
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic primary alcohol oxidation on WO3 nanoplatelets†
Kori D. McDonald and
Bart M. Bartlett *
*
Department of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan 48109-1055, USA. E-mail: bartmb@umich.edu
First published on 11th September 2019
Abstract
With the aid of direct heating through microwave irradiation in non-aqueous media, nanocrystalline tungsten(VI) oxide is achievable in 30 minutes at 200 °C, faster and at a lower temperature than conventional synthesis methods. Forming in a platelet morphology, these particles are as small as 20 nm with a BET surface area of 37 m2 g−1 WO3. These nanoplatelets are active for the photocatalytic oxidation of the 1° alcohols benzyl alcohol (rate constant, k of 2.6 × 10−3 h−1) and 5-(hydroxymethyl)-2-furfural (k of 0.01 h−1) using 10 mg of WO3 with 2 mL of 0.250 M substrate in acetonitrile and a 150 mW cm−2 460 nm blue LED source. As expected, these rate constants are larger than those observed for commercially prepared, micron-sized WO3. XPS analysis shows that during catalysis, the concentration of W5+ on the surface increases, but the nanoplatelets are stable under these reaction conditions. The overall morphology and size of the particles are retained through the reactions. Moreover, the nanoplatelets are recyclable—showing no loss in activity for four reaction cycles.
Introduction
Transforming organic substrates into value-added chemicals has long been researched through forming carbon–carbon and carbon–nitrogen bonds. Using sources of visible light, these reactions have improved with the help of many homogeneous transition metal photocatalysts.1–3 While these routes prove feasible for achieving high reactivity, limitations arise as many known catalysts contain toxic and expensive metals such as ruthenium or iridium. In addition, their homogeneity, and thus complex removal, hinders any larger scale utility. As such, heterogeneous metal oxide catalysts present themselves as an alternative route to achieving the same reactivity.4,5 Many offer long-term photostability and easy filtering to prevent the leaching and loss of expensive metals.Many metal oxides are highly reactive towards photocatalyzed transformations of organic substrates. Among them, tungsten oxides have emerged as a prominent photocatalyst capable of performing various energy and environmentally relevant transformations. Our group and many others have explored the usefulness of tungsten(VI) oxide (WO3), a well-studied visible light absorbing semiconductor with a band gap of 2.7–2.8 eV, for its ability to photochemically6,7 and photoelectrochemically8–10 oxidize water and degrade organic dyes. Using the knowledge gained from these known reactions as a guide, we are now beginning to explore the controlled oxidation of organic molecules.
In addition to implementing metal oxide catalysts, recent work has shown that for the same catalyst composition, using nanoscale particles can lead to improved rates of photocatalytic reactions.11,12 The rate enhancement can be attributed to the increase in the specific surface area of the material and the decrease in electron–hole transport distance photogenerated charge carriers must diffuse before reaching the particle surface. Attempts have been made to synthesize many binary oxides on the nanoscale, though many routes are time consuming, energy demanding, and require a post-synthesis heat treatment to achieve a crystalline phase, which often results in sintering (and thus increased particle size and potential loss of important morphology).13–16 To avoid these drawbacks, we adapt a known microwave-assisted synthesis of crystalline monoclinic WO3 nanoparticles prepared in benzyl alcohol solvent.17 The direct heating afforded by microwave irradiation results in faster heating rates and a more homogeneous heat distribution in the reaction solution to impart control over the nucleation and growth of the desired material.18–24
Moreover, the non-aqueous solvents act as surfactants and control particle size and shape during heating in microwave reactions.22,25,26 Efforts have already been directed towards forming metal oxides through this combined route, though directly comparing how the synthesis conditions affect organic photocatalysis has seldom been explored.
Herein, we show that under visible light illumination, the photocatalytic oxidation of benzyl alcohol and 5-(hydroxymethyl)-2-furfural (HMF) proceeds at faster rates on the surface of WO3 nanoplatelets than on the bulk material. Then, the HMF substrate shows the promise of WO3 for transforming biomass-derived feedstocks in value-added chemicals.
Experimental section
Materials and methods
Tungsten(VI) chloride (99.9%) was purchased from Strem Chemicals. Chlorobenzene (99+%, for spectroscopy), and tungsten(VI) oxide (99.9%) were purchased from Sigma-Aldrich. Acetonitrile (Certified ACS), dichloromethane (Certified ACS), ethanol (200 proof), benzyl alcohol (Certified ACS), and 5-(hydroxymethyl)-2-furfural (98%) were purchased from Fisher Scientific. Acetonitrile-d3 was purchased from Cambridge Isotope Laboratories. Benzyl alcohol was vacuum distilled and stored on sieves under N2. All other reagents were used as received. Nylon syringe filters (25 mm, 0.2 μm membrane) were purchased from VWR or Macherey-Nagel.Microwave synthesis
In a typical synthesis, 0.5 mmol of tungsten hexachloride was added to 10 mL of stirring benzyl alcohol in a glass microwave vial with an inner volume of 30 mL in an N2-filled glovebox (Vacuum Atmospheres). The dissolved powder formed a red solution that proceeded to turn blue with further stirring. The glass microwave vessel was then sealed with a PTFE-lined cap and taken out of the glovebox. The microwave experiments were conducted using an Anton Paar Monowave 400 synthesis reactor. During microwave heating, stirring was maintained with a magnetic stir bar in the reaction mixture at 800 rpm. A movie of the reaction at 10 frames per s is included as Movie S1.† The organic reaction was induced by automatic adjustments to the microwave power to rapidly heat the solution to 200 °C (Fig. S1†). This temperature is held constant for 30 minutes before being quenched with compressed air and cooled to room temperature. The temperature and pressure are controlled and maintained by an internal IR thermometer and pressure sensor, respectively. After microwave irradiation, the resulting precipitate is collected by centrifugation and thoroughly washed with ethanol (2×) and chloroform (2×) under ambient conditions. Yield: 0.43 mmol WO3, 87% based on starting WCl6. The initial reaction mixture is saved for GC-MS analysis to observe the organic by-products. The powders were dried in a vacuum oven (Fisher Scientific) at 60 °C overnight.Material characterization
Powder X-ray diffraction (XRD) data were collected on a Panalytical Empyrean diffractometer at a power of 1.8 kW (45 kV, 40 mA) with Cu Kα (λ = 1.5418 nm) radiation. The detector was a X'Celerator Scientific, a position sensitive 1D detector equipped with Bragg–Brentano HD X-ray optic delivering only Kα radiation. Patterns were collected with a sampling step of 0.020° and a scan rate of 0.080° s−1 while spinning the sample stage at a rate of 0.25 Hz. Transmission electron microscopy (TEM) was performed on a JEOL 3011 high-resolution electron microscope operated at 300 kV. The samples were ground with a mortar and pestle and then suspended in ethanol. One drop of this suspension was deposited on a carbon grid. The ethanol was allowed to evaporate before sample imaging. Gas chromatography coupled to mass spectrometry (GC-MS) was performed on a Shimadzu QP-2010 GCMS quadrupole. Brunauer–Emmett–Teller (BET) surface area measurements of the catalysts were obtained on a Quantachrome NOVA4200e. A Varian Cary 5000 spectrophotometer equipped with an external diffuse reflectance accessory was used for UV-vis measurements. These reflectance data were transformed into absorbance using the Kubelka–Munk function. Barium sulfate was used as a 100% reflectance standard. Photoluminescence (PL) spectra were collected using a Horiba Quanta Master fluorometer with a Xe-arc lamp as the excitation source. Electron paramagnetic resonance (EPR) spectroscopy was performed using a Bruker EMX electron spin resonance spectrometer with a Bruker 4102-ST cavity. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Axis Ultra using a monochromatic Al source. Collected spectra were corrected for charging by referencing the C(1s) peak to 284.8 eV. All peaks were fitted in Casa XPS with the Shirley-type background.Photocatalytic activity tests
All photocatalytic reactions, unless otherwise stated, were carried out using narrow linewidth blue LEDs (460 nm) with an irradiance of ∼150 mW cm−2 (1.5-sun). Reactions were run in 4 mL glass dram vials containing 10 mg of catalyst and 2 mL of 250 mM substrate solution in acetonitrile. Reactions were capped with a rubber septum and kept under an O2 atmosphere using balloons. A fan was used to keep the reaction at room temperature, measured with thermometer in a control acetonitrile solution (Fig. S2†). Prior to irradiation, the solutions were stirred for an hour in the dark to establish an adsorption–desorption equilibrium before having an aliquot taken out to determine the initial concentration of substrate in each solution. To confirm the importance of an O2 atmosphere, control experiments were performed in an N2-filled glovebox (Vacuum Atmospheres), using the above described LED set-up. Recyclability experiments were conducted in observation of the photocatalytic oxidation of benzyl alcohol. In each successive cycle, the reaction solution was illuminated for 24 hours. Afterwards, the solution was separated by centrifugation and the remaining powder was washed with acetonitrile (3×) to remove any residual benzyl alcohol. The powder was finally allowed to dry in a 60 °C vacuum oven before being used further. Reaction progression was monitored by GC with a flame ionization detector on a Trace 1310 GC-FID system with a TG-5MS Amine column. Argon was used as the carrier gas. Data were collected with chlorobenzene as an internal standard and were then converted to first order rate plots. Selectivity data was collected using 1H NMR spectroscopy with dichloromethane as an internal standard in acetonitrile-d3. Before NMR analysis, each reaction was filtered through a nylon syringe filter to remove the catalyst. 1H NMR data was collected on a Varian MR400 NMR spectrometer equipped with a Varian 5 mm PFG AutoX Dual Broadband probe.Results and discussion
(A) Synthesis, phase, and morphology
Similar to a previously reported example,17 crystalline WO3 is formed through rapid heating of soluble tungsten hexachloride in benzyl alcohol solvent at 200 °C for 30 minutes with stirring under microwave irradiation with no further annealing. This material is denoted as MW-WO3. The powder X-ray diffraction pattern for the as-synthesized powder is shown in Fig. 1a.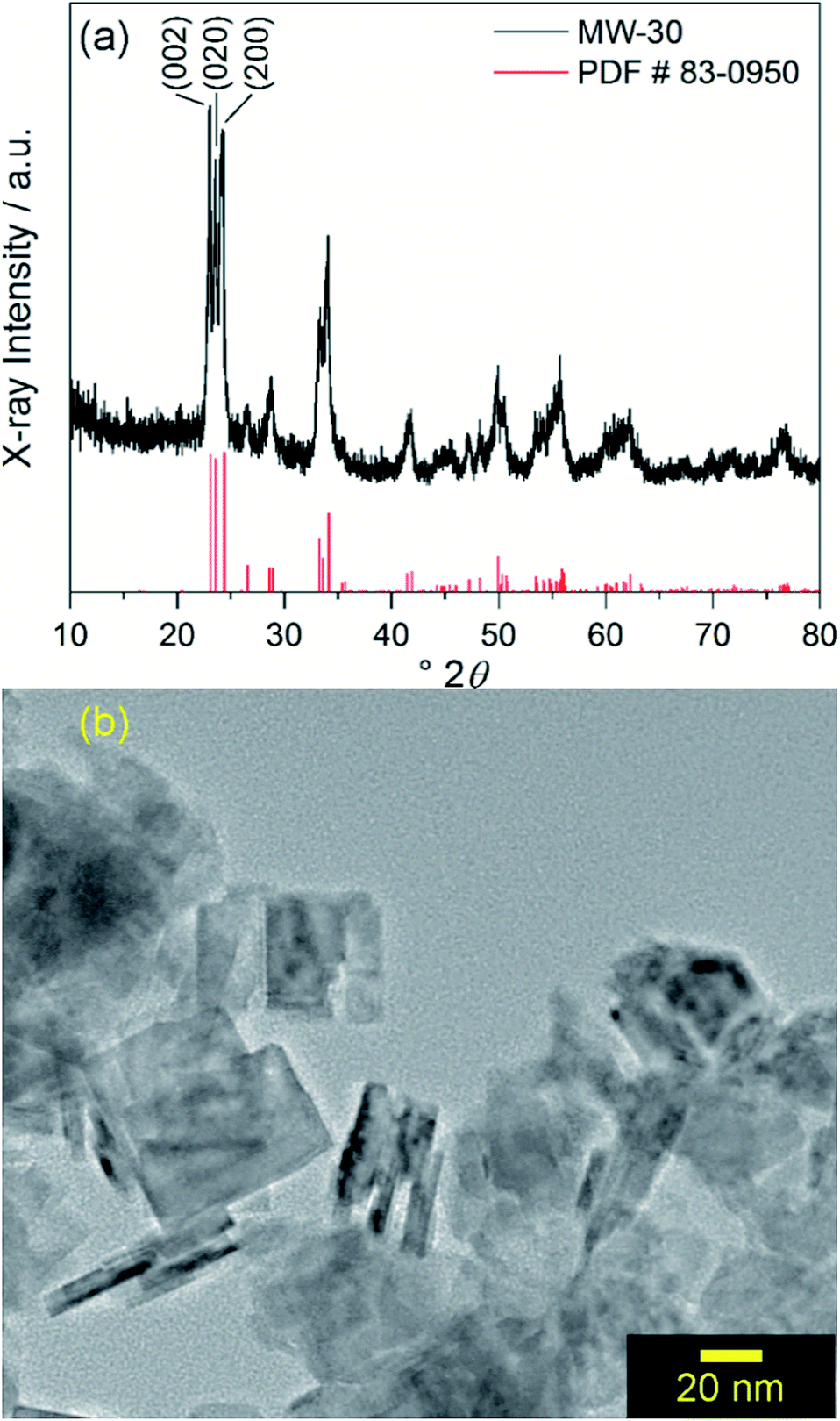 | ||
| Fig. 1 (a) Powder XRD pattern of MW-WO3 nanoplatelets (black) and reference pattern (red) corresponding to monoclinic WO3. (b) TEM image of MW-WO3 nanoplatelets. | ||
All diffraction peaks can be assigned to the monoclinic phase of WO3 (PDF #83-0950). The average domain size was calculated to be 20 nm with the Scherrer equation using the (002) reflection. Transmission electron microscopy (TEM) shows that the particle size is consistent with this domain size, with a platelet morphology of the particles, along with a 5–10 nm thickness (Fig. 1b). Slight agglomeration from the lack of stabilizing surfactants is observed, though the individual grains are clear. Nitrogen sorption isotherms reveal a BET surface area of 37 m2 g−1 for the particles, which is notably larger than that of commercially available nanocrystalline WO3 (only 2.6 m2 g−1).
We observe oxide formation from benzyl alcohol, which acts as solvent and reactant, analogous to results found in previous solvent assisted syntheses.17,22,25 GC-MS analysis (Fig. S3†) performed on the reaction solution after removal of the suspended particles supports the common mechanisms of halide elimination and either elimination with benzyl chloride, residual benzyl alcohol, and dibenzyl ether observed at retention times 5.72 min, 5.95 min, and 10.43 min respectively. The identity of each by-product was confirmed and referenced by the corresponding mass spectra for each (Fig. S4†). The reaction proceeds through the mechanisms illustrated in Scheme 1. Integrating the GC data shows that these two by-products form in a ratio of 12.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dibenzyl ether to benzyl chloride.
:1 dibenzyl ether to benzyl chloride.
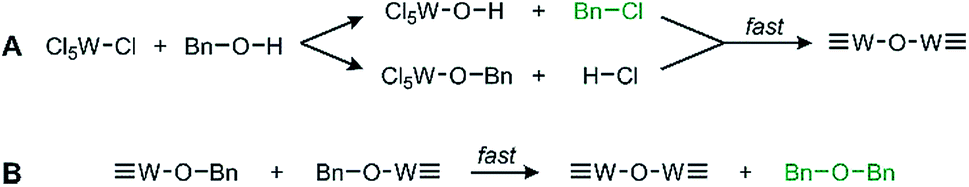 | ||
| Scheme 1 General proposed routes of WCl6 and BnOH reacting via (A) halide elimination and (B) ether elimination. The observed by-products are in green text. | ||
(B) Electronic structure before photocatalysis
Our goal is to probe photocatalytic activity on MW-WO3, and it is therefore imperative that charge separation and transfer to the oxidizable substrate are efficient. As such, the charge separation was observed for the presently studied material. With the obtained photoluminescence spectra, the movement of the photogenerated charge carriers is observed. From the presented spectra in Fig. 2, a prominent blue emission is displayed at 460 nm, commonly found for WO3 materials.27 This feature is attributed to the radiative recombination of conduction band electrons and valence band holes, and the energy of this transition is close to the observed band gap of the oxide, 2.7 eV (Fig. S5†). This data also agrees with the spectra collected for the commercially available WO3, which shows a similar blue emission and recombination (Fig. S6†).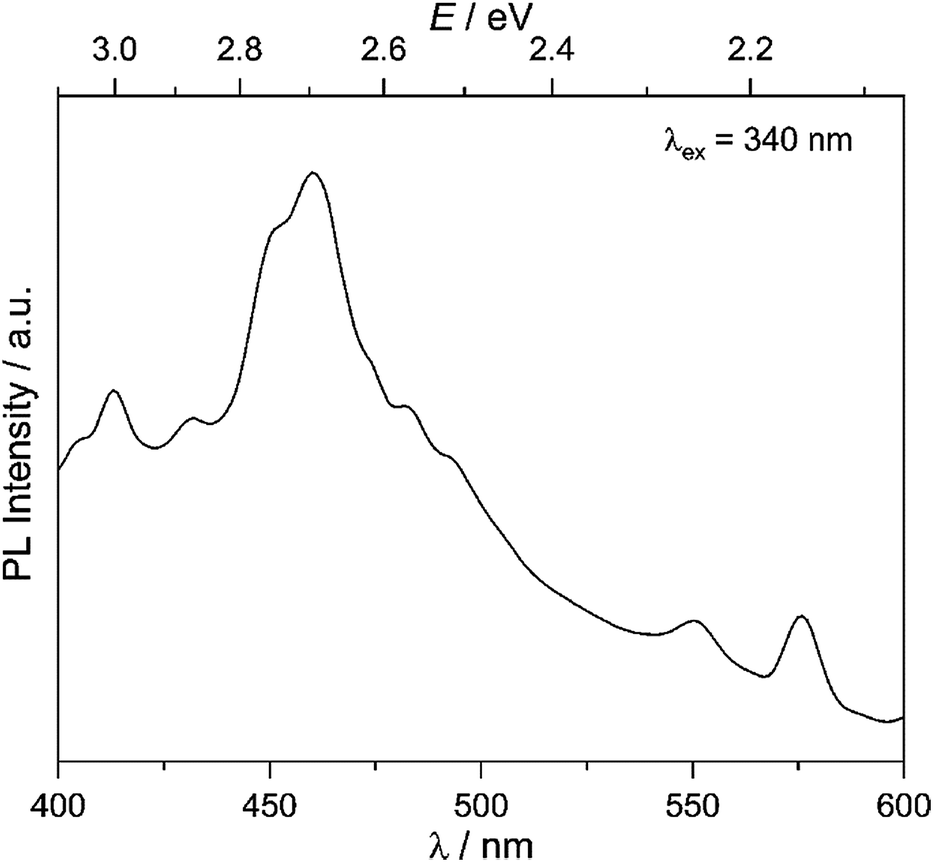 | ||
| Fig. 2 Room temperature photoluminescence spectra for the as-synthesized MW-WO3 particles. Spectra were collected with an excitation wavelength of 340 nm. | ||
We further characterized MW-WO3 to probe the defect structure in the synthesized material. To start, EPR spectroscopy was performed on neat powder, and the spectrum is presented in Fig. 3. The observed broad resonances with Landé g factors at 4.22 and 2.02 can be assigned to the presence of W5+ impurities. There is also a very sharp feature centered at g = 2.00, indicative of F+ centers in the form of electrons trapped on oxygen vacancies.28 These defects are those typically found in n-type WO3.
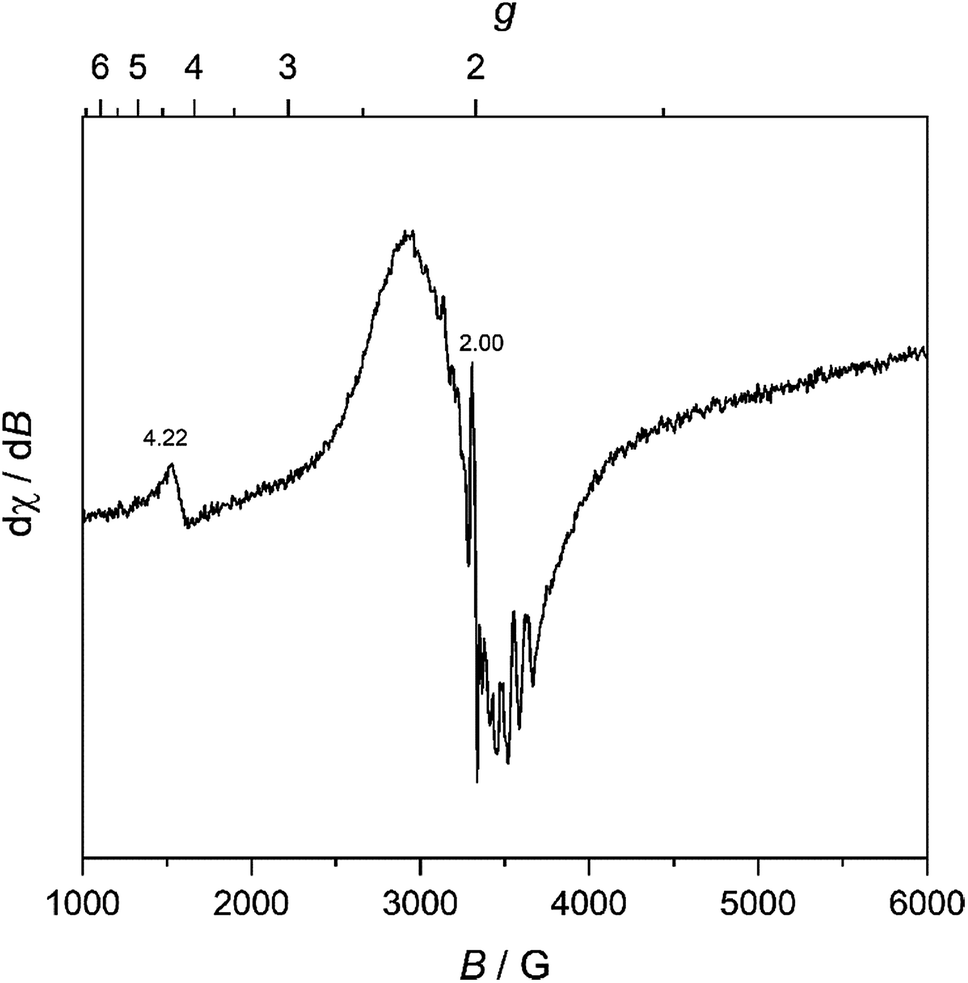 | ||
| Fig. 3 Room temperature EPR spectra for the as-synthesized MW-WO3 particles. | ||
X-ray photoelectron spectroscopy (XPS) was then used to confirm surface reduction of tungsten. From the spectrum in Fig. 4a of the 4f orbitals on tungsten, the main spin–orbit coupled 2F lines seen at 35 eV and 37.5 eV can be attributed to W6+ in the oxide. However, small shoulders appear at lower binding energy, which corroborates having reduced W5+ in the material. Integrating the deconvoluted peaks reveals only a 4.6 mol% W5+ content. Such an occurrence is not uncommon, as seen in our previous report, among others, on the reduction of tungsten in the synthesis of WO3.29 This minimal W5+ content is also observed in the commercially available WO3 powder with 2.6% W5+, calculated from the deconvoluted W 4f XPS spectra (Fig. S7†). In MW-WO3, further reduction likely arises from the known strong reducing power of alcohols under solvothermal conditions wherein W6+ is easily reduced as a small quantity of benzyl alcohol solvent is oxidized to benzaldehyde, as observed by GC-MS. Further mechanistic analysis is also revealed in the O 1 s spectra (Fig. 4b). Deconvolution of the O 1s spectra to give peaks at 530 eV and 531 eV, respectively, and correspond to W–O–W bonds and W–O–H bonds respectively, indicating hydroxy termination at the surface of the synthesized material.30
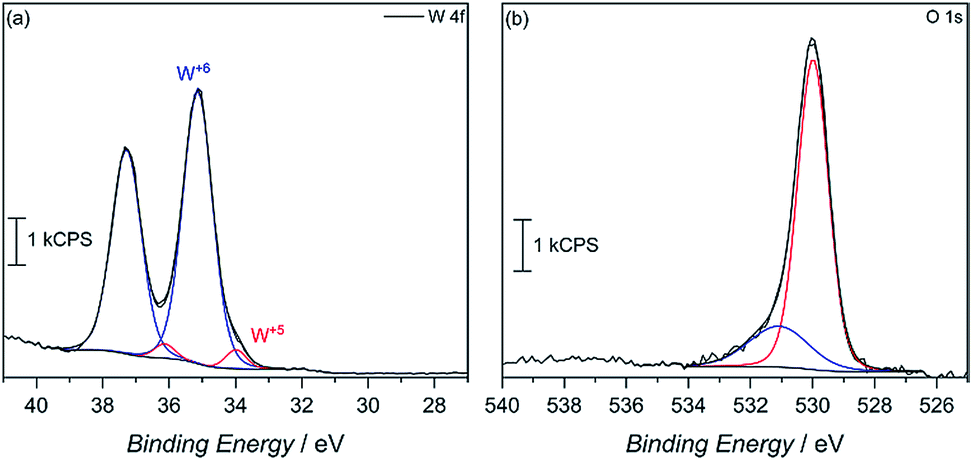 | ||
| Fig. 4 (a) W 4f and (b) O 1s XPS spectra for the as-synthesized MW-WO3 particles. | ||
(C) Photocatalytic reactions with 1° alcohols
In looking to address the state of energy-relevant catalytic conversions, a model substrate that has proven successful in being oxidized on metal oxides is benzyl alcohol.31,32 As such, we note kinetics for photocatalytic benzyl alcohol oxidation to benzaldehyde as a test case with visible light illumination according to the proposed balanced chemical equation: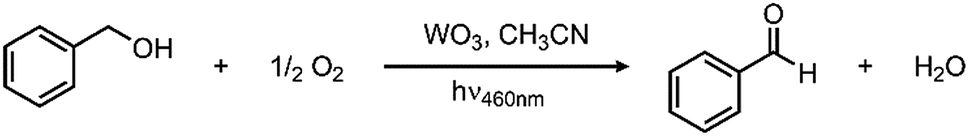
To demonstrate how particle size alters the reaction rate, we obtained larger particles from a commercial source. The commercially available particles show an average grain size of 100 nm and a surface area that is much lower, only 2.63 m2 g−1. Initial efforts to oxidize benzyl alcohol on MW-WO3 proved successful. When illuminated under O2, a conversion of 16% was achieved after 3 days of illumination, corresponding to a rate constant of 2.6 × 10−3 h−1. When this same reaction is performed on bulk WO3 particles, the rate is much slower. By GC analysis, the disappearance of benzyl alcohol could not be measured as well as for the microwave particles (Fig. S8†). However, the appearance of the single product benzaldehyde could be measured for this process. A trend is still observed in Fig. 5, wherein the appearance of benzaldehyde is much faster for our microwave synthesized particles than on their commercial counterparts. At the end of the photocatalytic process there is a 3.5-fold increase in the yield of benzaldehyde for conversions performed on the smaller microwave synthesized particles than for the WO3 of a larger scale (Fig. S9†). Notable is that the control experiment—carrying out the reaction under an air-free N2-filled atmosphere—results in no substrate conversion after 24 hours, showing the need for O2 as a reductant in the reactions. Over the course of the 3 day reaction, the temperature varies from 22–25 °C (Fig. S2†), which is not different than the variation in room temperature.
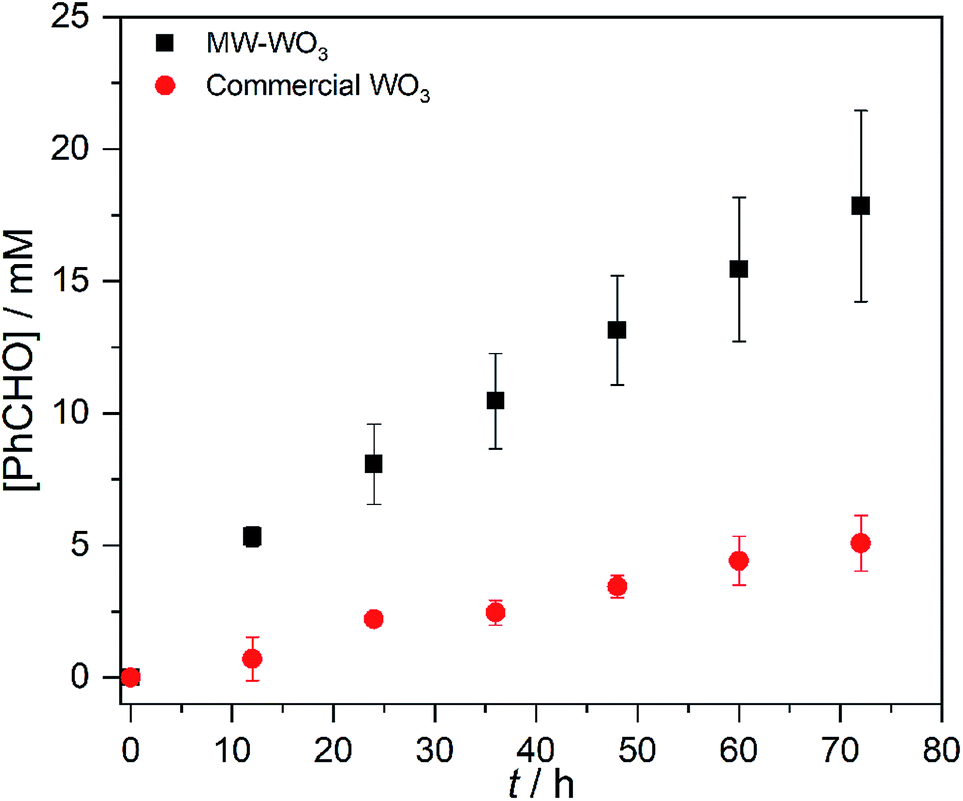 | ||
| Fig. 5 Benzaldehyde production during the photocatalytic oxidation of benzyl alcohol on MW-WO3 (black squares) and bulk WO3 (red circles) using 460 nm blue LEDs. | ||
This result confirms the expected relationship that smaller particles with larger surface areas lead to faster photocatalysis. In addition to an increase in reaction rate as the particle size decreases, we observe high chemoselectivity for the benzaldehyde product (Fig. S10†); both catalysts have a selectivity of >99% benzaldehyde. After the 3 day reaction, the catalyst stability was also confirmed by recycling the catalyst. Over the course of five three-day reaction cycles, the recycled catalytic conversion presented in Fig. 6 shows sustained stability and similar conversion over the first four successive catalytic cycles. At cycle 5, we observe the same average yield, but a larger variance in the conversion. We attribute this behavior to changes in the surface composition of the WO3 during photocatalysis (vide infra).
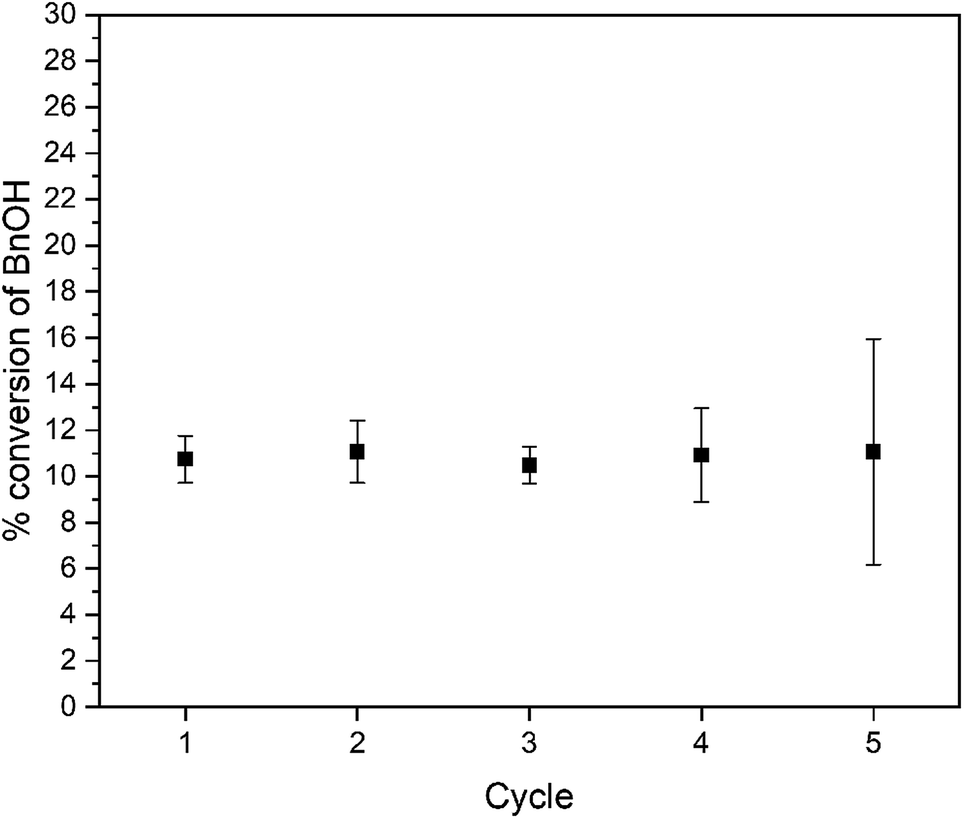 | ||
| Fig. 6 Recyclability of WO3 for photocatalytic oxidation of benzyl alcohol. | ||
In the same regime of energy-relevant conversions, initial test reactions were performed in the oxidation of a common biomass derived compound. 5-(Hydroxymethyl)-2-furaldehyde (HMF) is a well-known biomass derived product from sugarcane bagasse. As well, unlike the substrates presented above, there are limited investigations into its further transformations into value-added chemicals like 2,5-diformylfuran (DFF)33,34 and 2,5-furandicarboxylic acid (FDCA). As such, we present our initial finding on the photocatalytic oxidation of HMF into DFF on the microwave synthesized WO3 illuminated by blue LEDs. Monitored by GC-MS, the first order rate data presented in Fig. 7 shows a conversion rate constant, k of 0.01 h−1. With a much faster reaction, the direct rate comparison can be made between our nanoscale particles and the larger particles purchased commercially. When compared to the bulk, commercial WO3 counterpart we see the same trend as before; the increased particle size is accompanied by a decrease in observed photocatalytic rate. For these commercial particles, the photocatalytic oxidation of HMF into DFF proceeds with the rate constant, k of 6.3 × 10−3 h−1.
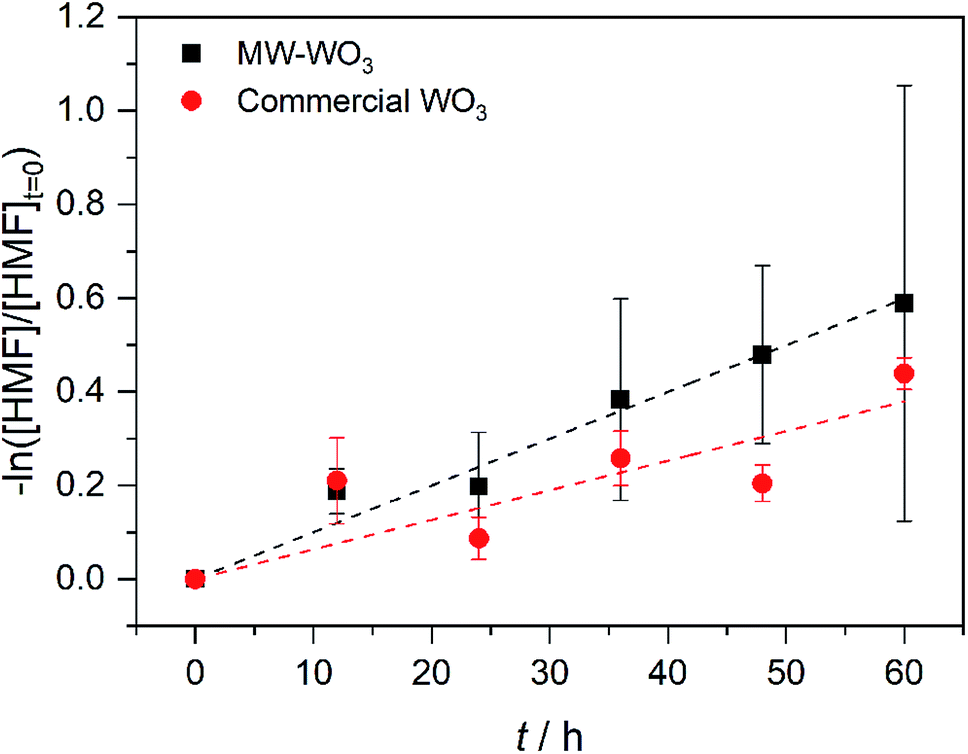 | ||
| Fig. 7 First order rate plot for the photocatalytic oxidation of HMF on MW-WO3 (black squares) and bulk WO3 (red circles) using 460 nm blue LEDs. | ||
(D) Phase, morphology, and electronic structure after photocatalysis
After photocatalysis, the suspended particles were separated from the reaction solution by centrifugation, washed with acetonitrile to remove any surface bound species, and dried overnight. The X-ray diffractograms remain unchanged from those of the as-synthesized materials. In addition, TEM imaging in Fig. 8a reveals a retention of particle morphology with no observable change in particle size. The platelets are less agglomerated which allows for analyzing the particle size distribution here. The 23(9) nm × 19(7) nm dimensions are consistent with the diffraction data in Fig. 1. The single crystalline nature of the WO3 particles after photocatalysis remains clear from the TEM image in Fig. 8b. The 0.385 nm lattice spacing corresponds to the (002) Bragg reflection of WO3. The lack of particle degradation combined with consistent kinetics with recycling suggests that WO3 synthesized by the described microwave-assisted method is photochemically stable for visible-light driven primary alcohol photocatalysis. | ||
| Fig. 8 (a) TEM images of MW-WO3 particles after photocatalysis on benzyl alcohol. (b) High resolution TEM image of a WO3 nanoplatelet after photocatalysis. | ||
Key changes to the material post photocatalysis reside in the distribution of electrons in W5+ and F+ center defects. The post-photocatalysis EPR spectrum reveals an increase in the signals with g factors attributed to the presence of W5+ (Fig. S11†), suggesting that during photocatalysis, more W6+ gets reduced to W5+. This result is corroborated by XPS analysis (Fig. S12†), where the W5+ content increases to 21.7%. This reduction is likely due to trapping conduction band electrons (formally W5+) that cannot diffuse to the surface in order to participate in the coupled oxygen reduction reaction needed to complete the catalytic cycle. Accompanying this increase in W5+ content, XPS analysis of the O(1s) spectrum reveals the emergence of a new lower binding energy feature ascribed to weaker binding interactions between the oxygen and the W5+ cation.35 This weak binding creates an environment with increased electron density around the oxygen, and results in a peak shift to lower binding energy. To corroborate this claim, quantification of the oxygen content post photocatalysis was performed on this data wherein it is found that this feature comprises 17.2% of the oxygen in this material. This is in strong agreement with our presently reported results showing a 17.1% increase in the W5+ content from the as-synthesized material to the material post photocatalysis. We propose that under illumination, F+ centers (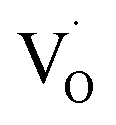 in Kröger–Vink notation) which are EPR active, transfer an electron to the W(5d) conduction band
in Kröger–Vink notation) which are EPR active, transfer an electron to the W(5d) conduction band 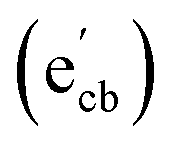 to give a non-EPR active oxygen vacancy
to give a non-EPR active oxygen vacancy 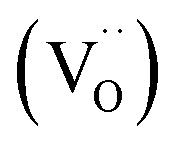 with an increase in W5+ according to the reaction:
with an increase in W5+ according to the reaction: 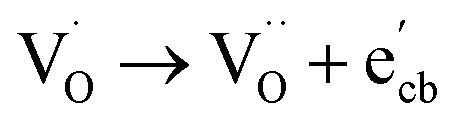 .
.
To support this proposed reaction further, we illuminated WO3 in presence of O2, but in the absence of the 1° alcohol substrate. In agreement, we still see a disappearance of the sharp EPR signal indicative of an F+ center (Fig. S13†). With no substrate around to oxidize, the photogenerated carriers simply recombine in this case resulting in no changes to the tungsten valency or oxygen binding from the material pre-photolysis, as confirmed by XPS (Fig. S14†). Comparatively, the commercial particles show no major change in W5+ content, only 0.7% after photocatalysis by XPS (Fig. S15†). Therefore, we conclude that the improved rates of photocatalysis are not solely due to the diminished particle size. Rather, the rate improvement also results from the increased W5+ and oxygen vacancy content that couples the oxidation of the primary alcohol substrate.
Conclusions
We show a fast and low-temperature synthesis of crystalline WO3 nanoplatelets without the need for further heat treatment or additional surfactants beyond the solvent. By combining what is known about non-aqueous sol–gel chemistry and microwave heating techniques, particles on a nanoscale are easily achieved. Compared to their bulk counterparts, WO3 nanoplatelets show high photocatalytic activity towards the oxidation of benzyl alcohol and HMF upon illumination by visible light. The increased surface area affords faster catalytic rates without sacrificing stability. Though limitations in the efficiency of visible light absorption are present for WO3, this route proves viable for investigating other small oxide photocatalysts. As well, these particles present themselves as a strong candidate for further exploration into the photocatalytic oxidation of biomass derived substrates into value chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a grant from the U.S. Department of Energy, Basic Energy Sciences, Catalysis Science Program, under Award No. DE-SC0006587. K. D. M. thanks the University of Michigan for a Rackham Merit Fellowship. We thank the Michigan Center for Materials Characterization for use of the TEM and Drs Kai Sun, Haiping Sun, and Bobby Kerns for assistance, as well as the University of Michigan College of Engineering and NSF Grant No. DMR-0420785. We would also like to thank Dr Antek Wong-Foy for assistance with BET surface-area analysis.Notes and references
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem.–Eur. J., 2012, 18, 2624–2631 CrossRef CAS.
- V. Augugliaro, G. Camera-Roda, V. Loddo, G. Palmisano, L. Palmisano, J. Soria and S. Yurdakal, J. Phys. Chem. Lett., 2015, 6, 1968–1981 CrossRef CAS.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS.
- D. P. Depuccio, P. Botella, B. O'Rourke and C. C. Landry, ACS Appl. Mater. Interfaces, 2015, 7, 1987–1996 CrossRef CAS.
- C. R. Lhermitte, J. Garret Verwer and B. M. Bartlett, J. Mater. Chem. A, 2016, 4, 2960–2968 RSC.
- Y. Liu, J. Li, W. Li, Y. Yang, Y. Li and Q. Chen, J. Phys. Chem. C, 2015, 119, 14834–14842 CrossRef CAS.
- P. Dias, T. Lopes, L. Meda, L. Andrade and A. Mendes, Phys. Chem. Chem. Phys., 2016, 18, 5232–5243 RSC.
- T. K. Townsend, E. M. Sabio, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2011, 4, 4270–4275 RSC.
- E. M. Sabio, R. L. Chamousis, N. D. Browning and F. E. Osterloh, J. Phys. Chem. C, 2012, 116, 3161–3170 CrossRef CAS.
- M. Niederberger, M. H. Bartl and G. D. Stucky, J. Am. Chem. Soc., 2002, 124, 13642–13643 CrossRef CAS.
- C. M. Magdalane, K. Kaviyarasu, J. J. Vijaya, B. Siddhardha and B. Jeyaraj, J. Photochem. Photobiol., B, 2016, 163, 77–86 CrossRef CAS.
- X. Sun, Y. W. Zhang, R. Si and C. H. Yan, Small, 2005, 1, 1081–1086 CrossRef CAS.
- K. Zhou, R. Wang, B. Xu and Y. Li, Nanotechnology, 2006, 17, 3939–3943 CrossRef CAS.
- S. Hilaire, M. J. Süess, N. Kränzlin, K. Bieńkowski, R. Solarska, J. Augustyński and M. Niederberger, J. Mater. Chem. A, 2014, 2, 20530–20537 RSC.
- Microwaves in Organic Synthesis, ed. A. de la Hoz and A. Loupy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 3rd edn, 2012 Search PubMed.
- S. Horikoshi and N. Serpone, Microwaves in Nanoparticle Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358 RSC.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 886–888 RSC.
- Z. Moorhead-Rosenberg, K. L. Harrison, T. Turner and A. Manthiram, Inorg. Chem., 2013, 52, 13087–13093 CrossRef CAS.
- S. Baek, S. H. Yu, S. K. Park, A. Pucci, C. Marichy, D. C. Lee, Y. E. Sung, Y. Piao and N. Pinna, RSC Adv., 2011, 1, 1687–1690 RSC.
- M. Niederberger and G. Garnweitner, Chem.–Eur. J., 2006, 12, 7282–7302 CrossRef CAS.
- I. Bilecka, P. Elser and M. Niederberger, ACS Nano, 2009, 3, 467–477 CrossRef CAS.
- G. Ma, Z. Chen, Z. Chen, M. Jin, Q. Meng, M. Yuan, X. Wang, J.-M. Liu and G. Zhou, Materials Today Energy, 2017, 3, 45–52 CrossRef.
- C. Di Valentin and G. Pacchioni, Acc. Chem. Res., 2014, 47, 3233–3241 CrossRef CAS.
- A. G. Breuhaus-Alvarez, J. L. DiMeglio, J. J. Cooper, C. R. Lhermitte and B. M. Bartlett, J. Phys. Chem. C, 2019, 123, 1142–1150 CrossRef CAS.
- J. Meng, Q. Lin, T. Chen, X. Wei, J. Li and Z. Zhang, Nanoscale, 2018, 10, 2908–2915 RSC.
- S. Higashimoto, K. Okada, T. Morisugi, M. Azuma, H. Ohue, T.-H. Kim, M. Matsuoka and M. Anpo, Top. Catal., 2010, 53, 578–583 CrossRef CAS.
- C. J. Li, G. R. Xu, B. Zhang and J. R. Gong, Appl. Catal., B, 2012, 115–116, 201–208 CrossRef CAS.
- S. Yurdakal, B. S. Tek, O. Alagöz, V. Augugliaro, V. Loddo, G. Palmisano and L. Palmisano, ACS Sustainable Chem. Eng., 2013, 1, 456–461 CrossRef CAS.
- H. Zhang, Q. Wu, C. Guo, Y. Wu and T. Wu, ACS Sustainable Chem. Eng., 2017, 5, 3517–3523 CrossRef CAS.
- F. Liu, X. Chen, Q. Xia, L. Tian and X. Chen, RSC Adv., 2015, 5, 77423–77428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Microwave heating profile observed during microwave heating. GC trace of the organic by-products for the MW-WO3 particles, Sample mass spectra for the observed GC-MS by-product peaks, Tauc plot for MW-WO3 particles, photoluminescence and XPS for commercial WO3, sample 1H-NMR spectra obtained after the photochemical oxidation of benzyl alcohol, post photocatalysis EPR and XPS of MW-WO3, a movie of the microwave reaction. See DOI: 10.1039/c9ra04839a |
| This journal is © The Royal Society of Chemistry 2019 |