DOI:
10.1039/C9RA05034E
(Paper)
RSC Adv., 2019,
9, 31370-31377
Kinetics and pathways of diclofenac degradation by heat-activated persulfate
Received
3rd July 2019
, Accepted 23rd September 2019
First published on 2nd October 2019
Abstract
In this study, the degradation of diclofenac (DCF) by heat-activated persulfate (HAP) was investigated. It was found that DCF could be degraded efficiently by HAP. The degradation of DCF followed the pseudo-first-order kinetic model, and the highest observed degradation rate constant (kobs) was obtained at pH 3. The sulfate radical was mainly responsible for DCF removal at pH < 7, whereas it was the hydroxyl radical at high pH. The elimination of DCF was enhanced with the increase in temperature or initial dosage of persulfate. Presence of Cu2+ and CO32− could improve DCF degradation, while an inhibition effect was observed in the presence of natural organic matter. According to the identified nine transformation products, the potential DCF degradation mechanism was proposed revealing five different reaction pathways, including hydroxylation, decarboxylation, formylation, dehydrogenation and C–N bond cleavage. This study indicates that HAP can effectively oxidize and degrade DCF, especially under acidic conditions.
1. Introduction
Huge amounts of pharmaceuticals are used every year by humans throughout the world.1,2 For example, 836 tons acetylsalicylic acid, 622 tons paracetamol, 345 tons ibuprofen and 86 tons diclofenac were used in Germany in 2001.3 Diclofenac (DCF), a widely used non-steroidal anti-inflammatory drug, was frequently detected in groundwater, treated sewage and surface water in recent years.4,5 It could hardly be removed completely by the conventional wastewater treatment plants. Thus, the effluent of raw and treated sewage was considered to be its primarily pathway entering the environment.6,7 Oaks et al.8 reported that DCF could cause a high death rate among three species of vulture in India and Pakistan. The biological toxicity of DCF may also induce a risk to the other organisms or even human health. As a result, its risk should be paid urgent attention though its concentration in the natural environment is very low.
Advanced oxidation processes (AOPs) have received significant attention for the degradation of emerging contaminants in recent years.9–11 AOPs are characterized by the production of active radicals, such as hydroxyl radical (HO˙), sulfate radical (SO4˙−) and carbonate radical (CO3˙−), which are very reactive and even can mineralize organic pollutants thoroughly. Hydroxyl radical-based AOPs (E0 = 2.8 V), including Fenton, photo-Fenton, UV/H2O2 and O3/H2O2, are widely investigated in last decades.11–13 Recently, considerable papers about the degradation of DCF by hydroxyl radical-based AOPs were reported, such as UV/H2O2,12 photo-Fenton,14 UV/TiO2,15etc. In addition, sulfate radical (E0 = 2.5–3.1 V) and carbonate radical (E0 = 1.78 V at pH 7) also drew public attention because of their selectivity and high oxidation capability.9,16,17 Particularly, sulfate radical (SR)-based AOPs have caused more attention, because SO4˙− can be readily generated through transition metal, UV or heat activation of cheap oxidants (e.g., persulfate (PS) and peroxymonosulfate (PMS)) and these technologies have achieved success in the elimination of many emerging contaminants. Of which heat-activated persulfate (HAP) is a relatively green SR-AOPs which will not bring secondary pollution though it needs energy input. Therefore, HAP has been widely studied to degrade amounts of organic compounds such as duron,18 carbamazepine,19p-nitrophenol,20 benzoic acid,21 triclosan22 and so on. The degradation of DCF by HAP was also investigated by Chen et al.,23 but its reaction mechanism in this system is still unclear until now. Besides, although the influence of several water matrix (e.g., Cl−, HCO3−, and natural organic matter (NOM)) on DCF removal were explored in their study, the effect of the other constituents such as CO32−, SO42−, NO3−, Fe3+, and Cu2+ were not investigated. Accordingly, the removal of DCF by HAP was systematically investigated in this study including degradation kinetics, influence factors and transformation mechanism.
The main objectives of this work were: (i) to investigate the effect of common water matrix including metal cations (e.g., Fe3+ and Cu2+), inorganic anions (i.e., CO32−, SO42− and NO3−) and NOM on DCF degradation; (ii) to detect the transformation products of DCF for speculating its degradation mechanism by HAP; and (iii) to identify the dominant reactive radical species and quantify their contribution to DCF removal in HAP system at different pH values through the radical scavenging experiments based on the fact that the radical species is dependent on the solution pH in SR-AOPs.
2. Materials and methods
2.1 Materials
Diclofenac sodium (99%) was obtained from Aladdin (China). Methanol, acetic acid, isopropanol and tert-butanol were all HPLC grade and purchased from Kelong (Chengdu, China). All the other reagents were AR grade and used without further purification. Milli-Q water (18 MΩ cm) was used for preparing all aqueous solutions.
2.2 Experimental setup
All experiments were carried out in a 1 L glass beaker containing 600 mL reaction solution. Before adding DCF and the other chemicals, the reactor was pre-heated in a thermostat water bath for about 30 min to reach to the desired temperature. Then, DCF standard solution was added into the reaction solution and its initial concentration was 1 μM. After that, the solution was stirred rapidly for several minutes by a mechanical agitator, ensuring a complete mixing of solution. Finally, appropriate volume of 6 mM persulfate was added into the solution, and the reaction began. At given time intervals, 1.5 mL reaction solution was sampled and fleetly quenched with 0.45 M Na2S2O3. All the experiments were conducted in triplicate. The error bars in the figures represent the standard error of the mean.
2.3 Chemical analysis
The concentration of DCF was determined by a high performance liquid chromatograph (HPLC, Waters 2695, USA) equipped with a symmetry C18 column (5 μm, 4.6 × 150 mm) and a UV detector (Waters 2966, USA) at 276 nm. The mobile phase consisted of methanol and 1‰ acetic acid water solution (75/25, v/v) with a flow rate of 1.0 mL min−1. The column temperature was 30 °C and the sample injection volume was 20 μL. pH was measured by a pH meter (INESA Scientific Instrument Co., Ltd, China). The transformation products of DCF were detected using an ultra performance liquid chromatograph coupled with a quadrupole-time of flight-mass spectrometry (UPLC-QTOF/MS, Waters Xevo G2-XS QT, USA). The chromatographic separation was performed on a C18 column (1.7 μm, 2.1 × 100 mm) with the sample injection volume of 10 μL. The mobile phase contained A (0.1% formic acid in water) and B (acetonitrile) at the flow rate of 0.3 mL min−1. The gradient was 10% B in the initial 0.5 min, linearly increasing to 100% B for 6.5 min, and decreasing back to 10% B for 3 min. The mass spectrum (m/z 50–800) was analyzed in a positive ion mode by electrospray ionization (ESI) with the drying gas temperature of 400 °C and capillary voltage of 2.5 kV. Data were analyzed through Masslynx 4.1 software (Waters, USA).
3. Results and discussion
3.1 Degradation of DCF by HAP
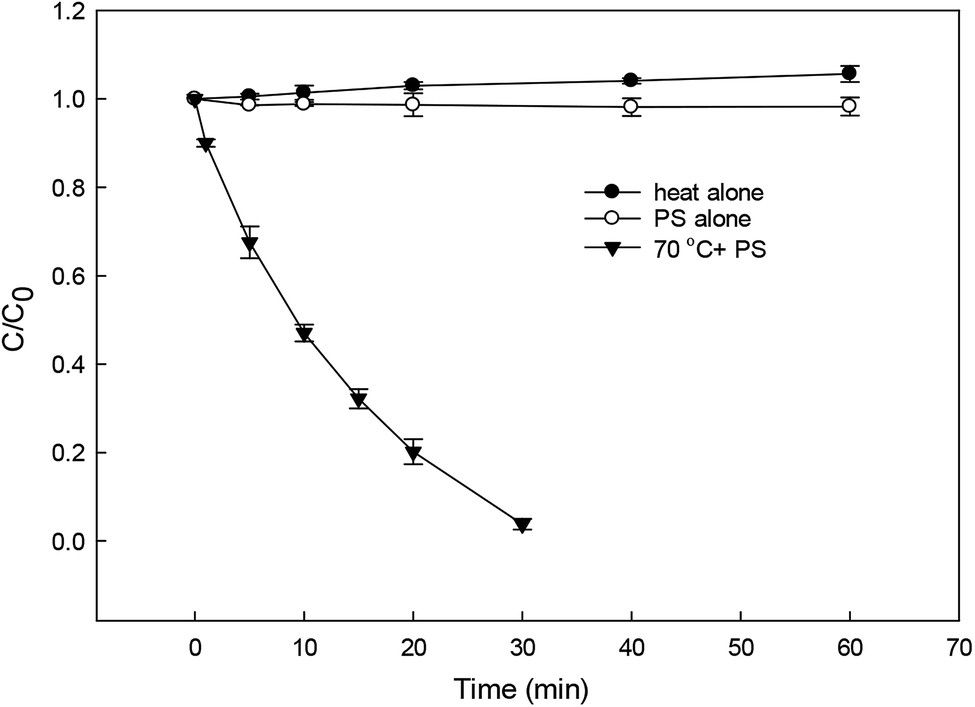
Fig. 1 shows the degradation of DCF by heat alone, PS alone and HAP. DCF was hardly degraded after 60 min by heat alone and PS alone, while about 96% DCF was removed after 30 min with the addition of persulfate at 70 °C which was probably due to the role of the formed radicals from the activated PS by heat. The degradation of DCF in HAP system followed a pseudo-first-order kinetic model, and the observed degradation rate constant was thus calculated to be 0.103 min−1 according to eqn (1).| | | −d[DCF]/dt = kobs[DCF] | (1) |
where kobs is the pseudo-first-order rate constant of DCF by HAP, and [DCF] is the molar concentration of DCF at any time.
 |
| | Fig. 1 Degradation of DCF by different reaction systems. Experimental conditions: [DCF]0 = 1 μM. [PS]0 = 50 μM, no buffer, T = 25 °C for PS alone system, T = 70 °C for heat alone and HAP systems. | |
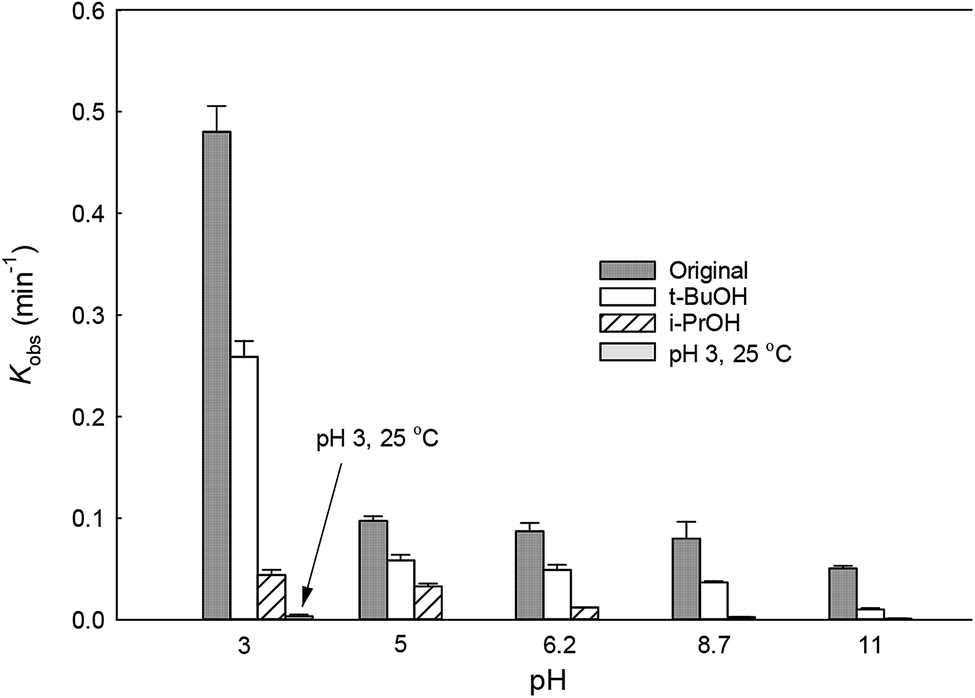
3.2 Effect of initial pH
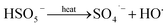
The solution pH can affect the species and concentration of active radicals (i.e., HO˙ and SO4˙−) in SR-AOPs, which may influence the degradation of organic contaminants.24,25 The effect of pH (3.0–11.0) on DCF degradation is shown in Fig. 2. The removal rate of DCF at pH 3 (kobs = 0.48 min−1) was remarkably higher than those at the other pH values. At the strong acid condition, Caro's acid (H2SO5) might be formed, as shown in eqn (2), and its redox potential was reported to be 1.50 V.26,27 To clarify if DCF could be oxidized by the formed H2SO5, the removal of DCF in the presence of persulfate under the room temperature (25 °C) at pH 3 was investigated. As shown in Fig. 2, DCF could hardly be degraded in this reaction condition, indicating that it could not be removed by H2SO5 oxidation. Therefore, the fastest DCF degradation at pH 3 might be ascribed to the increase in the steady-state concentration of reactive radicals in this system, because the structure of Caro's acid was similar to that of peroxymonosulfate, leading to its possible activation by heat to produce HO˙ and SO4˙−, as presented in eqn (3).28 To verify the above speculation, two different alcohols, i.e., tert-butanol (t-BuOH, 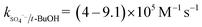 , kHO˙/t-BuOH = (4.2–7.6) × 108 M−1 s−1) and isopropanol (i-PrOH,
, kHO˙/t-BuOH = (4.2–7.6) × 108 M−1 s−1) and isopropanol (i-PrOH, 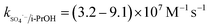 , kHO˙/i-PrOH = (1.6–2.3) × 109 M−1 s−1), were added into this system, respectively.29–31 As shown in Fig. 2, due to the scavenging of t-BuOH for HO˙, the degradation of DCF at pH 3 was inhibited significantly with its addition. While the inhibition effect was more obvious in the presence of i-PrOH, because it could also react with SO4˙− except quenching HO˙. This radical scavenging experiment indicated that SO4˙− and HO˙ were both present and responsible for DCF removal by HAP at pH 3 and their contribution to DCF degradation were 54% and 46%, respectively.
, kHO˙/i-PrOH = (1.6–2.3) × 109 M−1 s−1), were added into this system, respectively.29–31 As shown in Fig. 2, due to the scavenging of t-BuOH for HO˙, the degradation of DCF at pH 3 was inhibited significantly with its addition. While the inhibition effect was more obvious in the presence of i-PrOH, because it could also react with SO4˙− except quenching HO˙. This radical scavenging experiment indicated that SO4˙− and HO˙ were both present and responsible for DCF removal by HAP at pH 3 and their contribution to DCF degradation were 54% and 46%, respectively.| | | S2O82− + H+ + H2O → H2SO5 + HSO4− | (2) |
| |  | (3) |
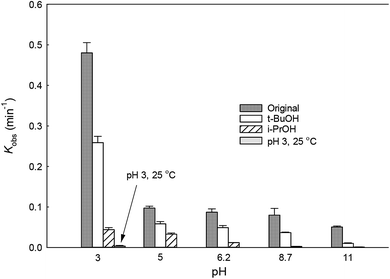 |
| | Fig. 2 Variation of kobs as a function of initial pH for DCF degradation. Experimental conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, [t-BuOH]0 = [i-PrOH]0 = 5 mM, T = 70 °C (T = 25 °C for verifying group at pH 3), 5 mM phosphate buffer. | |
With the increase of initial pH from 5 to 11, the concentration of OH− in the solution increased gradually. It could react with SO4˙−, as presented in eqn (4), lowering the contribution of this radical to DCF removal,32 which was also confirmed by the radical scavenging experiments where the contribution of SO4˙− to DCF degradation were 60.4%, 56.4%, 46.2% and 20.1% at pH 5, 6.2, 8.7 and 11, respectively, as described in Fig. 2 and Table 1. Although HO˙ could be formed from the above reaction, it could also be scavenged by OH− and SO4˙−, as shown in eqn (5) and (6).32 Hence, the degradation rate of DCF declined from 0.097 to 0.050 min−1 when the initial pH changed from 5 to 11. According to these results, we could conclude that except pH 3, SO4˙− was the dominant radical species at pH < 7; while HO˙ became the main radical at high pH condition in HAP system. Similar conclusion was also obtained by Tan et al.33 Overall, HAP was considered to be an efficient treatment technology for the degradation of DCF because of its effectiveness in a broad range of pH, especially at pH ≤ 3.
| | | SO4˙− + OH− → SO42− + HO˙ k = 7.3 × 107 M−1 s−1 | (4) |
| | | HO˙ + OH− → O˙− + H2O k = 1.2 × 1010 M−1 s−1 | (5) |
| | | SO4˙− + HO˙ → HSO4− + 0.5O2k = 1.2 × 109 M−1 s−1 | (6) |
Table 1 Contribution of reactive radical species to DCF degradation in HAP system at different pH values
| pH |
k
obs (min−1) |
SO4˙− (%) |
HO˙ (%) |
| 3 |
0.480 |
54 |
46 |
| 5 |
0.097 |
60.4 |
39.6 |
| 6.2 |
0.087 |
56.4 |
43.6 |
| 8.7 |
0.080 |
46.2 |
53.8 |
| 11 |
0.050 |
20.1 |
79.9 |
3.3 Effect of reaction temperature
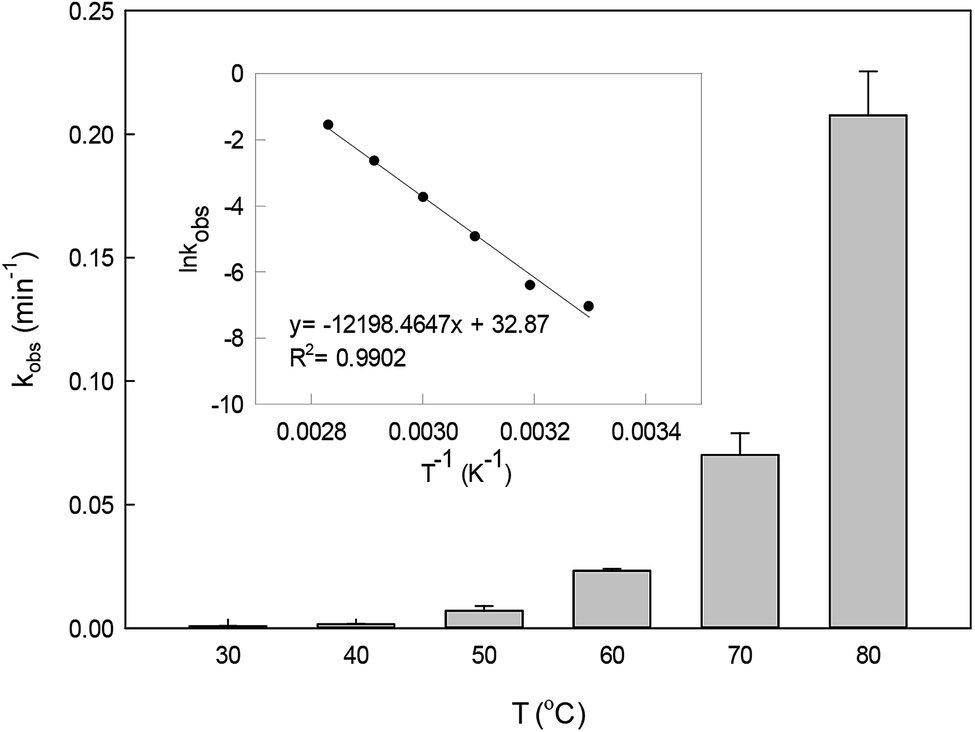
The effect of temperature on DCF degradation in a range of 30–80 °C is shown in Fig. 3. When the reaction temperature was 30 °C and 40 °C, DCF could hardly be degraded. As the temperature increased, the persulfate could gradually be activated readily to produce SO4˙− leading to the enhancement of its steady-state concentration. Meanwhile, the collision probability between DCF and the reactive radicals might be improved with the increasing temperature based on the thermodynamics principle.34 Therefore, the kobs increased from 0.007 to 0.207 min−1 when the temperature changed from 50 to 80 °C. Most of the published papers on HAP system all found that the degradation of organic contaminant was faster in the higher temperature.33,35,36
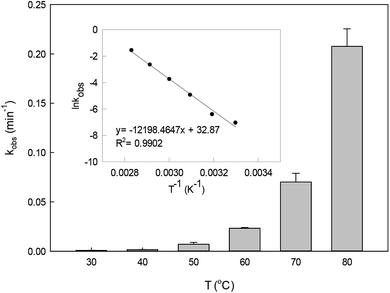 |
| | Fig. 3 Effect of temperature on kobs in HAP system. The inset is the Arrhenius plot for DCF degradation. Experimental conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, 5 mM phosphate buffer at pH 6.2. | |
The plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kobsvs. 1/T (the insert in Fig. 3) showed an excellent fit with the Arrhenius type model (eqn (7)). The activation energy of DCF degradation by HAP was thus calculated to be 101.4 kJ mol−1 in this study. However, Chen et al.23 reported that the activation energy of this reaction was 157.63 kJ mol−1, which was higher than that obtained by this study. This disagreement might be due to the difference of the reaction conditions between their study (pH = 7) and ours (pH = 6.2).
kobsvs. 1/T (the insert in Fig. 3) showed an excellent fit with the Arrhenius type model (eqn (7)). The activation energy of DCF degradation by HAP was thus calculated to be 101.4 kJ mol−1 in this study. However, Chen et al.23 reported that the activation energy of this reaction was 157.63 kJ mol−1, which was higher than that obtained by this study. This disagreement might be due to the difference of the reaction conditions between their study (pH = 7) and ours (pH = 6.2).
| | 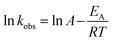 | (7) |
where
A is the pre-exponential factor,
EA is the apparent activation energy of the reaction,
R is the universal gas constant (8.314 J mol
−1 K
−1), and
T is the absolute temperature.
3.4 Effect of initial PS dosage
The dosage of PS is an important parameter in SR-AOPs, which can influence the degradation of target pollutants. Many researches have found that within the definite range of PS concentration, the degradation of organic contaminants enhanced with the increase in its dosage. Similar result was also obtained in our study on DCF removal by HAP, as shown in Fig. 4. The degradation rate of DCF enhanced from 0.003 to 0.087 min−1 as the concentration of PS increased from 1 to 50 μM. The increase of PS dosage could produce more active radicals in HAP system, leading to the improvement on DCF degradation.
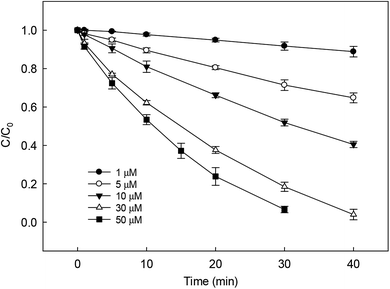 |
| | Fig. 4 Effect of initial PS dosage on DCF removal by HAP. Experimental conditions: [DCF]0 = 1 μM, 5 mM phosphate buffer at pH = 6.2. | |
3.5 Effect of water matrix
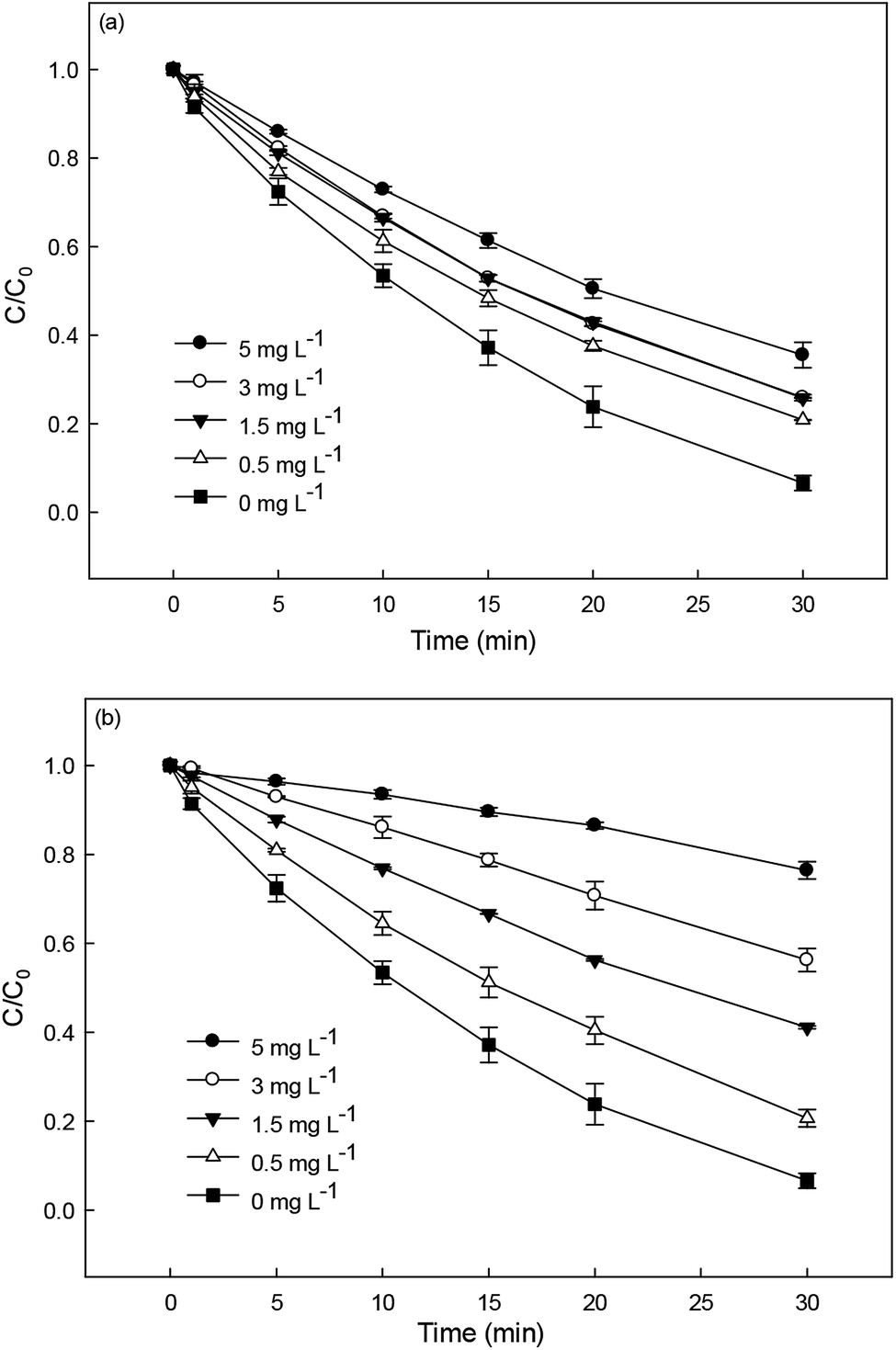
3.5.1 Effect of metal cations.
The effect of Fe3+ and Cu2+ (0–10 μM) on DCF degradation in HAP system at 70 °C was investigated. As shown in Fig. 5a, Fe3+ was found to be no obvious impact on the degradation of DCF. Anipsitakis et al.37 also reported that the persulfate could not be activated by Fe(III). However, the addition of Cu2+ could significantly improve DCF degradation (Fig. 5b), which was in agreement with the results reported by Deng et al.19 and Zhang et al.20 The improvement effect enhanced gradually with the increase of Cu2+ concentration. Possible explanations for this result include: (1) PS could be activated by Cu2+ to produce SO4˙−, as shown in eqn (8), increasing the steady-state concentration of SO4˙− in HAP system;38 (2) the formed Cu3+ possessed a high oxidation capability and might be responsible for DCF degradation at a certain extent.19| | | S2O82− + Cu2+ → SO42− + SO4˙− + Cu3+ | (8) |
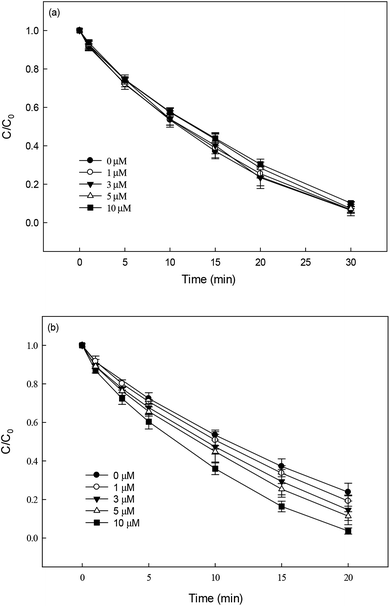 |
| | Fig. 5 Effect of Fe3+ (a) and Cu2+ (b) on DCF degradation in HAP system. Experimental conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, 5 mM phosphate buffer at pH 6.2. | |
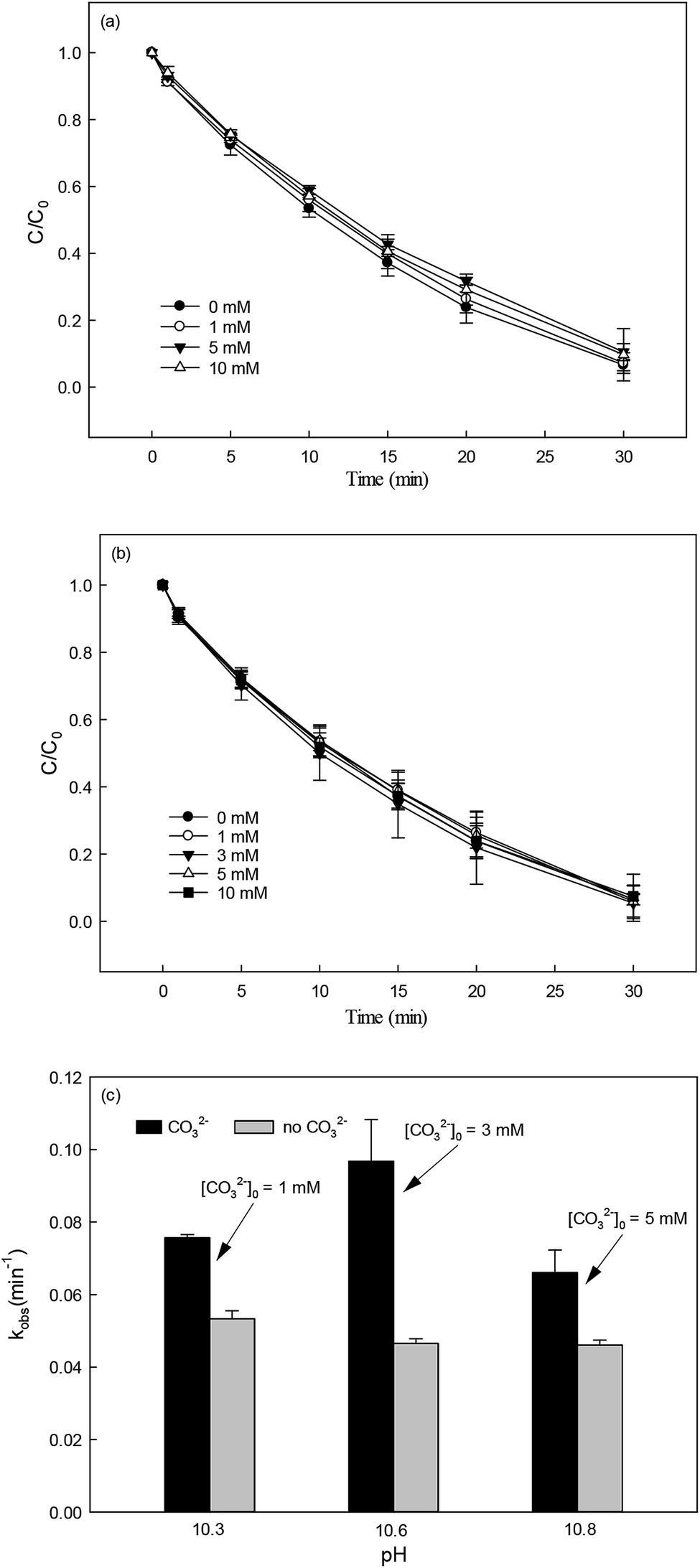
3.5.2 Effect of inorganic anions.
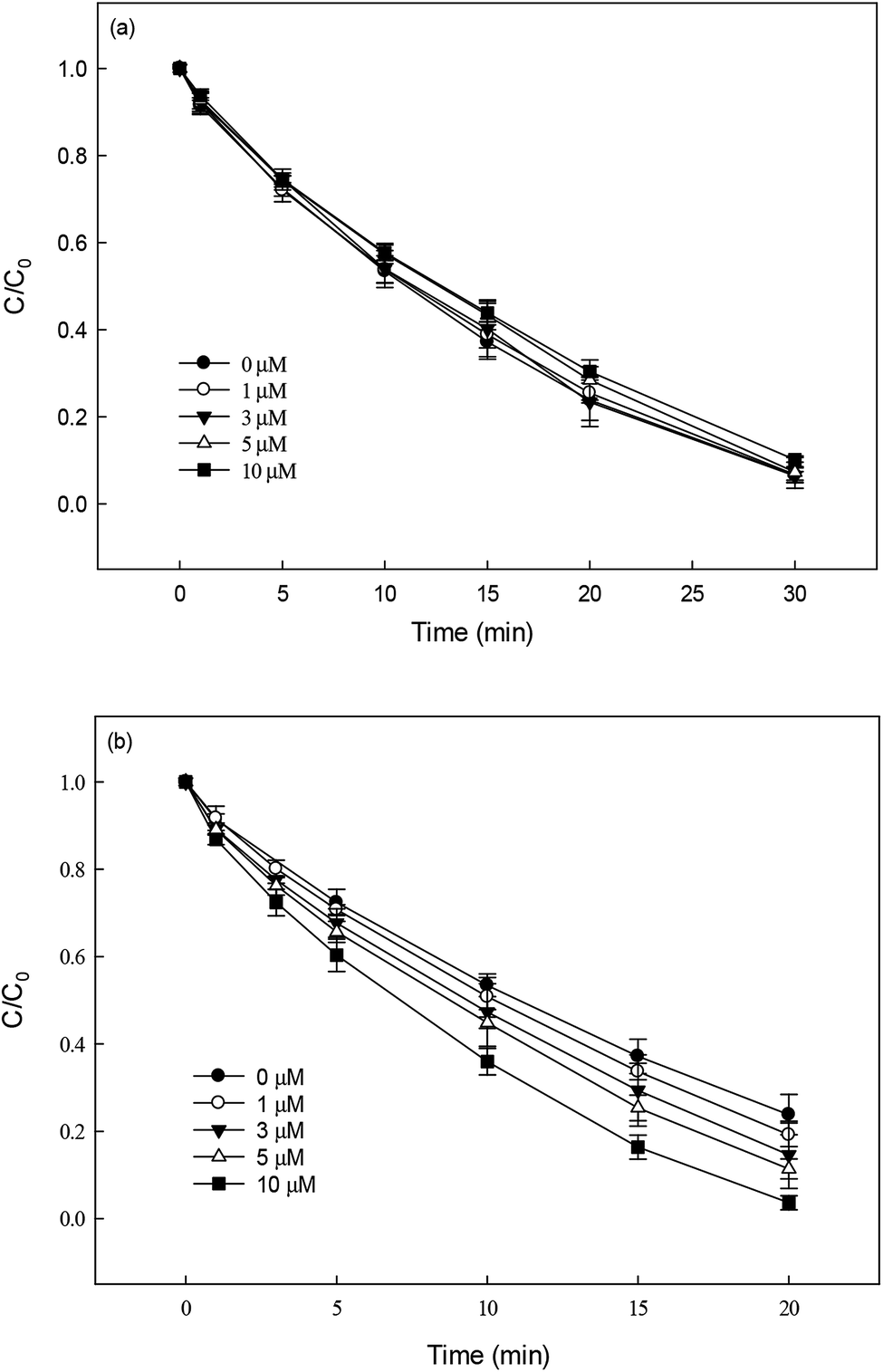
Nitrate (NO3−), sulfate (SO42−) and carbonate (CO32−) are common anions in natural water and may influence the degradation of target contaminant by SR-AOPs. Hence, their effect on DCF removal in HAP system was investigated, respectively. As shown in Fig. 6a and b, the degradation of DCF could hardly be influenced in the presence of various concentrations of NO3− and SO42−, because they can scarcely react with SO4˙−.19 CO32− is known to be an excellent scavenger for HO˙ and SO4˙−, which may inhibit the removal of organic pollutants in AOPs.18,39 However, an enhancement effect on DCF degradation by HAP was observed in our study, as presented in Fig. 6c. The presence of CO32− could change the pH of the solution, and reached 10.3, 10.6 and 10.8 when the concentration of carbonate was 1, 3 and 5 mM, respectively. To avoid the influence of pH on DCF removal, the degradation of DCF by HAP at these pH values was conducted as a control system. Compared with the respective control system, the removal of DCF in the presence of CO32− was obviously improved, which was probably attributed to the role of carbonate radical (CO3˙−) formed through the reactions of CO32− with SO4˙− and HO˙, as presented in eqn (9) and (10).40,41 Although the second-order rate constant of DCF with CO3˙− (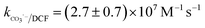 42) was lower than those with SO4˙− (
42) was lower than those with SO4˙− (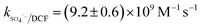 43) and HO˙ (kHO˙/DCF = (7.5 ± 1.5) × 109 M−1 s−111), its steady-state concentration might be higher in the reaction system due to its selectivity,44 resulting in a positive effect of CO32− on DCF degradation. Similar finding was also reported by Huang et al.42 in DCF degradation by UV/NO3− in the presence of HCO3−.
43) and HO˙ (kHO˙/DCF = (7.5 ± 1.5) × 109 M−1 s−111), its steady-state concentration might be higher in the reaction system due to its selectivity,44 resulting in a positive effect of CO32− on DCF degradation. Similar finding was also reported by Huang et al.42 in DCF degradation by UV/NO3− in the presence of HCO3−.| | | SO4˙− + CO32− → SO42− + CO3˙−k = (6.2 ± 0.4) × 106 M−1 s−1 | (9) |
| | | HO˙ + CO32− → OH− + CO3˙−k = 3.9 × 108 M−1 s−1 | (10) |
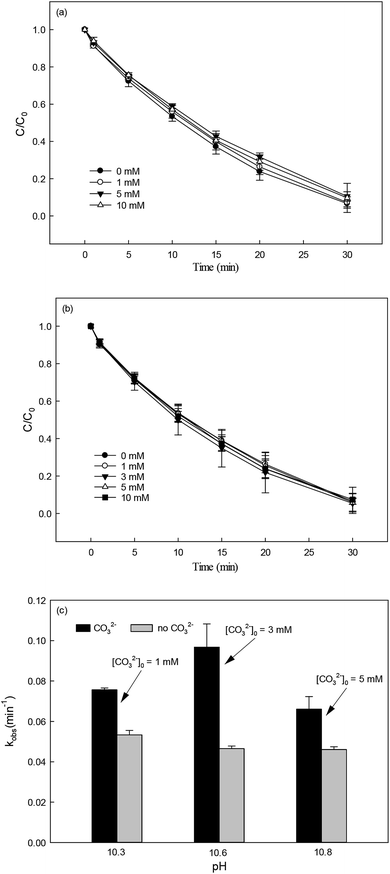 |
| | Fig. 6 Effect of NO3− (a), SO42− (b) and CO32− (c) on DCF degradation in HAP system. Experimental conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, 5 mM phosphate buffer at pH = 6.2 for NO3− and SO42− groups, no buffer for CO32− group. | |
3.5.3 Effect of NOM.
Natural organic matter is a common constituent in natural water. It can react with SO4˙− and HO˙ due to its electron-rich moieties, resulting in an inhibition effect on the removal of target contaminant.25,45,46 In this study, humic acid (HA) and fulvic acid (FA) were chosen to represent NOM for investigating their effect on DCF degradation in HAP system. As shown in Fig. 7a and b, the presence of HA and FA could both inhibit DCF degradation and the inhibition effect was more obvious with the increase of their concentrations. This was probably due to the competition of NOM with DCF for the reactive radicals. Compared with HA, the addition of FA showed a stronger inhibition on the degradation of DCF. However, it needs further investigation, because there is very limited available information on the reactivity of NOM toward SO4˙−.
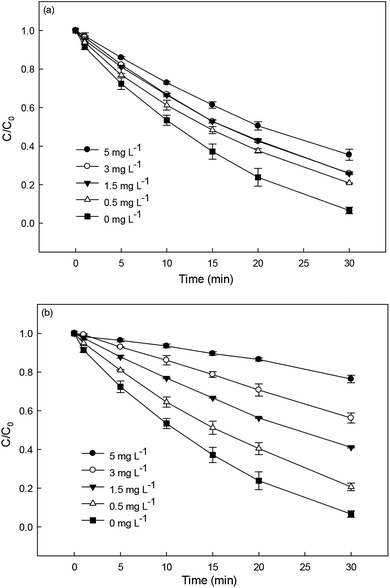 |
| | Fig. 7 Effect of HA (a) and FA (b) on DCF degradation in HAP system. Experimental conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, 5 mM phosphate buffer at pH = 6.2. | |
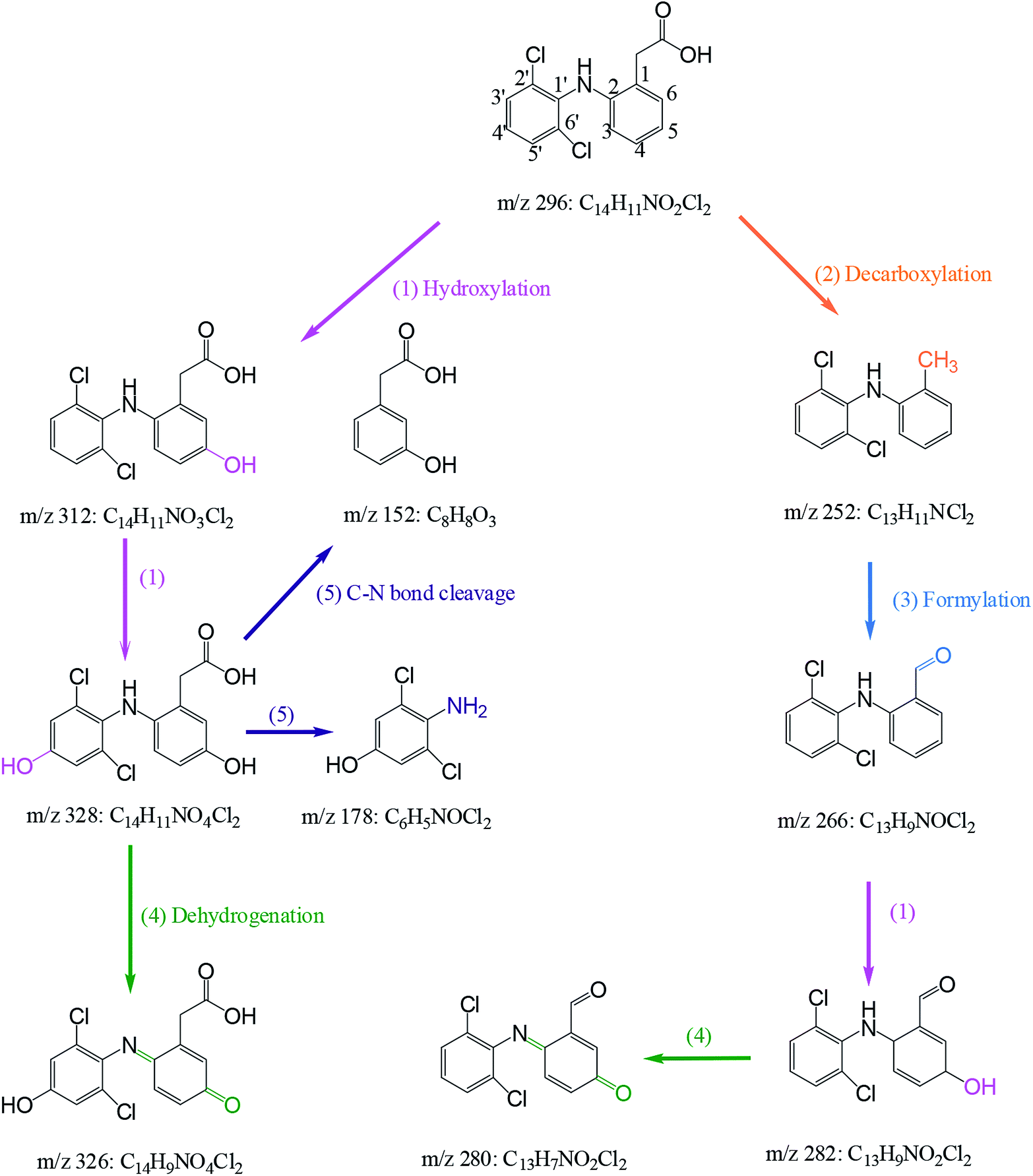
3.6 Degradation mechanism of DCF by HAP
Nine transformation products (TPs) were detected during the degradation of DCF by HAP in this study. Based on these identified TPs, the potential DCF degradation mechanism was proposed, exhibiting five different transformation pathways including hydroxylation, decarboxylation, formylation, dehydrogenation and C–N bone cleavage, as depicted in Scheme 1.
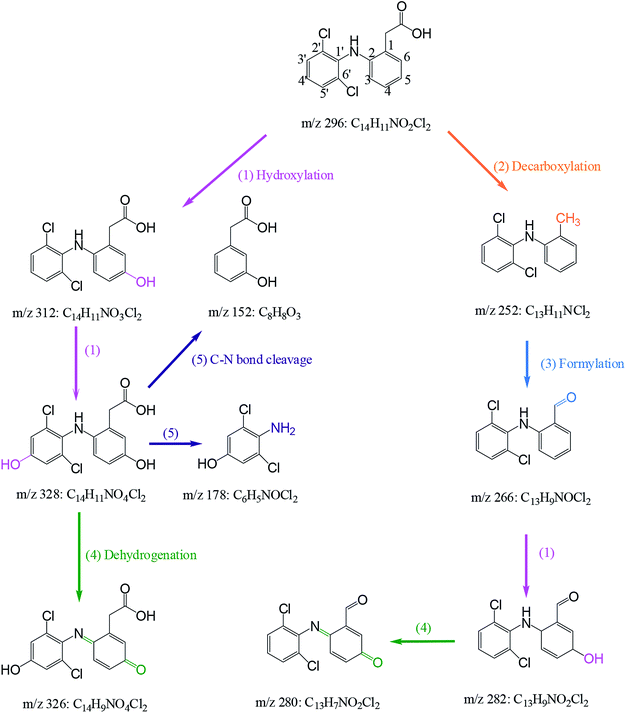 |
| | Scheme 1 Proposed degradation mechanism of DCF by HAP: (1) hydroxylation, (2) decarboxylation, (3) formylation (4) dehydrogenation and (5) C–N bond cleavage. Experiment conditions: [DCF]0 = 1 μM, [PS]0 = 50 μM, no buffer. | |
(i) Hydroxylation (pathway (1) in Scheme 1) was an important pathway where DCF could be hydroxylated by the reactive radicals in HAP system. It is believed that SO4˙− and HO˙ have the similar reaction mechanisms, including hydrogen abstraction, hydroxyl addition and electron transfer.32,47 In terms of DCF structure, aromatic ring might be a target site which was easily attacked by SO4˙− due to its electron-rich nature. Therefore, monohydroxylation product 5-hydroxy-DCF (m/z 312) and dihydroxylation product 5,4′-hydroxy-DCF (m/z 328) were produced.
(ii) Decarboxylation (pathway (2) in Scheme 1) meant an elimination of –COOH group from DCF structure. Phenylacetic acid structure on DCF might be attacked by SO4˙− resulting in the transfer of an electron from aromatic ring to sulfate radical, and subsequently a –COOH group could be removed from this structure via intramolecular electron transfer producing a decarboxylation product m/z 252 (2-(2′,6′-dichlorphenylamino)methylbenzene).48 This product was also detected by Zhao et al.49 and Liu et al.50 in DCF degradation using ferrate(VI) and photoelectrocatalytic system, respectively.
(iii) Formylation (pathway (3) in Scheme 1) might result from the oxidation of the methyl group in the product m/z 252 leading to the formation of the product m/z 266 (2-(2′,6′-dichlorophenylamino)benzaldehyde). The product m/z 282 might be further generated from the formed m/z 266 by hydroxylation.
(iv) Dehydrogenation (pathway (4) in Scheme 1) engaged in the oxidation of the –OH group at C5 and the –NH2 group at C2 in the formed hydroxylation products. Zhou et al.51 reported that –OH and –NH2 groups in the aromatic ring are two electron-donating groups, which increased the electron density in the ortho- and para-positions of aromatic ring and therefore attracted the attacks of SO4˙− and HO˙. As a result, two quinone imine products m/z 326 and 280 were generated from the products m/z 328 and 282 through dehydrogenation, respectively.
(v) C–N bond cleavage (pathway (5) in Scheme 1) occurred between two aromatic rings of the product m/z 328 by the attack of reactive radicals producing two products m/z 178 and 152. Similar transformation pathway was also reported by Chong et al.52 in the degradation of DCF using FeCeOx catalyzed H2O2.
4. Conclusions
This study systematically investigated the degradation of DCF under different experimental conditions by HAP. Compared to heat alone and PS alone systems, the degradation of DCF was significantly enhanced in HAP system at 70 °C and about 96% DCF was removed after 30 min. Its degradation in HAP system fitted with a pseudo-first-order kinetic model. The degradation rate of DCF decreased gradually with increasing pH and the highest kobs was obtained at pH 3 probably due to the formation of Caro's acid. The radical scavenging experiments suggested that except pH 3, SO4˙− was the dominant radical species at pH < 7; while HO˙ was mainly responsible for DCF degradation at high pH condition in HAP system. With the increase in the reaction temperature, the removal of DCF was improved, and the activation energy of this reaction was calculated to be 101.4 kJ mol−1. Increasing initial dosage of PS could enhance the elimination of DCF. Presence of Cu2+ and CO32− could improve DCF degradation, while an inhibition effect was observed in the presence of NOM. The other water constituents such as Fe3+, SO42− and NO3− could hardly influence DCF removal. Nine degradation products of DCF were detected and identified using UPLC-QTOF/MS. Hence, the probable DCF transformation mechanism was proposed showing five different reaction pathways, including hydroxylation, decarboxylation, formylation, dehydrogenation and C–N bond cleavage.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by Sichuan Science and Technology Programs (No. 2017SZ0175 and No. 2018SZDZX0026). Yiqing Liu is also thankful to the financial support from the Fundamental Research Funds for the Central Universities (No. 2682018CX32).
References
- A. Jelic, M. Gros, A. Ginebreda, R. Cespedes-Sánchez, F. Ventura, M. Petrovic and D. Barcelo, Water Res., 2011, 45, 1165–1176 CrossRef CAS.
- M. Carballa, F. Omil, J. M. Lema, M. Llompart, C. García-Jares, I. Rodríguez, M. Gómezc and T. Ternes, Water Res., 2004, 38, 2918–2926 CrossRef CAS.
- K. Fent, A. A. Weston and D. Caminada, Aquat. Toxicol., 2006, 76, 122–159 CrossRef CAS.
- N. M. Vieno, H. Harkki, T. Tuhkanen and L. Kronberg, Environ. Sci. Technol., 2007, 41, 5077–5084 CrossRef CAS.
- Y. Zhang, S. U. Geiβen and C. Gal, Chemosphere, 2008, 73, 1151–1161 CrossRef CAS.
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319–2364 CrossRef CAS.
- I. S. Ruhoya and C. G. Daughton, Environ. Int., 2008, 34, 1157–1169 CrossRef PubMed.
- J. L. Oaks, M. Gilbert, M. Z. Virani, R. T. Watson, C. U. Meteyer, B. A. Rideout, H. L. Shivaprasad, S. Ahmed, M. J. I. Chaudhry, M. Arshad, S. Mahmood, A. Ali and A. A. Khan, Nature, 2004, 427, 630–633 CrossRef CAS.
- J. Wang and S. Z. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- Y. Q. Liu, X. X. He, Y. S. Fu and D. D. Dionysiou, Chem. Eng. J., 2016, 284, 1317–1327 CrossRef CAS.
- M. M. Huber, S. Canonica, G. Y. Park and U. von Gunten, Environ. Sci. Technol., 2003, 37, 1016–1024 CrossRef CAS.
- D. Vogna, R. Marotta, A. Napolitano, R. Andreozzi and M. d’lschia, Water Res., 2004, 38, 414–422 CrossRef CAS.
- M. Ravina, L. Campanella and J. Kiwi, Water Res., 2002, 36, 3553–3560 CrossRef CAS.
- L. A. Pérez-Estrada, S. Malato, W. Gernjak, A. Agüera, E. M. Thurman, I. Ferrer and A. R. Fernández-Alba, Environ. Sci. Technol., 2005, 39, 8300–8306 CrossRef.
- A. Achilleos, E. Hapeshi, N. P. Xekoukoulotakis, D. Mantzavinos and D. Fatta-Kassinos, Chem. Eng. J., 2010, 161, 53–59 CrossRef CAS.
- P. Mazellier, C. Busset, A. Delmont and J. De Laat, Water Res., 2007, 41, 4585–4594 CrossRef CAS.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- C. Q. Tan, N. Y. Gao, Y. Deng, N. An and J. Deng, Chem. Eng. J., 2012, 203, 294–300 CrossRef CAS.
- J. Deng, Y. S. Shao, N. Y. Gao, Y. Deng, S. Q. Zhou and X. H. Hu, Chem. Eng. J., 2013, 228, 765–771 CrossRef CAS.
- M. Zhang, X. Q. Chen, H. Zhou, M. Murugananthan and Y. R. Zhang, Chem. Eng. J., 2015, 264, 39–47 CrossRef CAS.
- N. Zrinyi and A. L. T. Pham, Water Res., 2017, 120, 43–51 CrossRef CAS.
- H. P. Gao, J. B. Chen, Y. L. Zhang and X. F. Zhou, Chem. Eng. J., 2016, 306, 522–530 CrossRef CAS.
- J. B. Chen, Y. J. Qian, H. M. Liu and T. Y. Huang, Environ. Sci. Pollut. Res., 2016, 23, 3824–3833 CrossRef CAS.
- C. J. Liang, Z. S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS.
- Y. Fan, D. Y. Kong, J. H. Lu and Q. S. Zhou, J. Hazard. Mater., 2015, 300, 39–47 CrossRef CAS.
- M. Spiro, Electrochim. Acta, 1979, 24, 313–314 CrossRef CAS.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055–3059 CrossRef CAS.
- S. Y. Yang, P. Wang, X. Yang, L. Shan, W. Y. Zhang, X. T. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552–558 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- C. L. Clifton and R. E. Huie, Int. J. Chem. Kinet., 1989, 21, 677–687 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- Y. Q. Liu, X. X. He, Y. S. Fu and D. D. Dionysiou, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS.
- C. Q. Tan, N. Y. Gao, Y. Deng, W. L. Rong, S. D. Zhou and N. X. Lu, Sep. Purif. Technol., 2013, 109, 122–128 CrossRef CAS.
- Y. F. Ji, C. X. Dong, D. Y. Kong, J. H. Lu and Q. S. Zhou, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2012, 197, 483–492 CrossRef CAS.
- R. H. Waldemer, P. G. Tratnyek, R. L. Johnson and J. T. Nurmi, Environ. Sci. Technol., 2007, 41, 1010–1015 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- C. S. Liu, K. Shih, C. X. Sun and F. Wang, Sci. Total Environ., 2012, 416, 507–512 CrossRef CAS.
- J. Deng, Y. S. Shao, N. Y. Gao, S. J. Xia, C. Q. Tan, S. Q. Zhou and X. H. Hu, Chem. Eng. J., 2013, 222, 150–158 CrossRef CAS.
- Y. Q. Liu, X. X. He, X. D. Duan, Y. S. Fu and D. D. Dionysiou, Chem. Eng. J., 2015, 276, 113–121 CrossRef CAS.
- Y. Q. Liu, X. X. He, X. D. Duan, Y. S. Fu, D. Fatta-Kassinos and D. D. Dionysiou, Water Res., 2016, 95, 195–204 CrossRef CAS.
- Y. Huang, M. B. Kong, D. Westerman, E. G. Xu, S. Coffin, K. H. Cochran, Y. Q. Liu, S. D. Richardson, D. Schlenk and D. D. Dionysiou, Environ. Sci. Technol., 2018, 52, 12697–12707 CrossRef CAS.
- M. M. Ahmed, S. Barbati, P. Doumenq and S. Chiron, Chem. Eng. J., 2012, 197, 440–447 CrossRef.
- J. P. Huang and S. A. Mabury, Environ. Toxicol. Chem., 2009, 19, 2181–2188 CrossRef.
- P. Westerhoff, S. P. Mezyk, W. J. Cooper and D. Minakata, Environ. Sci. Technol., 2007, 41, 4640–4646 CrossRef CAS.
- P. M. D. Gara, G. N. Bosio, M. C. Gonzalez and D. O. Martire, Int. J. Chem. Kinet., 2008, 40, 19–24 CrossRef CAS.
- X. X. He, A. A. de la Cruz, K. E. O’shea and D. D. Dionysiou, Water Res., 2014, 63, 168–178 CrossRef CAS.
- V. Madhavan, H. Levanon and P. Neta, Radiat. Res., 1978, 76, 15–22 CrossRef CAS.
- J. F. Zhao, Y. Q. Liu, Q. Wang, Y. S. Fu, X. H. Lu and X. F. Bai, Sep. Purif. Technol., 2018, 192, 412–418 CrossRef CAS.
- S. S. Liu, X. Zhao, H. B. Zeng, Y. B. Wang, M. Qiao and W. Guan, Chem. Eng. J., 2017, 320, 168–177 CrossRef CAS.
- Z. W. Zhou and J. Q. Jiang, J. Pharm. Biomed. Anal., 2015, 106, 37–45 CrossRef CAS.
- S. Chong, G. M. Zhang, N. Zhang, Y. Liu, T. Huang and H. Chang, J. Hazard. Mater., 2017, 334, 150–159 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yongsheng
Fu
,
Hongbin
Wang
and
Peng
Wu
*,
Yongsheng
Fu
,
Hongbin
Wang
and
Peng
Wu
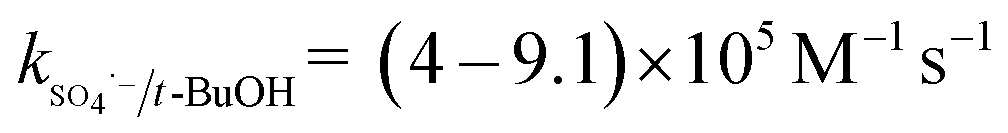 , kHO˙/t-BuOH = (4.2–7.6) × 108 M−1 s−1) and isopropanol (i-PrOH,
, kHO˙/t-BuOH = (4.2–7.6) × 108 M−1 s−1) and isopropanol (i-PrOH, 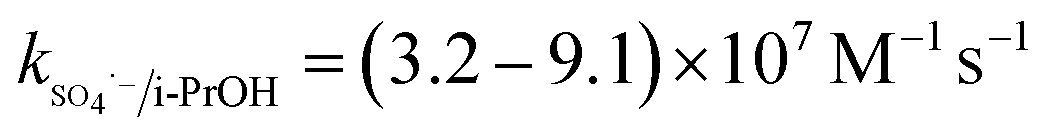 , kHO˙/i-PrOH = (1.6–2.3) × 109 M−1 s−1), were added into this system, respectively.29–31 As shown in Fig. 2, due to the scavenging of t-BuOH for HO˙, the degradation of DCF at pH 3 was inhibited significantly with its addition. While the inhibition effect was more obvious in the presence of i-PrOH, because it could also react with SO4˙− except quenching HO˙. This radical scavenging experiment indicated that SO4˙− and HO˙ were both present and responsible for DCF removal by HAP at pH 3 and their contribution to DCF degradation were 54% and 46%, respectively.
, kHO˙/i-PrOH = (1.6–2.3) × 109 M−1 s−1), were added into this system, respectively.29–31 As shown in Fig. 2, due to the scavenging of t-BuOH for HO˙, the degradation of DCF at pH 3 was inhibited significantly with its addition. While the inhibition effect was more obvious in the presence of i-PrOH, because it could also react with SO4˙− except quenching HO˙. This radical scavenging experiment indicated that SO4˙− and HO˙ were both present and responsible for DCF removal by HAP at pH 3 and their contribution to DCF degradation were 54% and 46%, respectively.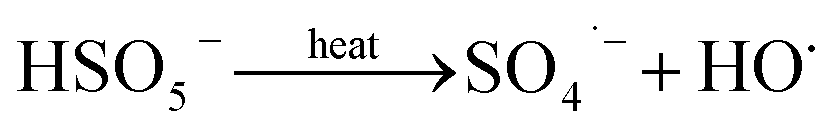
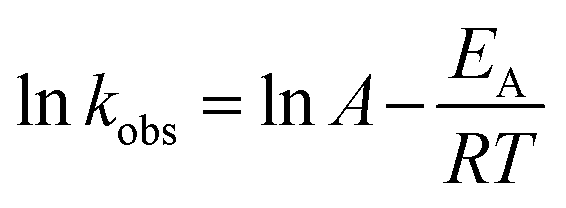
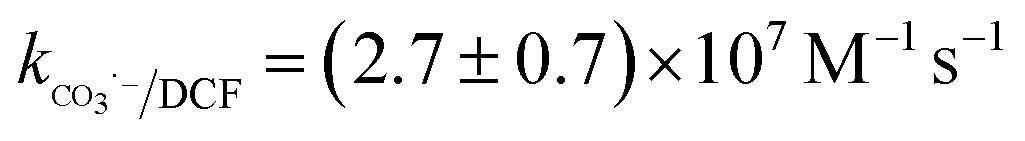 42) was lower than those with SO4˙− (
42) was lower than those with SO4˙− (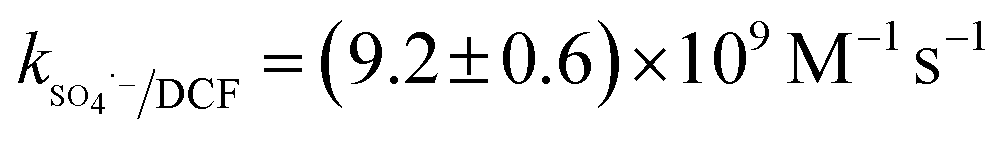 43) and HO˙ (kHO˙/DCF = (7.5 ± 1.5) × 109 M−1 s−111), its steady-state concentration might be higher in the reaction system due to its selectivity,44 resulting in a positive effect of CO32− on DCF degradation. Similar finding was also reported by Huang et al.42 in DCF degradation by UV/NO3− in the presence of HCO3−.
43) and HO˙ (kHO˙/DCF = (7.5 ± 1.5) × 109 M−1 s−111), its steady-state concentration might be higher in the reaction system due to its selectivity,44 resulting in a positive effect of CO32− on DCF degradation. Similar finding was also reported by Huang et al.42 in DCF degradation by UV/NO3− in the presence of HCO3−.