alcohol, sinapyl alcohol and p-coumaryl alcohol (seen Fig. 1). Based on its structure, lignin can be considered a source of value-added chemicals, such as aromatic aldehydes, ketones, acids and esters. The annual output of lignin in nature is 150 billion tons. More than 70 million tons of lignin are annually produced by the paper industry, and historically, most of this material is burned to provide energy.3 As a nonedible and renewable biomass, lignin is a potentially valuable feedstock to produce organic compounds, especially high-value aromatics.4,5 Efficiently depolymerizing lignin into well-defined aromatics represents a key challenge that limits the valorization of lignin.6 In the past decade, increasing attention has been paid to the catalytic depolymerization of lignin in academia and industry.7–9 Many efforts have focused on utilizing and manipulating the inherent structure and functionality of lignin,10,11 especially the most abundant type, β-O-4 linkages, because they make up approximately 50% of all linkages, making them key to any depolymerization strategy.4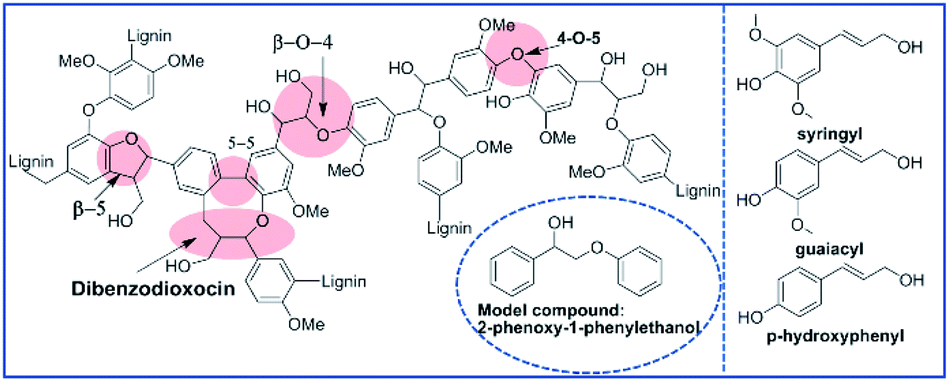 | ||
| Fig. 1 The basic structural units and connections in lignin. | ||
The best strategy for the depolymerization of lignin is still under debate. Traditional strategies have been based on catalytic cracking, hydrolysis, hydrogenolysis, reduction and oxidation.12–16 Of these strategies, catalytic oxidization has received substantial attention due to its potential for yielding more highly functionalized monomers or oligomers, which are widely applicable in the chemical industry. Several attempts have been made toward an economically practical catalytic oxidative decomposition of lignin to generate acids or aldehydes using O2 as the oxidant with a heterogeneous catalyst.17,18 In recent years, Au nanoparticles (NPs) supported on metal oxides (Au/MnO2, Au/TiO2, Au/CeO2, etc.) have been investigated to selectively catalyze the aerobic oxidation of alcohols to the corresponding carbonyl compounds.19–21 Very recently, Yang Song, etc. discussed a Au/hydroxide catalyst in which an alcohol was converted to the corresponding carbonyl by a two-electron process.22 To date, however, there has been little discussion of the performance of Au/CeO2 in the catalytic conversion of lignin.
In this paper, we introduce a heterogeneous catalyst, Au/CeO2, for the depolymerization of lignin under molecular oxygen. The high catalytic activity of Au/CeO2 with the lignin model compounds, the study of the reaction mechanism and the application of this catalyst to the conversion of lignin to aromatics are highlighted in this manuscript.
Experimental
Materials and methods
Preparation of CeO2 nanorods.23 First, 25.2 g of NaOH and 1.73 g of Ce (NO3)3·6H2O were dissolved in 70 and 10 mL of deionized water, respectively, in beakers. These two suspensions were then mixed together in a Teflon bottle and stirred for 1 h at room temperature. Then, the Teflon bottle was placed in an autoclave and heated in an oven at 373 K for 72 h. The solid products were recovered by centrifugation, washed, and dried at 343 K for 12 h. Finally, the solid sample was calcined at 873 K in air for 6 h.Preparation of Au/CeO2.24 First, 0.5 g of CeO2 was added to 50 mL of an aqueous solution of 2.2 × 10−3 M HAuCl4 with 0.22 M urea. The suspension was vigorously stirred for 4 h at 353 K and then centrifuged. The solid was washed with deionized water, dried at 343 K overnight and calcined at 573 K in air for 4 h, other metal oxides are treated in the same way.
Catalyst characterization
A 50 mg sample of the catalyst was transferred into a U-shaped quartz reactor and purged with He at 403 K for 3 h. Then, after cooling to ambient temperature, the flowing gas was switched to 5% H2/Ar, and the sample was heated from room temperature to 973 K at a rate of 10 K min−1. The X-ray diffraction (XRD) patterns of the catalyst were recorded using a Rigaku MiniFlex 600 diffractometer system. The morphology of the prepared Au/CeO2 was observed by TEM on a Jeol 2100F electron microscope. X-ray photoelectron spectroscopy (XPS) measurements of the Au/CeO2 were recorded on a Thermo SCIENTIFIC ESCALAB 250Xi. The binding energies were referenced to the C 1s level of adventitious carbon at 284.6 eV. The electron spin-resonance (ESR) signals were recorded on a Bruker EMX-10/12 spectrometer. The settings for the ESR spectrometer were as follows: center field, 3500.00 G; sweep width, 150.00 G; microwave frequency, 9.85 G; power, 3.99 mW; conversion time, 40.0 ms.Catalytic test
All catalytic reactions were carried out in an 80 mL autoclave reactor. First, 0.47 mmol or 0.1 g of substrate, 20 mg of catalyst and 25 mL of methanol were added to a stainless steel autoclave. Then, the reactor was pressurized with 1 MPa O2 and heated to the target temperature under mechanical stirring. The products were analyzed via HPLC and GC-MS (see the ESI†). The conversion and the selectivity of conversion of the substrate were calculated according to the following equations:| Conversion = The mole of converted substrate/The mole of total substrate × 100 (%) |
| Yield = The mole of the product/The total mole of the substrate × 100 (%) |
Results and discussion
Characterizations of the materials
The phase purity and crystal structure of the catalyst were determined by XRD, and the results are displayed in Fig. S1.† Diffraction peaks from the (111), (200), (220), (311), (222), (400), (331) and (420) planes of CeO2 are consistent with a cubic fluorite-type structure (JCPDS Card No. 65-2975). Obviously, the strong diffraction peaks confirm the high crystallinity of the sample. At the same time, no additional peaks were observed, which means that the prepared CeO2 has better phase purity. After loading the Au NPs on the CeO2 support, no Au peaks were observed in the spectrum of 0.88 wt% Au/CeO2, suggesting an ultralow loading of Au, a particle size less than 5 nm, and a good dispersion of the Au NPs on the support. (The actual loading weight content was 0.88 wt%, as determined by ICP-AES analysis).The transmission electron micrographs (Fig. S2†) show the presence of Au NPs with an average particle size of 5 nm on the as-prepared Au/CeO2. The observed lattice spacing of 0.23 nm and 0.31 nm lattice fringes, which correspond to the Au (111) and CeO2 (111) atomic planes, are in good agreement with the XRD results (Fig. S1†).25
The H2-TPR data from the CeO2 nanorods and Au/CeO2 catalysts are shown in Fig. S3.† The pure CeO2 nanorods do not exhibit a reduction peak at 293-773 K.26 For the Au/CeO2 catalyst, significant changes due to the reaction/absorption of hydrogen on the solid were observed. Four reduction peaks, at 398 K, 523 K, 661 K and 869 K, were present. The Au species in the Au/CeO2 catalyst were reduced at 373–473 K. The peak at 398 K is attributed to Au reduction, while the peaks at 523 K and 661 K are attributed to the reduction of surface oxygen by CeO2, and the reduction peak at 869 K is attributed to the lattice oxygen.27 This indicates that the deposition of the Au NPs on ceria remarkably alters the catalytic activity and reducibility of the catalyst.
To further clarify the chemical states of the Au NPs, XPS analysis was conducted, as shown in Fig. S4.† The XPS spectrum of Au 4f displays typical doublet peaks with binding energies of 83.9 eV (Au 4f7/2) and 87.6 eV (Au 4f5/2), indicating that Au is present in its elementary metallic form.28
Metal oxides catalyze the oxidative of lignin model compound
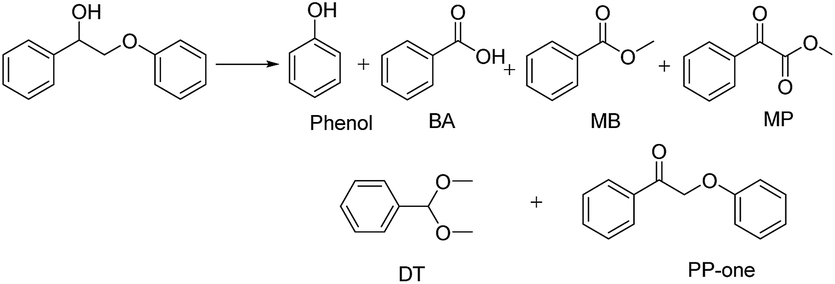
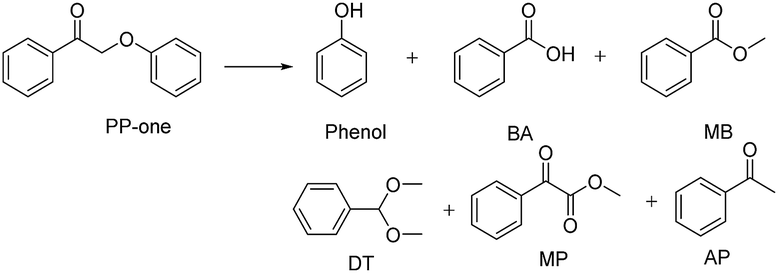
The diffusion limit was eliminated by optimizing the agitation speed before the catalytic experiments (Fig. S5†). We found that when the rotation speed reached 400 rpm, the internal and external diffusion phenomena were controlled.Based on this result, we tested the catalytic activities of several typical metal oxides and metal oxide-supported Au in the oxidative conversion of 2-phenoxy-1-phenylethanol (PP-ol). The results are shown in Table 1. When several of the typical metal oxides or no catalyst were used, the substrate displayed a very low conversion of 0.1–2.7% (Entries 1–5). When the Au NPs were loaded into metal oxides, the PP-ol was effectively converted to benzoic acid (BA), methyl benzoate (MB), phenol, dimethoxytoluene (DT), methyl phenylglyoxylate (MP) and uncleaved product 2-phenoxy-1- phenylethanone (PP-one) (Entries 6–9). Compared to several typical metal oxides (Entries 1–5), Au loaded on a metal oxide significantly accelerates the conversion of PP-ol. Au/CeO2 showed the highest PP-ol conversion (71.5%). Phenol, MB, PP-one and DT were the major products with yields of 45.3%, 31.4%, 20.8% and 6%, respectively (Entry 9). The appearance of MB also demonstrates that the cleavage of the Cα–Cβ bond is accompanied by the breakage of the β-O-4 linkage. Stahl et al. recently developed a novel methodology for the conversion of lignin into aromatic compounds under mild conditions.9,29 This method involves the catalytic conversion of a Cα–hydroxyl group into the ketonic group and the subsequent cleavage of the β-O-4 bond. This strategy was developed by Deng and coworkers.4 Because PP-one was also detected as a product (Table 1, Entry 6–9), we investigated its behavior under catalytic conditions. The results are shown in Table 2.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | Cons. (%) | Yield (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenol | DT | BA | MB | PP-one | MP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: substrate, 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; O2, 1 MPa; 453 K; 4 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Blank | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | SiO2 | <0.1 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Al2O3 | 2.9 | 0.5 | 0 | 0.24 | 2 | 0.9 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Pr6O11 | 1.7 | 0.7 | 0 | 0.1 | 0 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CeO2 | 2.7 | 0 | 0 | 1.7 | 1.3 | 1.7 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Au/SiO2 | 54.8 | 9.5 | 1.9 | 21.2 | 24.6 | 18 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Au/Al2O3 | 62.7 | 45.1 | 14.6 | 15.4 | 4.3 | 14.9 | 7.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Au/Pr6O11 | 44.8 | 25.1 | 6.7 | 26.2 | 4.3 | 17.4 | 14.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Au/CeO2 | 71.5 | 45.3 | 6 | 4 | 31.4 | 20.8 | 6.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalysis | Cons. (%) | Yield (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenol | DT | BA | MB | MP | AP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: PP-one, 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; O2, 1 MPa; 443 K; 2 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Blank | 21 | 15 | 0 | 9.2 | 3.8 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Al2O3 | 23 | 20 | 0 | 8.2 | 4.2 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | SiO2 | 26 | 19 | 0 | 13 | 4.6 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | CeO2 | 76 | 58 | 0 | 2.8 | 56 | 0 | 4.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Au/CeO2 | >99 | 71.1 | 1.5 | 12.3 | 58.6 | 6.8 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
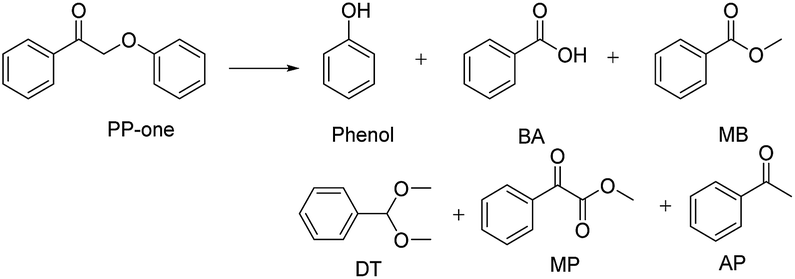
PP-one is transformed into the major products i.e., Phenol, BA and MB, with a conversion of 21% (Table 2) without a catalyst. Compared with PP-ol, it reacted more quickly under the same operating conditions because PP-one also contains a β-O-4 bond, but the Cα–OH moiety is converted to a Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in PP-ol. This reactivity is consistent with the idea that the breakage of the β-O-4 linkage is easier when a CαO is present instead of a Cα–OH moiety.29
O group in PP-ol. This reactivity is consistent with the idea that the breakage of the β-O-4 linkage is easier when a CαO is present instead of a Cα–OH moiety.29
Then, we investigated the oxidative activity of Au/CeO2 catalysts with different Au loadings (the actual loadings (weight content) were determined by ICP-AES analysis). As shown in Fig. 2, when the Au loading was increased from 0.08 wt% to 0.88 wt%, the conversion of PP-ol significantly increased from 25% to 71%. These results suggest that the Au NPs play a key role in the catalytic conversion of the substrate. Au NPs may enrich and activate molecular oxygen, greatly reducing the absorption energy of the reactants, as Wang reported.30 Because Au NPs provide active sites for PP-ol oxidation in the Au/CeO2 systems, increasing the number of Au active sites enhances the catalysis. However, it should be noted that the increase in the PP-ol conversion and the yield of Phenol and MB decreased as the Au loading exceeded 0.88 wt%. Moreover, the yield of DT increased significantly with increasing Au loading, and DT may be an intermediate in repolymerization, making it undesirable in the reaction system.
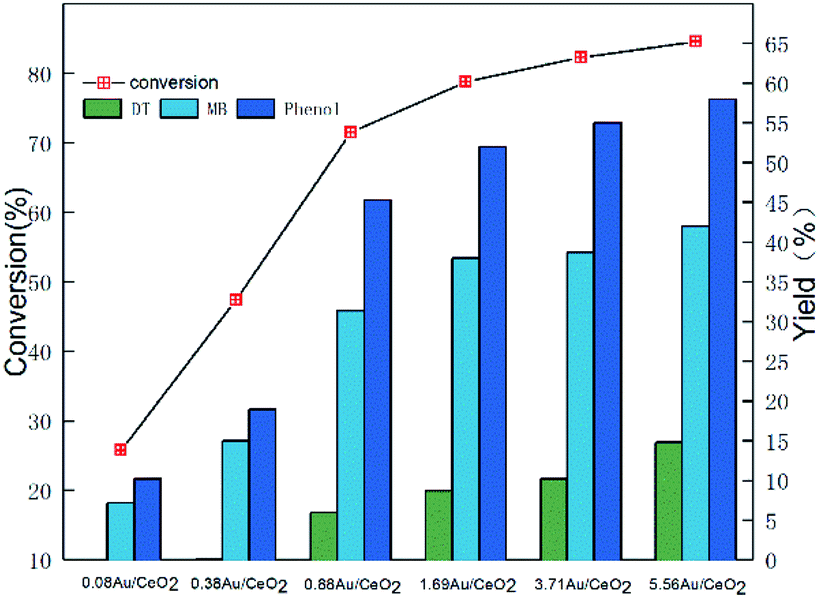 | ||
| Fig. 2 Effect of Au loadings on the catalytic behaviors of Au/CeO2 for the oxidation of PP-ol. Reaction conditions: PP-ol, 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; O2, 1 MPa; 453 K; 4 h. | ||
Both β-O-4 bonds and methoxy groups at various substitution positions are abundant in the aromatic units of lignin, and those methoxy groups may influence the activation of the β-O-4 linkages. Therefore, we investigated the oxidative performance of Au/CeO2 for the catalytic transformation of substituted PP-ol at 453 K. As displayed in Table 3, the substituted compound is more reactive than PP-ol. When a CH3O moiety was present, more than 90% the model compound was converted to the corresponding esters and phenols. This result indicates that Au/CeO2 performs well in the catalytic oxidative cleavage of β-O-4 in the lignin model compounds.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Cons. (%) | Yield (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MB | BA | Phenol | DB | DT | Ketone | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: substrate, 0.47 mmol; Au/CeO2, 20 mg; CH3OH, 25 mL; O2, 1 MPa; 453 K; 4 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
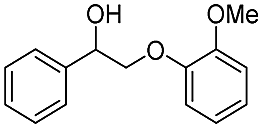
|
>99 | 82.5 | 1.7 | 85.3 | 0 | 1.9 | 6.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
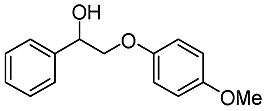
|
98.2 | 73.7 | 5.6 | 72 | 0 | 3.7 | 10.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
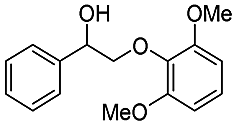
|
94.7 | 60.7 | 4.3 | 1.7 | 70.2 | 6.7 | 15.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The stability of the Au/CeO2 catalyst for the oxidative conversion of PP-ol was investigated (Fig. S7†). The catalyst could be recycled four times without a significant decline in its oxidative activity, and its product distribution remained stable. The reduction in the conversion may be attributed to an increase in the size of the Au NPs or a change in the catalyst surface properties, such as the presence of adsorbed products on the Au/CeO2 or the oxidization of the catalyst surface.
Reaction mechanism for the oxidation of PP-ol
To elucidate the reaction pathway, we examined the oxidation of PP-ol over time on the Au/CeO2 catalyst, and the results are shown in Fig. 3. As Fig. 3 shows, PP-one was the main product in the initial stage of the reaction with ∼80% selectivity. As the oxidation progressed, the selectivity for PP-one rapidly decreased, and the selectivities for Phenol, DT and MB increased. After 4 h, the difference in selectivity became less significant. Subsequently, we calculated the concentration of the products, and depicted the changing trend of products concentration vs. time, as Fig. S6† shows. These findings support the hypothesis that the reaction proceeded through PP-one as an intermediate, and PP-one was further oxidized over the Au/CeO2 catalyst. These results further indicate that the Au NPs play a key role in the oxidation of PP-ol by catalyzing the conversion of Cα-OH to Cα = =O. A carbonyl compound was formed, and cleavage of the β-O-4 and Cα−Cβ bonds subsequently occurred. Such a preoxidation process may reduce the energy barrier for the breakage of the β-O-4 linkage. Gregg T. Beckham et al. previously reported that by converting the Cα-hydroxyl group to a Cα-carbonyl, the β-O-4 bond dissociation enthalpy was reduced from 69.5 to 60.6 kcal mol−1.31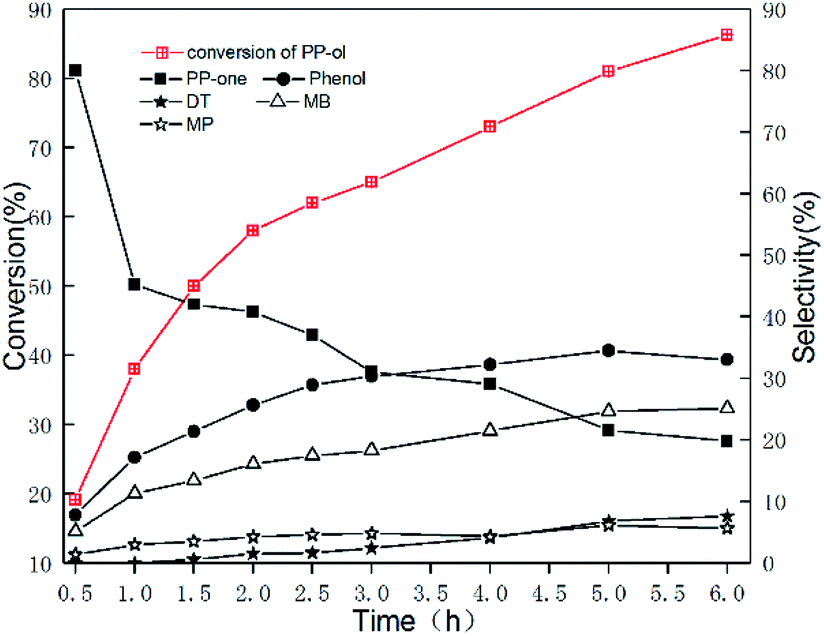 | ||
| Fig. 3 Time course for the oxidation of PP-ol catalyzed by Au/CeO2. Reaction conditions: 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; O2, 1 MPa; 453 K. | ||
To further investigate the role of O2, we probed the effect of O2 pressure in the catalytic system. We observed a 29% transformation of PP-ol with 0.1 MPa O2 (Table 4, entry 1). PP-one, Phenol and MB were the major products. An increase in O2 pressure efficiently promoted the conversion, suggesting that O2 facilitates the breakage of both the Cα–Cβ and β-O-4 bonds (Table 4, Entry 2).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalysis | Cons. (%) | Yield (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenol | DT | BA | MB | MP | PP-one | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: PP-ol, 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; 453 K; 4 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| O2 (0.1 Mpa) | 29 | 15.1 | 4.90 | 5.1 | 5.5 | 0 | 8.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| O2 (1 Mpa) | 71.5 | 45.3 | 2 | 4 | 31.4 | 6.7 | 20.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 (1 Mpa) | 0.7 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When N2 was employed, only a 0.7% conversion of PP-ol (Table 4, Entry 3) was observed. This finding suggests that Au/CeO2 cannot catalyze this reaction in the presence of N2, but the cleavage of the β-O-4 linkage could occur in the absence of O2.
It was also confirmed that PP-one could more readily than PP-ol undergo this transformation because PP-one contains a β-O-4 bond adjacent to a CαO moiety, while PP-ol has a Cα–OH group. However, we observed 17% conversion of PP-one in the presence of N2 (Table 5, Entry 3), which is obviously lower than the conversions (56%, >99%) under O2 (Table 5, Entry 1 and 2). Interestingly, phenol and acetophenone were the major products, and they were obtained in yields of 15.2 and 14%, respectively. The appearance of MB confirms that the β-O-4 linkage was cleaved.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalysis | Cons. (%) | Yield (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenol | DT | BA | MB | MP | AP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: PP-one, 0.1 g (0.47 mmol); catalyst, 20 mg; CH3OH, 25 mL; 443 K; 2 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| O2 (0.1 Mpa) | 56 | 39 | 0.7 | 16.9 | 24 | 5 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| O2 (1 Mpa) | >99 | 71.1 | 1.5 | 12.3 | 58.6 | 6.8 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 (1 Mpa) | 17 | 15.2 | 0 | 0 | 0 | 0 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Radical trapping experiments
p-Benzoquinone is a scavenger of superoxide radicals,32 and it was added to the reactor along with the substrate, and the results are presented in Table 6.As displayed in Table 6, the conversion of PP-ol reached 71.5% (entry 1) in the absence of p-benzoquinone. However, when benzoquinone was added to the system, the conversion of PP-ol dropped dramatically (Entry 2–4) from 10.7% to 0.1% with increasing loading of the radical scavenger. When PP-one was used as the substrate, similar results were observed. These results demonstrated that a radical species played a vital role in the catalytic oxidation of the lignin model compounds; thus, we speculate that the oxidation occurred via a free radical process.
To verify this hypothesis, more mechanistic information was obtained by identifying the surface intermediates using in situ liquid-phase ESR spin-trapping experiments, as Fig. 4 shows. Data were acquired from a suspension of 0.1 g Au/CeO2 sample in methanol with DMPO and oxygen. An obvious 6-fold signal characteristic of the superoxide radical anion was observed in the ESR data, suggesting that oxygen is activated by Au/CeO2 to form superoxide anion free radicals.28,33 Therefore, these experiments confirm that the Au/CeO2 catalyst can strongly activate oxygen.
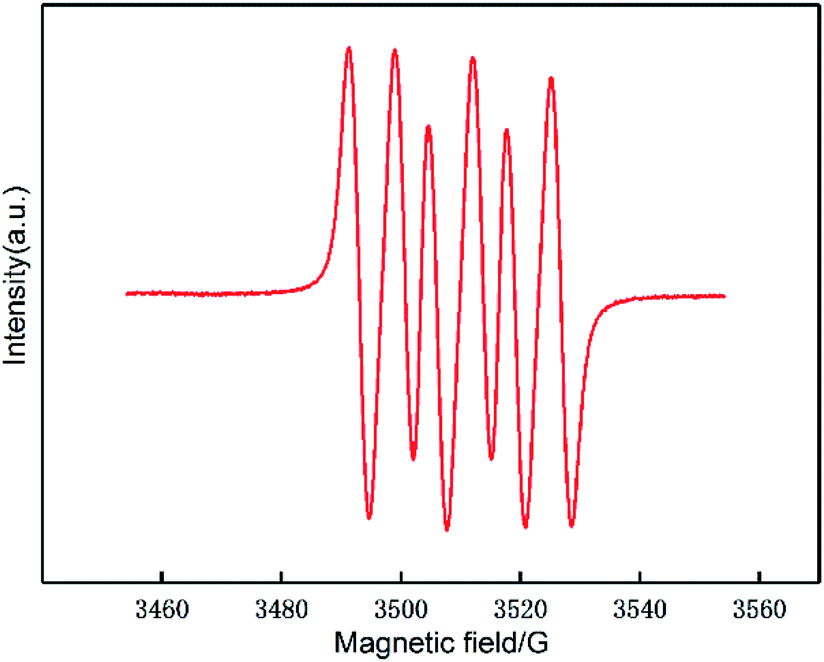 | ||
| Fig. 4 ESR spectrum of DMPO-O2˙− recorded in a system containing methanol, the catalyst and O2 at 453 k. | ||
Employing molecular oxygen as the oxidant is green, economical, and attractive from an environmental viewpoint.34 Efficiently catalyzing the reactions between inert, ground triplet-state O2 (3ΣgO2) and organic molecules (mainly in singlet-state) is key to this process. Substantial effort has been devoted to activating O2 into active oxygen species.35 Au NPs show a unique ability to generate superoxide radicals (O2˙−) through electron transfer processes.36
Tsukuda et al. reported that molecular oxygen is activated through negatively charged Au NPs to form a peroxo species or superoxide, which can then activate C–H bonds.37 As Woodham et al.38 demonstrated, electrons can be added to the O2 π* orbital of O2via electron donation from loaded Au NPs, generating elongated O–O bonds that closely resemble those of the superoxo-like (O2−) adsorbate state. The oxidation process is subsequently accomplished through a series of deprotonation and elimination steps, i.e., adsorbed molecular O2 obtains an electron from Au/CeO2 to generate Au/CeO2–O2˙−, which then reacts with PP-ol to form PP-one. Based on the results presented and discussed above, a possible mechanism for the oxidation of PP-ol over the Au/CeO2 catalyst is proposed in Scheme 1.
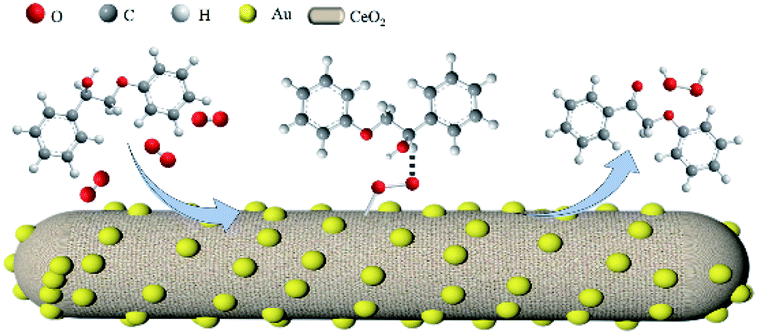 | ||
| Scheme 1 Proposed mechanism for the aerobic oxidation of PP-ol over Au/CeO2. | ||
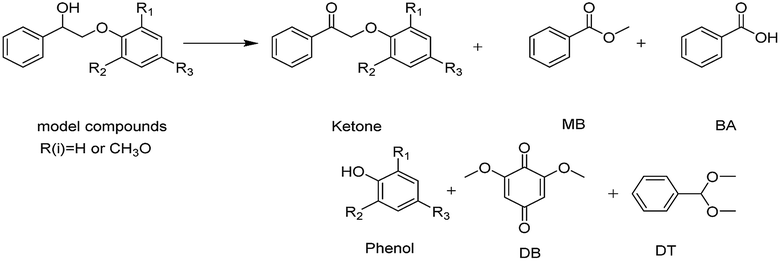
To investigate the breakage of the β-O-4 linkage, the oxidation of several potential intermediates by Au/CeO2 in an O2 atmosphere at 453 K was explored (Fig. 5). First, phenylglyoxal can be fully converted to the corresponding breakage products, i.e., Phenol, DT, BA and MB (eqn (1) in Fig. 5). These products are in good agreement with the products of the Au/CeO2-catalyzed oxidation of PP-ol (Table 1). Because phenylglyoxal was also detected as a minor product by GC-MS, phenylglyoxal may be the intermediate in the oxidation, and it would presumably be produced from the breakage of β-O-4 linkage. Second, benzaldehyde showed a low conversion (10.2%) to BA, MB and DT (eqn (2)). Benzaldehyde is stable under the reaction conditions, suggesting that little of its Cα–Cβ bond was broken. BA also showed a low transformation (24.4%) to MB (eqn (4)), indicating that MB was mainly formed from phenylglyoxal (eqn (1)). Third, methyl phenylglyoxylate showed a conversion of 32.9% under these conditions, and the major products were MB, MA, benzaldehyde and DT (eqn (3)). CO2 was also released by this reaction (Fig. S8†). These results indicate that Cβ is transformed via two pathways, including Cα–Cβ bond and Cβ–O bond breakage to CO2 and methyl formate. Methyl phenylglyoxylate is stable in the reaction, meaning that it is not a possible intermediate.
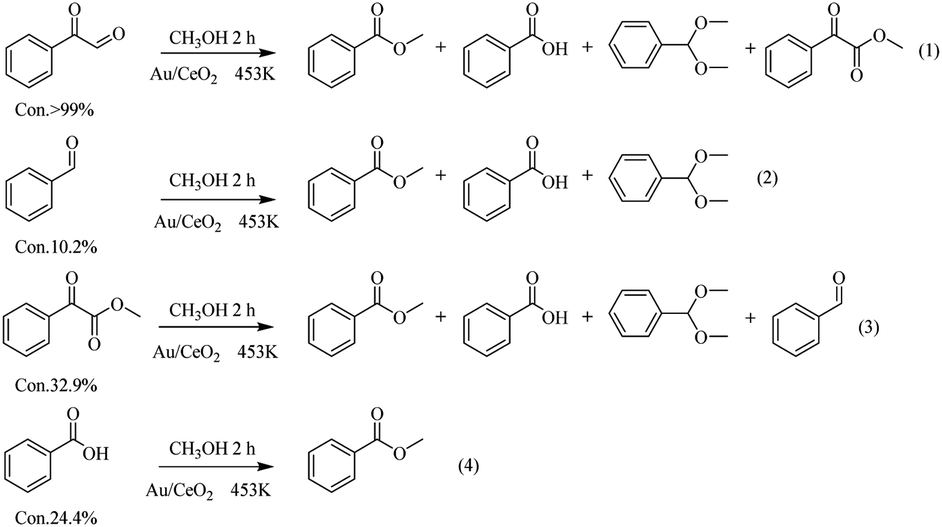 | ||
| Fig. 5 Catalytic conversions of the possible intermediates under an O2 atmosphere over Au/CeO2. | ||
Based on these results, a possible cleavage process for the oxidation was proposed (Scheme 2). In short, the Cα–hydroxyl group of PP-ol is primarily oxidized a Cα–carbonyl via the catalytic action of Au NPs. The subsequent oxidation of the β-O-4 linkage of PP-one over the Au/CeO2 catalyst via O2˙− could afford phenylglyoxal, methyl phenylglyoxylate and phenol. The catalytic breakage of the β-O-4 linkage over CeO2 affords an oxidized intermediate (e.g., phenylglyoxal) and phenol. A small amount of methyl phenylglyoxylate could also be produced in this process. The catalytically generated intermediate may undergo rapid oxidative cleavage of the C–C bond, generating BA and then MB, benzaldehyde and ultimately DT.
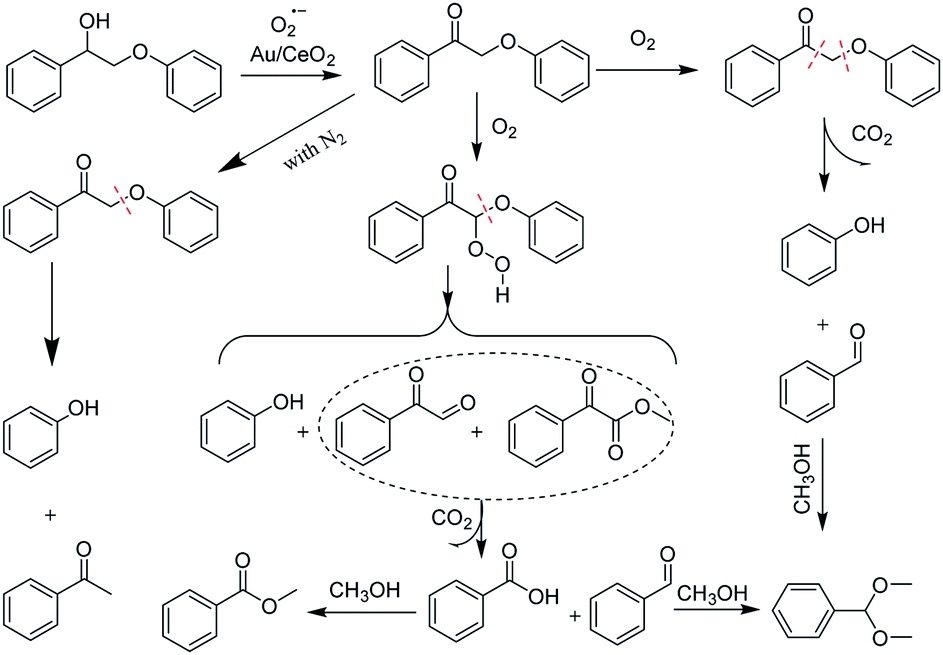 | ||
| Scheme 2 Possible reaction mechanism for the oxidative conversion of PP-ol. | ||
Our observations and mechanistic studies demonstrate the vital role of the preoxidation of the Cα–hydroxyl group. In our earlier research,4 we loaded Au NPs onto commercial CeO2 from Alfa Aesar. This material could also catalyze the transformation of PP-ol into monomeric aromatic compounds but a lower conversion than that achieved over Pd/CeO2. However, when we changed the support to CeO2 nanorods via a hydrothermal method and loaded Au NPs (<5 nm) on these rods, the prepared catalyst displayed excellent performance.
Au/CeO2-catalyzed conversion of real lignin
The oxidative conversion of organosolv lignin by the Au/CeO2 catalyst system was subsequently investigated. Lignin was extracted via the ethyl alcohol-based organosolv process from cotton stalk according to a previous report. Then, the organosolv lignin was subjected to catalytic oxidation in methanol under O2 with the Au/CeO2 catalyst at 453 K for 4 h.The results are displayed in Fig. 6. Several monomeric aromatic compounds, including vanillin (10.5 wt%), methyl vanillate (6.8 wt%), 2,6-dimethoxy-1,4-benzoquinone, methylene syringate (3.4 wt%) and a ring-opened compound, were detected and quantified by GC-MS. According to earlier reports, LaMnO3 and LaCoO3 are highly active for the catalytic oxidation of lignin to aromatic aldehydes, and they offer high yields of vanillin (∼5%) and syringaldehyde (∼10%).39,40 Perovskite-type LaFe0.8Cu0.2O3 materials have also been studied for the wet catalysis of lignin, and they provided maximum yields of vanillin and 4-hydroxylbenzaldehyde of 4.56 wt% and 2.49 wt%, respectively. However, an erosive strong base was required in these methods. Recently, a Pd/Al2O3 catalyst was reported for the oxidative conversion of alkaline lignin, affording only vanillin in a yield of 1.6 wt%.41 Yang et al. investigated Au/Li–Al LDH materials, which showed excellent activity in the oxidation of natural lignin, and a 40 wt% yield of aromatic compounds was recorded from GVLox, while KLox afforded 10 wt% of desired compounds.22 Our present results showed that Au/CeO2 is also a promising material for the depolymerization of lignin to aromatic monomers in the absence of an erosive strong base or acid.
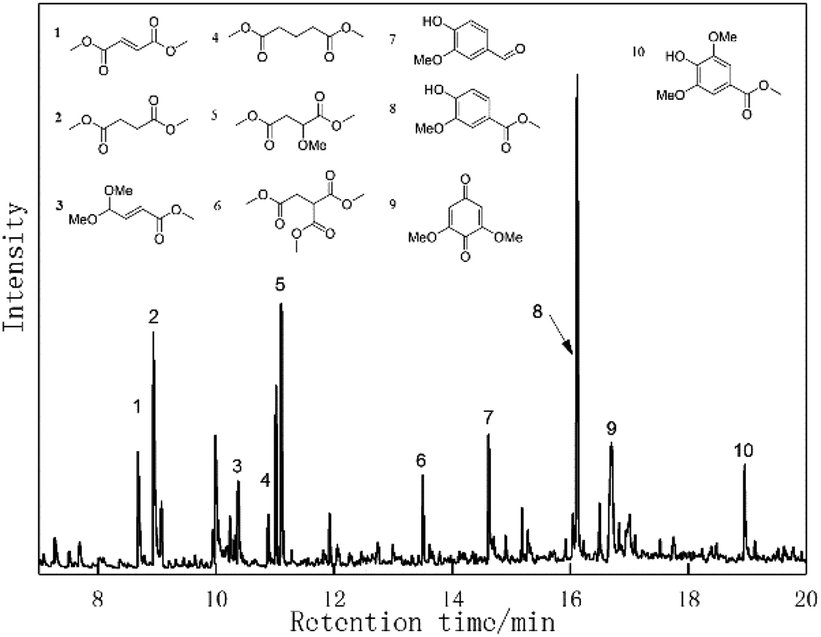 | ||
| Fig. 6 GC-MS of products from Au/CeO2-catalyzed organosolv lignin. | ||
Conclusions
Au/CeO2 was found to be a competent and environmentally friendly heterogeneous catalyst for the oxidation of model compounds and organosolv lignin using O2 as the oxidant and methyl alcohol as the solvent. The ESR data suggests that a superoxide anion plays a vital role in the catalytic system. The highest conversion (95.3%) was obtained for the oxidation of 2-phenoxy-1-phenylethanol at 453 K with 1 MPa O2 in 4 h. Phenol, methyl benzoate, 2-phenoxy-1-phenylethaone and dimethoxytoluene were the major products with yields of 45.3 wt%, 31.4 wt%, 20.8 wt% and 6 wt%, respectively. The Au/CeO2 materials could also be employed for the direct oxidation of lignin, producing vanillin, methyl vanillate and methylene syringate in relatively high yields. Based on the above results, Au/CeO2 is a promising catalyst system for the conversion of lignin to value-added low-molecular-weight aromatics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (21663002), and Education Department of Xinjiang Province (XJEDU2016I047).Notes and references
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- R. Gao, Y. Li, H. Kim, J. K. Mobley and J. Ralph, ChemSusChem, 2018, 11, 2045–2050 CrossRef CAS.
- H. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS.
- Y. Liu, W. Chen, Q. Xia, B. Guo, Q. Wang, S. Liu, Y. Liu, J. Li and H. Yu, ChemSusChem, 2017, 10, 1692–1700 CrossRef CAS.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- T. Yoshikawa, T. Yagi, S. Shinohara, T. Fukunaga, Y. Nakasaka, T. Tago and T. Masuda, Fuel Process. Technol., 2013, 108, 69–75 CrossRef CAS.
- A. Das, A. Rahimi, A. Ulbrich, M. Alherech, A. H. Motagamwala, A. Bhalla, L. da Costa Sousa, V. Balan, J. A. Dumesic and E. L. Hegg, ACS Sustainable Chem. Eng., 2018, 6, 3367–3374 CrossRef CAS.
- M. Nagy, K. David, G. J. Britovsek and A. J. Ragauskas, Holzforschung, 2009, 63, 513–520 CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. Bjelić, M. Grilc and B. Likozar, Chem. Eng. J., 2018, 333, 240–259 CrossRef.
- S. G. Yao, J. K. Mobley, J. Ralph, M. Crocker, S. Parkin, J. P. Selegue and M. S. Meier, ACS Sustainable Chem. Eng., 2018, 6, 5990–5998 CrossRef CAS.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. García, Tetrahedron, 2006, 62, 6666–6672 CrossRef CAS.
- L.-C. Wang, Y.-M. Liu, M. Chen, Y. Cao, H.-Y. He and K.-N. Fan, J. Phys. Chem. C, 2008, 112, 6981–6987 CrossRef CAS.
- L. M. Dias Ribeiro de Sousa Martins, S. A. C. Carabineiro, J. Wang, B. G. M. Rocha, F. J. Maldonado-Hódar and A. J. Latourrette de Oliveira Pombeiro, ChemCatChem, 2017, 9, 1211–1221 CrossRef.
- Y. Song, J. K. Mobley, A. H. Motagamwala, M. Isaacs, J. A. Dumesic, J. Ralph, A. F. Lee, K. Wilson and M. Crocker, Chem. Sci., 2018, 9, 8127–8133 RSC.
- J. He, T. Xu, Z. Wang, Q. Zhang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2012, 51, 2438–2442 CrossRef CAS.
- R. Zanella, L. Delannoy and C. Louis, Appl. Catal., A, 2005, 291, 62–72 CrossRef CAS.
- Y. Liu, B. Liu, Q. Wang, Y. Liu, C. Li, W. Hu, P. Jing, W. Zhao and J. Zhang, RSC Adv., 2014, 4, 5975–5985 RSC.
- M. F. Luo, Y. J. Zhong, X. X. Yuan and X. M. Zheng, Appl. Catal., A, 1997, 162, 121–131 CrossRef CAS.
- X. Hong, Y. Sun, T. Zhu and Z. Liu, Catal. Sci. Technol., 2016, 6, 3606–3615 RSC.
- Z. Cui, W. Wang, C. Zhao, C. Chen, M. Han, G. Wang, Y. Zhang, H. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 31394–31403 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS.
- Z. Wang, J. Qi, K. Zhao, L. Zong, Z. Tang, L. Wang and R. Yu, Mater. Chem. Front., 2017, 1, 1629–1634 RSC.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem. - Eur. J., 2006, 12, 4844–4857 CrossRef CAS.
- Y. Ren, Y. Che, W. Ma, X. Zhang, T. Shen and J. Zhao, New J. Chem., 2004, 28, 1464–1469 RSC.
- M. Liu and C. J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806–10810 CrossRef CAS.
- H. Su, K.-X. Zhang, B. Zhang, H.-H. Wang, Q.-Y. Yu, X.-H. Li, M. Antonietti and J.-S. Chen, J. Am. Chem. Soc., 2017, 139, 811–818 CrossRef CAS.
- A. Roldán, J. M. Ricart, F. Illas and G. Pacchioni, J. Phys. Chem. C, 2010, 114, 16973–16978 CrossRef.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2013, 47, 816–824 CrossRef.
- A. P. Woodham, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 4444–4447 CrossRef CAS.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, J. Li and S. Liu, Energy Fuels, 2008, 23, 19–24 CrossRef.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, Z. Li and S. Liu, Catal. Lett., 2008, 126, 106 CrossRef CAS.
- J. Zhang, H. Deng and L. Lin, Molecules, 2009, 14, 2747–2757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, XRD and XPS; lignin model compounds synthesis and cycling tests on Au/CeO2. See DOI: 10.1039/c9ra04838c |
| This journal is © The Royal Society of Chemistry 2019 |