
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerfluorinated phosphine and hybrid P–O ligands for Pd catalysed C–C bond forming reactions in solution and on Teflon supports†
Farzana Begumab,
Muhammad Ikramac,
Brendan Twamleya and
Robert J. Baker *a
*a
aSchool of Chemistry, University of Dublin Trinity College, Dublin 2, Ireland. E-mail: bakerrj@tcd.ie
bDepartment of Chemistry, Mirpur University of Science and Technology, Mirpur AJK, Pakistan
cDepartment of Chemistry, Abdul Wali Khan University, Mardan, Pakistan
First published on 13th September 2019
Abstract
The synthesis of two types of phosphine ligands that feature perfluorinated ponytails is reported. A bidentate (RfCH2CH2)2PCH2CH2P(CH2CH2Rf)2 (Rf = CF3(CF2)n; n = 5, 7) and an alkoxyphosphine made by ring opening a fluorous epoxide, RfCH2CH(OH)CH2PR2 (Rf = CF3(CF2)7), have been prepared and spectroscopically characterised. The electronic effects of the fluorous chains have been elucidated from either the 1JPt–P or 1JP–Se coupling constants in Pt(II) or phosphine selenide compounds. Whilst the bidentate phosphines do not give stable or active Pd catalysts, the hybrid ligand does allow Susuki, Heck and Sonogashira catalysis to be demonstrated with low catalyst loadings and good turnovers. Whilst a fluorous extraction methodology does not give good performance, the ligand can be adsorbed onto Teflon tape and for the Suzuki cross coupling reaction the catalytic system can be run 6 times before activity drops and this has been traced to oxidation of the ligand. Additionally the crystal structure of the hybrid phosphine oxide is reported and the non-covalent interactions discussed.
Introduction
Immobilisation of homogeneous catalysts is an attractive methodology for generating recoverable and recyclable catalysts and many methods have been exploited.1 The principle advantage is catalyst recoverability and recycling,2 especially where expensive metals are used. As an example, N-heterocyclic carbenes, which are prevalent in homogeneous catalysis, have been extensively studied and a plethora of immobilisation techniques reported.3 An interesting methodology has been in the use of fluorous groups as the solubility in organic solvents can be tuned by control of the number of fluorous groups or the choice of fluorous or organic solvent. This is due to the ‘thermomorphic’ behaviour of mixed solvent systems where at certain temperatures the fluorous and organic solvents are miscible, but phase separation occurs upon changing the temperature.4 Therefore if the metal complex can have significant solubility in the fluorous phase then homogenous catalysis and catalyst separation can be controlled by simply changing the temperature. The first example of this was reported by Horváth in the synthesis of a perfluorinated triphenylphosphine rhodium complex in hydroformylation reactions,5 and many modifications of phosphines decorated with fluorous ponytails of varying lengths have since been reported,6 with a wide scope in catalytic reactions. The uses of fluorous ponytails are not limited to phosphines and a range of ligand types have been prepared.7 An elegant use of the preferential solubility of fluorous phosphines in fluorous solvents has been using the concept of phase transfer activation by the modification of Grubbs II catalyst [(NHC)Ru(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR)(PRf3)Cl2]. The initiation step involves dissociation of the phosphine to form the vacant coordination site so when run under biphasic conditions the phosphine is “removed” from the reaction solvent and cannot re-coordinate, thus the overall rate of reaction can be increased.8 We have shown that using a fluorous alkoxide as a quenching agent we can recycle catalysts for the ring opening of caprolactone.9 However, by introducing the electron withdrawing perfluorinated ponytails, the electronic parameters of the phosphines can be significantly affected. Methods to combat this have included the use of aryl spacers6,10 or methylene groups11 that can attenuate this electronic impact. As an illustrative example, the ν(C
CHR)(PRf3)Cl2]. The initiation step involves dissociation of the phosphine to form the vacant coordination site so when run under biphasic conditions the phosphine is “removed” from the reaction solvent and cannot re-coordinate, thus the overall rate of reaction can be increased.8 We have shown that using a fluorous alkoxide as a quenching agent we can recycle catalysts for the ring opening of caprolactone.9 However, by introducing the electron withdrawing perfluorinated ponytails, the electronic parameters of the phosphines can be significantly affected. Methods to combat this have included the use of aryl spacers6,10 or methylene groups11 that can attenuate this electronic impact. As an illustrative example, the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) stretching frequency in Vaska's type complexes [IrCl(CO)(PR3)2] can be compared with electron poor (P(OPh)3) (ν(CO = 2003 cm−1)) or electron rich (PCy3) (ν(CO = 1931 cm−1)) traditional phosphines12 for catalysis and Rf3P (Rf = CH2CH2(CF2)5CF3); (ν(CO = 1976 cm−1)).11b
O) stretching frequency in Vaska's type complexes [IrCl(CO)(PR3)2] can be compared with electron poor (P(OPh)3) (ν(CO = 2003 cm−1)) or electron rich (PCy3) (ν(CO = 1931 cm−1)) traditional phosphines12 for catalysis and Rf3P (Rf = CH2CH2(CF2)5CF3); (ν(CO = 1976 cm−1)).11b
The major drawbacks of these methodologies are that the fluorous solvents and ponytails are not environmentally friendly and can persist in the environment causing long term adverse effects.13 Secondly, the syntheses of the fluorous ligands are typically prohibitively expensive for large scale applications and sometimes multi-step synthesis using experimentally difficult conditions,14 or formed in poor yields,15 although new synthetic pathways somewhat reduce this effect.16 Finally, as the fluorous chains are increased the solubility in all solvents tends to decrease, meaning characterisation becomes difficult. Light fluorous (i.e. <40% fluorine) chemistry has been used to circumvent some of these issues,17 most notably the use of fluorous silica for phase separation. These reagents are expensive and subsequent washing steps may degrade the catalyst, but several interesting applications have been reported.18 A medium fluorous approach (i.e. 40–60% fluorine) has been utilised successfully, but typically use protic solvents such as water, which is incompatible with some organometallic catalysts;19 however judicious use of fluorinated solvents can alleviate this problem.20 Given the observation that the temperature can control the solubility of the fluorinated ligands in both fluorous and organic solvent, the elimination of the expensive and environmentally unfriendly fluorinated solvent can be achieved by thermomorphic control for liquid/solid phase separation i.e. the fluorinated catalyst will dissolve in suitably chosen organic solvents at high temperatures but will precipitate upon lowering of the temperature.21 An emerging solution has been to use fluorous supports such as Teflon or Gore-Tex whereby the fluorous catalyst is presumably adsorbed onto the surface and provides an efficient vehicle for catalyst delivery and recovery,22 although catalyst leaching can still be of concern. The sorption process is not well understood, but we have shown that measurable, though rather weak, non-covalent C–F⋯F–C interactions could be involved.23 Herein we report on two synthetic pathways for the formation of phosphines and expand the idea of supporting these fluorinated ligands onto PTFE tape, commonly used in the laboratory, and their use in homogeneous catalysis, particularly targeted at the recovery and reuse of the expensive fluorous ligands in C–C cross coupling reactions, that avoids issues of catalyst decomposition and/or leaching. This “ligand-on-Teflon” has been characterised by thermal methods.
Results and discussion
We will first describe the synthesis of the ligands, followed by their use as traditional homogeneous catalysts under biphasic conditions, before describing the characterisation on Teflon and finally the catalysis using the ligand-on-Teflon approach.Synthesis and characterisation of fluorous phosphine ligands
The synthesis of the phosphine and P–O ligands with fluorous ponytails was achieved in good yields using two methodologies.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :96 for 1 and 2:98 for 2; when n = 9 a solid precipitated out of the reaction mixture and proved to be insoluble in all organic and fluorous solvents, even at elevated temperatures. In contrast, the reactions with 1,2-biphosphinobenzene were extremely sluggish and very low yielding (δP = −31 ppm) so further reactivity studies were not conducted.
:96 for 1 and 2:98 for 2; when n = 9 a solid precipitated out of the reaction mixture and proved to be insoluble in all organic and fluorous solvents, even at elevated temperatures. In contrast, the reactions with 1,2-biphosphinobenzene were extremely sluggish and very low yielding (δP = −31 ppm) so further reactivity studies were not conducted.
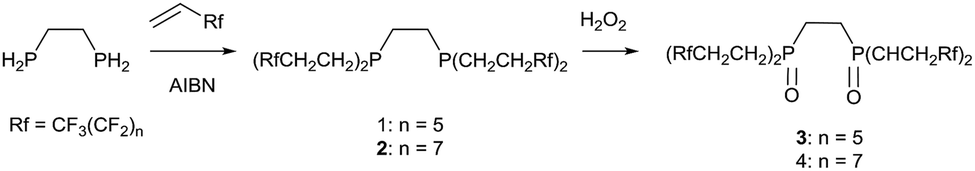 | (1) |
To understand the changes in the electronic effect of the ligand we sought to synthesise [(PP)PtCl2] as the magnitude of the 1JPt–P coupling constant has been used to evaluate the σ-donor ability of phosphines, specifically where a decrease in the coupling constant can be related to a decrease in the σ-donation from the phosphorus.27 Thus, an NMR tube was charged with one equivalent of 1 and one equivalent of [(COD)PtCl2] in the amphiphilic solvent 1,3-trifluoromethylbenzene and heated to 50 °C for 1 h. This afforded a shift in the 31P{1H} NMR spectrum from δP = −26 ppm to δP = +49 ppm with Pt satellites (1JPt–P = 3487 Hz). This can be compared to 3523 Hz for the electron rich [(dmpe)PtCl2]28 or 3362 Hz for the electron poor [(CF3CF2)2PCH2CH2P(CF2CF3)2PtCl2]29 indicating that the methylene spacers do attenuate the electron withdrawing nature of the fluorous groups to a degree, and in line with numerous other experimental studies.11 Interestingly, over an hour, a black precipitate formed and the 31P{1H} NMR spectrum showed several peaks in addition to free ligand, and we were unable to obtain analytically pure material for further analysis. One was identified as the phosphine oxide (3), by the deliberate oxidation of the ligand (δP = 31.6 ppm), was only soluble in fluorinated solvents (perfluorinated hexane or 1,3-trifluoromethylbenzene). This suggests that the metal complexes of this ligand are susceptible to decomposition and in line with data from some other fluorous phosphine palladium compounds.25,30
Preliminary investigations show that when [tBu2P]Li is added to the fluorous epoxide, followed by quenching with water, 31P{1H} NMR spectroscopy showed a single peak at δP = 19.3 ppm that can be assigned to the expected ring opened product. However when the smaller [Ph2P]Li was used, two peaks were observed at δP = −27.1 and −15.5 ppm indicating that the nucleophile ring opened at both positions; this has been previously observed in non-fluorous epoxides.31 To regain control of regioselectivity, we increased the size of the nucleophile by reacting the phosphine–borane adducts with nBuLi and the epoxide.32 Under these conditions only one peak in the 31P{1H} NMR spectrum was observed in all Li[R2P·BH3] adducts (R = Ph, δP = 12.8 ppm; R = iPr, δP = 32.5 ppm; R = tBu, δP = 40.6 ppm), indicating a regioselective ring opening. All spectroscopic data (1H, 13C{1H}, 31P{1H}, 7Li NMR and IR spectrosocopy) support the formulation of the ring opened salts 5–7 (ESI†). Deprotection of the borane by refluxing with TMEDA followed by quenching with degassed water gave ligands 11–13 in good yield; the order of the quenching and deprotection did not make a difference to the isolated yield but could not be done simultaneously as by-products from quenching the tmedaBH3 complex complicated purification.33 This reaction can be conveniently followed by 31P{1H} and 11B NMR spectroscopy and the shift in the 31P{1H} NMR spectra are accompanied by the loss of the 1J31P–11B coupling (11, δP = 19.3 ppm; 12, δP = 27.4 ppm; 13, δP = −22.6 ppm) and resonances in the 11B{1H} NMR spectrum ascribed to the TMEDA·BH3 complex.33b All other NMR spectroscopy confirm the formulations (ESI†). Importantly for catalysis, the partition coefficient between perfluoromethylcyclohexane and toluene were measured using 19F NMR spectroscopy4 for 11–13 and the results were all around 55:45 indicating that there is little preferential solubility in fluorous phases, as anticipated from the inclusion of the hydroxy and alkyl groups.
The phosphines are sensitive to oxygen, and the corresponding phosphine oxide can be readily prepared and isolated by simply exposing the phosphine to air (Scheme 1). In order to understand the electronic changes that occur in these three ligands, the phosphines 11–13 were reacted with elemental Se and the phosphine selenide 17–19 isolated and characterised by multinuclear NMR spectroscopy (Scheme 1). The 1JP–Se coupling constants have been used to give electronic information on the phosphorus34 and the coupling constants are 1JP–Se = 674 Hz for 17, 1JP–Se = 688 Hz for 18 and 1JP–Se = 705 Hz for 19, in line with the expected trends i.e. the lower the coupling constant the more electron rich the phosphine. Moreover we can compare the shift from R3PSe (R = Ph, 1JP–Se = 736 Hz;35 R = iPr, 1JP–Se = 686;36 R = tBu, 1JP–Se = 687 Hz)36 or Ph2PEt model compounds (1JP–Se = 725 Hz); these data show that the phosphines are not significantly affected by the fluorous ponytails.
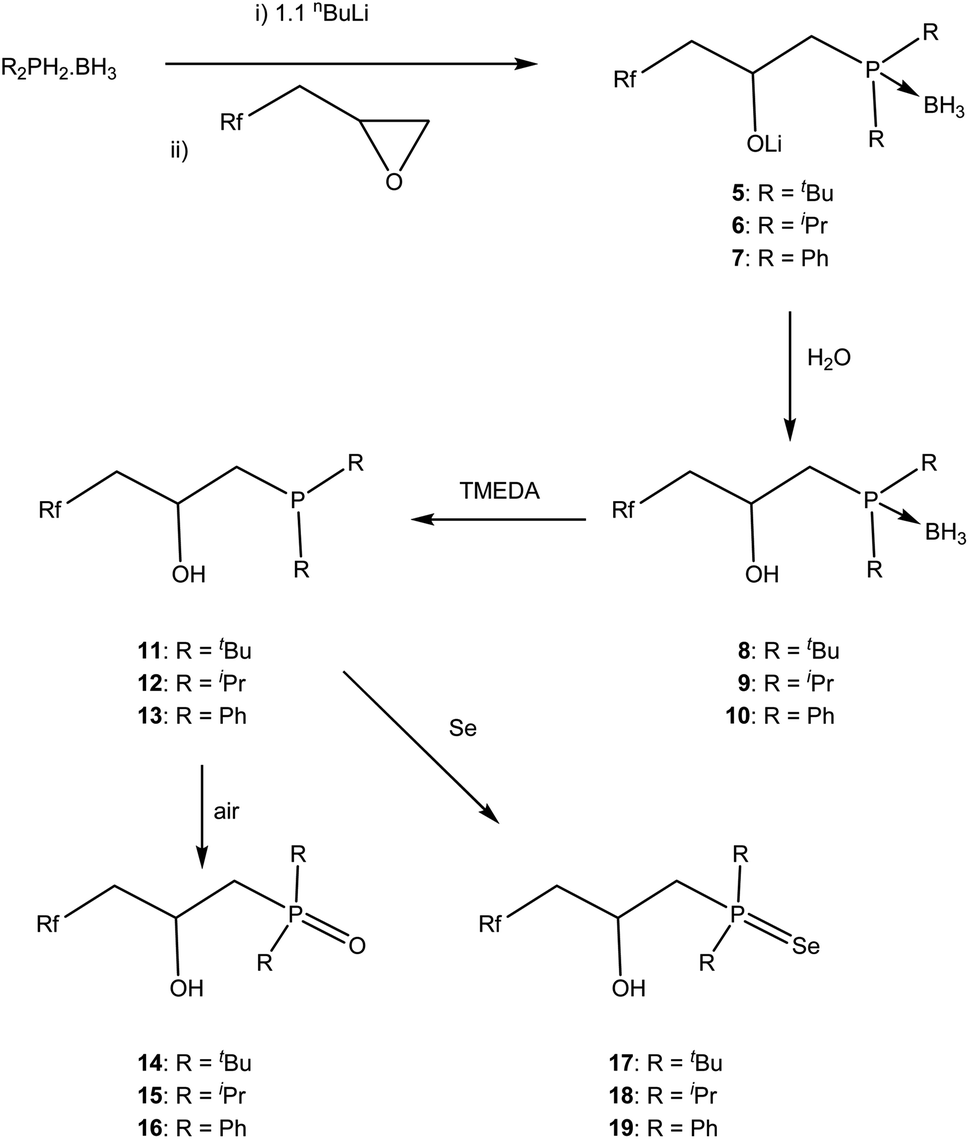 | ||
| Scheme 1 Synthesis of P–O ligands (Rf = CF3(CF2)7). | ||
We were able to grow single crystals of 16 from slow evaporation of DCM and the structure is shown in Fig. 1 (metric parameters are collated in Tables S1 and S2†).
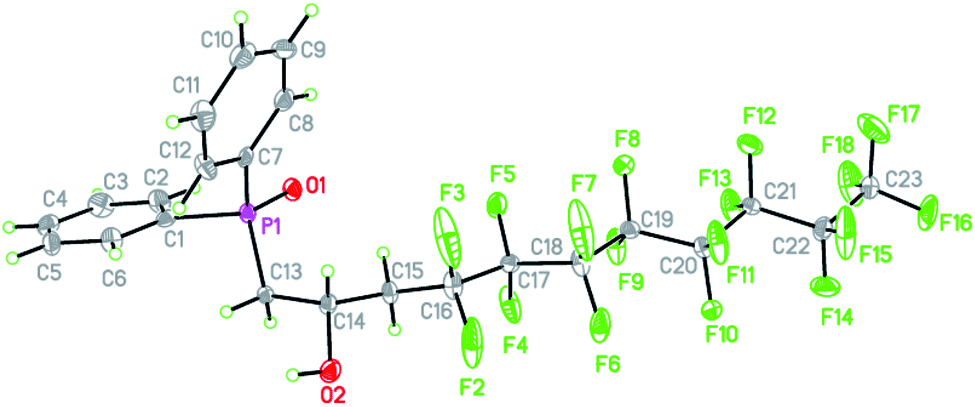 | ||
| Fig. 1 Molecular structure of 16 with atomic displacement parameters shown at 50% probability. | ||
The structure confirms the regioselectivity of the ring opening and the metric parameters are unexceptional. For example the PO = 1.486(3) Å is comparable to the PO bond length of 1.4871(15) Å in the hemihydrate of triphenylphosphine,37 (Ph3PO)(H2O)0.5 or to the PO bond length of 1.494(2) Å in Ph2MePO which features no hydrogen bonding.38 However, the packing and non-covalent interactions (Fig. 2) warrant comment. There are strong intermolecular O–H⋯OP interactions (O(1)⋯O(2) = 2.698(4) Å, Fig. 2(a)) and a longer intramolecular C–H⋯O–P (C(13)–H(13A)⋯O(1) = 3.351(5) Å, Fig. 2(b)); the increased acidity of these protons have been shown computationally previously.23 To explore and quantify the fluorous based non-covalent interactions, Hirshfeld surface39 can be conveniently used and close interactions are labelled in Fig. 2(c) as red spots. Fig. 2(c) highlights the C–F⋯H–Csp2 interactions40 (dC(10)⋯F(5) = 3.449(5) Å and dC(5)⋯F(14) = 3.107(5) Å) and numerous C–F⋯F–C interactions ranging from 2.744(4) to 2.934(4) Å (sum of the van der Waals radii41 = 2.92 Å).
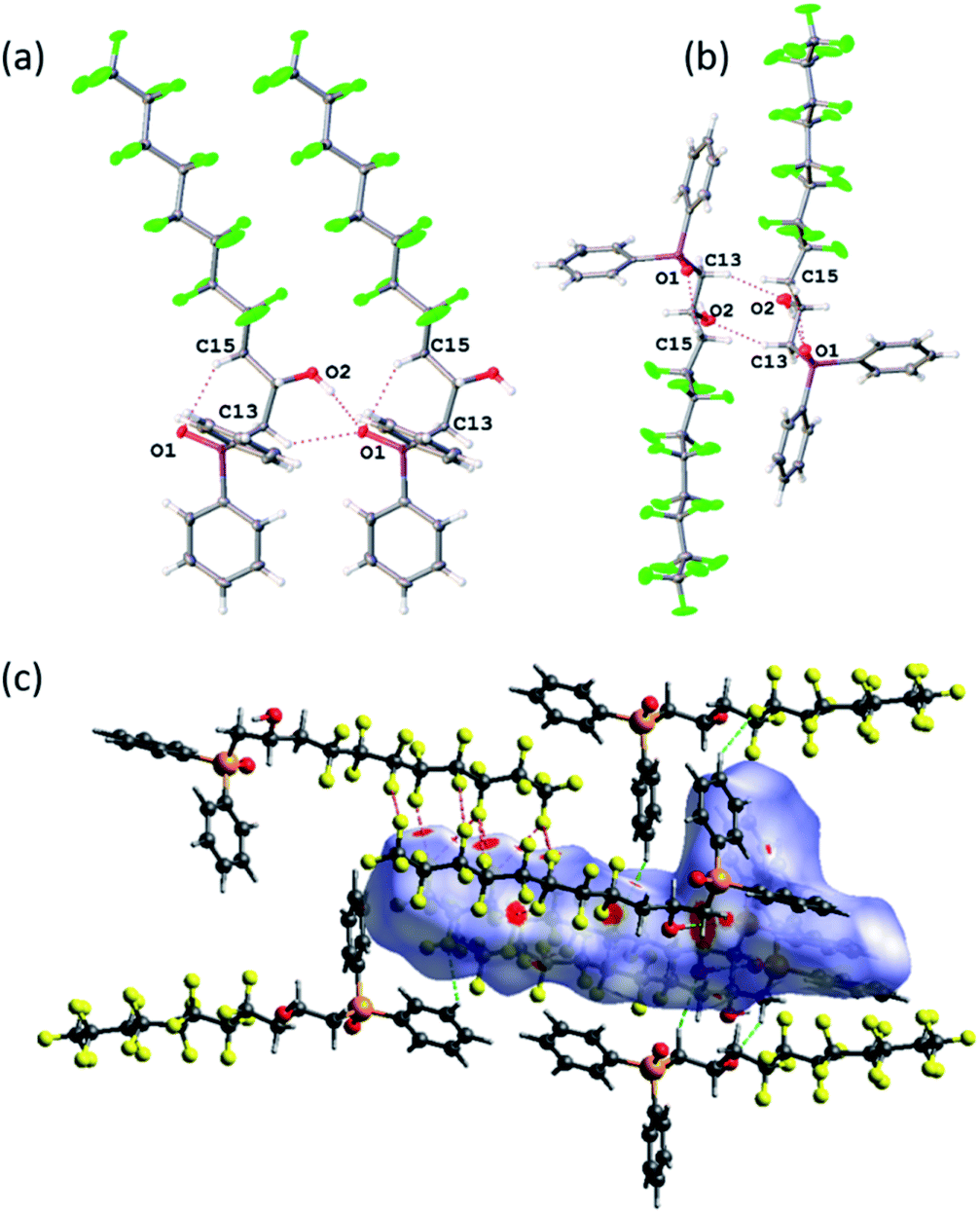 | ||
| Fig. 2 Non-covalent bonding patterns in 1: hydrogen bonding (a) normal to the a-axis showing the connectivity along layers; (b) normal to the b-axis showing the connectivity between layers; (c) Hershfield analysis showing the F⋯F (red lines) and H⋯F (green lines) interactions. | ||
Bifurcated three-point interactions (F⋯F⋯F = 54.43°) are also present holding chains together. Finally, the Hirschfeld surfaces can give quantitative information and the H⋯F close contacts account for 30.0%, while the F⋯F = 24.9% and H⋯H only 22.2%.
Catalytic studies in solution
To assess the use of the fluorous phosphines 11–13 in catalysis we chose to explore their Pd complexes in the Heck, Suzuki and Sonogashira C–C coupling reactions. These important reactions have been extensively studied42 and fluorous ligands examined, thus providing a benchmark. The Heck reaction is typically used as a testbed for new catalytic systems,43 but all intimate a highly reactive undercoordinated Pd(0) that is intrinsically unstable outside the catalytic cycle and the formation of Pd nanoparticles can also effectively catalyse these reactions.44 These can present challenges for effective recycling protocols.The fluorous bidentate phosphines 1 and 2 give immediate precipitation of a black powder upon addition of any source of Pd(II), or Pd(0) and 31P{1H} NMR analysis of the mixture showed numerous peaks indicating decomposition of the Pd ligand complex. No further catalytic studies were conducted with this ligand, although we note that it can form catalytically competent rhodium complexes for hydroformylation.24 Conversely, reaction of ligands 11–13 with palladium sources afforded active catalysts for Heck, Suzuki and Sonogashira C–C coupling reactions (Scheme 2) using 0.5–1 mol% of the catalyst and the results are summarised in Table 1. The purpose of this study was not to fully optimise conditions nor demonstrate scope of the reaction, but as a proof of principle that the reactions work so that the ligand-on-Teflon approach can be then tested and compared. Therefore the yields of the reaction, whilst high, have not been optimised. However, we note that the Sonogashira reaction required 2 mol% of the catalyst and the yields were low, with long reaction times.
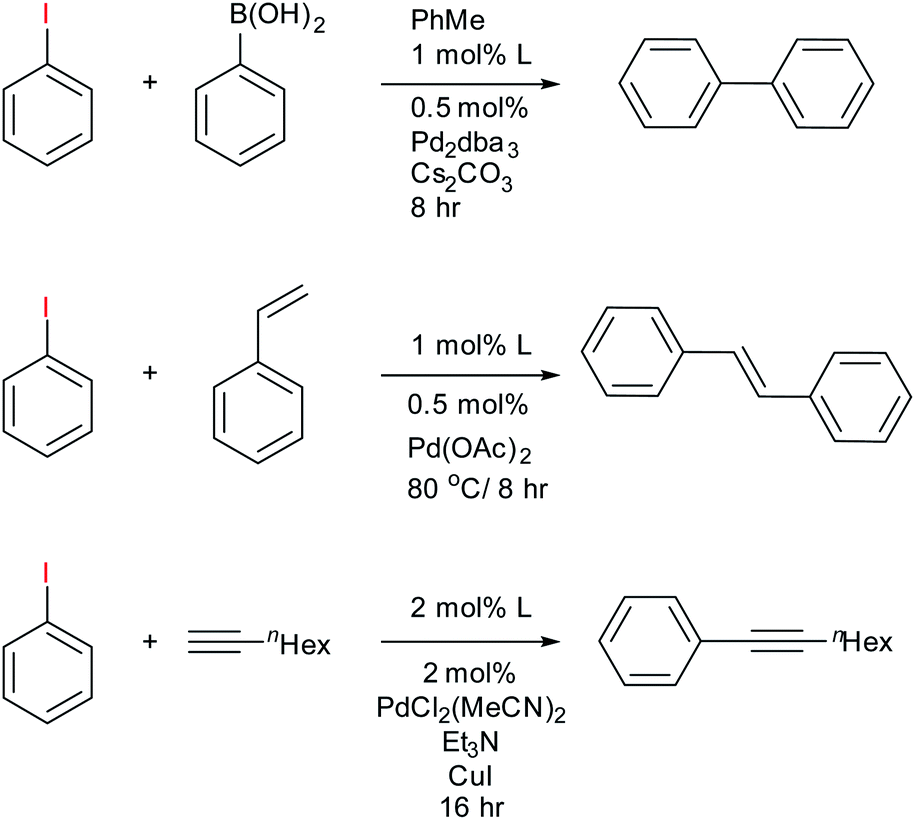 | ||
| Scheme 2 Summary of catalytic experiments from ligands 11–13 with results reported in Table 1. | ||
| Reaction | Ligand | Yield (%) | TON | TOF (h−1) |
|---|---|---|---|---|
| Suzuki | 11 | 95 | 9500 | 1187 |
| 12 | 91 | 9100 | 1137 | |
| 13 | 75 | 7500 | 937 | |
| Heck | 11 | 81 | 8100 | 1012 |
| 12 | 72 | 7157 | 894 | |
| 13 | 68 | 6713 | 839 | |
| Sonogashira | 11 | 48 | 2460 | 151 |
| 12 | 36 | 1772 | 111 | |
| 13 | 25 | 1423 | 89 |
Moreover, in the Heck reaction we observe only the E isomer by NMR spectroscopy. Because of the electron rich nature of the tBu substituted phosphine, we were able to also use bromobenzene in the Suzuki cross coupling reaction, albeit in reduced yield (yield = 23%; TON = 2300) and only traces of product formed with chlorobenzene (yield = <5%). For context, a number of fluorous phosphines have been developed for cross coupling reactions and our yields are similar to those observed for the complexes [PdCl2(n-C10F21PPh2)]16c or a perfluoroalkylated PCP45 or perfluoroarylated SCS46 pincer palladium complex for the heck reaction that could be recycled by fluorous solid-phase extraction. However, Gladysz and co-workers have shown that in perfluoroalkylated SCS pincer compounds of Pd, the catalyst is actually Pd nanoparticles.47 We do not compare to the state of the art NHC based catalysts48 where TON of 104–106 are obtained using very low catalyst loadings. To illustrate the concept of electron richness further, the Suzuki reaction was followed by 1H NMR spectroscopy using ligand 11 and 12 (ligand 13 gave overlapping peaks in the 1H NMR spectrum that proved impossible to deconvolute) and the conversion to biphenyl measured over time (Fig. 3). It is clear that the most electron rich phosphine enhances the rate of the reaction. Also apparent is that there is no initiation step within the timeframe of our measurements.
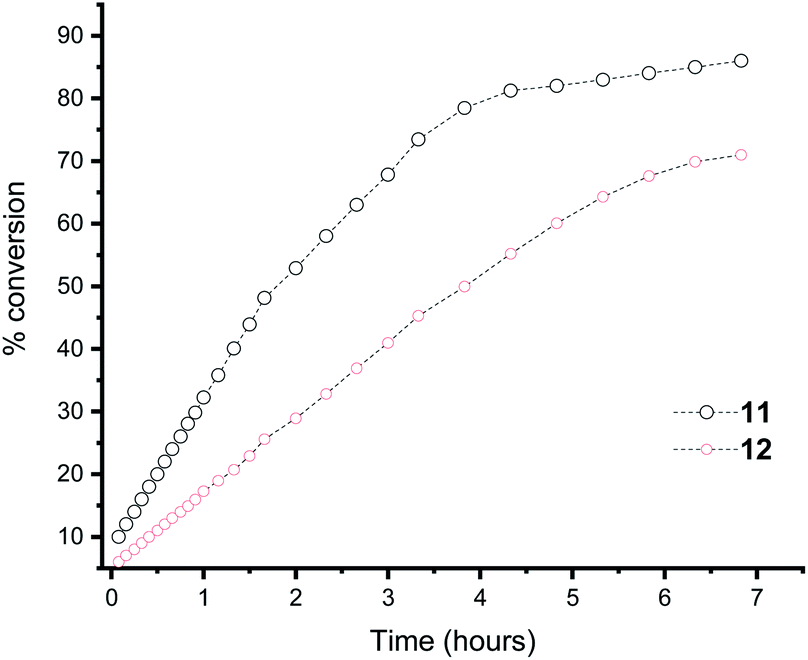 | ||
| Fig. 3 Plot of the % conversion of biphenyl using ligands 11 and 12, as monitored by NMR spectroscopy. | ||
Some recycling studies were carried out in solution by quenching the reaction and then extracting the ligand in fluorinated solvents. Whilst we did recover some of the ligand, the NMR studies showed this was as the oxide and, given the rather low partition coefficients, in variable yields. This approach clearly does not hold any benefit for an efficient catalyst recycling strategy.
Supported ligands on Teflon
We next turned our attention to supporting the ligands 11–13 on Teflon tape. The P–O ligands were dissolved in acetone and a piece of Teflon tape of ca. 1 cm length added and this was stirred for 10 minutes. Removal of the Teflon tape and drying under a stream of N2 gas afforded a brownish coloured material (ESI†). 31P{1H} NMR analysis of the solution revealed no ligand present. This reaction was also followed in an NMR tube and, without adjusting any instrument parameters, the intensity of the ligand peak deceases to ca 5% in just a few minutes. IR spectroscopy of the Teflon tape was not informative, but TGA (Fig. 4) shows the presence of the ligands which are lost at ca 300 °C; Teflon decomposes at 600 °C. Qualitatively, 13 appears to sorp more than the other two ligands. It is worth noting that the surface of Teflon is undefined as the porosity and chemical permeability has been previously studied,49 especially for uses as phase vanishing reactions.50 Whether our compounds are surface sorped or entrained inside the pores was not thoroughly investigated in this study, but the high temperatures of ligand loss from the TGA experiments and the much enhanced stability to air, points to an entrainment process; we are investigating this adsorption process in more detail and will report in due course. In passing, we also note that though the stirrer bars we used were PTFE coated, and could act as similar sources for sorption, the Teflon did not noticeably discolour in any of our experiments.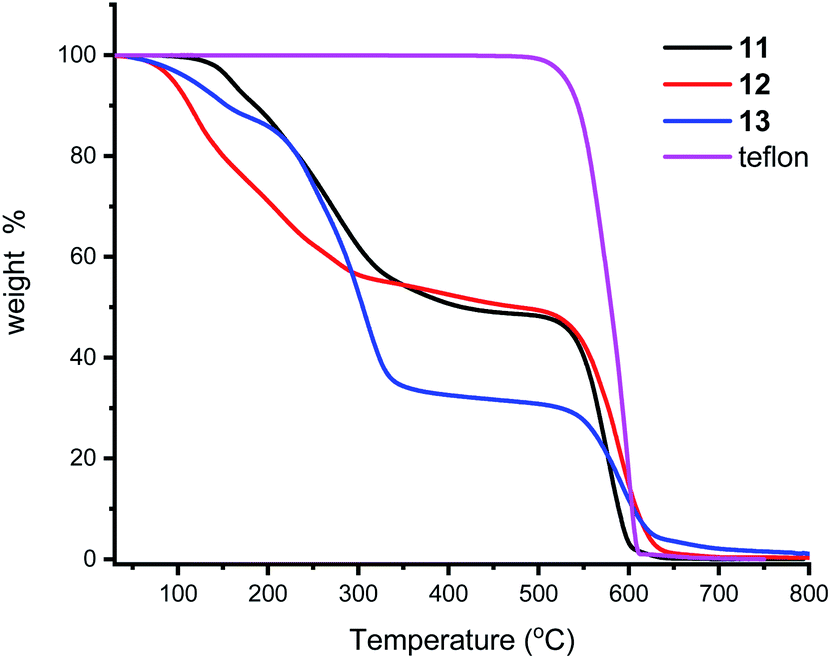 | ||
| Fig. 4 TGA of phosphine ligands 11–13 adsorbed onto Teflon tape. | ||
Ligand-on-Teflon studies
The next step in our study was to observe if the ligand-on-Teflon approach could be used as a recycling study. Our initial attempts with the Pd catalyst did not generate reproducible results, and NMR studies showed that the ligand–metal complex was present in solution as well as on the Teflon tape, in line with the partition coefficients measured for the ligand. However, it is well known that in homogeneous catalysis the price of the ligand is orders of magnitude more than the precious metal,51 so recycling the ligand may give significant cost savings as well as negating the issue of metal leaching during multiple recycles. Moreover, the generally high molecular weights of the ligands mean that relatively large amounts of catalysts are needed to obtain high reaction rates and/or selectivity. We used a model reaction to examine the recyclability of the ligand that gave the most active catalyst (11 in this experiment), the coupling of iodobenzene with phenylboronic acid to form biphenyl and Fig. 5 reports the isolated yields and TON of biphenyl. In this case, the ligand was not present in the solution at the end of the reaction, as judged by 19F and 31P{1H} NMR spectroscopy (including a fluorous standard) and can be recycled multiple times before activity appreciably drops off. The presence of 14 was then observed by 31P{1H} NMR spectroscopy, pointing to an oxidative decomposition pathway.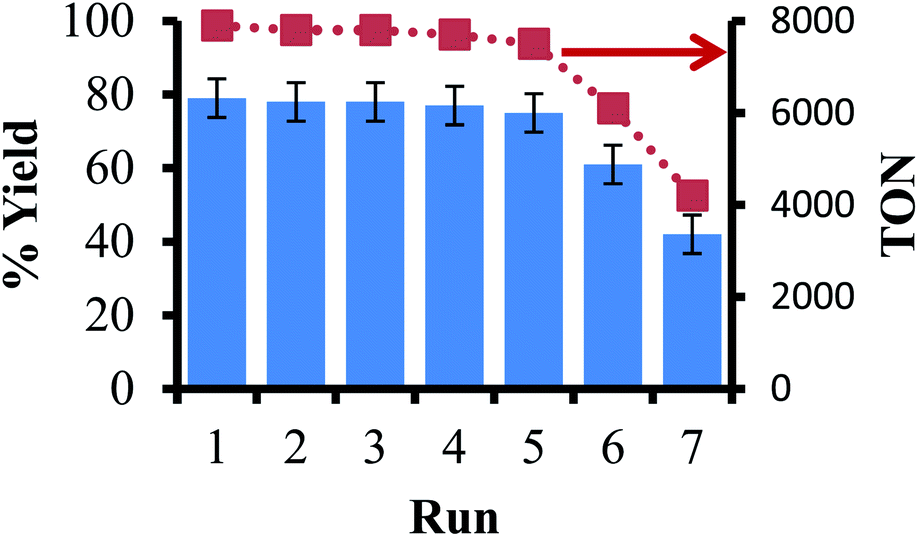 | ||
| Fig. 5 Recycling study of the coupling of iodobenzene and phenylboronic acid using ligand-on-Teflon method. | ||
Conclusions
The synthesis and applicability of two electronically different phosphine ligands with fluorous ponytails in a variety of C–C bond forming reactions have been shown. Whilst a fluoroalkyl phosphine (RfCH2CH2)2PCH2CH2P(CH2CH2Rf)2 does not give catalytically competent palladium complexes, a β-hydroxyphosphine with the fluorous chain further away from the phosphine centre does. The catalysis can be run with low loadings and reasonable turnovers, but because of the hydroxy group cannot be recycled with conventional fluorous solvent recovery methods. However, we have shown that the ligands can be sorped onto Teflon tape and used for the Suzuki cross coupling reaction of simple substrates with 6 recycles before activity starts to drop off. The ligand on Teflon approach add to the growing numbers of reactions that can be catalysed by fluorous immobilisation, but further optimisation could include precise catalyst loading as this approach does not require metals on the tape and the downside of metal leaching is avoided. More generally, this work also shows that ligand effects in recycling strategies are very important to consider.Experimental
General
1H, 13C{1H}, 31P{1H}, 77Se{1H} and 7Li NMR spectra were recorded on a Bruker AV400 spectrometer operating at 400.23 MHz, 155.54 MHz, 161.98 MHz 76.33 MHz and 156 MHz respectively, and were referenced to the residual 1H and 13C resonances of the solvent used or external H3PO4, Me2Se or LiCl. IR spectra were recorded on a PerkinElmer Spectrum One spectrometer with attenuated total reflectance (ATR) accessory. All thermogravimetric analysis were measured on the PerkinElmer Pyris 1 TGA heating at 10 °C per minute in a nitrogen atmosphere. Data for 11 were collected on a Bruker D8 Quest ECO using Mo Kα (λ = 0.71073 Å). The sample was mounted on a MiTeGen microloop and data collected at 100(2) K using an Oxford Instruments Cryostream low temperature device. Bruker APEX52 software was used to collect and reduce data and determine the space group. The structure was solved using direct methods (XT)53 and refined with least squares minimization (XL)54 in Olex2.55 Absorption corrections were applied using SADABS.56 Crystal data, details of data collection and refinement are given in Table S1.† The hydrogen H2a on O2 was located on the difference map and refined with restraints (DFIX). The fluorine atoms are prolate and were modelled with restraints to minimize this (ISOR). CCDC 1912783 contains the supplementary crystallographic data for this paper.All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of a high purity dry argon. THF and Hexane was distilled over potassium, C6D6 and toluene over sodium whilst DCM, acetonitrile, CDCl3 and all fluorous solvents and catalyst precursors were distilled over CaH2 and degassed immediately prior to use. The Teflon® tape (PTFE thread seal tape BS 7786: 1995 Grade L) was obtained from commercial sources. 1 and 2 were made by the literature procedure.24 The phosphine boranes were prepared by the reduction of the corresponding dialkylchlorophosphines with NaBH4.57 Pd2dba3,58 [PdCl2(MeCN)2]59 were made via literature procedures. The concentration of nBuLi was verified via a Gilman double titration before use. All other chemicals and solvents were obtained from commercial sources and used as received. The syntheses of 14–19 and catalytic studies can be found in the ESI.†
Synthesis of 3 and 4
To solid (Rf)2PCH2CH2P(Rf)2 was added 2.5 equivalents of H2O2 (30 wt% solution in water) under a nitrogen atmosphere. The mixture was stirred for 3 hours and then the excess peroxide decomposed by heating to 90 °C under an ambient atmosphere until all the water had been evaporated. The residue was extracted into 1,4-bis(trifluoromethyl) benzene and dried over MgSO4. Removal of the solvent afforded a white microcrystalline air stable powder.3: yield 78%; mp: 134–138 °C; 1H NMR (FC-72): δH = 1.08 (m, 6H, CH2) 1.29 (m, 6H, CH2), 3.14 (m, 4H, CH2CH2); 19F NMR (FC-72): δF = −84.5 (t, 4JFF = 15 Hz, CF3), −103.1 (m CF2CH2), −118.7 (CF2), −119.2 (CF2), −120.9 (CF2), −122.5 (CF2); 31P{1H} NMR (FC-72): δP = 31.6 (s); IR (cm−1): 2949 (w), 1530 (w), 1444 (w), 1364 (w), 1234 (s), 1184 (s), 1141 (s), 1122 (m), 1068 (m), 1017 (w), 996 (w), 943 (w), 928 (w), 847 (w), 770 (w), 721 (m), 708 (m), 645 (m), 566 (w), 529 (m); ms (EI): 1511.7 [40%, M+].
4: yield 45%; mp: 162–168 °C; 1H NMR (FC-72): δH = 1.10 (m, 6H, CH2) 1.32 (m, 6H, CH2), 3.18 (m, 4H, CH2CH2); 19F NMR (FC-72): δF = −84.7 (t, 4JFF = 14 Hz, CF3), −102.7 (m CF2CH2), −118.4 (CF2), −119.1 (CF2), −120.7 (CF2), −122.5 (CF2); 31P{1H} NMR (FC-72): δP = 32.8 (s); IR (cm−1): 2949 (w), 1444 (w), 1370 (w), 1332 (w), 1197 (s), 1184 (s), 1115 (s), 1081 (m), 959 (m), 932 (w), 872 (w), 737 (m), 705 (m), 652 (m), 558 (w), 528 (m);
Synthesis of 5–7
R2PHBH3 (2.82 mmol) in hexane (5 cm3) was cooled to −78 °C and nBuLi (1.45 cm3 of a 2.37 M solution in hexane, 3.1 mm) was added dropwise with stirring. After warming to room temperature 3-(perfluorooctyl)-1,2-propenoxide (0.78 ml, 2.8 mm) was added dropwise and stirred overnight. The solvent was removed under vacuum to give a yellowish-brown oil.5: IR ν (cm−1); 2958 (w, CH), 2390 (s, BH), 1432, 1364 (w, CH), 1232, 1194, 1143, 1122 (s, CF), 1061, 1075 (s, CF), 1074 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.07 (18H, d, 3JH–P = 12.5 Hz, 6CH3), 1.47 (3H, d, 1JB–H = 2.43 Hz, BH3), 1.75 (1H, m, 2JP–H = 77.14 Hz, 1JP–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.84 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 2.23 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.59 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.73 (1H, q, CHOH); 13C{1H} NMR (100 MHz, C6D6): δC = 27.95 (d, 3JC–P = 3 Hz, CH3), 31.53 (d, 1JC–P = 30.5 Hz, P–CH2), 38.75 (d, 1JC–P = 31.1 Hz, CCH3), 45.10 (m, 2JC–F = 22.8 Hz, CF2CH2), 63.5 (CHOH), 105 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 110 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 113 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, 4CF2), 115 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 118.5 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −81.85(CF3), −112.90 (CF2), −122.30(CF2), −123.16 (CF2), −123.73 (CF2), −126.79 (CF2); 7Li NMR (156 MHz, C6D6): δLi = 1.07; 11B NMR (128 MHz, C6D6): δB = −42.92 (m, 1JB–H = 2.43 Hz, 1JB–P = 60 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 40.61 (d, 1JP–B = 60 Hz).
6: IR ν (cm−1); 2966 (w, CH), 2377 (s, BH), 1465, 1370 (w, CH), 1238, 1201, 1145, 1114 (s, CF), 1065, 1047 (s, CF), 1036 (s, CO); 1H NMR (400 MHz, C6D6): δH = 0.77 (12H, d, 3JH–P = 12.5 Hz, (CH3)), 0.90 (2H, m, 2JH–P = 69.5 Hz, 3JH–H = 10.5 Hz, CHCH3), 1.35 (3H, d, 1JB–H = 2.45 Hz, BH3), 1.37 (1H, m, 2JP–H = 75.5 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.44 (1H, m, 2JP–H = 68.2 Hz, 1JH–H = 15.1, 3JH–H = 10.2, CH2P), 2.01 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.37 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.43 (1H, q, CHOH); 13C{1H} NMR (100 MHz, C6D6): δC = 16.5 (d, 3JC–P = 3 Hz, CH3), 22.1 (d, 1JC–P = 30.5 Hz, P–CH2), 28.1 (d, 1JC–P = 31.1 Hz, CCH3), 45.66 (m, 2JC–F = 22.8 Hz, CF2CH2), 57.70 (CHOH), 108 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 112 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 115 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, 4CF2), 116 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 118 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −81.45 (CF3), −112.73 (CF2), −122.30 (CF2), −123.08 (CF2), −123.71 (CF2), −126.52 (CF2); 7Li NMR (156 MHz, C6D6): δLi = 0.98; 11B NMR (128 MHz, C6D6): δB = −43.23 (m, 1JB–H = 2.43 Hz, 1JB–P = 60 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 32.50 (d, 1JP–B = 60 Hz). MS(ES+) m/z: found for C17F17H23LiOBP: 615.1450 [M + H+], calculated 615.1468.
7: IR ν (cm−1); 2955 (w, CH), 2382 (s, BH), 1669 (s, CC, Ar), 1469, 1394 (w, CH), 1238, 1202, 1148, 1114 (s, CF), 1022 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.71 (3H, d, 1JB–H = 2.43 Hz, BH3), 2.31 (1H, m, 2JP–H = 77.14 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 2.60 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 3.66 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.58 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.87 (1H, q, CHOH), 7.54 (2H, m, 4JH–P = 1.2 Hz, 2JH–H = 7.5 Hz, ArH), 7.75 (4H, m, 3JH–P = 8.4 Hz, 3JH–H = 7.5 Hz, ArH); 13C{1H} NMR (100 MHz, C6D6): δC = 34.74 (d, 1JC–P = 30.5 Hz, P–CH2), 45.35 (m, 2JC–F = 22.8 Hz, CF2CH2), 61.76 (CHOH), 108 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 111 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 113 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 116 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 119 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF2), 128.80 (m, ArC), 131.18 (m, ArC), 132.17 (m, ArC); 19F NMR (376 MHz, C6D6): δF = −81.73 (CF3), −112.86 (CF2), −122.18 (CF2), −123.09 (CF2), −123.59 (CF2), −126.62 (CF2); 7Li NMR (156 MHz, C6D6): δLi = 0.88 ppm; 11B NMR (128 MHz, C6D6): δB = −38.52 (m, 1JB–H = 2.43 Hz, 1JB–P = 54 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 12.81 (d, 1JP–B = 54 Hz).
Synthesis of 8–10
Solid samples of 5, 6 or 8 were quenched with degassed water (5 cm3) and DCM (10 cm3) added. The organic phase was separated, dried over MgSO4 and filtered. The solvent removed in vacuo to yield yellow oil.8: IR ν (cm−1): 3298 (s, OH) 2955 (w, CH), 2387 (s, BH), 1474, 1395, 1370 (w, CH), 1232, 1194, 1143, 1122 (s, CF), 1061, 1075 (s, CF), 1074 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.07 (18H, d, 3JH–P = 12.5 Hz, CH3), 1.65 (1H, m, 2JP–H = 77.14 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.84 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 2.01 (3H, d, 1JB–H = 2.43 Hz, BH3), 2.22 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.53 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.02 (1H, s, CHOH), 4.63 (1H, q, CHOH); 13C{1H} NMR (100 MHz, C6D6): δC = 27.34 (d, 3JC–P = 3 Hz, CH3), 29.51 (d, 1JC–P = 30.5 Hz, PCH2), 32.43 (d, 1JC–P = 31.1 Hz, CCH3), 45.68 (m, 2JC–F = 22.8 Hz, CF2CH2), 63.02 (CHOH), 105 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 110 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 113 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 115 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 118.5 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −81.39 (CF3), −112.75 (CF2), −122.07 (CF2), −122.96 (CF2), −123.56 (CF2), −126.69 (CF2); 11B NMR (128 MHz, C6D6): δB = −43.36 (m, 1JB–H = 2.43 Hz, 1JB–P = 60 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 40.20 (d, 1JP–B = 60 Hz); MS (MALDI+) m/z: found for C19H27F17OPB 636.1736 calculated 636.16211.
9: IR ν (cm−1); 3495 (s, OH), 2963 (w, CH), 2403 (s, BH), 1471, 1427, 1371, 1352, 1332 (w, CH), 1239, 1196, 1128, 1107 (s, CF), 1075, 1047 (s, CF), 1029 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.24 (12H, d, 3JH–P = 12.5 Hz, CH3), 1.26 (2H, m, 2JH–P = 69.5 Hz, 3JH–H = 10.5 Hz, CHCH3), 1.40 (3H, t, 1JB–H = 2.45 Hz, BH3), 2.08 (1H, m, 2JP–H = 75.5 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 2.20 (1H, m, 2JP–H = 68.2 Hz, 1JH–H = 15.1, 3JH–H = 10.2, CH2P), 2.44 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.64 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.43 (s, OH), 4.41 (1H, q, CHOH); 13C{1H} NMR (100 MHz, C6D6): δC = 17.06 (d, 3JC–P = 3 Hz, CH3), 19.06 (d, 1JC–P = 30.5 Hz, P–CH2), 36.13 (d, 1JC–P = 31.1 Hz, CCH3), 45.49 (m, 2JC–F = 22.8 Hz, CF2CH2), 60.88 (CHOH), 108 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 111 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 114 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 115 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 119 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −80.63 (CF3), −111.38 (CF2), −121.65 (CF2), −122.53 (CF2), −123.36 (CF2), −126.15 (CF2); 11B NMR (128 MHz, C6D6): δB = −43.74 (m, 1JB–H = 2.43 Hz, 1JB–P = 67 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 31.96 (d, 1JP–B = 67 Hz); MS (ES+) m/z: found for C17F17H22OBP: 607.1248 [M + H+] calculated 607.1230.
10: IR ν (cm−1): 3299 (s, OH) 2955 (w, CH), 2387 (s, BH), 1668 (s, CC, Ar), 1474, 1395, 1370 (w, CH), 1236, 1200, 1144, 1133 (s, CF), 1022 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.01 (1H, m, 2JP–H = 77.14 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.16 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 1.5 (3H, d, 1JB–H = 2.43 Hz, BH3), 1.9 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.3 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 3.67 (1H, q, CHOH), 4.67 (1H, s, CHOH), 7.20 (6H, m, 3JH–P = 1.2 Hz, 2JH–H = 7.5 Hz, ArH), 7.26 (4H, m, 3JH–P = 8.4 Hz, 3JH–H = 7.5 Hz, ArH); 13C{1H} NMR (100 MHz, C6D6): δC = 35.06 (d, 1JC–P = 30.5 Hz, P–CH2), 46.00 (m, 2JC–F = 22.8 Hz, CF2CH2), 61.44 (CHOH), 108 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 111 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 113 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 116 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 118 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF3), 129 (m, ArC), 131 (m, ArC), 132 (m, ArC); 19F NMR (376 MHz, C6D6): δF = −80.79 (CF3), −112.28 (CF2), −121.77 (CF2), −122.74(CF2), −123.33 (CF2), −126.17 (CF2); 11B NMR (128 MHz, C6D6): δB = −39.33 (m, 1JB–H = 2.43 Hz, 1JB–P = 60 Hz); 31P{1H} NMR (162 MHz, C6D6): δP = 11.69 (d, 1JP–B = 60 Hz); MS(ES−) m/z: found for C23F17H18OBP: 675.0920 [M − H−], calculated 675.0917.
Synthesis of 11–13
To a solution of 8–10 in DCM (5 cm3), TMEDA (2 cm3) was added and the reaction was stirred for 3 hours and followed by 31P{1H} NMR spectroscopy until the complete deprotection had occurred. The solvents were removed in vacuo until all TMEDA·BH3 had been removed.11: IR ν (cm−1); 3495 (s, OH) 2963 (w, CH), 1471, 1427, 1392, 1371, 1332 (w, CH), 1239, 1198, 1107 (s, CF), 1075 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.04 (18H, d, 3JH–P = 12.5 Hz, CH3), 1.66 (1H, m, 2JP–H = 77.14 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.83 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 2.25 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.51 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.04 (1H, s, CHOH), 4.63 (1H, q, CHOH); 13C{1H} NMR (100 MHz, C6D6): δC = 27.26 (d, 3JC–P = 3 Hz, CH3), 31.84 (d, 1JC–P = 30.5 Hz, P–CH2), 38.60 (d, 1JC–P = 31.1 Hz, CCH3), 44.39 (m, 2JC–F = 22.8 Hz, CF2CH2), 62.73 (CHOH), 105 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 108 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 113 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 116 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 118 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −80.78 (CF3), −112.38 (CF2), −121.77 (CF2), −122.74 (CF2), −123.33 (CF2), −126.17 (CF2); 31P{1H} NMR (162 MHz, C6D6): δP = 19.32; MS(ES−) m/z: found for C19F17H23OP: 621.1225 [M − H−] calculated 621.1215, MS(MALDI+) m/z: found for C19F17H25OP: 623.1402 [M + H+] calculated 623.1372.
12: IR ν (cm−1): 3495 (s, OH), 2963 (w, CH), 1471, 1427, 1371, 1332 (w, CH), 1239, 1195, 1146, 1108 (s, CF), 1075 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.19 (12H, d, 3JH–P = 12.5 Hz, CH3), 1.23 (2H, m, 2JH–P = 69.5 Hz, 3JH–H = 10.5 Hz, CHCH3), 1.87 (1H, m, 2JP–H = 75.5 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 2.06 (1H, m, 2JP–H = 68.2 Hz, 1JH–H = 15.1, 3JH–H = 10.2, CH2P), 2.31 (1H, m, 3JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.55 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 4.38 (s, OH), 4.51 (1H, q, CHOH); 13C{1H}NMR (100 MHz, C6D6): δC = 17.05 (d, 3JC–P = 3 Hz, CH3), 22.84 (d, 1JC–P = 30.5 Hz, PCH2), 27.98 (d, 1JC–P = 31.1 Hz, CCH3), 46.32 (m, 2JC–F = 22.8 Hz, CF2CH2), 62.08 (CHOH), 105 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 107 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 110 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 112 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 115 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3); 19F NMR (376 MHz, C6D6): δF = −81.29 (CF3), −112.40 (CF2), −121.73 (CF2), −122.89 (CF2), −123.30 (CF2), −126.27 (CF2); 31P{1H} NMR (162 MHz, C6D6): δP = 27.44. MS(MALDI+) m/z: found for C17F17H21OP: 595.1061 [M + H+] calculated 595.1059.
13: IR ν (cm−1): 3361 (s, OH), 2959 (w, CH), 1638 (s, CC, Ar), 1468, 1368, (w, CH), 1238, 1202, 1145 (s, CF), 1021 (s, CO); 1H NMR (400 MHz, C6D6): δH = 1.01 (1H, m, 2JP–H = 77.14 Hz, 1JH–H = 15.24 Hz, 3JH–H = 10.41, CH2P), 1.16 (1H, m, 2JP–H = 69.2 Hz, 1JH–H = 15.4, 3JH–H = 10.3, CH2P), 1.90 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 2.30 (1H, m, 2JH–F = 15.0 Hz, 1JH–H = 15.0 Hz, 3JH–H = 6.2 Hz, CH2CF2), 3.67 (1H, q, CHOH), 4.67 (1H, s, CHOH), 7.20 (2H, m, 3JH–P = 1.2 Hz, 2JH–H = 7.5 Hz, ArH), 7.76 (4H, m, 3JH–P = 8.4 Hz, 3JH–H = 7.5 Hz, ArH); 13C{1H}NMR (100 MHz, C6D6): δC = 36.03 (d, 1JC–P = 30.5 Hz, PCH2), 44.39 (m, 2JC–F = 22.8 Hz, CF2CH2), 61.44 (CHOH), 108 (m, 1JC–F = 270 Hz, 2JC–F = 32.9 Hz, CF2CF3), 111 (tt, 1JC–F = 270 Hz, 2JC–F = 32.9, CF2), 116 (tt, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2), 119 (tt, 1JC–F = 288 Hz, 2JC–F = 32.9 Hz, CF2CH2), 120 (tt, 118.7, 1JC–F = 257 Hz, 2JC–F = 32.9 Hz, CF2CF3), 128 (6C, m, ArC), 131 (2C, m, ArC), 132 (4C, m, ArC);19F NMR (376 MHz, C6D6): δF = −81.73 (CF3), −112.86 (CF2), −122.18 (CF2), −123.09 (CF2), −123.59 (CF2), −126.62 (CF2); 31P{1H} NMR (162 MHz, C6D6): δP = −22.61; MS(ES+) m/z: found for C23F17H17OP: 663.0721 [M + H+] calculated 663.0746.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FB and MI thank the Higher Education Commission for Pakistan for funding.Notes and references
- (a) P. Barbaro and F. Liguori, Heterogenized Homogeneous Catalysis for Fine Chemicals Production: Materials and Processes, Springer, Heidelberg, 2010 CrossRef; (b) A. Kirschning, Immobilized Catalysis: Solid Phases, Immobilization and Applications, Springer, Berlin, 2004 CrossRef.
- Á. Molnár and A. Papp, Coord. Chem. Rev., 2017, 349, 1 CrossRef.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970 CrossRef CAS.
- L. P. Barthel-Rosa and J. A. Gladysz, Coord. Chem. Rev., 1999, 190–192, 587 CrossRef CAS.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72 CrossRef.
- C.-K. E. Law and I. T. Horváth, Org. Chem. Front., 2016, 3, 1048 RSC.
- (a) J.-M. Vincent, M. Contel, G. Pozzi and R. H. Fish, Coord. Chem. Rev., 2019, 380, 584 CrossRef CAS; (b) W.-B. Yi, J.-J. Ma, L.-Q. Jiang, C. Cai and W. Zhang, J. Fluorine Chem., 2014, 157, 84 CrossRef CAS.
- (a) J. Balogh, A. R. Hlil, H.-L. Su, Z. Xi, H. S. Bazzi and J. A. Gladysz, ChemCatChem, 2016, 8, 125 CrossRef CAS; (b) Z. Xi, H. S. Bazzi and J. A. Gladysz, Catal. Sci. Technol., 2014, 4, 4178 RSC; (c) R. Tuba, R. Correa da Costa, H. S. Bazzi and J. A. Gladysz, ACS Catal., 2012, 2, 155 CrossRef CAS; (d) Z. Xi, H. S. Bazzi and J. A. Gladysz, Org. Lett., 2011, 13, 6188 CrossRef CAS; (e) R. Corrêa da Costa and J. A. Gladysz, Adv. Synth. Catal., 2007, 349, 243 CrossRef; (f) R. Corrêa da Costa and J. A. Gladysz, Chem. Commun., 2006, 2619 RSC.
- M. Ikram and R. J. Baker, J. Fluorine Chem., 2012, 139, 58 CrossRef CAS.
- P. Bhattacharyya, D. Gudmunsen, E. G. Hope, R. D. W. Kemmitt, D. R. Paige and A. M. Stuart, J. Chem. Soc., Perkin Trans. 1, 1997, 1, 3609 RSC.
- (a) H. Jiao, S. Le Stang, T. Soós, R. Meier, K. Kowski, P. Rademacher, L. Jafarpour, J.-B. Hamard, S. P. Nolan and J. A. Gladysz, J. Am. Chem. Soc., 2002, 124, 1516 CrossRef CAS; (b) L. J. Alvey, R. Meier, T. Soós, P. Bernatis and J. A. Gladysz, Eur. J. Inorg. Chem., 2000, 1975 CrossRef CAS; (c) I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rábai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef.
- M. R. Wilson, H. Liu, A. Prock and W. P. Giering, Organometallics, 1993, 12, 2044 CrossRef CAS.
- (a) J. W. Washington and T. M. Jenkins, Environ. Sci. Technol., 2015, 49, 14129 CrossRef CAS; (b) K. Li, C. Li, N.-Y. Yu, A. L. Juhasz, X.-Y. Cui and L. Q. Ma, Environ. Sci. Technol., 2015, 49, 150 CrossRef CAS; (c) C. M. Butt, D. C. G. Muir and S. A. Mabury, Environ. Toxicol. Chem., 2014, 33, 243 CrossRef CAS; (d) A. Arakaki, Y. Ishii, T. Tokuhisa, S. Murata, K. Sato, T. Sonoi, H. Tatsu and T. Matsunaga, Appl. Microbiol. Biotechnol., 2010, 88, 1193 CrossRef CAS; (e) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany, 2013 CrossRef.
- L. J. Alvey, D. Rutherford, J. J. J. Julietteand and J. A. Gladysz, J. Org. Chem., 1998, 63, 6302 CrossRef CAS.
- S. Benefice-Malouet, H. Blancou and A. Commeyras, J. Fluorine Chem., 1985, 30, 171 CrossRef CAS.
- (a) S. K. Ghosh, C. C. Cummins and J. A. Gladysz, Org. Chem. Front., 2018, 5, 3421 RSC; (b) S.-I. Kawaguchi, Y. Minamida, T. Okuda, Y. Sato, T. Saeki, A. Yoshimura, A. Nomoto, A. Akihiro and A. Ogawa, Adv. Synth. Catal., 2015, 357, 2509 CrossRef CAS; (c) S.-I. Kawaguchi, Y. Minamida, T. Ohe, A. Nomoto, M. Sonoda and A. Ogawa, Angew. Chem., Int. Ed., 2013, 52, 1748 CrossRef CAS.
- (a) D. P. Curran, Aldrichimica Acta, 2006, 39, 3 CAS; (b) Light Fluorous Chemistry-A User’s Guide, in Handbook of Fluorous Chemistry, ed. J. Gladysz, I. Horvath and D. P. Curran, Wiley- VCH, Weinheim, Germany, 2004, pp. 128−155 Search PubMed; (c) A. Studer, S. Hadida, R. Ferritto, S. Y. Kim, P. Jeger, P. Wipf and D. P. Curran, Science, 1997, 275, 823 CrossRef CAS.
- (a) R. Roychoudhury and N. L. B. Pohl, in Modern Synthetic Methods in Carbohydrate Chemistry, ed. D. B. Werz, and S. Vidal, 2014, p. 221 Search PubMed; (b) W. Zhang, in Modern Tools for the Synthesis of Complex Bioactive Molecules, ed. J. Cossy and S. Arseniyadis, 2012, p. 335 Search PubMed.
- (a) Y. Kobayashi, S. Inukai, N. Kondo, T. Watanabe, Y. Sugiyama, H. Hamamoto, T. Shioiri and M. Matsugi, Tetrahedron Lett., 2015, 56, 1363 CrossRef CAS; (b) M. Matsugi, M. Suganuma, S. Yoshida, S. Hasebe, Y. Kunda, K. Hagihara and S. Oka, Tetrahedron Lett., 2008, 49, 6573 CrossRef CAS.
- J. Hošek, O. Šimůnek, P. Lipovská, V. Kolaříková, K. Kučnirová, A. Edr, N. Štěpánková, M. Rybáčková, J. Cvačka and J. Kvíčala, ACS Sustainable Chem. Eng., 2018, 6, 7026 CrossRef.
- Selected examples: (a) N. Lu, W.-C. Chung, H.-F. Chiang, Y.-C. Fang and L.-K. Liu, Tetrahedron, 2016, 72, 8508 CrossRef CAS; (b) A. E. C. Collisand and I. T. Horvath, Catal. Sci. Technol., 2011, 1, 912 RSC; (c) C. Gimbert, V. Carolina, A. Vallribera, J. A. Gladysz and M. Jurisch, Tetrahedron Lett., 2010, 51, 4662 CrossRef CAS; (d) N. Lu, S.-C. Chen, T.-C. Chen and L.-K. Liu, Tetrahedron Lett., 2008, 49, 371 CrossRef CAS; (e) Y.-Y. Huang, Y.-M. He, H.-F. Zhou, L. Wu, B.-L. Li and Q.-H. Fan, J. Org. Chem., 2006, 71, 2874 CrossRef CAS; (f) M. Contel, P. R. Villuendas, J. Fernandez-Gallardo, P. J. Alonso, J.-M. Vincent and R. H. Fish, Inorg. Chem., 2005, 44, 9771 CrossRef CAS; (g) M. Wende and J. A. Gladysz, J. Am. Chem. Soc., 2003, 125, 5861 CrossRef CAS; (h) C. Rocaboy and J. A. Gladysz, Org. Lett., 2002, 4, 1993 CrossRef CAS; (i) M. Wende, R. Meier and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490 CrossRef CAS.
- Selected references: (a) L. V. Dinh, M. Jurisch, T. Fiedler and J. A. Gladysz, ACS Sustainable Chem. Eng., 2017, 5, 10875 CrossRef CAS; (b) Y. Kobayashi, S. Inukai, N. Kondo, T. Watanabe, Y. Sugiyama, H. Hamamoto, T. Shioiri and M. Matsugi, Tetrahedron Lett., 2015, 56, 1363 CrossRef CAS; (c) A. Motreff, G. Raffy, A. Del Guerzo, C. Belin, M. Dussauze, V. Rodriguez and J.-M. Vincent, Chem. Commun., 2010, 46, 2617 RSC; (d) F. O. Seidel and J. A. Gladysz, Adv. Synth. Catal., 2008, 350, 2443 CrossRef CAS; (e) D. Mandal, M. Jurisch, C. S. Consorti and J. A. Gladysz, Chem.–Asian J., 2008, 3, 1772 CrossRef CAS; (f) L. V. Dinh and J. A. Gladysz, Angew. Chem., Int. Ed., 2005, 44, 4095 CrossRef CAS.
- R. J. Baker, P. E. Colavita, D. Murphy, J. A. Platts and J. D. Wallis, J. Phys. Chem. A, 2012, 116, 1435 CrossRef CAS.
- M. F. Sellin, I. Bach, J. M. Webster, F. Montilla, V. Rosa, T. Avilés, M. Poliakoff and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 4569 RSC.
- C. S. Consorti, F. Hampel and J. A. Gladysz, Inorg. Chim. Acta, 2006, 359, 4874 CrossRef CAS.
- R. Tuba, V. Tesevic, L. V. Dinh, F. Hampel and J. A. Gladysz, Dalton Trans., 2005, 2275 RSC.
- (a) E. G. Hope, R. D. W. Kemmitt, D. R. Paige, A. M. Stuart and D. R. W. Wood, Polyhedron, 1999, 18, 2913 CrossRef CAS; (b) J. Fawcett, E. G. Hope, R. D. W. Kemmitt, D. R. Paige, D. R. Russell and A. M. Stuart, J. Chem. Soc., Dalton Trans., 1998, 3751 RSC; (c) C. J. Cobley and P. G. Pringle, Inorg. Chim. Acta, 1997, 265, 107 CrossRef CAS.
- G. K. Anderson and G. J. Lumetta, Inorg. Chem., 1987, 26, 1518 CrossRef CAS.
- R. K. Merwin, R. C. Schnabel, J. D. Koola and D. M. Roddick, Organometallics, 1992, 11, 2972 CrossRef CAS.
- J. J. M. de Pater, B.-J. Deelman, C. J. Elsevier and G. van Koten, J. Mol. Catal. A: Chem., 2006, 258, 334 CrossRef CAS.
- See for example: (a) H. Fernándéz-Pérez, P. Etayo, J. L. Núñez-Rico, B. Balakrishna and A. Vidal-Ferran, RSC Adv., 2014, 4, 58440 RSC; (b) T. Koch, S. Blaurock, F. Somoza, Jr. and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2000, 21672 Search PubMed; (c) H. Brunner and A. Sicheneder, Angew. Chem., Int. Ed. Engl., 1988, 27, 718 CrossRef; (d) K. Issleib and H. R. Roloff, Chem. Ber., 1965, 98, 2091 CrossRef CAS.
- P. Pellon, Tetrahedron Lett., 1992, 33, 4451 CrossRef CAS.
- (a) F. Begum, B. Twamley and R. J. Baker, J. Chem. Crystallogr., 2019 DOI:10.1007/s10870-019-00775-8; (b) F. Begum, M. A. Choudhary, M. A. Mirza, B. Twamley and R. J. Baker, J. Chem. Crystallogr., 2018, 48, 209 CrossRef CAS.
- (a) D. J. Adams, J. A. Bennett, D. Duncan, E. G. Hope, J. Hopewell, A. M. Stuart and A. J. West, Polyhedron, 2007, 26, 1505 CrossRef CAS; (b) J. A. S. Howell, N. Fey, J. D. Lovatt, P. C. Yates, P. McArdle, D. Cunningham, E. Sadeh, H. E. Gottlieb, Z. Goldschmidt, M. B. Hursthouse and M. E. Light, J. Chem. Soc., Dalton Trans., 1999, 3015 RSC.
- P. A. W. Dean, Can. J. Chem., 1979, 57, 754 CrossRef CAS.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610 CrossRef CAS.
- S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1431 CrossRef CAS.
- F. D. M. Bolte, H.-W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2006, 5138 Search PubMed.
- (a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC; (c) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- (a) R. Shukla and D. Chopra, CrystEngComm, 2015, 17, 3596 RSC; (b) A. G. Dikundwar, R. Sathishkumar and T. N. Guru Row, Z. Krist, 2014, 229, 609 CAS and refs therein.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- For recent reviews see: (a) L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3 CrossRef CAS; (b) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS.
- Recent reviews see: (a) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC; (b) A. Fihri, M. Bouhrara, B. Nekoueisharki, J. M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC; (c) D. Astruc, Inorg. Chem., 2007, 46, 1884 CrossRef CAS.
- D. Duncan, E. G. Hope, K. Singh and A. M. Stuart, Dalton Trans., 2011, 40, 1998 RSC.
- D. P. Curran, K. Fischer and G. Moura-Letts, Synlett, 2004, 1379 CrossRef CAS.
- R. Corrêada Costa, M. Jurisch and J. A. Gladysz, Inorg. Chim. Acta, 2008, 361, 3205 CrossRef.
- J.-Q. Liu, X.-X. Gou and Y.-F. Han, Chem.–Asian J., 2018, 13, 2257 CrossRef CAS.
- (a) A. Motreff, C. Belin, R. Correa da Costa, M. El Bakkaria and J.-M. Vincent, Chem. Commun., 2010, 46, 6261 RSC; (b) B. A. Parsons, O. L. Smith, M. Chae and V. Dragojlovic, Beilstein J. Org. Chem., 2015, 11, 980 CrossRef CAS.
- N. J. Van Zee and V. Dragojlovic, Chem.–Eur. J., 2010, 16, 7950 CrossRef CAS PubMed.
- For example, ligand 13 has a calculated cost of €14000 per mole, whilst Pd(OAc)2 is €5904 per mole, calculated on the basis of Ph2PHBH3 (Sigma Aldrich €295.00 for 5 g; cat no. 449563) and 3-(perfluorooctyl)-1,2-propenoxide (fluorochem £28.00 for 5 g; cat no: 007145) prices correct 22/04/2019.
- Bruker APEX 2 v2012.12–0, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, SADABS, Bruker AXS Inc., University of Göttingen, Germany, Madison, Wisconsin, USA, 2014 Search PubMed.
- T. Imamoto, T. Oshiki, T. Onozawa, T. Kusumoto and K. Sato, J. Am. Chem. Soc., 1990, 112, 5244 CrossRef CAS.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- G. K. Anderson and M. Lin, Inorg. Synth., 1990, 28, 60 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1912783. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04863d |
| This journal is © The Royal Society of Chemistry 2019 |