
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of solvent nature on propylene glycol oxidation with tert-butyl hydroperoxide over metal–organic framework Cr-MIL-101†
Viktoriia Torbina,
Mikhail Salaev and
Olga Vodyankina *
*
National Research Tomsk State University, Tomsk, Russian Federation. E-mail: vodyankina_o@mail.ru
First published on 20th August 2019
Abstract
The effect of solvent nature on conversion, product yields and reaction kinetics of selective propylene glycol oxidation with tert-butyl hydroperoxide over porous chromium terephthalate Cr-MIL-101 used as a heterogeneous catalyst is considered. Differences in hydrogen bonding of propylene glycol molecules in different solvents and adsorption of components of the reaction mixture on the active sites of the catalyst are studied by Fourier-transformed infrared spectroscopy. The characteristics of the solvent are shown to play a key role in the process under consideration. In the case of aprotic solvents, the oxidant utilization efficiency, the propylene glycol conversion and the product yields are significantly higher in comparison with those in protic solvents. The protic solvents can adsorb on the active sites of the catalyst which leads to a decrease of their accessibility for the reagents. The initial rate of propylene glycol oxidation decreases linearly with the increasing of the diameter of molecules of the protic solvents. DFT calculations support the competitive adsorption of the molecules of protic solvents on Cr active sites of MIL-101. In the aprotic solvents, the reactivity and distribution of propylene glycol molecules in the solution are determined by the involvement of hydroxyl groups of the substrate into the intermolecular interactions. The ability of the aprotic solvents to break the hydrogen bond network in the associates of propylene glycol is responsible for the concentration of the substrate molecules in the pores of Cr-MIL-101. The highest selectivities towards hydroxyacetone for Cr-MIL-101 catalyst are obtained in solvents, where the initial rates of propylene glycol oxidation are the lowest ones.
Introduction
Propylene glycol (PG) is one of the products of biomass conversion1 that can be oxidized into a variety of valuable products.2–4 However, the investigation of the mechanisms and factors that affect the PG reactivity is a challenge due to its high polarity and two α-hydrophilic groups. These groups are prone to forming strong hydrogen bonds resulting in the formation of associates and solvates with different structures,5–7 which possess different reactivities. Moreover, the presence of the highly polar substances often leads to the catalyst deactivation due to the leaching of the active component or blocking of the active sites through strong adsorption of the substrate or products.8Recently, we demonstrated that the PG oxidation over Cr- and Zr-based metal–organic frameworks (MOFs) as catalysts proceeds with high regioselectivity and leads to the formation of hydroxyacetone.9,10 It was observed that solvent nature strongly affects the activity of the MOFs in this reaction. However, the systematic studies of the reasons as well as reaction kinetics studies for a wide range of different classes of solvents were not carried out.
The application of MOFs as heterogeneous catalysts has increased rapidly due to their unique combination of properties, such as open crystal structure, high surface area and pore volume, and a large variety of accessible metal sites.11–14 However, the understanding of the fundamentals of the effects of different parameters, particularly, the nature of the solvent used, on the reaction kinetics with MOF catalysts is one of the most important open questions.15 Communications on the application of MOFs in catalysis usually have state-of-art character discussing the activity of the certain MOFs in different kinds of reactions to produce valuable compounds,16,17 synthetic or post-synthetic modification of MOFs to improve the catalytic properties18,19 or to control their activity by engineering the defects.20 Tunability of phosphorescence and luminescence properties of MOFs by guest inclusion and energy transfer in the host–guest system was thoroughly studied by DFT calculations and experimentally.21–23 However, there are very few reports discussing the control of adsorption properties or catalytic activity of MOFs due to the changing of the solvent nature.10,24–27 Meanwhile, it is known that the solvent nature can influence significantly on the rate, direction, diffusion, etc., due to different solvation of the reagents and products, stabilization of transition states, interaction with the active particles, changing in the adsorption of the reaction components on the catalyst, etc.28–30 Due to a significant difference between the nature of the active sites, their spatial arrangement, energy distribution, etc., in the MOFs and traditional supported catalysts, the regularities of the influence of solvent nature on the catalytic activity can be different for these kinds of materials. Thus, the studies discussing the effect of solvent on catalysis of different kinds of reactions by MOFs in the context of interaction of the solvent molecules with the active sites of the catalyst, accessibility of the internal space of the MOF structure for solvated components of the reaction mixture, etc., have a significant fundamental and practical importance.
The present work is focused on the main features of the effect of solvent on the selective PG oxidation over MOF Cr-MIL-101. The interpretation of the nature of the reaction processes is proposed. The porous chromium terephthalate Cr-MIL-101 ([Cr3X(H2O)2O(BDC)3]; X = F, OH; BDC = 1,4-benzenedicarboxylate) consists of the quasi-spherical mesoporous cages of two types (34 and 29 Å) accessible through the microporous windows with the diameters of 16 and 12 Å.31
Experimental
Cr-MIL-101 was synthesized according to the procedure described in ref. 31. The formation of the MOF structure was confirmed by the XRD (RigukuMiniflex 600) and FT-IR (Nicolet 6700, Thermo Fisher Scientific). Prior to the use and characterization, the catalyst was activated under vacuum at 180 °C. The BET surface area was 3200 m2 g−1 and the pore volume was 1.6 cm3 g−1.Catalytic experiments were carried out in glass reactors at 50 °C. The reaction was initiated by the addition of 0.25 mmol tert-butyl hydroperoxide (TBHP) to the reaction mixture containing 1 mmol of PG, 1 ml of solvent and 3 mg of Cr-MIL-101. The PG conversion and product yields were analyzed by gas chromatography (Chromatec Crystal 5000.1 equipped with the flame ionization detector, capillary column ZB-WAX). The amount of TBHP in the reaction mixture was determined by the iodometric titration.
For FT-IR studies of TBHP solutions inside the pores of Cr-MIL-101, 150 μL of TBHP solution (5.0 M in decane) were added to 3 mL of the corresponding solvent. Then the resulting solution was added to 20 mg Cr-MIL-101. The suspension was stirred vigorously for 15 min at room temperature and then centrifuged. After separation of the liquid, the FT-IR spectra of the paste-like solid were taken immediately.
Computational details
To estimate the binding energies (BE) of the solvents (except acetonitrile) and TBHP with MIL-101, a model of active Cr site was used. The model comprised the building unit of MIL-101 containing three Cr atoms bound through one oxygen and surrounded by six terephthalate linkers (Fig. 1).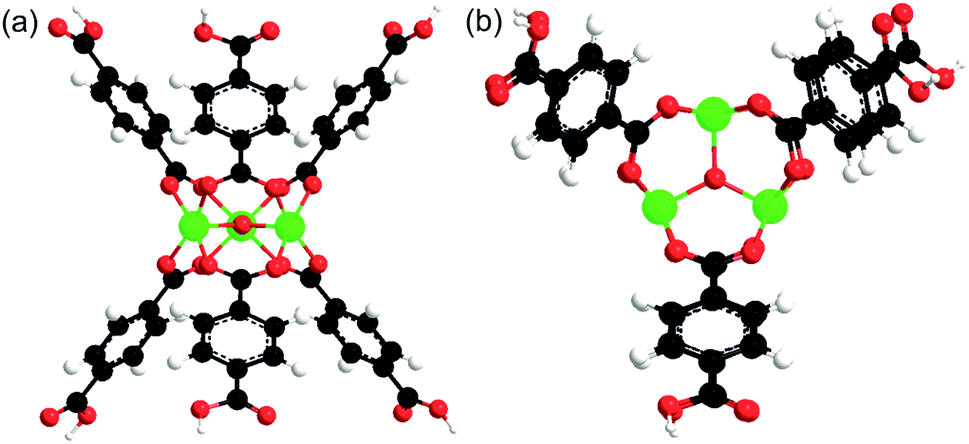 | ||
| Fig. 1 A model of active site of MIL-101, (a) side view, (b) top view. Black, white, red, and green atoms correspond to carbon, hydrogen, oxygen, and chromium, respectively. | ||
The calculations were carried out using density functional theory approach with B3LYP functional32–34 and Lanl2DZ and 6-31G* basis sets for Cr and light atoms (C, H, O), respectively, as implemented in the Gaussian'09 program package (Revision C.01) installed at SKIF “Cyberia” supercomputer of Tomsk State University.35 Firstly, the geometries of the building unit and the solvent and TBHP molecules were fully optimized. The mean Cr–O bond length in the MOF secondary unit was found to be 1.961 Å, which is consistent with the experimental value (1.971 Å) obtained in ref. 36 using EXAFS. Then the structures reflecting the solvent or TBHP binding with one of the Cr atoms of the building unit were optimized. The results for TBHP are consistent with those obtained in ref. 37. The geometry parameters of substrates are consistent with published data (e.g., ethoxy ethanol (r(C–O)exp = 1.203 Å),38 acetone (r(C–O)exp = 1.22 Å),39 methanol (r(C–O)exp = 1.427 Å),40 etc.). All calculated geometries showed the stationary character that was confirmed by the absence of imaginary frequencies in the calculated vibrational spectra. The predicted change in energy below −1.0D-08 was the convergence criterion. Only gas-phase optimization was considered given that the BEs obtained using the polarizable continuum model are somewhat reduced as compared to the gas phase values.41 The results obtained were used for comparison purposes. The BEs were estimated as BE = TE(building unit+solvent/TBHP) − TE(building unit) − TE(solvent/TBHP), where TE(building unit+solvent/TBHP) was a total energy of a complex of building unit with adsorbed solvent or TBHP molecule, TE(building unit) and TE(solvent/TBHP) were the total energies of individual building unit and adsorbed solvent or TBHP molecule, respectively.
Results and discussion
In order to investigate the effect of the solvent nature on the catalytic activity of Cr-MIL-101 in PG oxidation with TBHP the kinetic experiments in four protic and four aprotic solvents were carried out. The main reaction products in all cases were hydroxyacetone (HA) and acetic acid (AA) formed through the C–C bond cleavage (Table 1).| Solvent | PG conversion, % | Selectivity, % | TBHP utilization efficiency, % | |
|---|---|---|---|---|
| HA | AA | |||
| Butyl acetate | 11.5 | 66 | 12 | >100 |
| Ethyl acetate | 12 | 51 | 32 | >100 |
| Acetone | 9 | 86 | 14 | >100 |
| Acetonitrile | 10 | 88 | 10 | 72 |
| tert-Butanol | 6 | 87 | 5 | 80 |
| n-Butanol | 4 | 85 | 12.5 | 53 |
| Methanol | 5 | 62 | 6 | 50 |
| Ethoxyethanol | 4 | 80 | 12.5 | 42 |
PG conversion and TBHP utilization efficiency were almost two times higher in the case of aprotic solvents. The formal TBHP utilization efficiency >100% is connected with the radical-chain process that involves molecular oxygen.9
The polarity of the solvent is known to affect the rate and direction of the reaction through the solvation of the reagents, products or stabilization of the transition state.42 Moreover, during the radical-chain oxidation of substances with dipole moment > 0, the stages of propagation and termination of the chains were shown to be considered as an interaction of two dipoles. Thus, the reaction constants for these processes should depend on the dielectric constant of solvent, if there is no specific solvation.43 For the PG oxidation with TBHP over Cr-MIL-101, the dependence of the initial reaction rate on the dielectric constant has a different character for protic and aprotic solvents (Table 2). In solvents with close dielectric constant (acetonitrile and methanol), the initial reaction rate differs almost seven times. Similar effect was observed in catalytic hydrogenation of 1-octene with homogeneous Ni(II) complexes.44
In the PG oxidation over Cr-MIL-101, the TBHP activation on the metal sites of the catalyst to yield radicals that further react with the substrate molecules was shown to be the rate-limiting step.9 However, the initial rate of TBHP decomposition in the absence of PG was close in the cases of ethyl acetate and acetonitrile (Fig. 2). At the same time, the initial rate of the substrate oxidation differed significantly under the same conditions (Table 2). This fact indicated the difference in further ways of radical reactions in the solution.26 In particular, the intermolecular interactions may play a significant role.
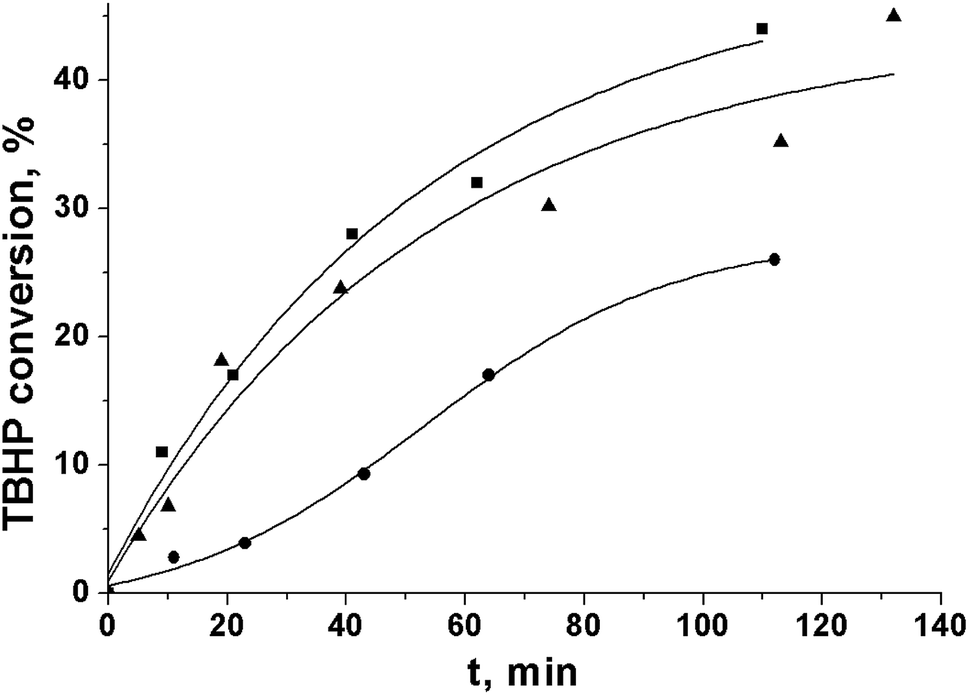 | ||
| Fig. 2 Kinetic curves of tert-butylhydroperoxide decomposition over Cr-MIL-101 in ethyl acetate (■), acetonitrile (▲), and tert-butanol (●). | ||
Polyhydric alcohols are highly prone to the formation of both intra- and intermolecular hydrogen bonds resulting in the formation of the associates of various sizes depending on the concentration of alcohols and the solvent nature.47 The presence of specific intermolecular interactions of functional groups of the substrate molecules between themselves and with the solvent molecules can influence significantly on the alcohol reactivity.43 In case of PG, a blue shift of the IR absorption band of O–H vibration in acetonitrile in comparison with the one in ethyl acetate and pure PG (Fig. 3) indicates the involvement of hydroxyl groups of PG in the hydrogen bonding with lower energy in this solvent.
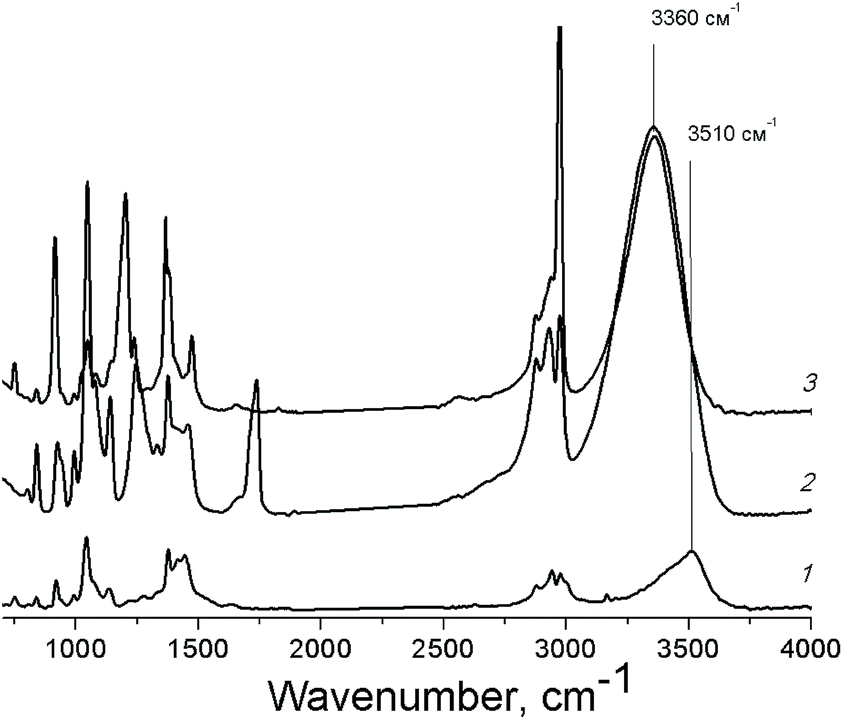 | ||
| Fig. 3 FT-IR spectra of PG solutions (1 M) in acetonitrile (1), ethyl acetate (2) and pure PG (3). | ||
For the solvents with high dielectric constants, it is preferable to build the dependence of the rate constant on the logarithm of the dielectric constant, since this function has no limit.45 An almost linear dependence of lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) W0 of PG oxidation over Cr-MIL-101 on lgε is observed in the case of aprotic solvents (Fig. 4).
W0 of PG oxidation over Cr-MIL-101 on lgε is observed in the case of aprotic solvents (Fig. 4).
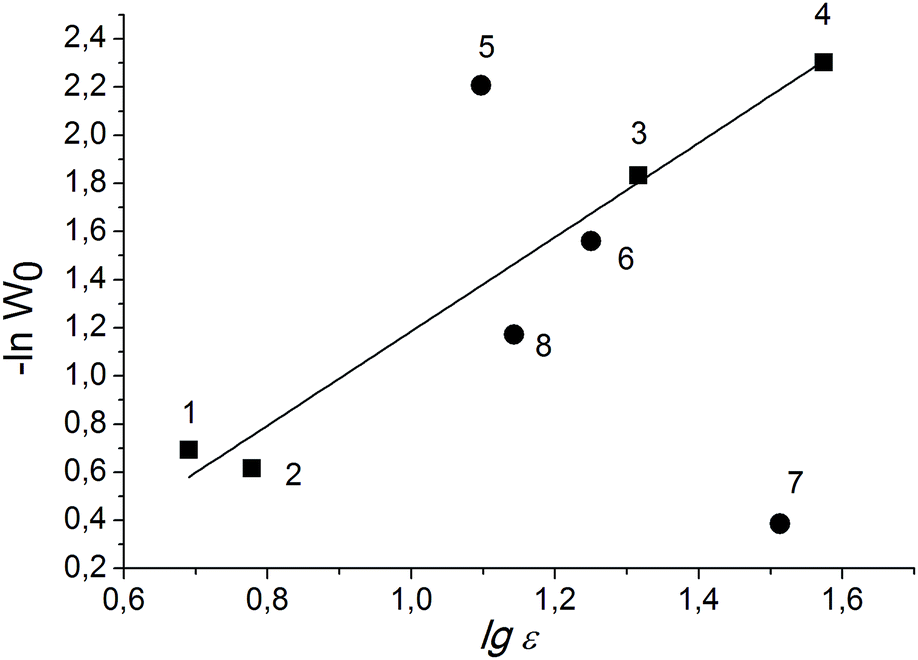 | ||
| Fig. 4 Dependence of initial rate of PG oxidation on solvent nature. Protic and aprotic solvents are labeled with circles and squares, respectively. Numbers correspond to solvents as in Table 2. | ||
Thus, the reaction rate increases with both increasing of the dielectric constant and the ability of the solvent to break the hydrogen bond network in the PG associates. We suppose that the change in the size of the associates and solvates leads to changing of the concentrations of substrate molecules inside the pores of the MOF, where the oxidant is preferentially activated, and the required availability of the PG molecules is ensured. Meanwhile, we cannot also exclude the effect of the solvent nature on the rates of continuation and termination of the chain due to the interaction of the radicals with the solvent molecules. This issue is under further investigation.
The initial rate of TBHP decomposition over Cr-MIL-101 in the protic solvent tert-butanol differs significantly from the corresponding rates in the aprotic solvents (Fig. 2). Moreover, in this case a small inductive effect is observed. This fact allows us to propose that the molecules of protic solvents are competitively adsorbed on the active sites of the catalyst leading to a decrease in their accessibility for TBHP molecules. One of the confirmations of this assumption is a linear decreasing of the initial rate of PG oxidation with the increasing of the diameter of the solvent molecule (Fig. 4 and Table 2). The lower reaction rate in tert-butanol in comparison with the one in n-butanol at close diameters of the molecules can be explained by a higher branching of tert-butanol molecule leading to higher steric hindrance. At the same time, there is no similar dependence in the case of aprotic solvents (Fig. 5). It is noteworthy that there is no specific effect of ethoxy group in ethoxyethanol on the PG oxidation over Cr-MIL-101. The catalyst showed similar activity in ethoxyethanol and butanol (Tables 1 and 2) due to close molecule sizes of these solvents.
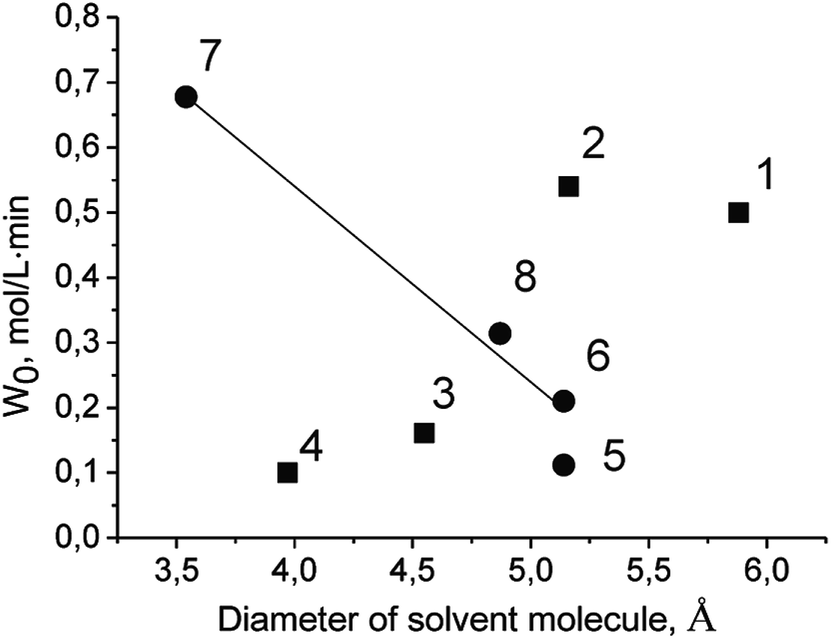 | ||
| Fig. 5 Dependence of initial rate of PG oxidation on diameter of solvent molecule. Protic and aprotic solvents are labeled with circles and squares, respectively. Numbers correspond to solvents as in Table 2. | ||
A shift of the absorption band of C–O vibrations in the solvent molecule was observed in the IR spectrum of the butanol solution of TBHP in the pores of Cr-MIL-101 (Fig. 6(a)). This fact also confirmed the adsorption of the protic solvents on active sites of the catalyst. In the IR spectra of TBHP solution in aprotic solvents after the adsorption on Cr-MIL-101, there are changes only in the region of –OO–H vibrations in the TBHP molecule (Fig. 6(b)).
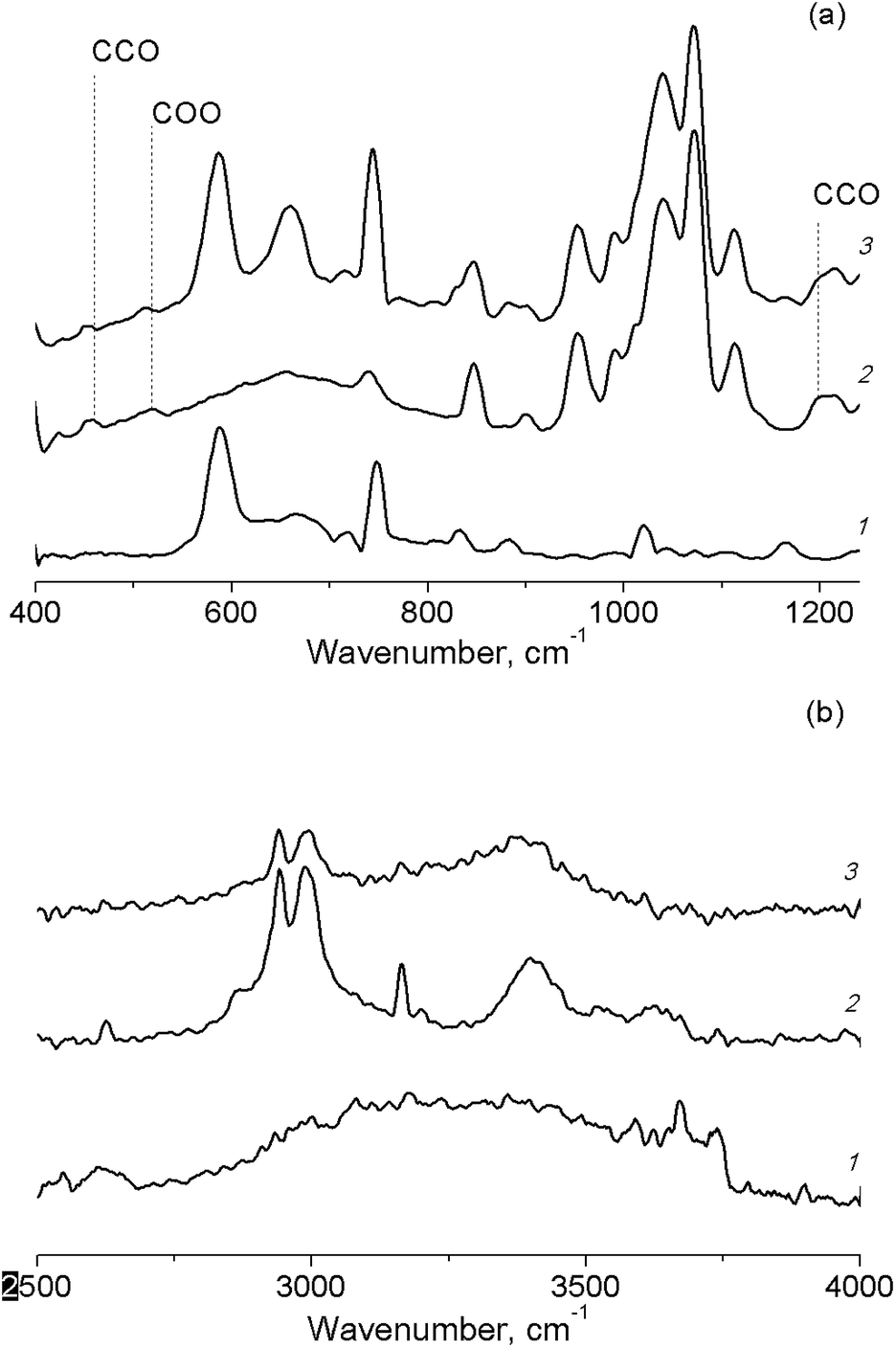 | ||
| Fig. 6 FT-IR spectra of TBHP solutions in n-butanol (a) and in acetonitrile (b) in pores of Cr-MIL-101. Cr-MIL-101 (1), TBHP solution (2), TBHP solution in Cr-MIL-101 (3). | ||
The BEs for aprotic solvents ethyl acetate and butyl acetate with active sites of Cr-MIL-101 are lower or equal to the one of TBHP (Table 3), while the opposite situation is observed in case of protic solvents, which supports the hypothesis about the competitive adsorption of this kind of solvent molecules on chromium active sites. Close BEs of acetone and TBHP (−77 and −71 kJ mol−1) lead to much lower initial reaction rate of PG oxidation with TBHP over Cr-MIL-101 in acetone in comparison with W0 in ethyl acetate and butyl acetate (Table 2). The initial reaction rates of PG oxidation in protic solvents do not strongly depend on BEs of the corresponding solvents indicating that Cr sites do not adsorb and activate TBHP in this case.
| Bound molecule | Binding energy, kJ mol−1 |
|---|---|
| TBHP | −71 |
| PG | −64 |
| Butyl acetate | −62 |
| Ethyl acetate | −59 |
| Acetone | −77 |
| tert-Butanol | −74 |
| n-Butanol | −89 |
| Methanol | −91 |
| Ethoxyethanol | −76 |
During adsorption of TBHP on Cr active sites, the relative stretching of the O–H bond is observed (Table S1 in ESI†), suggesting the classical Haber–Weiss mechanism of TBHP activation over transition metal catalysts.48 However, when MeOH-containing model of building unit adsorbs TBHP (Fig. S1 in ESI†), the O–H bond in the solvent molecule is stretched. Thus, it is possible to assume that there is another mechanism of TBHP activation on the active Cr sites occupied by the solvent molecule (Scheme 1). TBHP molecule interacts with adsorbed solvent through the hydrogen bond, and O–H bond in the solvent molecule and O–O bond in TBHP are activated to form two RO˙ radicals. Similar activation mechanism for hydroperoxides in the homogeneous media in the presence of alcohols was proposed in ref. 43 and 49. Such a decomposition was suggested to be more energetically beneficial, then monomolecular decomposition of ROOH, due to the exothermicity of the reaction.49 However, additional studies are required to check the processes that occur in this mechanism.
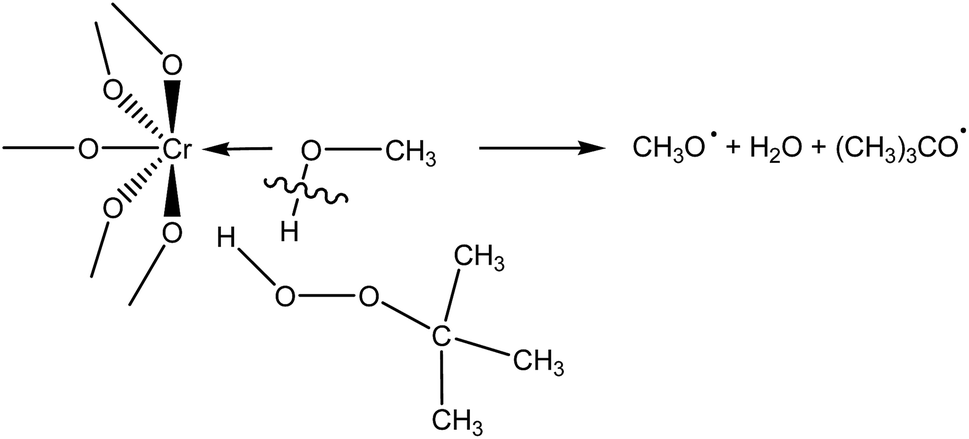 | ||
| Scheme 1 Tentative mechanism of TBHP activation over Cr-MIL-101 in methanol. | ||
The formation of two radicals capable of rapid recombination prior to the reaction with the substrate molecule may lead to the experimentally observed low oxidant utilization efficiency in case of protic solvents (Table 1).
It is noteworthy that the energy of PG molecule binding with chromium active sites of MOF is much lower than those for TBHP and most solvents (Table 3) supporting the fact that there is no PG adsorption on Cr-MIL-101 found in ref. 9 as well as proving the role of Cr-MOFs as catalysts for TBHP decomposition.
Conclusions
In the case of aprotic solvents the oxidant utilization efficiency, propylene glycol conversion and product yields are significantly higher in comparison with those in protic solvents. The reason is the interaction of the solvent molecules with the active sites of the catalyst and the intermolecular interactions in solution. Protic solvent molecules are able to competitively adsorb on the active sites of the catalyst, which leads to a decreasing of their accessibility for reagents. The initial rate of PG oxidation decreases linearly with the increasing of the diameter of the molecules of protic solvents. In the aprotic solvents, the PG molecule distribution in the solution is determined by the involvement of the hydroxyl groups of the substrate in the intermolecular interactions and plays a key role in its reactivity and concentration in the pores of the catalyst. The highest selectivities to hydroxyacetone were obtained in the solvents, where the initial rates of PG oxidation were the lowest ones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Russian Science Foundation (project no. 19-73-30026).References
- F. Cavani, S. Albonetti, F. Basile, and A. Gandini, Chemicals and Fuels from Bio-Based Building Blocks, Wiley, New Jersey, 2016 Search PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- N. H. Mohamad, R. Awang and W. M. Z. W. Yunus, Am. J. Appl. Sci., 2011, 11, 1135–1139 CrossRef.
- F. A. C. Martinez, E. M. Balciunas, J. M. Salgado, J. M. D. González, A. Converti and R. P. S. Oliveira, Trends Food Sci. Technol., 2013, 30, 70–83 CrossRef CAS.
- K. Z. Haufa and M. A. Szarnecki, Vib. Spectrosc., 2009, 51, 80–85 CrossRef CAS.
- A. M. Tolmachev, A. V. Kucherov and G. O. Khondar, Moscow Univ. Chem. Bull., 2012, 67, 45–49 CrossRef.
- S. Kumar, P. Periyasamy and P. Jeevanandham, Int. J. ChemTech Res., 2011, 3, 369–375 CAS.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001, 212, 175–187 CrossRef CAS.
- V. V. Torbina, I. D. Ivanchikova, O. A. Kholdeeva, I. Yu. Skobelev and O. V. Vodyankina, Catal. Today, 2016, 278, 97–103 CrossRef CAS.
- V. V. Torbina, N. S. Nedoseykina, I. D. Ivanchikova, O. A. Kholdeeva and O. V. Vodyankina, Catal. Today, 2019, 333, 47–53 CrossRef CAS.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- Y.-S. Kang, Y. Lu, K. Chen, Y. Zhao, P. Wang and W.-Y. Sun, Coord. Chem. Rev., 2019, 378, 262–280 CrossRef CAS.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- W. Xu, K. B. Thapa, Q. Ju, Z. Fang and W. Huang, Coord. Chem. Rev., 2017, 373, 199–232 CrossRef.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2540 RSC.
- Z. Hu and D. Zhao, CrystEngComm, 2017, 19, 4066–4081 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- Y. Zhang, X. Yang and H.-C. Zhou, Polyhedron, 2018, 154, 189–201 CrossRef CAS.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fisher, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- D. Yan, Y. Tang, H. Lin and D. Wang, Sci. Rep., 2014, 4, 4337 CrossRef PubMed.
- Y. Tang, W. He, Y. Lu, J. Fielden, X. Xiang and D. Yan, J. Phys. Chem. C, 2014, 118, 25365–25373 CrossRef CAS.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
- B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC.
- R. Limvorapitux, H. Chen, M. L. Mendonca, M. Liu, R. Q. Snurr and S. T. Nguyen, Catal. Sci. Technol., 2019, 9, 327–335 RSC.
- K. Gedrich, M. Heitbaum, A. Notzon, I. Senkovska, R. Frohlich, J. Getzschmann, U. Mueller, F. Glorius and S. Kaskel, Chem.–Eur. J., 2011, 17, 2099–2106 CrossRef CAS PubMed.
- M. Opanasenko, A. Dhakshinamoorthy, Y. K. Hwang, J.-S. Chang, H. Garcia and J. Čejka, ChemSusChem, 2013, 6, 865–871 CrossRef CAS PubMed.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- J. J. Varghese and S. H. Murshri, React. Chem. Eng., 2019, 4, 165–206 RSC.
- D. Y. Murzin, Catal. Sci. Technol., 2016, 6, 5700–5713 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski,G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- K.-S. Lin, A. K. Adhikari, Y.-H. Su, C.-L. Chiang and K. Dehvari, Chin. J. Phys., 2012, 50, 322–331 CAS.
- A. Y. Kosnikov, V. L. Antonovskii, S. V. Lindeman, M. Y. Antipin, Y. T. Struchkov, N. A. Turovskii and I. P. Zyat'kov, Theor. Exp. Chem., 1989, 25, 73–77 CrossRef.
- K. Kuchitsu, Structure of Free Polyatomic Molecules - Basic Data, Springer, Berlin, 1998 Search PubMed.
- L. E. Sutton, Tables of Interatomic Distances and Configuration in Molecules and Ions, London, The Chemical Society, 1958 Search PubMed.
- P. Venkateswarlu and W. Gordy, J. Chem. Phys., 1955, 23, 1200 CrossRef CAS.
- D. C. Cantu, B. P. McGrail and V.-A. Glezakou, Chem. Mater., 2014, 26, 6401–6409 CrossRef CAS.
- C. Reichardt, Solvent effects in organic chemistry, Wiley-VCH, Marburg, Germany, 2011 Search PubMed.
- N. M. Emanuel, G. E. Zaikov, Z. K. Maizus, Oxidation of Organic Compounds, Medium Effects in Radical Reactions, Pergamon Press Ltd., Oxford, 1984 Search PubMed.
- I. M. Angulo, S. M. Lok, V. F. Quiroga Norambuena, M. Luiz, A. L. Spek and E. Bouwman, J. Mol. Catal. A: Chem., 2002, 187, 55–67 CrossRef CAS.
- R. Schmid, and V. N. Sapunov, Non-formal kinetics: in search for chemical reaction pathways, Wiley-VCH, Weinheim, Germany, 1982 Search PubMed.
- M. Kodaka, J. Phys. Chem. B, 2004, 108, 1160–1164 CrossRef CAS.
- G. A. Krestov, Thermodynamics of ion processes in solutions, Khimya, Leningrad, 1984, in Russian Search PubMed.
- F. Haber and J. Weiss, Proc. R. Soc. London, Ser. A, 1934, 147, 332–351 CrossRef CAS.
- E. T. Denisov, Russ. J. Phys. Chem., 1964, 38, 2085 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05003e |
| This journal is © The Royal Society of Chemistry 2019 |