DOI:
10.1039/C9RA04966E
(Paper)
RSC Adv., 2019,
9, 25423-25428
The effect of steric repulsion between highly branched hydrophilic blocks on inverse cubic mesophase formation in block copolymers†
Received
1st July 2019
, Accepted 8th August 2019
First published on 14th August 2019
Abstract
The solution-based self-assembly of block copolymers (BCPs) into nanoparticulate or microparticulate inverse cubic mesophases, or polymer cubosomes, is of growing interest. This phenomenon could yield new polymeric mesoporous materials with three-dimensionally organized mazes of large water channels. In addition to the ratio of its hydrophilic and hydrophobic blocks, the architecture of a BCP critically influences self-assembly. BCP bilayers having triply periodic minimal surfaces with cubic lattice structures are formed. Here we report the synthesis and self-assembly of BCPs with T8 polyhedral oligomeric silsesquioxane (POSS), an inorganic cage molecule. POSS can be asymmetrically functionalized to host structural modules as branching units of the hydrophilic block that contain various spatial and chemical environments. BCPs, each containing seven poly(ethylene glycol) chains and the hydrophobic polystyrene block PEG3507-POSS-b-PS, were prepared to investigate the effect of the highly branched hydrophilic block architecture on self-assembly. We found that the BCPs self-assembled into polymer cubosomes. Regardless of the block ratio, only the primitive cubic phase (Schwarz P surface, Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) was observed in the cubosomes. This is in contrast to the self-assembly of conventional BCPs into inverse mesophases, in which the internal lattice preferentially transforms into double-diamond lattices to yield a Schwarz D surface as a consequence of block ratio changes. Our results suggest that BCP architecture may be related to the symmetry of the lattice formed via self-assembly in solution.
m space group) was observed in the cubosomes. This is in contrast to the self-assembly of conventional BCPs into inverse mesophases, in which the internal lattice preferentially transforms into double-diamond lattices to yield a Schwarz D surface as a consequence of block ratio changes. Our results suggest that BCP architecture may be related to the symmetry of the lattice formed via self-assembly in solution.
Introduction
The formation of nanostructures with three-dimensional (3D) order via molecular self-assembly is an efficient means by which to confer the chemical properties of the building block molecular structures to nanoscale or microscale materials with large surface areas. Atomic or molecular properties are amplified across the length of the materials when functional groups are added in specific locations through the self-assembly of specially designed building blocks.1 3D structures with reticulated mesoporous networks comprised of well-defined cubic lattices are particularly important materials. They can be utilized in a wide variety of applications in the fields of organic electronics, solid-state electrolysis, separations, catalysis, and nanotemplating.2–6 Nanostructures with 3D periodic porosity can be prepared via the self-assembly of block copolymers (BCPs) in the bulk, followed by extraction of labile domains from the continuous matrix.7,8 In contrast, amphiphilic BCPs composed of hydrophilic and hydrophobic blocks can self-assemble into inverse bi-continuous cubic mesophases in solution. This results in the formation of highly ordered mesoporous materials that contain nanochannel networks reticulated within a cubic crystalline order.9–17 The lattices of bi-continuous cubic mesophases can be controlled by adjusting BCP structural properties, such as the ratio of the hydrophilic and hydrophobic blocks and the architecture of the constituent polymer blocks.9,18–21
The architecture of a BCP influences its self-assembly in solution to yield bi-continuous cubic mesophases. We previously reported that steric repulsion between BCPs upon self-assembly could be increased by adding a branched architecture to the hydrophilic block. This limits stretching of the hydrophobic polymer block chains and increases the packing parameter (P).18 P is equal to V/al, where V is the volume of the hydrophobic block of a BCP with a bulky, branched hydrophilic block; a is the area of its hydrophilic bloc; and l is the length of the hydrophobic block. This influences self-assembly to favor the formation of cubic mesophases. To increase steric repulsion, dendritic scaffolds can be introduced into the hydrophilic blocks, which tether the hydrophilic chains at the periphery of the BCP. However, bond rotation within the scaffolds makes them compressible, which can alter the interfacial area of the hydrophilic block. This affects the BCP by reducing the value of P.
Molecular nanoparticles (MNPs) are shape-persistent molecules that can serve as structural motifs for the construction of shape-persistent amphiphiles that mimic the well-defined chemical structures of low-molecular-weight amphiphiles. The shape-persistent amphiphiles, or giant amphiphiles, have been shown to form well-defined nanostructures in bulk and in solution. Polyhedral oligomeric silsesquioxane (POSS) is the smallest silica nanoparticle, with an inorganic core diameter of about 1.0 nm.22,23 The vertices of POSS can be functionalized to prepare organic–inorganic hybrid molecules,24,25 Octakis(3-propyl methacrylate) octasilsesquioxane (T8) POSS has a cubic framework with Oh symmetry. The site-selective functionalization of T8 POSS vertices could lead to the formation of giant amphiphiles capable of self-assembly into complex nanostructures in bulk and in solution.26–29
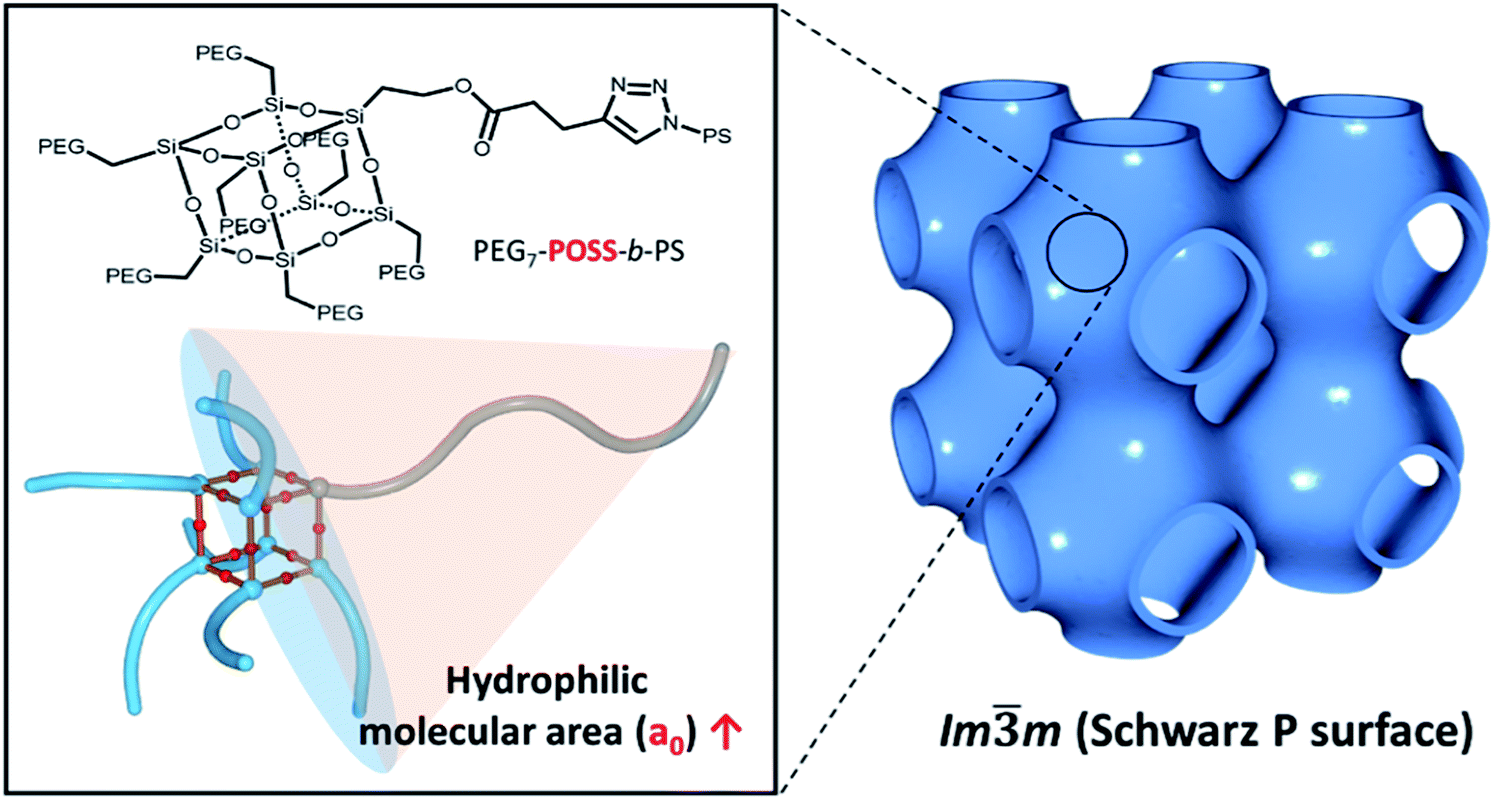
In this work, mono-alcohol heptavinyl POSS was used as a scaffold for the hydrophilic module to host poly(ethylene glycol) (PEG). Seven PEG chains were introduced to all the vertices of POSS with the exception of the alcohol-functionalized vertex. Hydrophobic PS chains were introduced via click chemistry. The shape of the resulting BCP, PEG3507-POSS-b-PS, was persistent due to the incompressibility of the POSS scaffold. During the self-assembly of these BCPs into polymer cubosomes (Fig. 1), primitive cubic mesophases with Imm symmetry were formed preferentially. This suggested that upon formation of the inverse cubic mesophases, increased steric repulsion between the hydrophilic blocks of the BCPs reduced bilayer curvature.
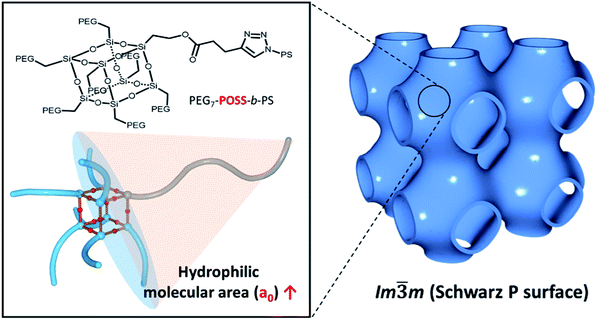 |
| | Fig. 1 Schematic representation showing the synthesis of PEG3507-POSS-b-PS with a large hydrophilic molecular area. Self-assembled cubosomes with exclusively primitive cubic mesophases (Imm symmetry) are shown on the right side of the figure. | |
Results and discussion
Design and synthesis of BCPs with hydrophilic POSS block
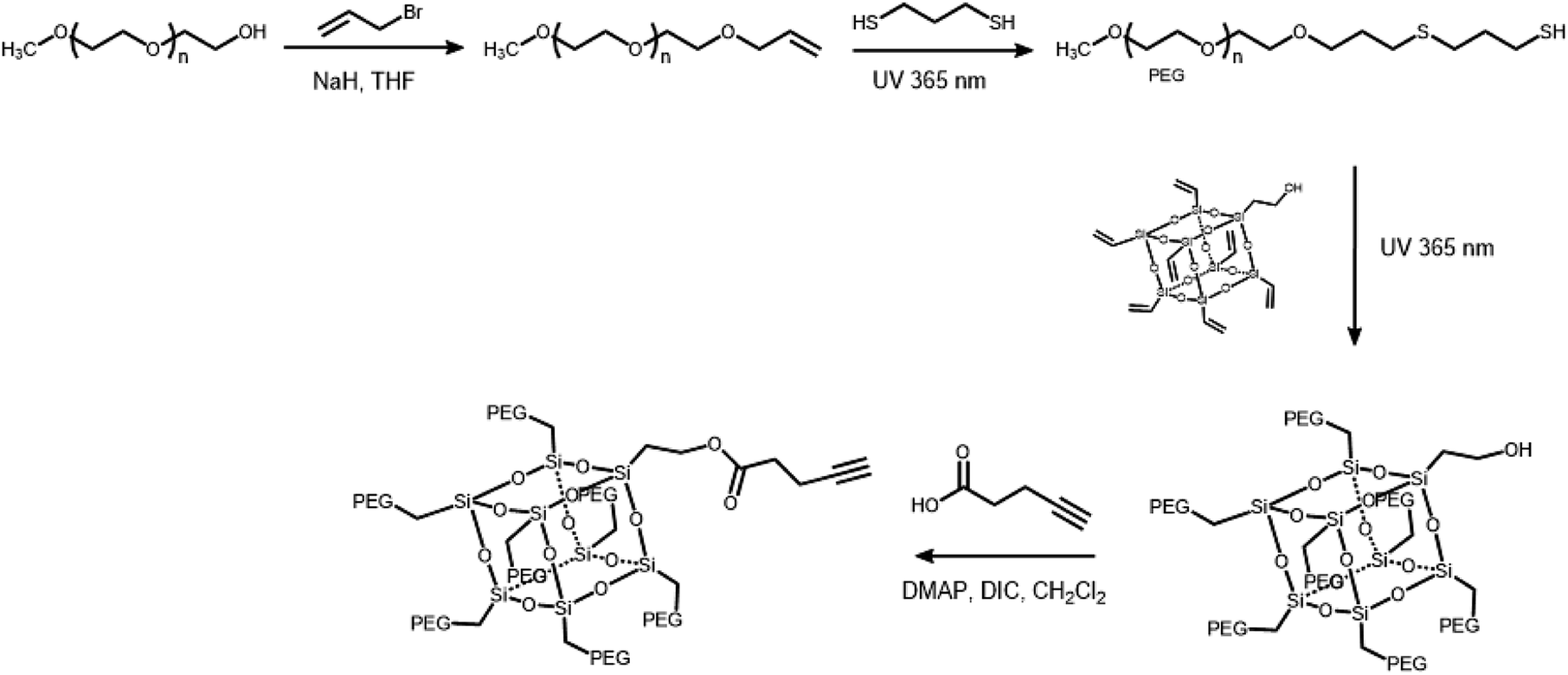
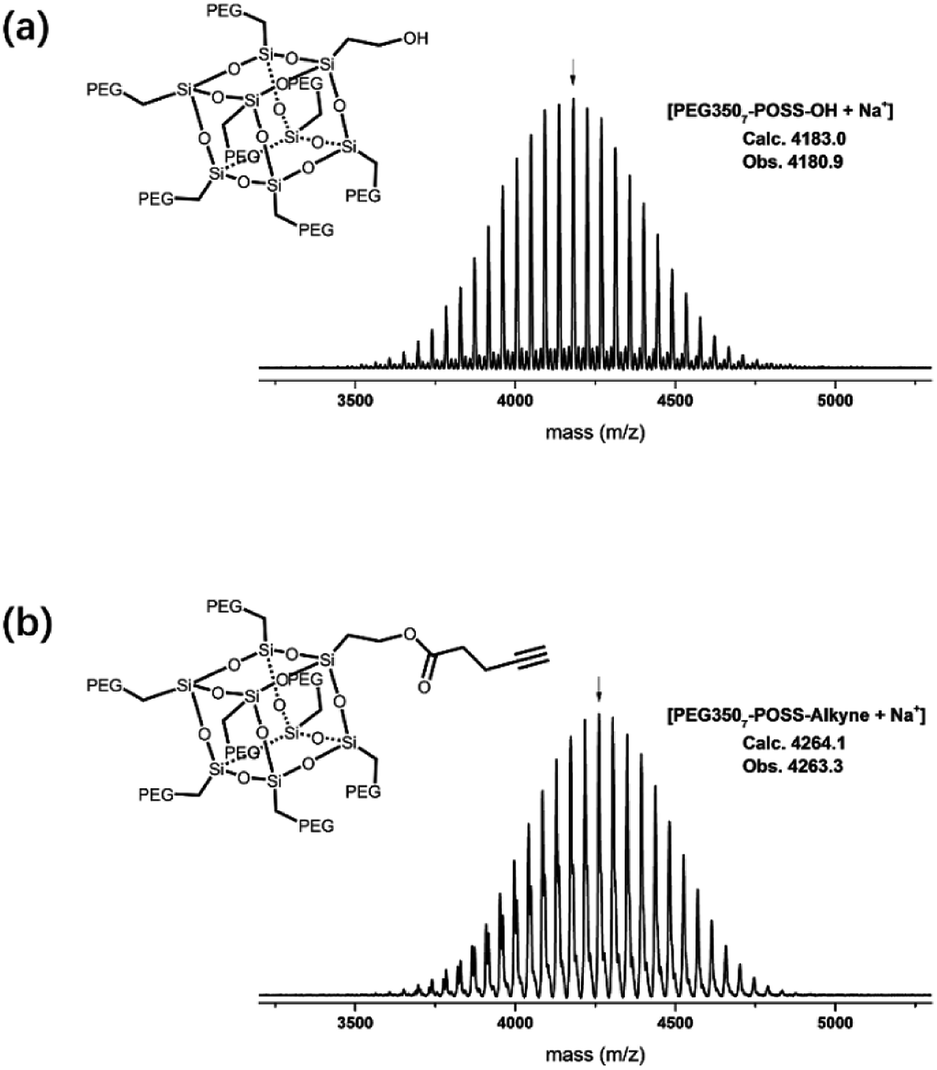
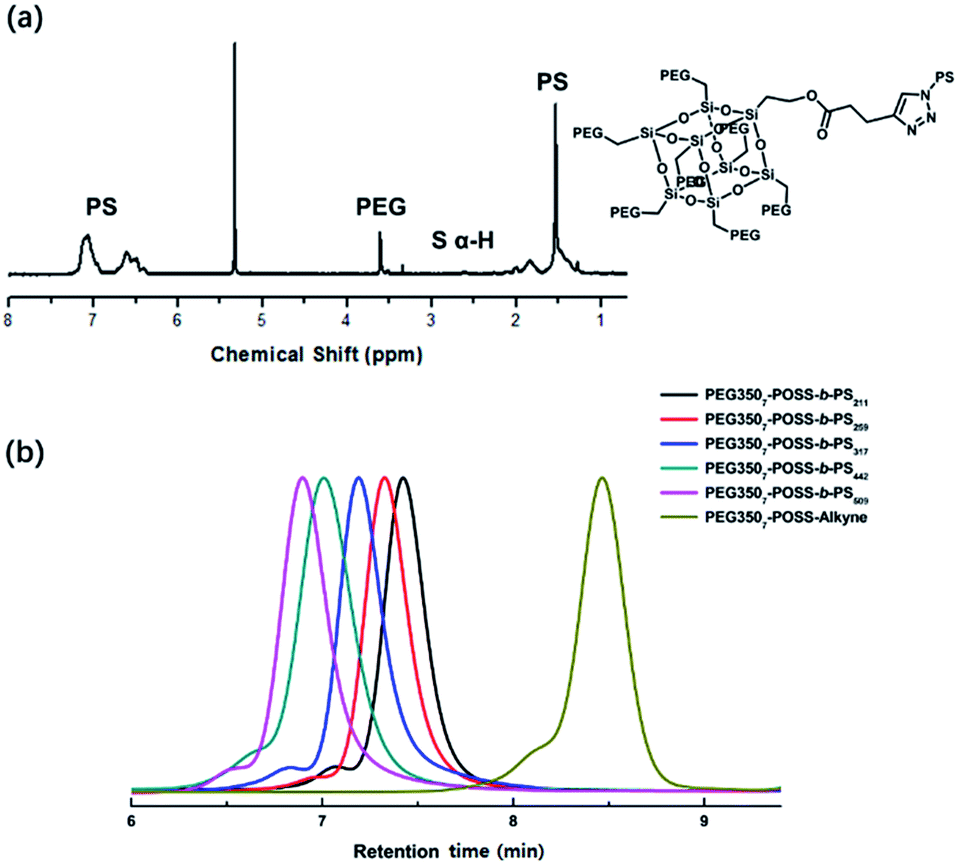
The branched hydrophilic modules, referred to as PEG3507-POSS-alkynes, were synthesized using 3-hydroxylpropylheptanol-POSS (HO-V7-POSS) as a scaffold to host water-soluble poly(ethylene glycol) (PEG) chains at the vertices of the POSS. The single-site oxidation of pendant vinyl groups in octavinyl-POSS was carried out through hydrolysis using 1.2 equivalents of triflic acid for every equivalent of POSS. PEG with a thiol terminal group (PEG-SH) was prepared by reacting methoxy-PEG (Mn = 350 g mol−1) with allyl bromide, followed by the addition of propanedithiol (Scheme 1). Water-soluble PEG-SH was introduced to vinyl groups at the vertices of HO-V7-POSS via thiol–ene click chemistry under UV irradiation at 365 nm, and PEG3507-POSS-OH was produced in high yield (71%). Successful introduction of PEG chains to POSS was confirmed with 1H nuclear magnetic resonance (NMR) spectroscopy and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Fig. 2, S4 and S8†). The hydroxyl group of PEG3507-POSS-OH was converted to afford the hydrophilic module via esterification with propargylic acid.
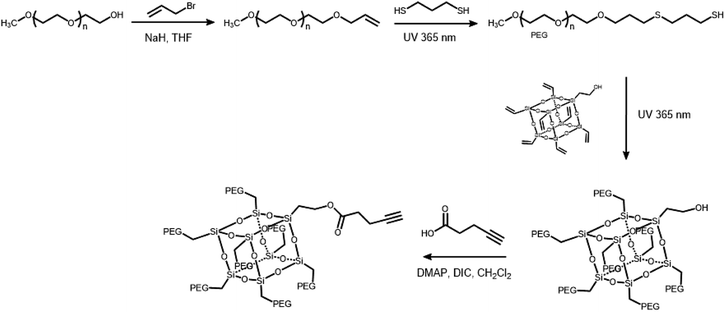 |
| | Scheme 1 Synthetic route of PEG3503-POSS hydrophilic modules. | |
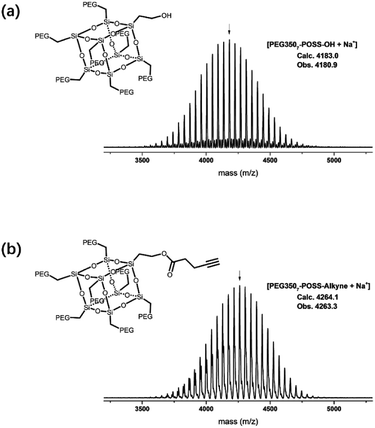 |
| | Fig. 2 MALDI-TOF-MS spectra of (a) PEG3507-POSS and (b) PEG3507-POSS-alkyne. | |
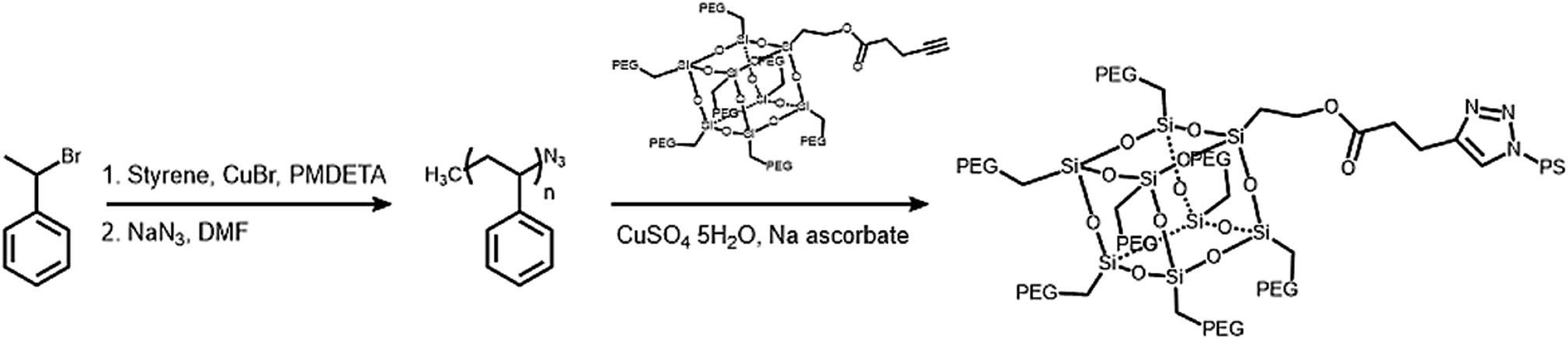
The hydrophobic modules were produced through the atom-transfer radical polymerization (ATRP) of the styrene functionalities in azido terminated polystyrene (PS–N3) using 1-bromoethylbenzene as an initiator. This was followed by end-group conversion with NaN3 in DMF. The hydrophilic and hydrophobic modules were joined through Cu-catalyzed azide–alkyne click chemistry (CuAAC) (Scheme 2). Residual PS–N3 was removed via flash column chromatography on silica using a dichloromethane/hexane mixture as an eluent. The resulting BCPs, PEG3507-POSS-b-PSn, were analyzed with 1H NMR and gel permeation chromatography (GPC) with PS standards to confirm synthesis was successful (Fig. 3). GPC was performed at 35 °C using THF as an eluent at a flow rate of 1 mL min−1. 1H NMR was used to determine the block ratios of the BCPs. The block ratios were defined by the molecular-weight (MW) fraction of their PEG chains (wPEG). The results of molecular characterization are shown in Table 1.
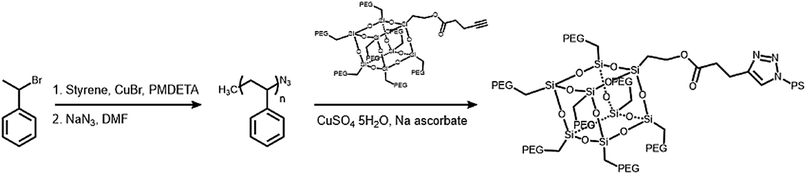 |
| | Scheme 2 Synthesis of polystyrene (PS) hydrophobic modules and PEG3507-POSS-b-PSn. | |
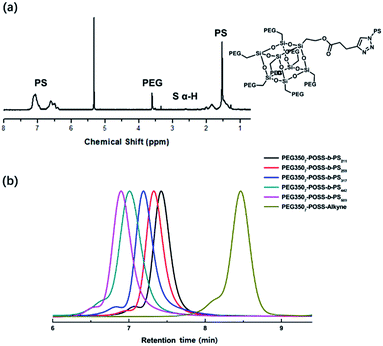 |
| | Fig. 3 (a) 1H NMR result of PEG3507-POSS-b-PS442, (b) GPC profiles of PEG3507-POSS-b-PS211, PEG3507-POSS-b-PS259, PEG3507-POSS-b-PS317, PEG3507-POSS-b-PS442, PEG3507-POSS-b-PS509 and PEG3507-POSS-alkyne. | |
Table 1 Molecular characterization of giant amphiphiles
| Sample |
Mn PS–N3a (kg mol−1) |
PS–N3 dispersity (Ð)b |
Mnc (kg mol−1) |
Ðd |
wPEGe (wt%) |
| Mn: number molecular weight of azido end-functionalized polystyrenes. Ð: dispersity of azido end-functionalized polystyrenes. Mn of PEG3507-POSS-b-PS. Ð of PEG3507-POSS-b-PS. Weight percent of PEG (wPEG) calculated from the overall Mn and MW of the PEG chains. |
| PEG3507-POSS-b-PS211 |
22.4 |
1.06 |
27.7 |
1.04 |
11.1 |
| PEG3507-POSS-b-PS259 |
26.5 |
1.08 |
31.1 |
1.08 |
9.1 |
| PEG3507-POSS-b-PS317 |
32.5 |
1.10 |
37.1 |
1.14 |
7.4 |
| PEG3507-POSS-b-PS442 |
45.5 |
1.11 |
53.2 |
1.12 |
5.3 |
| PEG3507-POSS-b-PS509 |
53.2 |
1.16 |
61.1 |
1.17 |
4.6 |
BCP self-assembly into inverse cubic mesophases
To investigate the effect of the hydrophilic block architecture on PEG3507-POSS-b-PSn self-assembly, the BCPs were allowed to self-assemble and form aggregates in water using a cosolvent method. Dioxane was employed as a common solvent to dissolve PEG and the PS blocks of the amphiphile (5 mg mL−1). An equal volume of water was added to the dioxane solution to induce self-assembly. The medium was exchanged with water by dialysis, and the morphology of the aggregates in the aqueous suspension was studied with scanning electron microscopy (SEM) and transmission electron microscopy (TEM).
Regardless of the block ratio (wPEG) in the BCP, only polymer vesicles (polymersomes) were observed in TEM images of the dried aqueous suspension (Fig. 4). This differed from the self-assembly of conventional BCPs with highly asymmetric block ratios in solution. In that case, formation of inverse mesophases, such as inverse cubic or hexagonal structures, predominates due to the increased P of the BCP.
 |
| | Fig. 4 TEM images of polymer vesicles in (a) PEG3507-POSS-b-PS442 and (b) PEG3507-POSS-b-PS509 self-assembled in dioxane. | |
We screened common solvents to steer the self-assembly of the BCPs into inverse mesophases. A mixture of acetone and dioxane was found to be an optimal common solvent for the self-assembly of inverse mesophases in giant amphiphiles. We studied the possible self-assembled morphologies of PEG3507-POSS-b-PS442 in solution using the acetone and dioxane mixture as the common solvent to dissolve the amphiphile. When the volume fraction of acetone was increased to approximately 80%, the structure of self-assembled PEG3507-POSS-b-PS442 changed. Instead of being comprised solely of polymer vesicles, polymer sponges coexisted with polymer vesicles in the BCPs. Polymer cubosomes were obtained when 90 vol% acetone was used (Fig. 5). Polymer hexosomes were observed when pure acetone was employed as the solvent. This was attributed to the lower solubility of the PS blocks in acetone, which limiting stretching of the hydrophobic PS chains during self-assembly. Therefore, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of acetone and dioxane was used as the common solvent for amphiphile dissolution in subsequent experiments.
:1 (v/v) mixture of acetone and dioxane was used as the common solvent for amphiphile dissolution in subsequent experiments.
 |
| | Fig. 5 TEM images showing the morphology of PEG3507-POSS-b-PS442 following self-assembly in (a) dioxane, (b) 80:20 acetone/dioxane (v/v), (c) 90:10 acetone/dioxane (v/v), and (d) acetone. | |
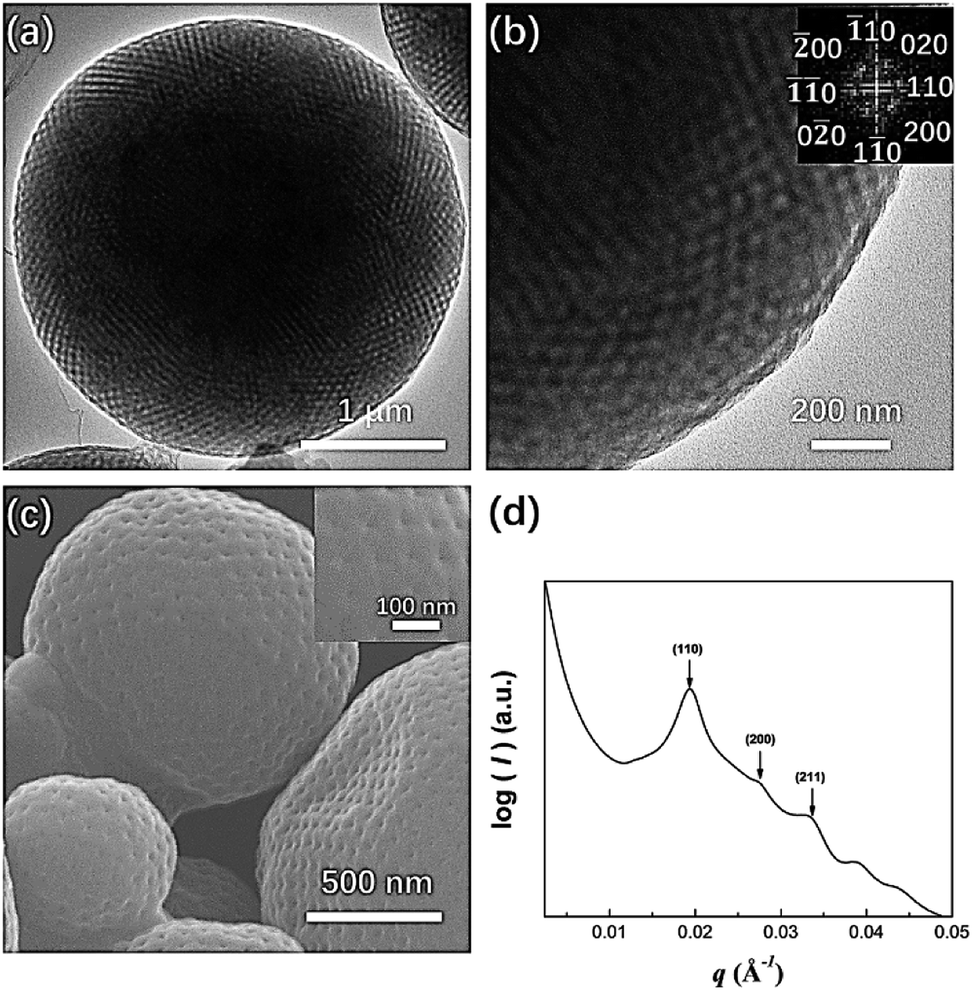
In the 9:1 mixture of acetone and dioxane, PEG3507-POSS-b-PS317 (fPEG = 7.4%) self-assembled into polymer cubosomes (Fig. 6). The internal inverse cubic mesophase of the PEG3507-POSS-b-PS317 polymer cubosomes was examined with TEM. The image (Fig. 6a) clearly showed a highly ordered cubic arrangement, and fast Fourier transform (FFT) of the TEM image indicated a Schwarz P surface (Fig. 6b). This was confirmed with small-angle X-ray scattering (SAXS) analysis of the dried PEG3507-POSS-b-PS317 polymer cubosomes. The SAXS scattering profile was assigned to the Schwarz P minimal surface of PEG3507-POSS-b-PS317. The primitive cubic phase lattice belonged to the Imm symmetry group with a lattice constant (a) of 46 nm.
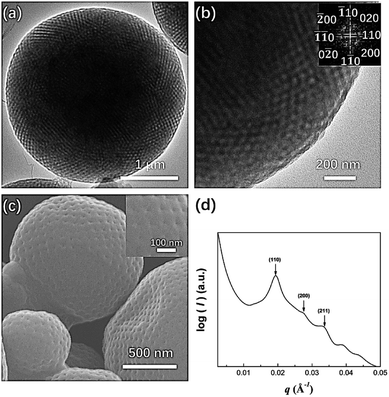 |
| | Fig. 6 (a) TEM image showing the morphology of PEG3507-POSS-b-PS317 following self-assembly in acetone; (b) fast Fourier transform (FFT) of the TEM image; (c) SEM image showing the morphology of PEG3507-POSS-b-PS317 following self-assembly in acetone; (d) SAXS profile of dried PEG3507-POSS-b-PS317 polymer cubosomes. | |
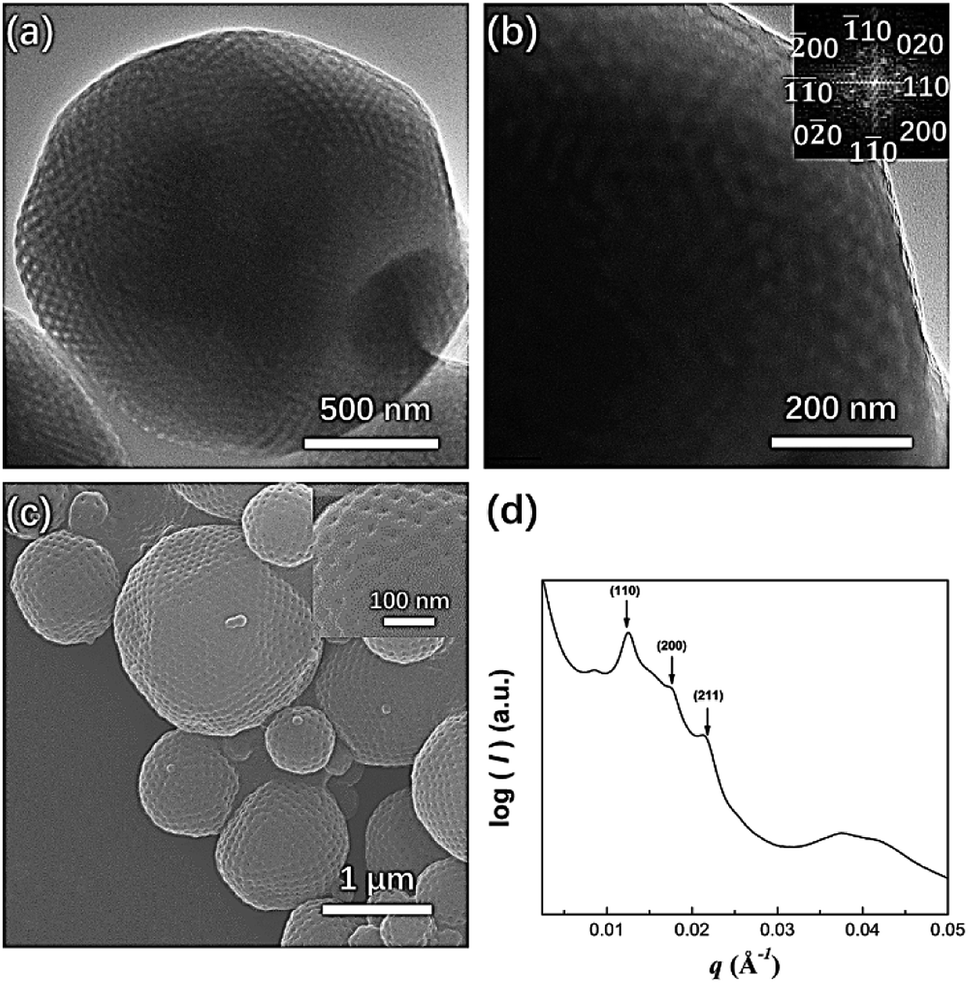
In our previous study, mesophases within branched-linear BCP polymer cubosomes transformed from a Schwarz P surface to a Schwarz D surface with Pnm symmetry as the hydrophilic polymer chain weight fraction decreased. Further reduction of the water-soluble chain weight fraction induced a phase change in the cubic mesophases, which were transformed into an inverse hexagonal phase. When we examined PEG3507-POSS-b-PS442 (fPEG = 5.3%) polymer cubosomes following self-assembly in the optimized common solvent, we found that the internal minimal surface of the polymer cubosome was a Schwarz P surface and retained Imm symmetry (a = 71 nm). This was confirmed through FFT of the TEM images and SAXS analysis (Fig. 7). PEG3507-POSS-b-PS509 self-assembled into polymer hexosomes with an internal inverse hexagonal structure. The observed transformation of the inverse mesophases differed from previously reported phase behavior, in which BCPs formed inverse mesophases in solution.
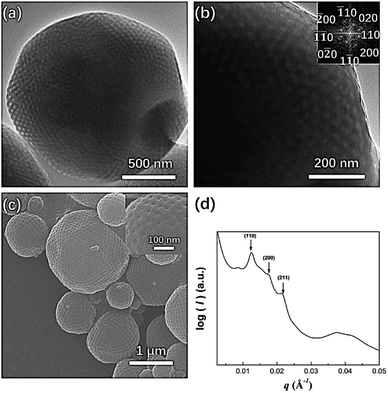 |
| | Fig. 7 (a) TEM image showing the morphology of PEG3507-POSS-b-PS442 following self-assembly in the optimized common solvent; (b) fast Fourier transform (FFT) of the TEM image; (c) SEM image showing the morphology of PEG3507-POSS-b-PS442 following self-assembly in the optimized common solvent; (d) SAXS profile of dried PEG3507-POSS-b-PS442 polymer cubosomes. | |
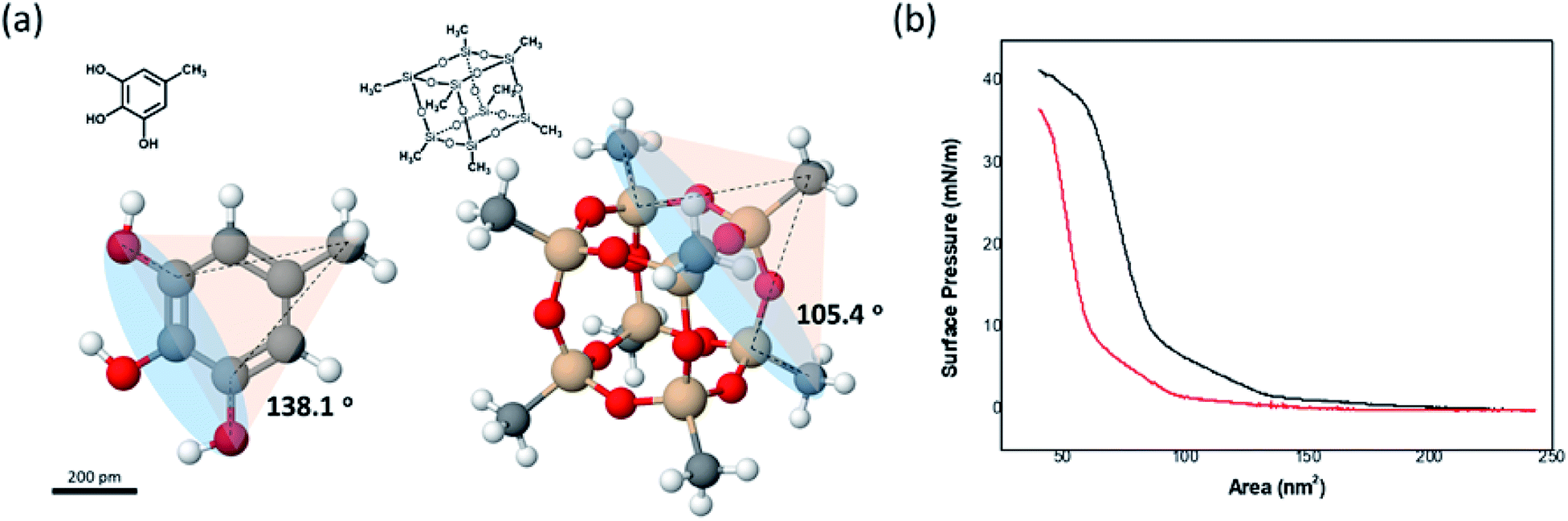
The disappearance of the Schwarz D surface from the self-assembled BCP structures as fPEG decreased was attributed to steric repulsion between the hydrophilic blocks due to the incompressibility of the POSS cage. We built three-dimensional models of (PEG750)3-benzyl alcohol and PEG3507-POSS-OH (Fig. 8a) to demonstrate high steric repulsion between the hydrophilic blocks of BCPs that contained POSS. The cone angles of (PEG750)3-benzyl alcohol and PEG3507-POSS-OH were estimated using the hydroxyl groups as focal points. We simplified cone angle estimation by omitting the PEG chains. We found that the angle between the focal point and the nearest substituted atom in (PEG750)3-benzyl alcohol was about 138°, while the cone angle of PEG3507-POSS-OH was approximately 105°. The surface pressure–area (π–A) isotherms of branched molecules containing PEG domains with comparable molecular weights were recorded at the air–water interface (Fig. 8b). The molecular areas of (PEG750)3-benzyl alcohol and PEG3507-POSS-OH were determined from the π–A isotherms and found to be 73 nm2 and 98 nm2, respectively. These results suggested that steric hindrance in the hydrophilic block of PEG3507-POSS-b-PS was higher than that observed in simple tri-PEG branched-linear BCPs upon self-assembly in solution.
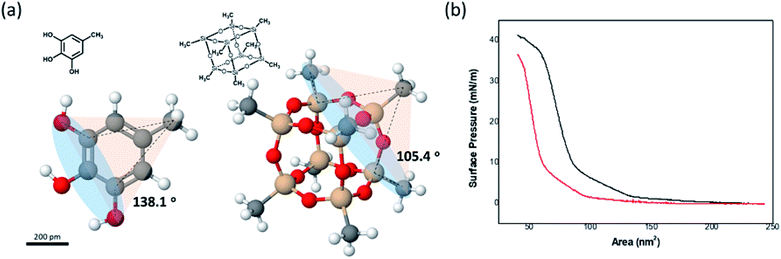 |
| | Fig. 8 (a) 3-D molecular models of PEG3-phenyl-b-PS and PEG3507-POSS-b-PS. (b) π–A isotherms of PEG7503 phenyl blocks (red) and PEG3507-POSS blocks (black) dispersed at the air–water interface. | |
Conclusion
In this work, a series of POSS-based giant amphiphiles (PEG3507-POSS-b-PSn) with different molecular weights was designed and synthesized. The giant amphiphiles self-assembled in a mixture of acetone and dioxane, which served as the common solvent. POSS afforded hydrophilic modules with large molecular areas, and the self-assembled morphologies strongly suggested increased steric repulsion between the hydrophilic blocks. Formation of BCP bilayers with low curvatures was favored over the generation effect observed during the solution-based self-assembly of dendritic-linear BCPs. In contrast to conventional amphiphilic block copolymers, PEG3507-POSS-b-PSn self-assembled into polymer cubosomes that contained only a primitive cubic phase (Schwarz P surface) in the Imm space group, regardless of the overall block ratio. The self-assembled morphology could be entirely altered by changing the composition of the organic solvent. In particular, the apparent packing parameter (P) could be significantly increased by increasing the proportion of acetone. Our results could make highly defined periodic nanostructures formed through the solution-based self-assembly of block copolymers more available. The resulting BCPs may have potential for applications in molecular separations and nanotemplating.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Science Foundation (NSF) of Korea (2019R1A2C3007541, 2016R1A2B3015089) and Seoul National University (SNU) for the support by Creative-Pioneering Researchers Program.
Notes and references
- W. B. Zhang, X. F. Yu, C. L. Wang, H. J. Sun, I. F. Hsieh, Y. W. Li, X. H. Dong, K. Yue, R. Van Horn and S. Z. D. Cheng, Macromolecules, 2014, 47, 1221–1239 CrossRef CAS.
-
(a) F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed;
(b) F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- M. L. Stefan and T. Mukundan, Macromolecules, 2004, 37, 8832–8835 CrossRef.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- S. D. Allen, S. Bobbala, N. B. Karabin and E. A. Scott, Nanoscale Horiz., 2019, 4, 258–272 RSC.
- C. Park, Y. La, T. H. An, H. Y. Jeong, S. Kang, S. H. Joo, H. Ahn, T. J. Shin and K. T. Kim, Nat. Commun., 2015, 6, 6392–6398 CrossRef PubMed.
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084–3096 CrossRef CAS PubMed.
- Y. La, T. H. An, T. J. Shin, C. Park and K. T. Kim, Angew. Chem., Int. Ed., 2015, 54, 10483–10487 CrossRef CAS PubMed.
- H. Yu, X. Qiu, S. P. Nunes and K. V. Peinemann, Nat. Commun., 2014, 5, 4110–4118 CrossRef CAS PubMed.
- L. E. Scriven, Nature, 1976, 263, 123–125 CrossRef CAS.
- S. J. Holder, G. Woodward, B. E. Mckenzie and N. A. Sommerdijk, RSC Adv., 2014, 4, 26354–26358 RSC.
- B. E. Mckenzie, H. Friedrich, M. J. Wirix, J. F. Visser, O. R. Monaghan, P. H. Bomans, F. Nudelman, S. J. Holder and N. A. Sommerdijk, Angew. Chem., Int. Ed., 2015, 54, 2457–2461 CrossRef CAS PubMed.
- A. Cho, Y. La, T. J. Shen, C. Park and K. T. Kim, Macromolecules, 2016, 49, 4510–4519 CrossRef CAS.
- K.-V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- A. Jung, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Macromol. Mater. Eng., 2012, 297, 790–798 CrossRef CAS.
- J. Hahn, V. Filiz, S. Rangou, J. Clodt, A. Jung, K. Buhr, C. Abetz and V. Abetz, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 281–290 CrossRef CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- Y. Li, X. H. Dong, Y. Zou, Z. Wang, K. Yue, H. J. Huang, H. Liu, Y. Y. Feng, Z. W. Lin, W. Zhang, W. B. Zhang and S. Z. D. Cheng, Polymer, 2017, 125, 303–329 CrossRef CAS.
- M. J. Huang, C. H. Hsu, J. Wang, S. Mei, X. H. Dong, Y. W. Li, M. X. Li, H. Liu, W. Zhang, T. Aida, W. B. Zhang, K. Yue and S. Z. D. Cheng, Science, 2015, 348, 424–428 CrossRef CAS PubMed.
- X. M. Wang, Y. Shao, J. Xu, X. Jin, R. H. Shen, P. F. Jin, D. W. Shen, J. Wang, W. Li, J. He, P. Ni and W. B. Zhang, Macromolecules, 2017, 50, 3943–3953 CrossRef CAS.
- X. H. Dong, B. Ni, M. J. Huang, C. H. Hsu, Z. Chen, Z. Lin, W. B. Zhang, A. C. Shi and S. Z. D. Cheng, Macromolecules, 2015, 48, 7172–7179 CrossRef CAS.
- D. L. Ho and C. J. Glinka, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 4337–4343 CrossRef CAS.
- R. Mieczkowski, Eur. Polym. J., 1988, 24, 1185–1189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04966e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence