
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long noncoding RNA ANRIL protects cardiomyocytes against hypoxia/reoxygenation injury by sponging miR-195-5p and upregulating Bcl-2†
Hui Zhao‡
a,
Li Meng‡b,
Chengyang Xuc,
Bin Lina,
Xiangming Zhengd,
Jiaxiang Wanga and
Deguang Feng *a
*a
aDepartment of Cardiac Surgery, The First Affiliated Hospital of Zhengzhou University, No. 1 Longhu Central Road, Jinshui District450052, Zhengzhou, China. E-mail: i3772442kuijia@163.com; Tel: +86-0371-66271808
bDepartment of Intensive Care Unit, The Henan Provincial Chest Hospital, Zhengzhou, China
cDepartment of International Medical Center, The Henan Provincial PeopleS Hospital, Zhengzhou, China
dDepartment of Anesthesiology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
First published on 4th November 2019
Abstract
Long noncoding RNAs have been widely accepted to play important roles in acute myocardial infarction (AMI). The dysregulation of cyclin-dependent kinase inhibitor 2B antisense RNA 1 (ANRIL) was discovered in AMI patients. Nevertheless, the detailed role and molecular mechanisms of ANRIL in AMI remain indistinct. The levels of ANRIL, miR-195-5p and Bcl-2 mRNA were determined by qRT-PCR. western blot was performed to assess the expression of Bcl-2, Bax, Cyclin D1 and p21. Cell proliferation was detected by CCK-8 assay, and cell apoptosis was measured by flow cytometry. The targeted correlation between ANRIL and miR-195-5p was confirmed by the dual-luciferase reporter and RNA pull-down assays. Our data revealed that ANRIL was downregulated and miR-195-5p was upregulated in the serum of AMI patients and hypoxia/reoxygenation (H/R)-induced myocardial cells. ANRIL upregulation or miR-195-5p knockdown alleviated H/R-induced myocardial cell injury. Moreover, ANRIL sequestered miR-195-5p by acting as a sponge of miR-195-5p. ANRIL upregulated Bcl-2 expression by sponging miR-195-5p. Additionally, ANRIL overexpression alleviated H/R-induced myocardial cell injury by upregulating Bcl-2. In conclusion, our study indicated that ANRIL upregulation alleviated H/R-induced myocardial cell injury partially through sponging miR-195-5p and upregulating Bcl-2, highlighting its role as a promising mediator for new therapies for AMI treatment.
1. Introduction
Acute myocardial infarction (AMI) is one of the most serious cardiovascular diseases, giving rise to high morbidity and mortality around the world.1 Hypoxia occurs in acute coronary occlusion and results in cardiac cell death, which is the first phase of AMI. Full reperfusion/reoxygenation takes place in primary angioplasty, but it is a nocuous event.2 Hypoxia/reoxygenation (H/R) induces local myocardial cell apoptosis, which in turn leads to nonreversible injure to the myocardium.1,3 Therefore, in the present study, we explored novel biomarkers for AMI prevention and therapy by using H/R-induced myocardial cells.For the last few years, the non-protein-coding portion of the genome has attracted increasing attention for its function in developmental processes, particularly emphasizing on mammalian development.4 Based on the size, noncoding RNAs can be divided into two major groups: long noncoding RNA (lncRNA, >200 nucleotides) and short noncoding RNA (<200 nucleotides mainly including microRNAs/miRNAs). Dysregulation of noncoding RNAs has been widely accepted to play important roles in human cardiovascular diseases, including AMI.5–8 More intriguingly, the competing endogenous RNA (ceRNA) concept proposes that lncRNA functions as a miRNA sponge to posttranscriptionally regulate the expression of target mRNAs, highlighting the importance of such interactions during AMI process.9,10
LncRNA cyclin-dependent kinase inhibitor 2B antisense RNA 1 (ANRIL also named CDKN2B-AS1) is a transcript of chromosome 9p21 region which is the strongest candidate for several important human diseases.11–14 The genetic variants of ANRIL gene have been demonstrated to be strongly associated with increased risk of coronary heart disease (CHD) and stroke.15 A low level of ANRIL impacts the expression of genes involved in coronary artery disease (CAD) and is correlated with CAD risk genotype.16 Moreover, a recent document reported that ANRIL expression was decreased in blood samples of AMI patients.6 Nevertheless, the detailed role and molecular mechanisms of ANRIL in AMT remain indistinct. In the present study, our data supported a significant downregulation of ANRIL in AMI patients. Moreover, our study indicated that ANRIL upregulation alleviated H/R-induced myocardial cell injury through sponging miR-195-5p and upregulating Bcl-2 expression.
2. Materials and methods
2.1. Samples collection
A total of 39 AMI patients and 39 healthy volunteers were included in this study and the clinical characteristics were presented in Table 1. AMI patients were diagnosed by The First Affiliated Hospital of Zhengzhou University based on the combination of several criteria: (1) ischemic symptoms; (2) increased cTnT level; (3) pathological Q wave; (4) increased creatine kinase-MB.17 The healthy volunteers were recruited at the same time. 1 ml of peripheral blood samples from AMI patients was collected at 12 h after the onset of symptoms. Serum samples were isolated by centrifugation and stored at −80 °C until use. All participators signed informed consent and the study was approved by The Ethics Committee of the First Affiliated Hospital of Zhengzhou University in accordance with the Declaration of Helsinki Principles. Informed consent was obtained from all participants.| Clinical characteristics | Normal (n = 39) | AMI (n = 39) | P-value |
|---|---|---|---|
| a BMI, body mass index; TC, total cholesterol; TG, triglycerides; HDL, high density lipoprotein; LDL, low density lipoprotein. | |||
| Age, years | 63.5 ± 8.9 | 52.4 ± 9.8 | <0.001 |
| Sex, male/female | 24/15 | 21/18 | 0.492 |
| Heart rate, beats per minute | 73.5 ± 8.5 | 76.7 ± 9.7 | 0.126 |
| BMI (kg m−2) | 25.6 ± 3.1 | 25.8 ± 3.3 | 0.783 |
| Systolic blood pressure, mmHg | 128 ± 16 | 125 ± 15 | 0.396 |
| Diastolic blood pressure, mmHg | 76 ± 9 | 79 ± 10 | 0.168 |
| Diabetes mellitus | 9/30 | 6/33 | 0.389 |
| Smoking | 20/19 | 16/23 | 0.364 |
| Drinking | 14/25 | 8/31 | 0.131 |
| Glucose, mmol L−1 | 4.9 ± 0.6 | 7.4 ± 1.2 | <0.001 |
| TC, mmol L−1 | 1.9 ± 0.4 | 4.2 ± 1.4 | <0.001 |
| TG, mmol L−1 | 1.8 ± 1.1 | 2.6 ± 1.2 | 0.003 |
| HDL, mmol L−1 | 0.8 ± 0.1 | 1.0 ± 0.1 | <0.001 |
| LDL, mmol L−1 | 3.1 ± 0.3 | 3.6 ± 0.5 | <0.001 |
2.2. Cell culture and treatment
Human myocardial cell line (AC16, which has the ability to contract and retain differentiated cardiac morphological, biochemical, and electrophysiological properties) was purchased from BeNa Culture Collection (Beijing, China) and maintained in DMEM medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, Gibco), 100 U ml−1 penicillin (Gibco) and 100 μg ml−1 streptomycin (Gibco) at 37 °C in a 5% CO2 humidified atmosphere. The basic marker cardiac troponin-I (cTNI) of AC16 cells was shown in ESI Fig. 1† using immunofluorescence microscopy, as described below.For H/R treatment, AC16 cells (1 × 106 each well) in serum-free medium were seeded in 96-well plates and placed in an incubator with 95% N2 and 5% CO2 for 4 h, followed by reoxygenation with 95% O2 and 5% CO2 for 20 min. Gene expression, cell proliferation and apoptosis changes were determined at the indicated time after H/R treatment. Cells maintained under normoxic conditions were used as control.
2.3. Cell transfection
The full sequence of ANRIL was cloned into pcDNA3.1(+) vector (CMV promoter, shown in ESI Fig. 2A†) to construct ANRIL overexpression vector (Vector-ANRIL, RiboBio, Guangzhou, China), as previously described,18 and nontarget vector (Vector-NC, RiboBio) was used as a negative control. To explore the effect of ANRIL, AC16 cells (1 × 106 each well) grown in 96-well plates were transfected with 100 ng of Vector-ANRIL or Vector-NC, 50 nM of silencer select predesigned small interfering RNA (si-ANRIL, Applied Biosystems, Foster City, CA, USA) or siRNA negative control (si-NC, Applied Biosystems) using Lipofectamine® RNAiMAX™ transfection reagent (Invitrogen, Waltham, MA, USA) following the protocols of manufacturers. To observe the function of miR-195-5p, cells (1 × 106 each well) were transfected with 50 nM of has-miR-195-5p mirVana miRNA mimic (miR-195-5p mimic, Applied Biosystems) or a scrambled negative sequence (miR-NC mimic, Applied Biosystems), 50 nM of miRIDIAN miR-195-5p inhibitor (anti-miR-195-5p, Applied Biosystems) or negative control inhibitor (anti-miR-NC, Applied Biosystems) using Lipofectamine® RNAiMAX™ transfection reagent. To study the effect of Bcl-2, 50 nM of siRNA against Bcl-2 (si-Bcl-2, Applied Biosystems) or si-NC was transfected into cells (1 × 106 each well) using the transfection reagent. The transfection efficiency was determined using quantitative real-time PCR (qRT-PCR) and fluorescence microscopy (ESI Fig. 3†).2.4. Determination of ANRIL, miR-195-5p and Bcl-2 expression
The expression levels of ANRIL, miR-195-5p and Bcl-2 were detected using qRT-PCR. Total RNA from serum samples and cells was extracted with the mirVANA miRNA isolation kit (Ambion Life Technologies, Carlsbad, CA, USA) following the instructions of manufacturers. For ANRIL and Bcl-2 mRNA detection, cDNA was synthesized by SuperScript-III reverse transcriptase (Invitrogen), and subjected to qRT-PCR using SYBR™ Green PCR Master Mix (Applied Biosystems) on an ABI 7500 real-time PCR system (Applied Biosystems). GAPDH mRNA level was measured for normalization. For detection for miR-195-5p, mature miR-195-5p and endogenous control U6 were analyzed using the TaqMan reverse transcription kit (Applied Biosystems) and TaqMan MicroRNA Assay kit (Applied Biosystems) according to the guidance of manufacturers. The relative gene expression was calculated by the 2−ΔΔCt method.2.5. Detection of LDH, MDA, SOD, ATP and GSH-PX
Glutathione peroxidase (GSH-PX) assay kit, superoxide dismutase (SOD) assay kit, malondialdehyde (MDA) assay kit, adenosine triphosphate (ATP) assay kit, and lactate dehydrogenase (LDH) release assay kit were obtained from Beyotime Biotechnology (Shanghai, China). The levels of GSH-PX, SOD, MDA and LDH release were determined using corresponding assay kits following the instructions of manufacturers and analyzed by a FLUOStar Omega microplate reader (BMG Labtech Gmbh, Ortenberg, Germany).2.6. Cell proliferation assay
Cell proliferation was measured using the Cell Counting kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) according to the protocols of manufacturers. Briefly, cells (1 × 106 each well) were seeded in a 96-well plate and treated or transfected. At the indicated time point, 10 μl of CCK-8 solution was added into each well and incubated at 37 °C for 2 h. The absorbance at 450 nm was measured by a FLUOStar Omega microplate reader.2.7. Cell apoptosis assay
Cell apoptosis was determined using flow cytometry with an Annexin V-FITC apoptosis detection kit (Thermo Fisher Scientific, Waltham, MA, USA) following the protocols of manufacturers. To be brief, cells were washed with ice-cold PBS and then stained with 5 μl of Annexin V-FITC and 10 μl of PI in darkness. Cell apoptosis was analyzed using the FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with CellQuest software and each analysis included about 1 × 105 cells.2.8. Assessment of Bcl-2, Bax, cyclin D1 and p21 levels
Cells were lysed using ice-cold RIPA lysis buffer (Beyotime Biotechnology) and total protein was quantified by a BCA protein assay kit (Thermo Fisher Scientific) according to the protocols of manufacturers. 50 μg of protein extracts was resolved on a 10% SDS polyacrylamide gel and electroblotted onto a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA). After blocking with 5% nonfat milk, the membranes were probed with primary antibodies including anti-Bcl-2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Cell Signaling Technology, Danvers, MA, USA), anti-Bax (1:1000, Cell Signaling Technology), anti-Cyclin D1 (1:1000, Cell Signaling Technology), anti-p21 (1:1000, Cell Signaling Technology) and anti-GAPDH (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), which were followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:2000, Cell Signaling Technology). Immunoreactivity signals were visualized using ECL chromogenic substrate (Bio-Rad Laboratories) by ImmunoStar LD (WAKO, Osaka, Japan) and gene expression was evaluated by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
:1000, Cell Signaling Technology, Danvers, MA, USA), anti-Bax (1:1000, Cell Signaling Technology), anti-Cyclin D1 (1:1000, Cell Signaling Technology), anti-p21 (1:1000, Cell Signaling Technology) and anti-GAPDH (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), which were followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:2000, Cell Signaling Technology). Immunoreactivity signals were visualized using ECL chromogenic substrate (Bio-Rad Laboratories) by ImmunoStar LD (WAKO, Osaka, Japan) and gene expression was evaluated by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.9. Determination of cTNI basic marker
AC16 cells and normal control H9C2 cardiomyoblast cells were fixed with 4% paraformaldehyde for 15 min, then permeablilized and washed three times in PBS before blocked with 1% goat serum at room temperature for 60 min. Afterwards, cells were incubated with anti-cTNI antibody (Cell Signaling Technology) at a dilution of 1:100 at 4 °C overnight, followed by the incubation with Alexa Fluor 488 conjugated goat anti-mouse IgG antibody (Cell Signaling Technology) at a dilution of 1:50. After being post-fixed in paraformaldehyde, cells were mounted on a pre-cleaned microscope slide with Prolong Gold Antifade Reagent (Invitrogen). Images were captured by a laser (Point) scanning confocal microscope (TCS SP5, Leica, Wetzlar, Germany) following the instructions of manufacturers.
2.10. Bioinformatics
Analysis for the targeted miRNAs of ANRIL was performed using online software starBase v.2.0 at http://starbase.sysu.edu.cn.2.11. Determination of targeted sequence
Dual-luciferase reporter assay was carried out to determine the targeted correlation between ANRIL and miR-195-5p. The partial sequence of ANRIL containing the targeted sequence for miR-195-5p was cloned into the pmirGLO Vector (SV40 promoter, shown in ESI Fig. 2B†) to construct ANRIL luciferase reporter vector (ANRIL-Wt, RibioBio), as previously described.19 Site-directed mutagenesis of the targeted sequence was performed using the GeneArt™ Site-Directed Mutagenesis System (Invitrogen) to generate ANRIL mutant-type reporter vector (ANRIL-Mut). AC16 cells (1 × 106 each well) grown in 96-well plates were cotransfected with 100 ng of ANRIL-Wt or ANRIL-Mut and 50 nM of miR-195-5p mimic or miR-NC mimic using Lipofectamine® RNAiMAX™ transfection reagent. At 48 h after transfection, firefly and Renilla luciferase activities were measured using the dual-luciferase assay system (Promega, Madison, WI, USA). The relative luciferase activity was normalized against Renilla luciferase activity.2.12. Confirmation of endogenous interaction between ANRIL and miR-195-5p
The endogenous interaction between ANRIL and miR-195-5p was confirmed by RNA pull-down assay. Biotin-labeled miR-195-5p mimic (Bio-miR-195-5p) and the mutation of ANRIL binding site (Bio-miR-195-5p-Mut), biotin-labeled ANRIL partial sequence (Bio-ANRIL) containing the miR-195-5p binding site and the mutation of the seeded region (Bio-ANRIL-Mut) were obtained from Thermo Fisher Scientific, and Bio-miR-NC or Bio-NC was used as the negative control. AC16 cells were harvested and lysed in RIPA lysis buffer. Then, cell lysates were incubated with Bio-miR-195-5p, Bio-miR-195-5p-Mut, Bio-miR-NC, Bio-ANRIL, Bio-ANRIL-Mut or Bio-NC at room temperature for 1 h, followed by the incubation with Streptavidin agarose beads (Thermo Fisher Scientific) for 1 h. Total RNA was extracted from the complex, and qRT-PCR was performed to detect the enrichment of ANRIL or miR-195-5p.2.13. Statistical analysis
Statistical analyses were performed using SPSS v.20.0 software (SPSS Inc., Chicago, IL, USA) and all data were described as mean ± standard deviation (SD) from at least three independent experiments. Differences between two groups were compared by Student's t-test or Mann–Whitney U test. One-way ANOVA was performed for comparison of more than two groups. Correlations between ANRIL, miR-195-5p and Bcl-2 were analyzed using Spearman's test. A value of P < 0.05 was considered statistically significant.3. Results
3.1. ANRIL was downregulated in the serum of AMI patients and H/R-induced myocardial cell
To explore the role of ANRIL on AMI, the expression of ANRIL was firstly determined by qRT-PCR in serum samples of AMI patients and healthy volunteers. These data revealed that AMI patients had serum lower ANRIL levels than healthy controls (Fig. 1A). Conversely, the serum amounts of cardiac troponin T (cTnT) were higher in AMI patients than those in healthy controls (Fig. 1B). More intriguingly, we found that ANRIL level was inversely correlated with cTnT amount in the serum of AMI patients (Fig. 1C). Then, ANRIL levels in AC16 cells were assessed at 12, 24 and 48 h after H/R treatment. In contrast to continual normoxic conditions, H/R treatment time-dependently weakened ANRIL expression in AC16 cells (Fig. 1D). These results together suggested the involvement of ANRIL in AMI progression.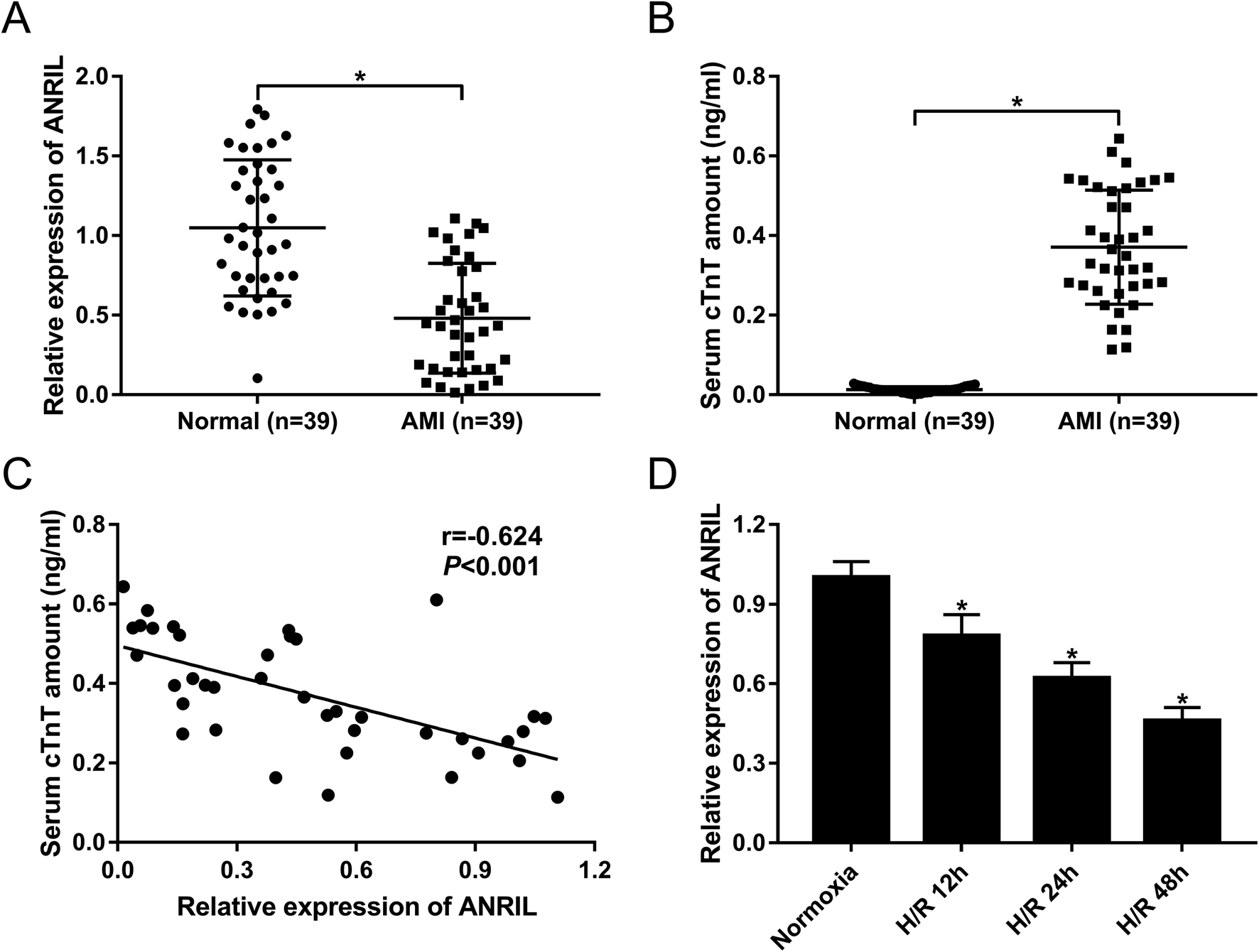 | ||
| Fig. 1 The expression of ANRIL in serum samples of AMI patients and H/R-induced myocardial cells. The expression of ANRIL by qRT-PCR (A), and cTnT amount (B) in serum samples of 39 AMI patients and 39 healthy volunteers. (C) The correlation between ANRIL level and cTnT amount in serum samples of 39 AMI patients. (D) ANRIL expression by qRT-PCR in AC16 cells at 12, 24 and 48 h after H/R treatment. *P < 0.05 vs. normal or normoxia. | ||
3.2. Upregulation of ANRIL alleviated H/R-induced myocardial cell injury
Myocardial injury induced H/R is demonstrated to be a significant pathological process in AMI.20,21 Herein, AC16 cells were firstly performed with H/R treatment to induce myocardial injury. These data showed that apart from the impact on ANRIL expression (Fig. 2A), H/R treatment resulted in increased levels of LDH release (Fig. 2B) and MDA (Fig. 2C), and decreased levels of SOD (Fig. 2D), GSH-PX (Fig. 2E) and ATP (Fig. 2F) compared with normoxia control. Moreover, H/R treatment led to a significant repression of cell proliferation (Fig. 2G) and a prominent enhancement of cell apoptosis (Fig. 2H and I). Additionally, the levels of Bcl-2 and Cyclin D1 in the H/R group were lower than those in the control group, while Bax and p21 levels were higher in H/R group (Fig. 2J), supporting the promotional effect of H/R treatment on cell apoptosis. All these data demonstrated that H/R treatment induced myocardial cell injury.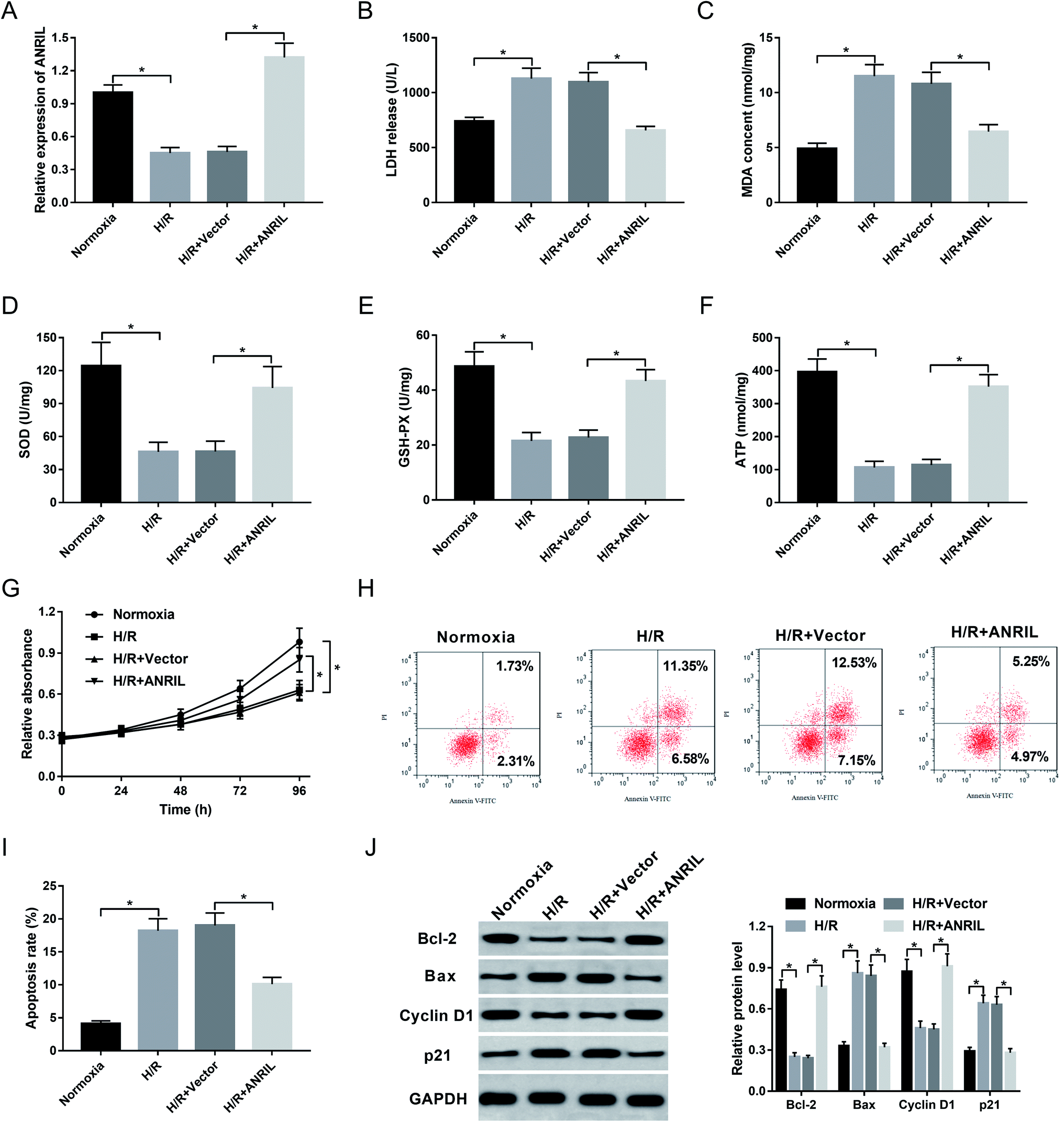 | ||
| Fig. 2 The effect of ANRIL upregulation on H/R-induced myocardial cell injury. AC16 cells were exposed to normoxic or H/R conditions, or AC16 cells were transfected with Vector-NC or Vector-ANRIL prior to H/R treatment. After H/R treatment for 48 h, ANRIL expression by qRT-PCR (A), LDH release (B), MDA content (C), SOD level (D), GSH-PX expression (E) and ATP level (F) by corresponding assay kits in treated cells. (G) At the indicated time point after H/R treatment, cell proliferation by CCK-8 assay in treated cells. At 48 h after H/R treatment, cell apoptosis by flow cytometry (H and I), and the expression of Bcl-2, Bax, Cyclin D1 and p21 by western blot (J) in treated cells. *P < 0.05 vs. normoxia or Vector-NC. | ||
Then, we observed the effect of ANRIL on H/R-induced myocardial cell injury by transfection of Vector-ANRIL prior to H/R treatment. As shown by qRT-PCR, transfection of Vector-ANRIL, but not a negative control vector, significantly increased ANRIL expression in H/R-induced AC16 cells (Fig. 2A). Subsequent experiments revealed that in contrast to the negative control, ANRIL overexpression strikingly reduced LDH release (Fig. 2B) and MDA content (Fig. 2C), and elevated the levels of SOD (Fig. 2D), GSH-PX (Fig. 2E) and ATP (Fig. 2F) in H/R-induced AC16 cells. Furthermore, ANRIL overexpression resulted in a promotion of cell proliferation (Fig. 2G) and a suppression of cell apoptosis (Fig. 2H–J) in H/R-induced AC16 cells. Together, these data indicated that ANRIL upregulation alleviated H/R-induced myocardial cell injury.
3.3. ANRIL directly targeted miR-195-5p
To further explore the underlying mechanism by which ANRIL protected the myocardial cells against H/R-induced injury, we carried out a detailed analysis for its targeted miRNAs. The predicted data by starBase software revealed that ANRIL contained a potential binding site for miR-195-5p (Fig. 3A). To determine whether ANRIL is a molecular sponge of miR-195-5p, we performed luciferase reporter assays with luciferase reporter (ANRIL-Wt) containing the targeted sequence for miR-195-5p. As demonstrated by qRT-PCR, transfection of miR-195-5p mimic significantly increased miR-195-5p expression in AC16 cells compared with miR-NC mimic (Fig. 3B). In contrast to the negative control, the luciferase activity of ANRIL-Wt was highly reduced by miR-195-5p overexpression (Fig. 3C). Whereas, the mutation of the targeted sequence (ANRIL-Mut) completely abolished the effect of miR-195-5p overexpression on reporter gene expression (Fig. 3C). After that, to validate the endogenous interaction between ANRIL and miR-195-5p, RNA pull-down assays were performed using Bio-miR-195-5p, Bio-miR-195-5p-Mut, Bio-ANRIL and Bio-ANRIL-Mut. In comparison to a corresponding control, the enrichment of ANRIL was substantially increased by incubation of Bio-miR-195-5p in AC16 cells (Fig. 3D). Likewise, miR-195-5p enrichment was highly elevated when incubated with Bio-ANRIL (Fig. 3E). However, these effects were completely abrogated by Bio-miR-195-5p-Mut or Bio-ANRIL-Mut (Fig. 3D and E). Further, we observed whether ANRIL modulated miR-195-5p expression in AC16 cells. In contrast to a corresponding control, ANRIL expression was significantly increased in the presence of Vector-ANRIL, while it was highly decreased by the transfection of si-ANRIL in AC16 cells (Fig. 3F). Moreover, miR-195-5p expression was markedly downregulated by ANRIL overexpression, and it was upregulated when ANRIL knockdown (Fig. 3G), indicating the negative modulation of ANRIL on miR-195-5p expression. These data demonstrated that ANRIL sequestered miR-195-5p through binding to miR-195-5p.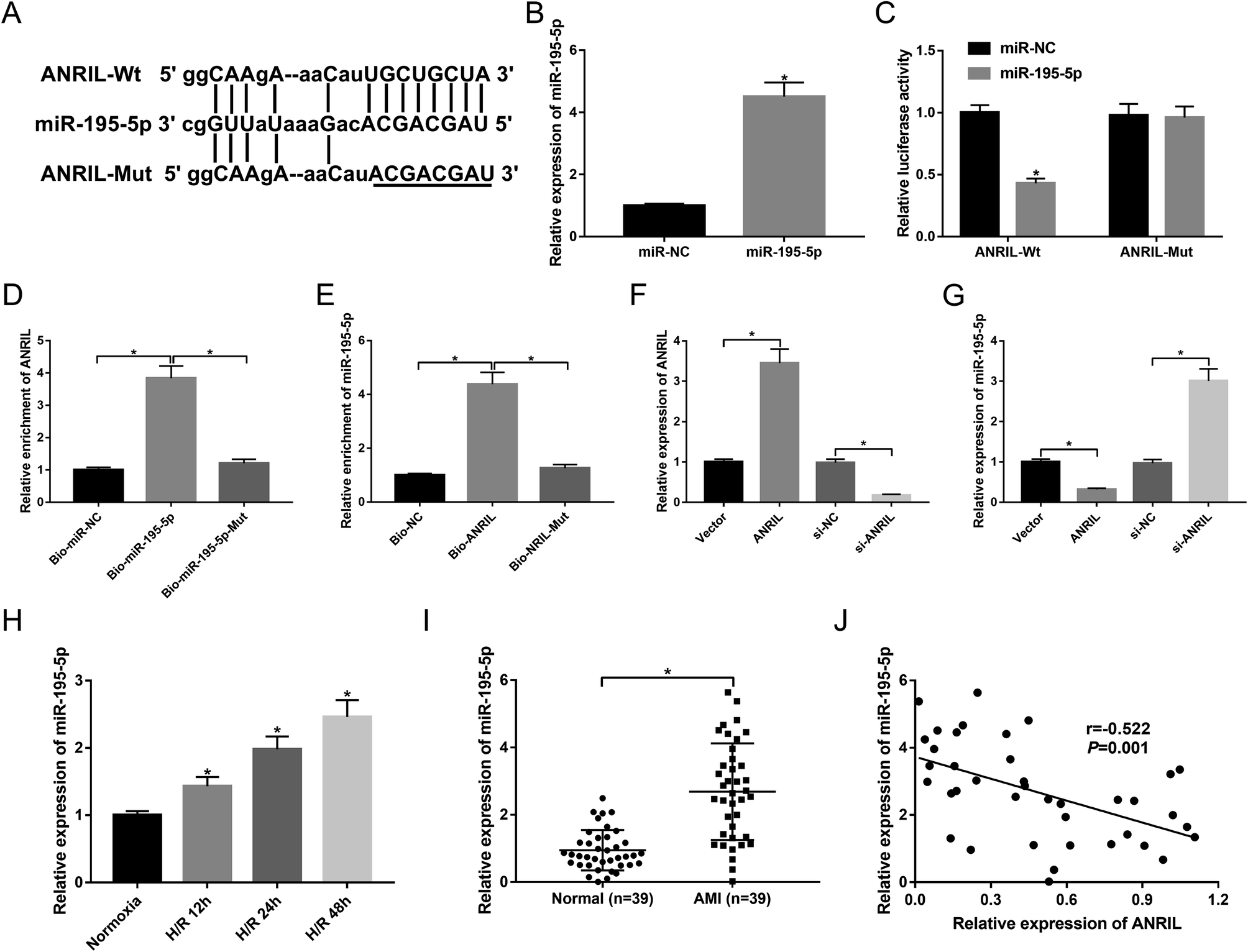 | ||
| Fig. 3 ANRIL directly targeted miR-195-5p in the myocardial cells. (A) Nucleotide resolution of the predicted miR-195-5p binding site in ANRIL and the mutated miR-195-5p binding site. (B) MiR-195-5p expression by qRT-PCR in AC16 cells transfected with miR-NC mimic or miR-195-5p mimic. (C) The luciferase activity in AC16 cells cotransfected with ANRIL-Wt or ANRIL-Mut and miR-NC mimic or miR-195-5p mimic. (D) The enrichment of ANRIL by qRT-PCR in AC16 cells incubated with Bio-miR-NC, Bio-miR-195-5p or Bio-miR-195-5p-Mut. (E) MiR-195-5p enrichment in AC16 cells incubated with Bio-NC, Bio-ANRIL or Bio-ANRIL-Mut. The expression of ANRIL (F) and miR-195-5p (G) in AC16 cells transfected with Vector-NC, Vector-ANRIL, si-NC or si-ANRIL. (H) MiR-195-5p expression in AC16 cells after H/R treatment for 12, 24 and 48 h. (I) MiR-195-5p expression in serum samples of 39 AMI patients and 39 healthy volunteers. (J) The correlation between miR-195-5p expression and ANRIL level in serum of AMI patients. *P < 0.05 vs. respective control. | ||
Next, we analyzed miR-195-5p expression in H/R-induced AC16 cells and the serum of AMI patients. As shown by qRT-PCR, H/R treatment elevated miR-195-5p expression in a time-dependent manner (Fig. 3H). Moreover, the serum levels of miR-195-5p were higher in AMI patients than those in healthy controls (Fig. 3I). More importantly, miR-195-5p expression was inversely correlated with ANRIL level in the serum of AMI patients (Fig. 3J).
3.4. Knockdown of miR-195-5p attenuated H/R-induced myocardial cell injury
Then, to understand the role of miR-195-5p on AMI, we manipulated miR-195-5p expression by the transfection of anti-miR-195-5p prior to H/R treatment. Transient introduction of anti-miR-195-5p, but not a scrambled control sequence, significantly reduced miR-195-5p expression in H/R-induced AC16 cells (Fig. 4A). Further experiments revealed that in comparison their counterparts, miR-195-5p knockdown resulted in a decrease of LDH release (Fig. 4B) and MDA content (Fig. 4C), and an increase of SOD level (Fig. 4D), GSH-PX expression (Fig. 4E) and ATP level (Fig. 4F), as well as a promotion of cell proliferation (Fig. 4G) and a repression of cell apoptosis (Fig. 4H and I). These data together indicated that miR-195-5p knockdown protected myocardial cell against H/R-induced injury.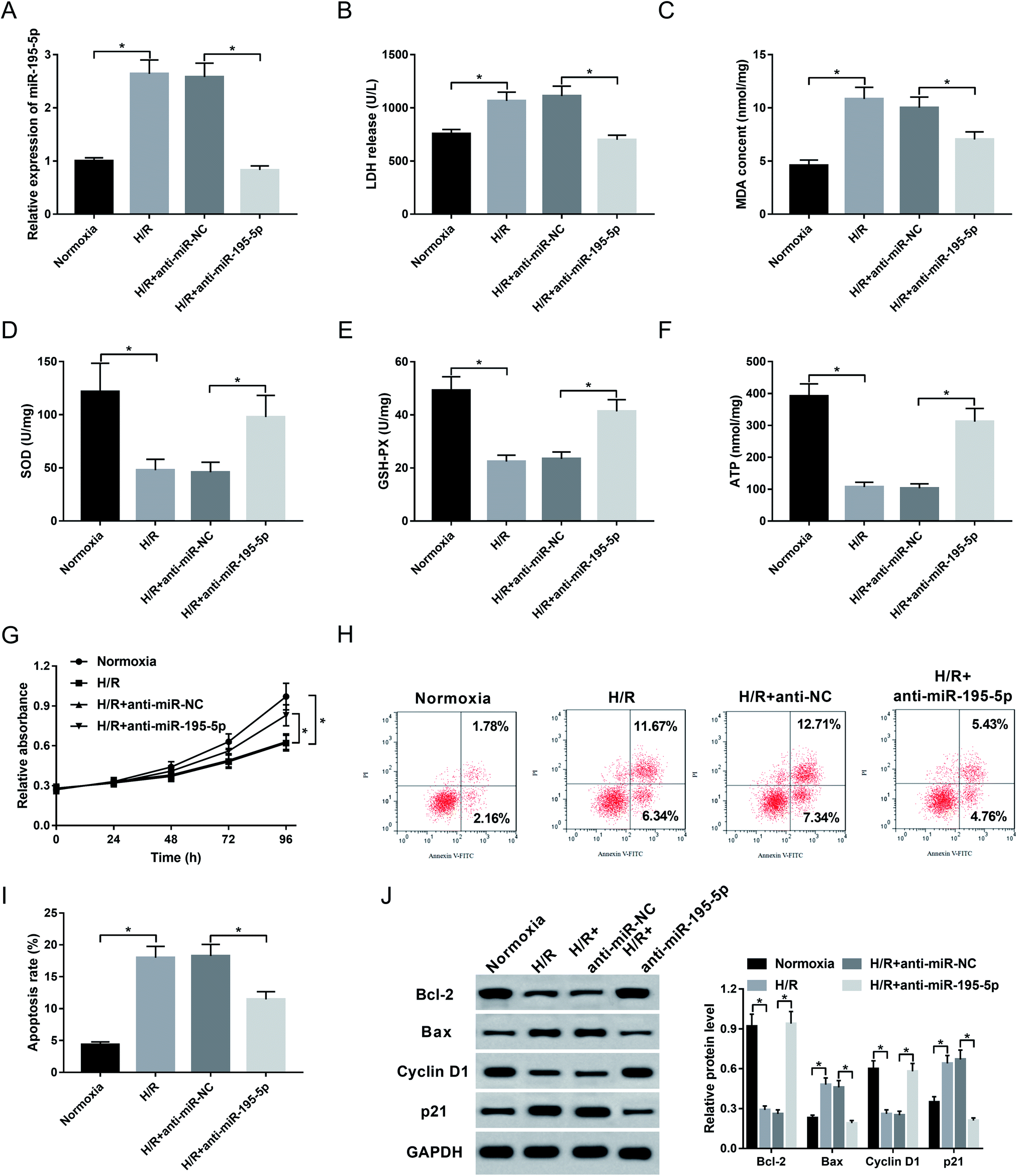 | ||
| Fig. 4 The role of miR-195-5p on H/R-induced myocardial cell injury. AC16 cells were transfected with anti-miR-NC or anti-miR-195-5p prior to H/R treatment. After H/R treatment for 48 h, miR-195-5p expression by qRT-PCR (A), LDH release (B), MDA content (C), SOD level (D), GSH-PX expression (E) and ATP level (F) by corresponding assay kits in treated cells. (G) At the indicated time point after H/R treatment, cell proliferation by CCK-8 assay in treated cells. 48 h after H/R treatment, cell apoptosis by flow cytometry (H and I), and the expression of Bcl-2, Bax, Cyclin D1 and p21 by western blot (J) in treated cells. *P < 0.05 vs. anti-miR-NC. | ||
3.5. ANRIL upregulated Bcl-2 expression by acting as a molecular sponge of miR-195-5p
MiRNAs exert biological functions through regulating their target genes.22 Bcl-2 had been manifested to be a functional target of miR-195-5p in human myocardial cells.23,24 As a result, in comparison to a corresponding control, the levels of Bcl-2 mRNA and protein were highly repressed by miR-195-5p overexpression, while they were significantly elevated after miR-195-5p depletion (Fig. 5A–C), indicating the negative regulation of miR-195-5p on Bcl-2 expression.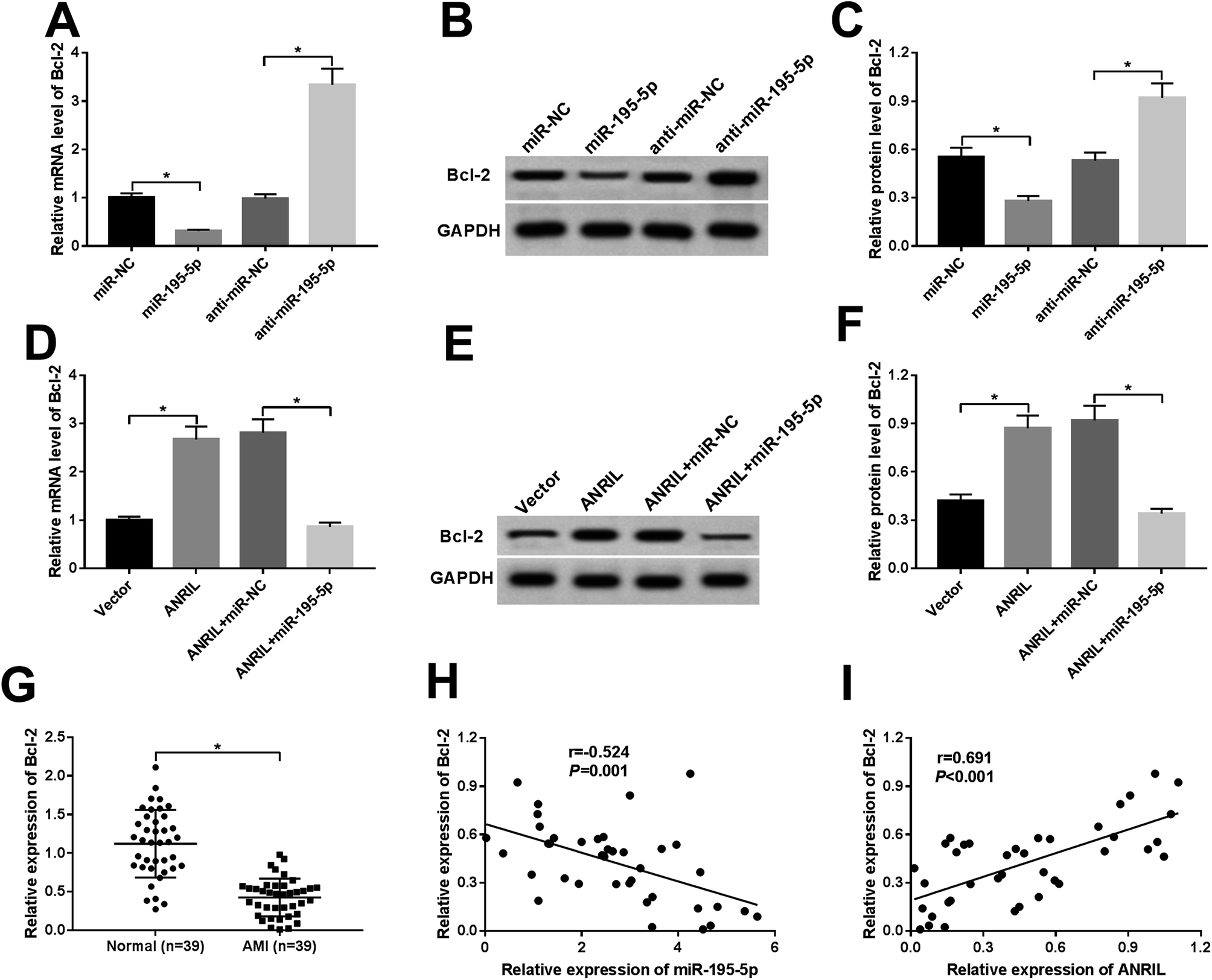 | ||
| Fig. 5 ANRIL upregulated Bcl-2 expression by sponging miR-195-5p in myocardial cells. Bcl-2 mRNA level by qRT-PCR (A) and Bcl-2 protein expression by western blot (B and C) in AC16 cells transfected with miR-NC mimic, miR-195-5p mimic, anti-miR-NC or anti-miR-195-5p. Bcl-2 mRNA (D) and protein (E and F) expression in AC16 cells transfected with Vector-NC, Vector-ANRIL, Vector-ANRIL + miR-NC mimic or Vector-ANRIL + miR-195-5p mimic. (G) Bcl-2 mRNA level in serum of AMI patients and healthy controls. The correlation between Bcl-2 expression and miR-195-5p (H) or ANRIL (I) level in serum of AMI patients. *P < 0.05 vs. corresponding control. | ||
Further, we observed whether ANRIL regulated Bcl-2 expression by sponging miR-195-5p in the myocardial cells. As expected, ANRIL upregulation resulted in increased Bcl-2 expression, while the effect was significantly abrogated by miR-195-5p overexpression (Fig. 5D–F). Moreover, Bcl-2 expression was highly downregulated in the serum of AMI patients compared with normal controls (Fig. 5G). More interestingly, Bcl-2 expression was negatively correlated with miR-195-5p level (Fig. 5H) and positively correlated with ANRIL level (Fig. 5I) in the serum of AMI patients. These results strongly pointed to the role of ANRIL as a molecular sponge for miR-195-5p to upregulate Bcl-2 expression.
3.6. Upregulation of ANRIL alleviated H/R-induced myocardial cell injury by upregulating Bcl-2
Given our data that ANRIL regulated Bcl-2 expression, we further explored whether ANRIL exerting its protective role was mediated by Bcl-2 in H/R-induced myocardial cells. As a result, cotransfection of si-Bcl-2, but not si-NC control, significantly reversed the regulatory effect of ANRIL upregulation on LDH release (Fig. 6A), MDA content (Fig. 6B), SOD expression (Fig. 6C), GSH-PX secretion (Fig. 6D) and ATP level (Fig. 6E) in H/R-induced AC16 cells. Moreover, Vector-ANRIL-mediated pro-proliferation and anti-apoptosis effects were highly abated by Bcl-2 depletion compared with negative control (Fig. 6F–I). Together, these data suggested that ANRIL upregulation protected myocardial cell against H/R-induced injury by upregulating Bcl-2.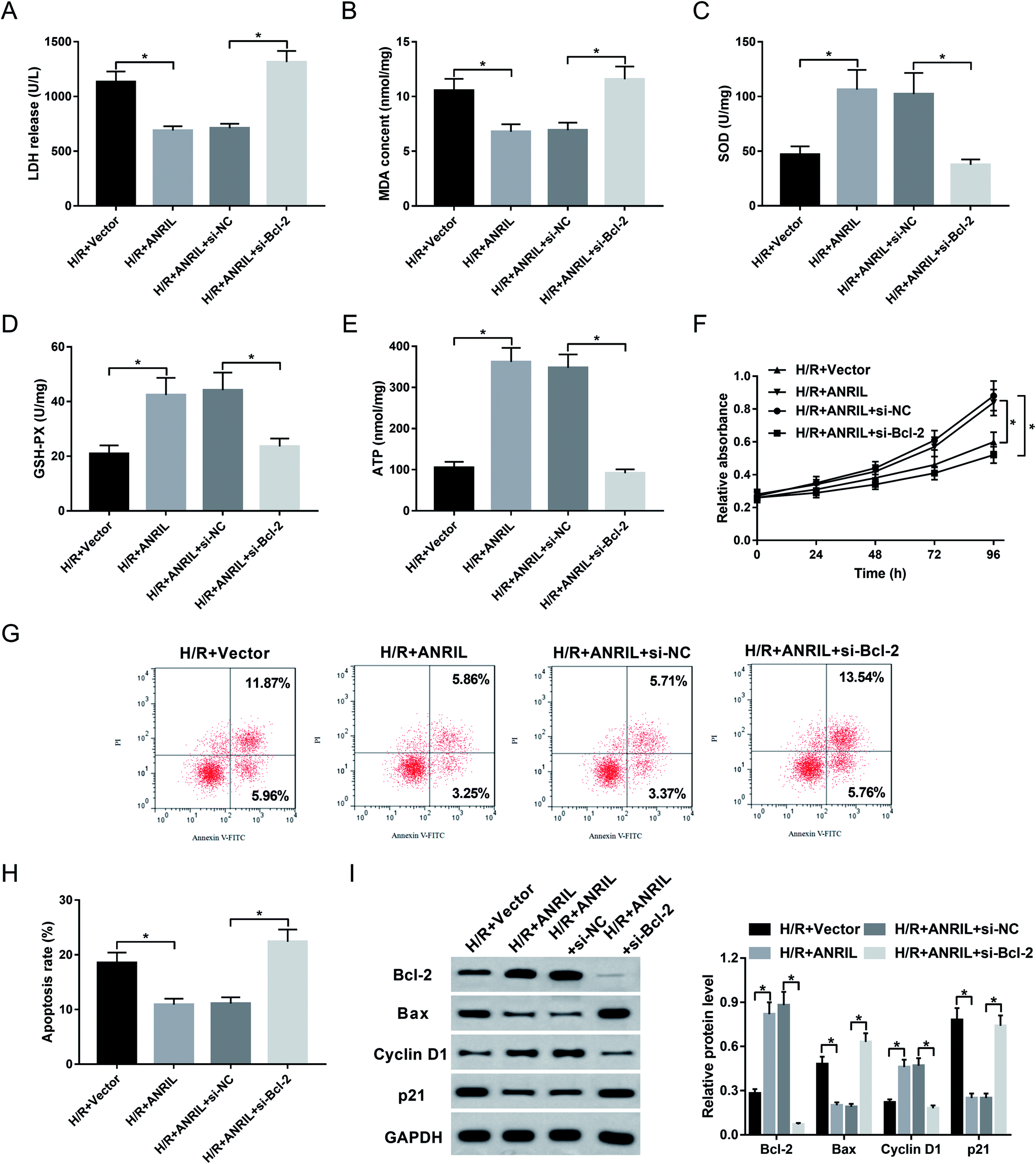 | ||
| Fig. 6 ANRIL exerted the protective role by upregulating Bcl-2 in H/R-induced myocardial cells. AC16 cells were transfected with Vector-NC, Vector-ANRIL, Vector-ANRIL + si-NC or Vector-ANRIL + si-Bcl-2 prior to H/R treatment. After 48 h H/R treatment, LDH release (A), MDA content (B), SOD level (C), GSH-PX expression (D) and ATP level (E) by corresponding assay kits in treated cells. (F) At the indicated time point after H/R treatment, cell proliferation by CCK-8 assay in treated cells. After H/R treatment for 48 h, cell apoptosis by flow cytometry (G and H), the expression levels of Bcl-2, Bax, Cyclin D1 and p21 by western blot (I) in treated cells. *P < 0.05 vs. Vector-NC or Vector-ANRIL + si-NC. | ||
4. Discussion
In recent years, AMI has become a major cause of morbidity and mortality worldwide. It is widely acknowledged that H/R is a hazardous factor, resulting in myocardial apoptosis and necrosis, which are two key cellular events in AMI.2,20 In the present study, our data supported that H/R induced myocardial cell injury, as evidenced by the repression of cell proliferation and the promotion of cell apoptosis, in accordance with previous studies.25,26 Subsequently, we further explored the effect and underlying mechanism of ANRIL on AMI by H/R-induced myocardial cells.ANRIL is an attractive lncRNA encoded in the 9p21 locus, which plays an important role in disease-associated polymorphisms. Increasing evidence has suggested that ANRIL is one of the best replicated genetic risk factors for cardiovascular disease, including CAD,12,16 CHD15,27 and atherosclerosis.28,29 Moreover, ANRIL is independently correlated with a series of other diseases such as endometriosis,30 diabetes31 and several human cancers.32–34 Additionally, the variants on ANRIL promoter and exons associated with ANRIL expression contribute to the risk of myocardial infarction.35 In the present study, our data supported that ANRIL was downregulated in the serum of AMI patients, consistent with a previous work.6 Moreover, we verified that H/R treatment time-dependently decreased ANRIL expression in myocardial cells. Thus, our study started with the hypothesis that ANRIL was associated with H/R-induced injury in myocardial cells. To confirm this, gain-of-function experiments were performed by transfection of Vector-ANRIL. As expected, ANRIL upregulation reduced the levels of LDH release, MDA, SOD and GSH-PX in H/R-induced myocardial cells. More importantly, ANRIL upregulation elevated cell proliferation and inhibited cell apoptosis, indicating that ANRIL upregulation alleviated H/R-induced myocardial cell injury. In short, ANRIL protected myocardial cell against H/R-induced injury, highlighting its role as a protective agent in AMI.
Then, bioinformatics software starBase was performed to predict the targeted miRNAs of ANRIL. Among these candidates, miR-195-5p was of interest in this study owing to its involvement in cardiomyocyte apoptosis induced by H/R injury.23,36 Subsequently, we firstly manifested that ANRIL directly targeted miR-195-5p and negatively regulated miR-195-5p expression. Moreover, our results demonstrated that miR-195-5p was upregulated in the serum of AMI patients. Similar to our finding, circulating increased miR-195-5p was reported to be a potential indicator for AMI.37 Additionally, our results demonstrated that H/R induced miR-195-5p expression in myocardial cells, in accordance with earlier studies.21,36 More interestingly, we firstly found that miR-195-5p knockdown reduced the levels of LDH rel5 apoptosis in H/R-induced myocardial cells, consistent with previous works.23,24,36,38 In a word, miR-195-5p knockdown alleviated H/R-induced myocardial cell injury.
It is widely accepted that lncRNA regulates gene expression by acting as a molecular sponge of miRNA. Bcl-2, an inhibitor of apoptosis, was reported to promote the therapeutic efficacy of myoblast sheet transplantation in AMI.39 Previous studies had manifested that Bcl-2 was a direct target of miR-195-5p, and it implicated in miR-195-5p-mediated apoptosis effect in human cardiomyocytes.23,24,38 In the present study, we firstly illuminated that ANRIL upregulated Bcl-2 expression by sponging miR-195-5p in myocardial cells. Moreover, we firstly demonstrated that ANRIL upregulation alleviated H/R-induced myocardial cell injury by upregulating Bcl-2. Similar to our findings, Zhang et al.24 elucidated that lncRNA SNHG1 functioned as a miR-195-5p sponge to regulate Bcl-2 expression, and SNHG1 regulated the viability and apoptosis through targeting the miR-195-5p/Bcl-2 axis in human cardiomyocytes. Additionally, Zhou et al.40 demonstrated that LINC00473 enhanced pancreatic cancer progression via miR-195-5p/Bcl-2 axis. Zhou and colleague41 reported that lncRNA PVT1 enhanced osteosarcoma development through sponging miR-195-5p and upregulating Bcl-2.
5. Conclusion
In conclusion, our study indicated that ANRIL upregulation alleviated H/R-induced myocardial cell injury through sponging miR-195-5p and upregulating Bcl-2. ANRIL is a probable mediator for new therapies for AMI management.Conflicts of interest
The authors have no financial conflicts to declare.Acknowledgements
Not applicable.References
- H. D. White and D. P. Chew, Lancet, 2008, 372, 570–584 CrossRef CAS.
- D. J. Hausenloy and D. M. Yellon, Nat. Rev. Cardiol., 2016, 13, 193–209 CrossRef CAS PubMed.
- H. M. Prentice, I. A. Moench, Z. T. Rickaway, C. J. Dougherty, K. A. Webster and H. Weissbach, Biochem. Biophys. Res. Commun., 2008, 366, 775–778 CrossRef CAS PubMed.
- J. H. Yoon, K. Abdelmohsen and M. Gorospe, Semin. Cell Dev. Biol., 2014, 34, 9–14 CrossRef CAS PubMed.
- Q. Shi and X. Yang, J. Cell. Physiol., 2016, 231, 751–755 CrossRef CAS PubMed.
- M. Vausort, D. R. Wagner and Y. Devaux, Circ. Res., 2014, 115, 668–677 CrossRef CAS PubMed.
- B. Meder, A. Keller, B. Vogel, J. Haas, F. Sedaghat-Hamedani, E. Kayvanpour, S. Just, A. Borries, J. Rudloff, P. Leidinger, E. Meese, H. A. Katus and W. Rottbauer, Basic Res. Cardiol., 2011, 106, 13–23 CrossRef CAS PubMed.
- E. van Rooij, L. B. Sutherland, J. E. Thatcher, J. M. DiMaio, R. H. Naseem, W. S. Marshall, J. A. Hill and E. N. Olson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13027–13032 CrossRef CAS PubMed.
- Y.-Y. Deng, W. Zhang, J. She, L. Zhang, T. Chen, J. Zhou and Z. Yuan, J. Am. Coll. Cardiol., 2016, 68, C51–C52 CrossRef.
- G. Zhang, H. Sun, Y. Zhang, H. Zhao, W. Fan, J. Li, Y. Lv, Q. Song, J. Li, M. Zhang and H. Shi, J. Am. Coll. Cardiol., 2018, 4, 35, DOI:10.1038/s41420-018-0036-7. eCollection 2018 Dec.
- R. McPherson, A. Pertsemlidis, N. Kavaslar, A. Stewart, R. Roberts, D. R. Cox, D. A. Hinds, L. A. Pennacchio, A. Tybjaerg-Hansen, A. R. Folsom, E. Boerwinkle, H. H. Hobbs and J. C. Cohen, Science, 2007, 316, 1488–1491 CrossRef CAS PubMed.
- N. J. Samani, J. Erdmann, A. S. Hall, C. Hengstenberg, M. Mangino, B. Mayer, R. J. Dixon, T. Meitinger, P. Braund, H. E. Wichmann, J. H. Barrett, I. R. Konig, S. E. Stevens, S. Szymczak and D. A. Tregouet, et al., N. Engl. J. Med., 2007, 357, 443–453 CrossRef CAS PubMed.
- A. Gschwendtner, S. Bevan, J. W. Cole, A. Plourde, M. Matarin, H. Ross-Adams, T. Meitinger, E. Wichmann, B. D. Mitchell, K. Furie, A. Slowik, S. S. Rich, P. D. Syme, M. J. MacLeod and J. F. Meschia, et al., Ann. Neurol., 2009, 65, 531–539 CrossRef CAS PubMed.
- L. J. Scott, K. L. Mohlke, L. L. Bonnycastle, C. J. Willer, Y. Li, W. L. Duren, M. R. Erdos, H. M. Stringham, P. S. Chines, A. U. Jackson, L. Prokunina-Olsson, C. J. Ding, A. J. Swift, N. Narisu and T. Hu, et al., Science, 2007, 316, 1341–1345 CrossRef CAS PubMed.
- M. S. Cunnington, M. Santibanez Koref, B. M. Mayosi, J. Burn and B. Keavney, PLoS Genet., 2010, 6, e1000899, DOI:10.1371/journal.pgen.1000899.
- L. Beeton, P. Chivers, J. Dawes, T. Kyriakou, A. Goel, J. Peden and F. Green, Heart, 2011, 97, e7 CrossRef.
- D. A. Morrow, C. P. Cannon, R. L. Jesse, L. K. Newby, J. Ravkilde, A. B. Storrow, A. H. Wu and R. H. Christenson, Circulation, 2007, 115, e356–375 CrossRef PubMed.
- X. Yu, W. Geng, H. Zhao, G. Wang, Y. Zhao, Z. Zhu and X. Geng, Med. Sci. Monit., 2017, 23, 5943–5950 CrossRef CAS PubMed.
- Y. Y. Hei, Y. X. Guo, C. S. Jiang, S. Wang, S. M. Lu and S. Q. Zhang, Biotechnol. Appl. Biochem., 2019, 66, 755–762 CrossRef CAS PubMed.
- S. Femmino, C. Penna, F. Bessone, F. Caldera, N. Dhakar, D. Cau, P. Pagliaro, R. Cavalli and F. Trotta, Polymers (Basel), 2018, 10, E211 CrossRef PubMed.
- G. Gong, Y. Li, X. Yang, H. Geng, X. Lu, L. Wang and Z. Yang, Anatolian J. Cardiol., 2017, 18, 168–174 CAS.
- Z. Li and T. M. Rana, Nat. Rev. Drug Discovery, 2014, 13, 622–638 CrossRef CAS PubMed.
- C.-K. Gao, H. Liu, C.-J. Cui, Z.-G. Liang, H. Yao and Y. Tian, J. Genet., 2016, 95, 99–108 CrossRef CAS PubMed.
- N. Zhang, X. Meng, L. Mei, J. Hu, C. Zhao and W. Chen, Cell. Physiol. Biochem., 2018, 50, 1029–1040 CrossRef CAS PubMed.
- W. Zhang, Y. Li and P. Wang, Braz. J. Med. Biol. Res., 2018, 51, e6555, DOI:10.1590/1414-431x20186555 . Epub 2018 Apr 23.
- H. Hu, J. Wu, D. Li, J. Zhou, H. Yu and L. Ma, Biomed. Pharmacother., 2018, 106, 738–746 CrossRef CAS PubMed.
- R. Murray, J. Bryant, P. Titcombe, S. J. Barton, H. Inskip, N. C. Harvey, C. Cooper, K. Lillycrop, M. Hanson and K. M. Godfrey, Clin. Epigenet., 2016, 8, 90, DOI:10.1186/s13148-016-0259-5. eCollection 2016.
- G. Bochenek, R. Hasler, N. E. El Mokhtari, I. R. Konig, B. G. Loos, S. Jepsen, P. Rosenstiel, S. Schreiber and A. S. Schaefer, Hum. Mol. Genet., 2013, 22, 4516–4527 CrossRef CAS PubMed.
- A. Congrains, K. Kamide, R. Oguro, O. Yasuda, K. Miyata, E. Yamamoto, T. Kawai, H. Kusunoki, H. Yamamoto, Y. Takeya, K. Yamamoto, M. Onishi, K. Sugimoto, T. Katsuya and N. Awata, et al., Atherosclerosis, 2012, 220, 449–455 CrossRef CAS PubMed.
- S. Uno, H. Zembutsu, A. Hirasawa, A. Takahashi, M. Kubo, T. Akahane, D. Aoki, N. Kamatani, K. Hirata and Y. Nakamura, Nat. Genet., 2010, 42, 707–710 CrossRef CAS PubMed.
- R. Saxena, B. F. Voight, V. Lyssenko, N. P. Burtt, P. I. de Bakker, H. Chen, J. J. Roix, S. Kathiresan, J. N. Hirschhorn, M. J. Daly, T. E. Hughes, L. Groop, D. Altshuler, P. Almgren and J. C. Florez, et al., Science, 2007, 316, 1331–1336 CrossRef CAS PubMed.
- F. Q. Nie, M. Sun, J. S. Yang, M. Xie, T. P. Xu, R. Xia, Y. W. Liu, X. H. Liu, E. B. Zhang, K. H. Lu and Y. Q. Shu, Mol. Cancer Ther., 2015, 14, 268–277 CrossRef CAS PubMed.
- D. Chen, Z. Zhang, C. Mao, Y. Zhou, L. Yu, Y. Yin, S. Wu, X. Mou and Y. Zhu, Cell. Immunol., 2014, 289, 91–96 CrossRef CAS PubMed.
- W. Deng, J. Wang, J. Zhang, J. Cai, Z. Bai and Z. Zhang, IUBMB Life, 2016, 68, 355–364 CrossRef CAS PubMed.
- J. Cheng, M. Y. Cai, Y. N. Chen, Z. C. Li, S. S. Tang, X. L. Yang, C. Chen, X. Liu and X. D. Xiong, Oncotarget, 2017, 8, 12607–12619 CrossRef PubMed.
- C. Chen, K. Y. Jia, H. L. Zhang and J. Fu, Riv. Eur. Sci. Med. Farmacol., 2016, 20, 3410–3416 CAS.
- G. Long, F. Wang, Q. Duan, S. Yang, F. Chen, W. Gong, X. Yang, Y. Wang, C. Chen and D. W. Wang, PLoS One, 2012, 7, e50926, DOI:10.1371/journal.pone.0050926 . Epub 2012 Dec 7.
- Z. Liu, D. Yang, P. Xie, G. Ren, G. Sun, X. Zeng and X. Sun, Cell. Physiol. Biochem., 2012, 29, 851–862 CrossRef CAS PubMed.
- K. Kitabayashi, A. Siltanen, T. Patila, M. A. Mahar, I. Tikkanen, J. Koponen, M. Ono, Y. Sawa, E. Kankuri and A. Harjula, Cell Transplantation, 2010, 19, 573–588 CrossRef PubMed.
- W. Y. Zhou, M. M. Zhang, C. Liu, Y. Kang, J. O. Wang and X. H. Yang, J. Cell. Physiol., 2019, 234, 23176–23189 CrossRef CAS PubMed.
- Q. Zhou, F. Chen, J. Zhao, B. Li, Y. Liang, W. Pan, S. Zhang, X. Wang and D. Zheng, Oncotaget, 2016, 7, 82620–82633 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04898g |
| ‡ Both of the authors contributed to this work equally as co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |