
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile one-step hydrothermal synthesis of α-Fe2O3/g-C3N4 composites for the synergistic adsorption and photodegradation of dyes†
Tao Wangab,
Manqi Huangab,
Xiawei Liuab,
Zhen Zhangc,
Yonghong Liuc,
Wei Tanga,
Shaopan Baoa and
Tao Fang *ab
*ab
aInstitute of Hydrobiology, Chinese Academy of Sciences, 7 Donghu South Road, Wuchang District, Wuhan 430072, China. E-mail: fangt@ihb.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Science, Huazhong Agricultural University, Wuhan 430070, China
First published on 17th September 2019
Abstract
With the expansion of industrialization, dye pollution has become a significant hazard to humans and aquatic ecosystems. In this study, α-Fe2O3/g-C3N4-R (where R is the relative percentage of α-Fe2O3) composites were fabricated by a one-step method. The as-prepared α-Fe2O3/g-C3N4-0.5 composites showed excellent adsorption capacities for methyl orange (MO, 69.91 mg g−1) and methylene blue (MB, 29.46 mg g−1), surpassing those of g-C3N4 and many other materials. Moreover, the ionic strength and initial pH influenced the adsorption process. Relatively, the adsorption isotherms best fitted the Freundlich model, and the pseudo-second-order kinetic model could accurately describe the kinetics for the adsorption of MO and MB by α-Fe2O3/g-C3N4-0.5. Electrostatic interaction and π–π electron donor–acceptor interaction were the major mechanisms for MO/MB adsorption. In addition, the photocatalytic experiment results showed that more than 79% of the added MO/MB was removed within 150 min. The experimental results of free-radical capture revealed that holes (h+) were the major reaction species for the photodegradation of MO, whereas MB was reduced by the synergistic effect of hydroxyl radicals (·OH) and holes (h+). This study suggests that the α-Fe2O3/g-C3N4 composites have an application potential for the removal of dyes from wastewater.
1. Introduction
With the rapid development of industrialization, dyes are being extensively used in the textile, paper and leather industries.1 Among them, methyl orange (MO) and methylene blue (MB) are the two most commonly used dyes.2 The intense use of MO/MB has inevitably increased their concentrations in environmental compartments including the aquatic environment. However, as reported, MO/MB in water is nonbiodegradable, neurotoxic and environmental persistent.3,4 As a result, the increased use of MO/MB has led to increasing concern related to their toxicity, and thus, it is a critical and urgent task to achieve the fast and highly efficient removal of dyes from wastewater.To date, many technologies, including adsorption, chemical oxidation, ion exchange, electrochemical degradation, and photocatalytic degradation, have been applied to remove dyes from aqueous solutions.5 Among these technologies, adsorption has generally been regarded as the most conventional method for removing dyes from aquatic ecosystems.6 The adsorbents used to remove dyes from wastewater mainly include activated carbon, natural minerals (bentonite, montmorillonite, etc.) and nanomaterials (graphene and carbon nanotubes).7 In addition to adsorption, photocatalytic reduction, as a new treatment, has attracted wide attention because of its high safety level and simple operation. For example, Lui et al.8 prepared graphene-wrapped hierarchical TiO2 nanoflower composites and found that the novel material showed excellent photocatalytic performances for the removal of MB. In recent years, coupled with photocatalytic degradation technology, adsorption has increasingly been used because it further guarantees the high removal efficiency of target dyes. Unfortunately, many materials that exist today cannot have both excellent adsorption performance and photocatalytic performance.9 Thus, it is essential to fabricate novel materials that present strong adsorption capacity, fast adsorption rate, and high photocatalytic degradation.
In recent years, two-dimensional (2D) materials have attracted extensive attention in adsorption and photocatalysis owing to their unique features, such as outstanding chemical stability, high surface area, and excellent visible light activity.10 Accordingly, 2D materials have been used as both adsorbents and catalysts to remove organic pollutants from wastewater. For example, Xiao et al.11 fabricated nitrogen-rich graphene-like carbon sheet composites, which not only showed high adsorption toward bisphenol A, but also exhibited excellent catalytic properties for bisphenol A. Among the 2D nanomaterials, g-C3N4 has gained immense scientific attention owing to its special physicochemical properties, such as thermal stability, low friction coefficient, rich functional groups, and metal-free elements, as well as nontoxicity.12 As a tri-s-triazine unit π-conjugated material, g-C3N4 has been used in many fields such as adsorption, CO2 capture, metal-free catalysts, fuel cells and photocatalysis.13 However, due to its single surface functional group and relatively small surface area, pure g-C3N4 exhibits a low adsorption performance for target dyes.14,15 Meanwhile, its relatively poor quantum efficiency and electron–hole recombination also limit its application in the field of photocatalysis.16 Therefore, increasing research has focused on the modification of pure g-C3N4 to improve the removal efficiency of target dyes, including stripping, semiconductor recombination, controlling its morphology, and element doping.17–19 α-Fe2O3 nanostructures have been widely used in environmental remediation in recent years due to their unique properties and development prospects.20 α-Fe2O3 nanostructures not only exhibit good performances in degrading organic pollutants, but also have effective adsorption capacity for organic dyes and heavy metals.21 α-Fe2O3/g-C3N4 composites not only increase the surface charge density of g-C3N4, but also improve its photocatalytic activity.
Herein, we prepared α-Fe2O3/g-C3N4-R composites (where R is the relative percentage of α-Fe2O3) through a one-step hydrothermal method and applied them as both adsorbents and catalysts for the removal of MO and MB from aqueous solution. The influence of different factors including adsorption time, temperature, initial pH, and ion strength on the uptake of MO and MB by the α-Fe2O3/g-C3N4 composites was systematically explored via batch adsorption experiments. The mechanisms for the adsorption of MO/MB on the as-prepared sample were analyzed by combining the adsorption experiment data and XPS characterization results. Then, the catalytic properties of the α-Fe2O3/g-C3N4 composites were assessed via photocatalytic experiments employing MO and MB. Finally, a series of experiments with different capture agents was performed to explore the possible catalytic reaction mechanisms. This study not only provides new perspectives for the removal of dyes, but also reveals the great application potential of the α-Fe2O3/g-C3N4 composites for environmental pollution control.
2. Experiment methods
2.1 Chemicals and materials
In this work, all reagents were of analytical grade as used as received without further purification. FeCl3·6H2O, H2NCONH2, CH3COONa, NaOH, HCl, C2H5OH, methyl orange (MO), methylene blue (MB), isopropanol (IPA), ethylenediaminetetraacetic acid (EDTA), and benzoquinone (BQ) were acquired from Sinopharm Chemical Reagent Co., Ltd. Superoxide dismutase (SOD) was obtained from Aladdin Chemical Reagent Co., Ltd.2.2 Synthesis of the composites
g-C3N4 was prepared using the method reported by Ma.22 Briefly, about 10 g of urea was placed in a 30 mL crucible and heated at 853 K for 180 min at a heating rate of 5 °C min−1, then cooled naturally to room temperature.The α-Fe2O3/g-C3N4 composites were obtained via a hydrothermal method. Typically, 200 mg of g-C3N4 was dissolved in 40 mL absolute ethyl alcohol and sonicated for 1 h. Then, 2.8 mL deionized water, 10 mg CH3COONa, and 3.4 mg FeCl3·6H2O were added and uniformly stirred for 20 min. Subsequently, the mixture was transferred to a 100 mL capacity Teflon-lined stainless autoclave and kept at 453 K for 10 h. The product was washed with alcohol and deionized water five times and freeze-dried. The final product was called α-Fe2O3/g-C3N4-0.5 (where 0.5 is the relative percentage of α-Fe2O3). In the present study, we changed the mass of FeCl3·6H2O to obtain materials with different relative percentages of α-Fe2O3 to explore the effect of α-Fe2O3 content on the properties of the composite.
2.3 Characterization
The morphologies and microstructure of the as-prepared samples were examined using transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HTEM), elemental mapping, and EDS (JEOL JEM-2010F). The textural properties of the as-prepared samples were tested by N2 adsorption–desorption at 77 K after degassing at 200 °C for 6 h using a surface area analyzer (Micromeritics Tristar ASAP 2020). FTIR spectra of the as-prepared samples (FTIR, Thermo Scientific, USA) were recorded from 4000 to 400 cm−1. X-ray diffraction (XRD) patterns were obtained on a Bruker D8 Advance X-ray diffractometer with CuK radiation (λ = 1.541781 Å). Zeta potential was measured utilizing a Zetasizer 3000 HSA system. X-ray photoelectron spectroscopy (XPS) data was obtained on a Thermo Scientific ESCALAB 250Xi VG. All the XPS tests were calibrated in reference to the C 1s peak (284.8 eV).2.4 Batch adsorption experiments
The adsorption of MO and MB by α-Fe2O3/g-C3N4-R was performed through a batch method. 5 mg α-Fe2O3/g-C3N4-R and 20 mL MO and MB solutions were transferred to a 50 mL polyethylene centrifuge tube. In addition, to explore the effect of pH on the adsorption of MO and MB by α-Fe2O3/g-C3N4-R, the initial pH of all the MO and MB solutions was adjusted to 3.0 ± 0.1 and 7.0 ± 0.1, respectively. The mixture was then shaken at 150 rpm in the dark. The pH of the solution was adjusted with a negligible volume of 0.1–1.0 mol L−1 NaOH or HCl solution. In the adsorption kinetic studies, α-Fe2O3/g-C3N4-0.5 was added to MO (50 mg L−1) and MB (18 mg L−1) solutions at different intervals (ranging from 5 to 720 min). For the adsorption isotherm and thermodynamic studies, the resulting series of α-Fe2O3/g-C3N4-R was added to MO and MB solutions with different concentrations (ranging from 1 to 60 mg L−1) at three different temperatures (298 K, 308 K, and 318 K) for 360 min. The study of the effect of ion strength on the MO and MB adsorption was executed in different concentrations of NaCl (0.001 mol L−1, 0.01 mol L−1, and 0.1 mol L−1). The recycling performance of the composite was also determined. After the adsorption, the desorption of MO/MB adsorbed on α-Fe2O3/g-C3N4-0.5 was achieved with 0.5 M NaOH solution and methanol solution, respectively. Then, α-Fe2O3/g-C3N4-0.5 was dried at 60 °C and reused. All the suspensions were filtered using a 0.45 μm filter membrane. The concentrations of MO and MB were determined using a UV-Vis spectrophotometer (UV-2450, Japan) at the wavelength of 464 and 664 nm, respectively. The α-Fe2O3/g-C3N4-0.5 sample after MO and MB adsorption for characterization was collected by centrifugation and dried at 353 K for 12 h and then characterized by XPS to examine its adsorption mechanisms. Three replicates were conducted in the adsorption experiments and the experiment data were averaged.The adsorption quantity of α-Fe2O3/g-C3N4-R, qe (mg g−1), was calculated using the following equation:
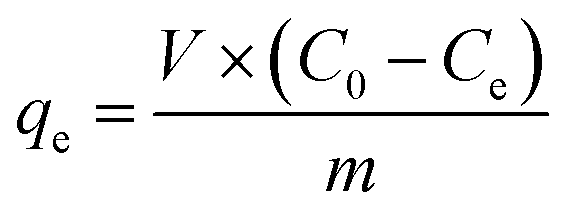 | (1) |
2.5 Photocatalytic degradation experiments
Consistent with the adsorption experiment, MO and MB were applied to study the photocatalytic degradation property of α-Fe2O3/g-C3N4-0.5. The photocatalyst (15 mg) was dispersed in 60 mL MO (50 mg L−1) and MB (18 mg L−1) solutions and their initial pH was adjusted to 3.0 ± 0.1 and 7.0 ± 0.1, respectively. Then, they were stirred in dark for 60 min to achieve adsorption/desorption equilibrium. Finally, the mixed suspension was exposed to a 300 W Xe lamp with a light filter (≥420 nm). At given time intervals, a mixed suspension of 5 mL was taken and filtered using a 0.45 μm filter membrane. To study the catalytic mechanism, three scavengers (IPA, TEOA, and SOD/BQ) were added into the solution. In the cycle experiments, the photocatalyst was washed with 0.5 mol L−1 NaOH and methanol solution, respectively. Then the product was dried at 353 K for 12 h. The concentrations of MO/MB were obtained using a UV-Vis spectrophotometer (UV-2450, Japan) at the wavelength of 464 and 664 nm, respectively.3. Results and discussion
3.1 Characterization of the as-prepared samples
The crystal structures of the as-prepared samples were identified by XRD. As shown in Fig. 1a, g-C3N4 has two main peaks located at 2θ = 13.1° and 27.4°, which can be indexed to the (100) and (002) diffraction planes of g-C3N4 (JCPDS 87-1526), respectively.23 The strong peak (2θ = 27.4°) and the relatively weak peak (2θ = 13.1°) corresponded to the interlayer stacking peak of conjugated aromatic systems and tri-s-triazine units, respectively.24 For α-Fe2O3/g-C3N4-R, the intensity of the diffraction peaks of the α-Fe2O3 species increased with an increase in the α-Fe2O3 content. The diffraction peaks at 33.2°, 35.5°, and 54.1° correspond to the (104), (110) and (116) planes of α-Fe2O3 (JCPDS 33-0664).25 These results demonstrate the existence of α-Fe2O3 and g-C3N4 components in the α-Fe2O3/g-C3N4-R composite. In addition, the FT-IR spectra of the as-prepared samples are shown in Fig. 1b. For g-C3N4, the series of peaks between 1200 and 1650 cm−1 is characteristic of s-triazine derivatives. Among them, the four strong peaks at 1407, 1462, 1570, and 1633 cm−1 are attributed to the stretching vibration of heptazine-derived rings.26 The adsorption bands at 1240 and 1317 cm−1 are ascribed to the secondary (2C–N) and tertiary (3C–N) amine fragments, respectively. The band at 808 cm−1 is a typical peak, corresponding to the heptazine ring of g-C3N4.27 Compared to g-C3N4, the new band at 541 cm−1 for α-Fe2O3/g-C3N4-1 is related to the stretching vibration of the Fe–O bond.28 These results confirm the existence of α-Fe2O3 and g-C3N4 in the as-prepared samples. The N2 adsorption–desorption isotherms and pore size distributions of the as-prepared samples are presented in Fig. S1† and the structural parameters of the adsorbents are summarized in Table S1.† The N2 adsorption–desorption isotherms were type IV with a distinct hysteresis loop at P/P0 above 0.4, implying the existence of a mesoporous structure in all the samples.29 In addition, the narrow pore size distribution for all the samples suggests that all the samples have strong adsorption capacity. As shown in Table S1,† the BET surface area of pure g-C3N4 is 69.6 m2 g−1, which is greater than that of α-Fe2O3/g-C3N4-0.25 (55.0 m2 g−1), α-Fe2O3/g-C3N4-0.5 (52.2 m2 g−1), and α-Fe2O3/g-C3N4-1 (45.4 m2 g−1). This is mainly ascribed to the relatively low BET surface area of α-Fe2O3.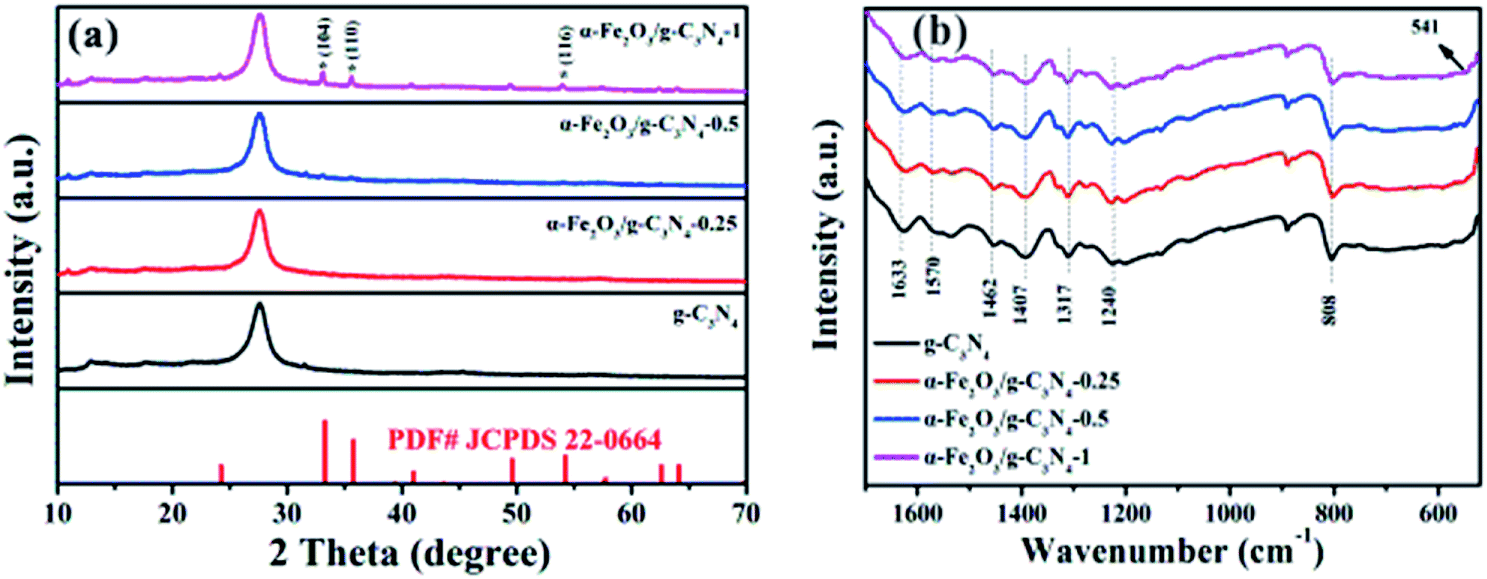 | ||
| Fig. 1 XRD patterns (a) and FTIR spectra (b) of the as-prepared samples. | ||
To further confirm the existence of α-Fe2O3, the microstructure and morphological characteristics of the as-prepared samples were examined by TEM, HRTEM, EDS, and element mapping. The TEM image of the as-prepared g-C3N4 sample shows a wrinkle in two-dimensional structure (Fig. 2a). As observed in Fig. 2b, the TEM images of α-Fe2O3/g-C3N4-0.5 display many nanometer particles well-dispersed on the surface of g-C3N4. The HRTEM image of α-Fe2O3/g-C3N4-0.5 (Fig. 2c) demonstrates the lattice fringe spacings of 0.333 nm, 0.274 nm and 0.252 nm, corresponding to the (002) crystal plane of g-C3N4 and the (104) and (110) crystal planes of α-Fe2O3, respectively25 (Xiao et al., 2015). Besides, the EDS and mapping in Fig. S2a and d† also indicate that α-Fe2O3/g-C3N4-0.5 contains C, H, O, Fe, and N elements. These results further demonstrate that α-Fe2O3 was successfully coupled with g-C3N4.
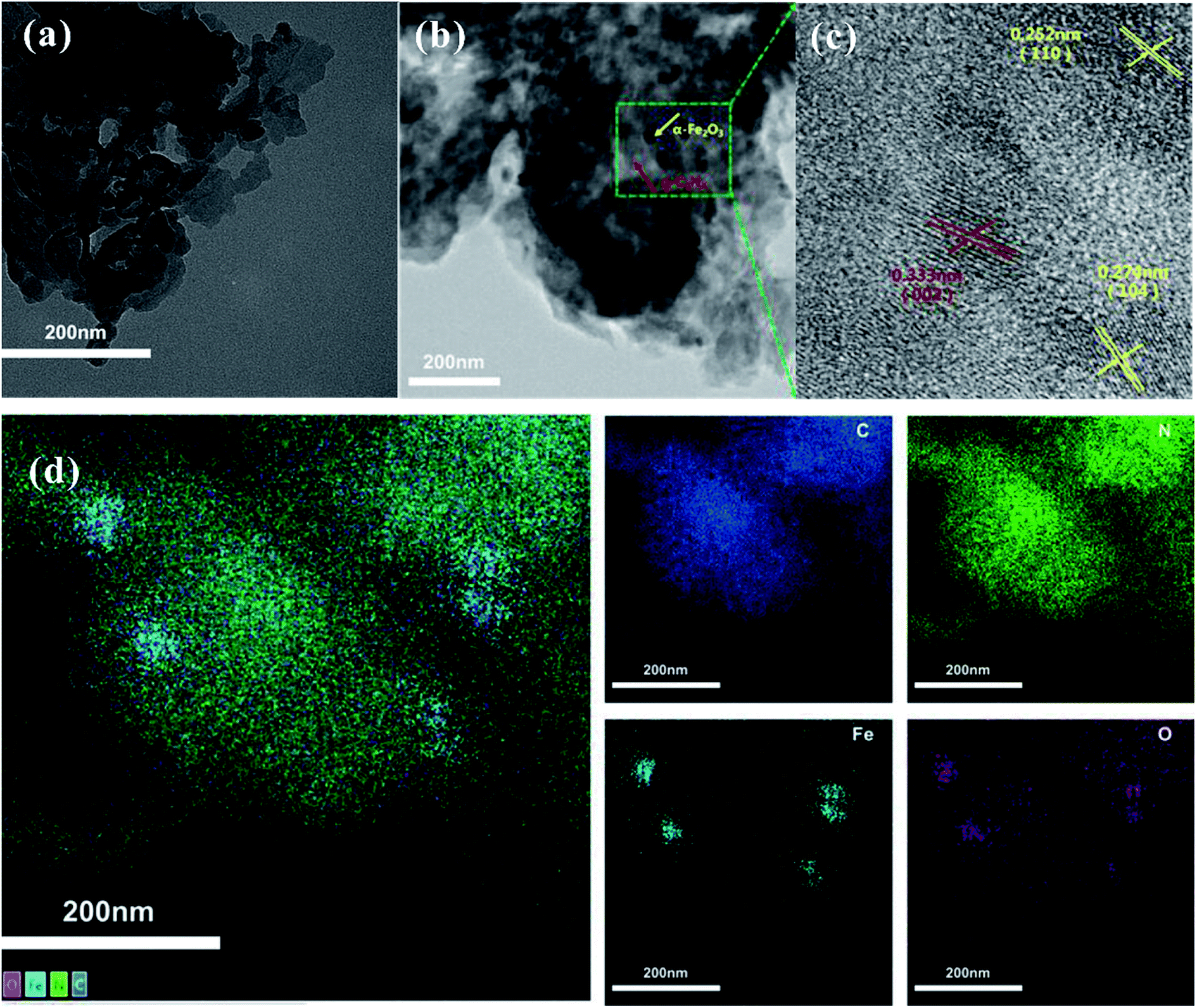 | ||
| Fig. 2 TEM images of g-C3N4 (a) and α-Fe2O3/g-C3N4-0.5 (b). HRTEM images of α-Fe2O3/g-C3N4-0.5 (c). Elemental mapping images of the different elements in α-Fe2O3/g-C3N4-0.5 (d). | ||
3.2 Adsorption isotherms of MO and MB on the as-prepared samples
To assess the adsorption properties of the as-prepared samples, MO and MB were selected as target pollutants to test the removal performance. The Langmuir and Freundlich isotherm models were used to fit the experimental data of the adsorption isotherms (Text S1†).The experimental data and fitting plot of the Langmuir and Freundlich isotherm models for MO and MB removal by the as-prepared samples are presented in Fig. 3, and the corresponding parameters are shown in Table 1. Compared with the two models, the Freundlich model was superior to the Langmuir model and it gave a better simulation to the experimental data, suggesting that the adsorption on the samples was multilayer adsorption.30 The values of the Freundlich constants, n, were found to be higher than 1 for all the samples at 25 ± 0.5 °C, confirming that the adsorption process of the as-prepared samples was favorable. According to the Langmuir adsorption simulation, the maximum adsorption capacity of MO on α-Fe2O3/g-C3N4-0.5 was 69.91 mg g−1, which is higher than that of MO on g-C3N4 (34.61 mg g−1), α-Fe2O3/g-C3N4-0.25 (69.24 mg g−1), and α-Fe2O3/g-C3N4-1 (67.92 mg g−1). Similar results (Table 1) were also obtained for the adsorption of MB on all the samples, which possessed impressive capacities of 9.66, 27.40, 29.46, and 26.56 mg g−1 for MB on g-C3N4, α-Fe2O3/g-C3N4-0.25, α-Fe2O3/g-C3N4-0.5, and α-Fe2O3/g-C3N4-1, respectively. Thus, the above results indicate that α-Fe2O3/g-C3N4-R has better adsorption properties for MO and MB than g-C3N4. In addition, the relatively poor adsorption performance of α-Fe2O3/g-C3N4-0.25 and α-Fe2O3/g-C3N4-1 may be ascribed to their relatively weak charge density and low BET surface area. Furthermore, the obtained maximum adsorption capacity of MO and MB on α-Fe2O3/g-C3N4-R was higher than that of the other reported materials (Table S2†). Therefore, α-Fe2O3/g-C3N4-R possesses excellent adsorption potential for dye pollution removal in real applications.
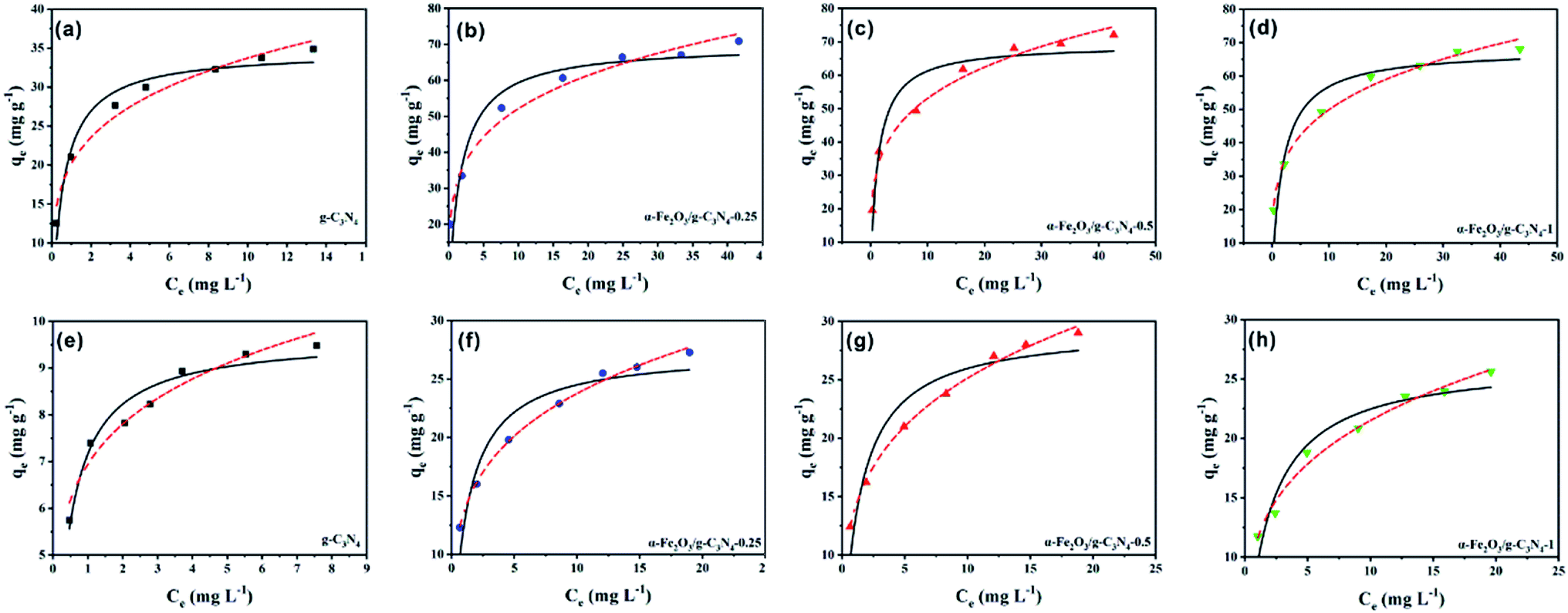 | ||
| Fig. 3 Adsorption isotherms of MO (a–d) and MB (e–h) on the as-prepared samples. pHMO = 3.0 ± 0.1, pHMB = 8.0 ± 0.1, m/v = 0.25 g L−1, and T = 298 ± 1 K. | ||
| Species | Sample | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|---|
| qmax (mg g−1) | b (L mg−1) | R2 | Kf (mg1−n Ln g−1) | n | R2 | ||
| MO | g-C3N4 | 34.61 | 1.752 | 0.960 | 20.17 | 4.48 | 0.966 |
| α-Fe2O3/g-C3N4-0.25 | 69.24 | 0.652 | 0.909 | 30.32 | 4.25 | 0.981 | |
| α-Fe2O3/g-C3N4-0.5 | 69.91 | 0.768 | 0.904 | 30.97 | 4.27 | 0.977 | |
| α-Fe2O3/g-C3N4-1 | 67.92 | 0.512 | 0.891 | 28.68 | 4.15 | 0.987 | |
| MB | g-C3N4 | 9.66 | 2.876 | 0.943 | 6.94 | 5.95 | 0.949 |
| α-Fe2O3/g-C3N4-0.25 | 27.40 | 0.846 | 0.901 | 13.68 | 4.17 | 0.996 | |
| α-Fe2O3/g-C3N4-0.5 | 29.46 | 0.744 | 0.899 | 13.84 | 3.86 | 0.996 | |
| α-Fe2O3/g-C3N4-1 | 26.56 | 0.548 | 0.924 | 11.56 | 3.71 | 0.983 | |
3.3 The effect of contact time and adsorption kinetics of MO and MB on α-Fe2O3/g-C3N4-0.5
The effects of contact time of MO and MB on α-Fe2O3/g-C3N4-0.5 are illustrated in Fig. 4a. The adsorption capacities of MO and MB on the as-prepared sample increased rapidly in the first 5 min and reached about 65% of the total sorption capacity. The main reason for this is that the higher driving force in the initial stage prompted MO and MB to transfer rapidly to the active sites of α-Fe2O3/g-C3N4-0.5. Then, the sorption rate increased slowly until equilibrium was achieved after 5 min. This was mainly caused by the reduction of vacant sites on α-Fe2O3/g-C3N4-0.5 and the repulsion between solute molecules on the solid surfaces and the solute molecules in the bulk phase.31 Compared with other adsorbents, the material prepared in this experiment is noteworthy since its sorption capacity in the first 5 min reached 65% of its total sorption capacity, which indicates that α-Fe2O3/g-C3N4-0.5 is an ideal adsorbent.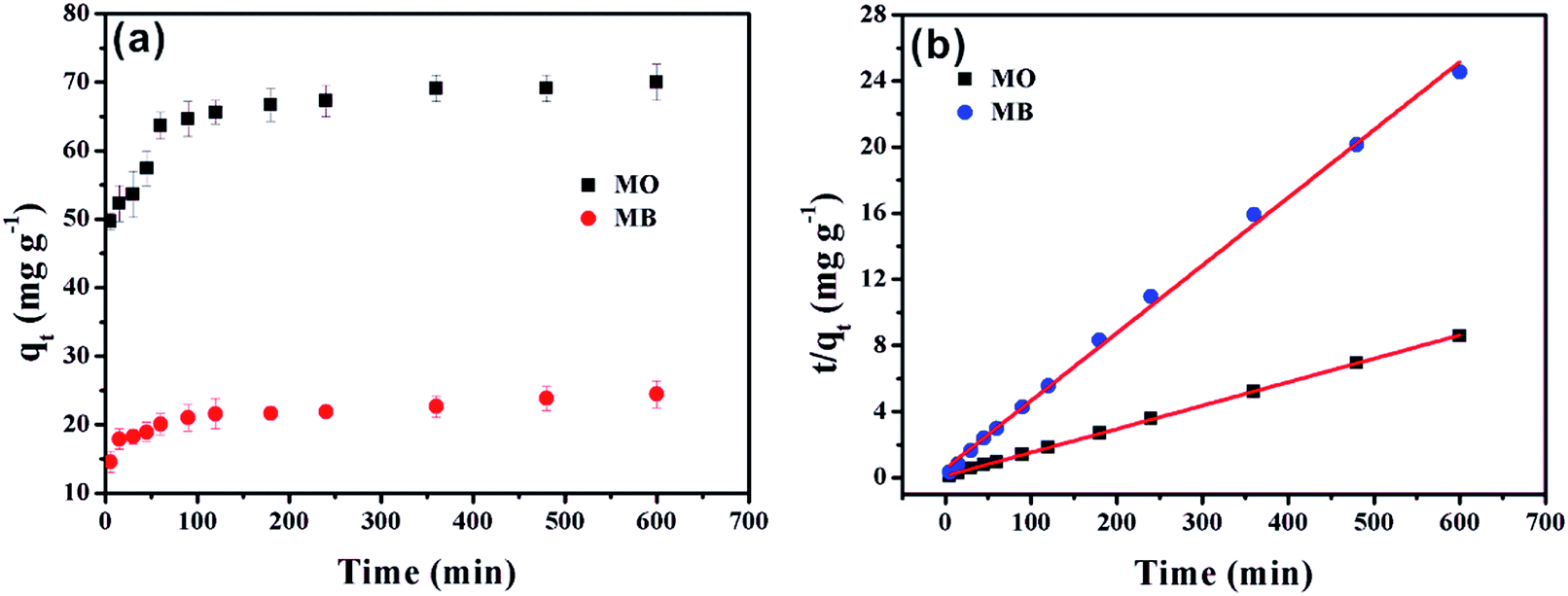 | ||
| Fig. 4 Effect of contact time of MO/MB on α-Fe2O3/g-C3N4-0.5 (a). Pseudo-second-order linear plots for the removal of MO/MB (b). pHMO = 3.0 ± 0.1, pHMB = 8.0 ± 0.1, m/v = 0.25 g L−1, and T = 298 ± 1 K. | ||
To the possible mechanism of adsorption, the pseudo-first-order and pseudo-second-order kinetic models were applied for the simulation of the data (Text S2†). The model curves are shown Fig. 4b and S3,† while the corresponding parameters are listed in Table S2.† The super-high correlation coefficient and the correspondence of qe,cal and qe,exp show that the experimental data is consistent with the pseudo-second-order model, which reveals that the adsorption is affected by chemical interactions.32
3.4 Adsorption thermodynamics of MO and MB on α-Fe2O3/g-C3N4-0.5
The thermodynamic parameters (ΔS0, ΔG0, and ΔH0) for MO and MB adsorption on α-Fe2O3/g-C3N4-0.5 were calculated from the temperature-dependent adsorption isotherms (Text S3†), and the results are presented in Table S4.† As shown in Table S4,† the ΔG0 values for MO and MB were negative, and therefore the adsorption process was spontaneous. However, the increase in ΔG0 indicates that α-Fe2O3/g-C3N4-0.5 had the most efficient adsorption for MO at a higher temperature, while the removal of MB showed the opposite trend.33 The negative value of ΔS0 for MO and the positive value of ΔS0 for MB exhibit the increase and decrease in the degree of disorder at the solid–liquid interface, respectively. The negative value of ΔH0 (−17.84 kJ mol−1) for MO adsorption and positive ΔH0 value (6.72 kJ mol−1) for MB show that the MO adsorption was an exothermic process, while MB adsorption was endothermic.343.5 Effect of initial pH and ion strength
The initial pH is a very important factor that affects the adsorption of pollutants.35 The effect of solution pH on MO and MB adsorption on α-Fe2O3/g-C3N4-0.5 is presented in Fig. 5a and b, respectively. The adsorption capacity of MO on α-Fe2O3/g-C3N4-0.5 obviously decreased from 3.0 to 5.0, then slowly decreased with an increase in the initial pH (Fig. 5a). The maximum adsorption capacity for MO at initial pH 3.0 reached 68.09 mg g−1. This result is consistent with a recent study, in which Robati et al. found that the optimum pH for the adsorption of the MO on synthesized GO was also 3.0.36 This result can be explained by the zeta potential of α-Fe2O3/g-C3N4-0.5 and the chemical structure of MO, as shown in Fig. S6 and S7.† In the pH range of 3.0–5.0, the surface charge of the as-prepared sample was positive and that of MO was negative. Thus, electrostatic attraction could occur, resulting in the high adsorption compared to that in the pH range of 6.0 to 10.0. The adsorption capacities of the adsorbent remained slow with a decrease in the initial pH in the range of 6.0–10.0, suggesting that the electrostatic attraction was converted to electrostatic repulsion and competitive adsorption between OH− and MO for the available sites.37 Compared to MO, the influence of initial pH on the removal of MB showed a different trend (Fig. 5b). The adsorption capacity revealed a rapid increase at initial pH 3.0–5.0, followed by a slow increase over the solution pH range of 6.0–9.0, and then the adsorption capacity finally increased rapidly at pH 10.0. This can be explained by the fact that the surface of α-Fe2O3/g-C3N4-0.5 was protonated and showed a positive charge in the pH range of 3.0–5.0, which was conducive to the immobilization of the positively charged MB by electrostatic repulsion. With an increase in the initial pH, the surface of the as-prepared sample became negative owing to deprotonation, and the electrostatic attraction between the positively charged MB and the negatively charged surface led an increase in adsorption capacity. Meanwhile, α-Fe2O3/g-C3N4-0.5 still adsorbed MB and MO under electrostatic repulsion. This may be dominated by the specific driving force between MO/MB and α-Fe2O3/g-C3N4-0.5.38 Moreover, the adsorption capacity of the adsorbent for MO and MB decreased and increased, respectively, at initial pH 2.0, compared with that at initial pH 3.0 due to the partial dissolution of the adsorbent. It is worth noting that the adsorbent had a certain buffering effect by comparing the initial pH value with the equilibrium pH value.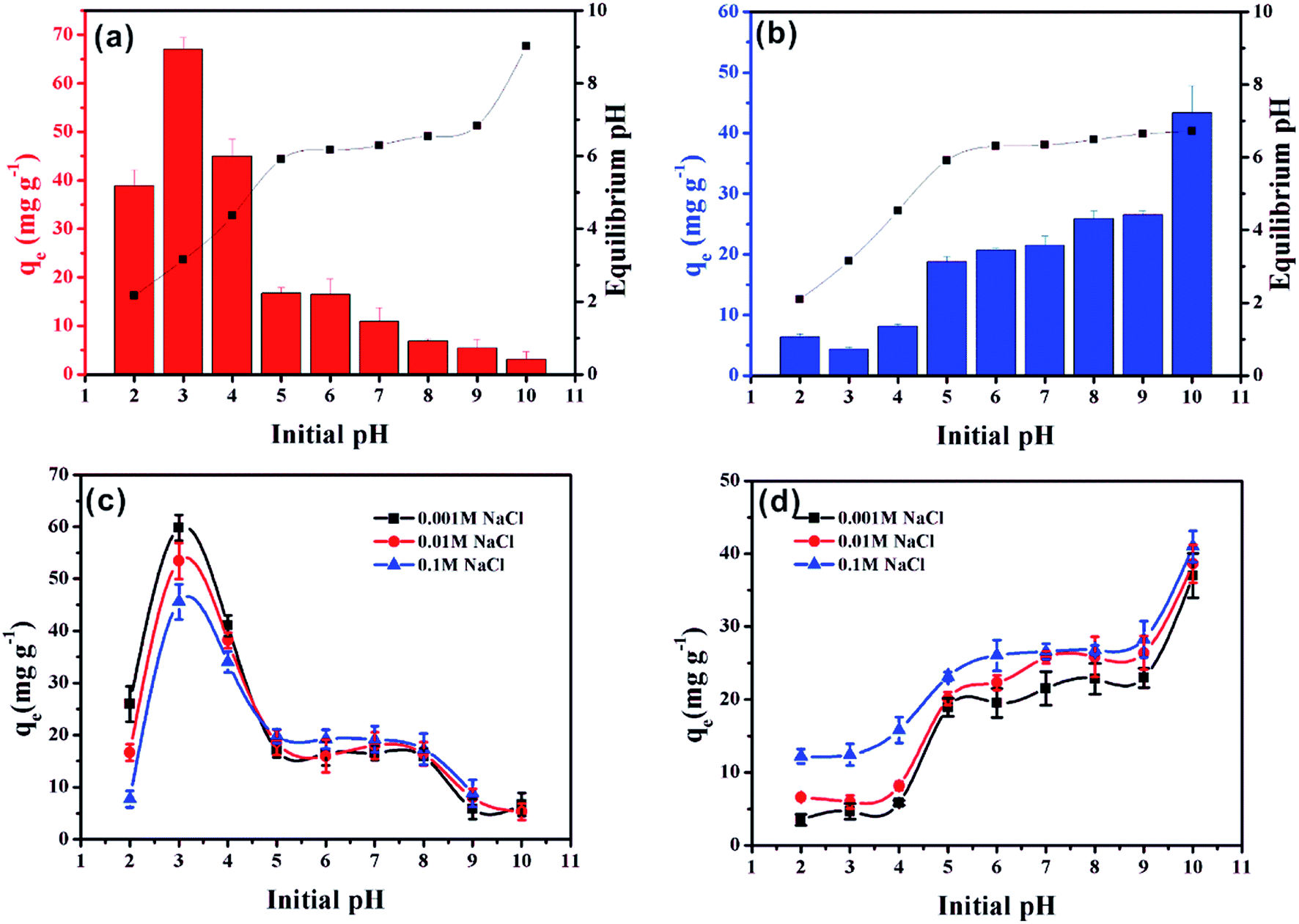 | ||
| Fig. 5 Effect of initial pH on MO (a) and MB (b) removal on α-Fe2O3/g-C3N4-0.5. Effect of ionic strength on MO (c) and MB (d) removal on α-Fe2O3/g-C3N4-0.5. m/v = 0.25 g L−1, and T = 298 ± 1 K. | ||
The adsorption of MO and MB on α-Fe2O3/g-C3N4-0.5 as a function of the initial pH in different NaCl concentrations (0.001, 0.01 and 0.1 mol L−1) was investigated (Fig. 5c and d, respectively). As shown in Fig. 5c and d, the adsorption of MO and MB was influence by ionic strength at all pH values, respectively. In the initial pH range of 2.0–4.0, with an increase in the concentration of NaCl from 0.001 M to 0.1 M, the adsorption capacity of MO decreased greatly. One possible explanation for this phenomenon is the competitive adsorption between the increased chloride ions in the solution and the negatively charged methyl orange, resulting in a decrease in effective adsorption sites.39 Hence, the adsorption within this initial pH range may be dominated by electrostatic interaction. However, with an increase in the initial pH from 5.0 to 10.0, the concentration of NaCl had little influence on the adsorption capacity of MO. This phenomenon indicates that the electrostatic interaction was no longer dominant in this initial pH region. In addition, the concentration of NaCl had a relatively large influence on the adsorption of MB at all pH values. This phenomenon can be explained by two aspects. On the one hand, in the initial pH range of 2.0–5.0, increasing ionic strength could reduce the electrostatic repulsion between MB and the adsorbent, thus increasing the adsorption capacity through other driving forces.40 On the other hand, in the initial pH range of 6.0–10.0, increasing ionic strength could promote the dissociation of MB and enhance its electrostatic interaction with the adsorbent surface, which is consistent with MB removal on a spherical CuO/meso-silica nano composite.41
3.6 Regeneration and reusability
The stability of adsorbents is considered an indispensable factor for their industrial application. Thus, the recycling of α-Fe2O3/g-C3N4-0.5 was also investigated (Fig. S8†). The adsorption capacity of the as-prepared sample only decreased slightly from 70.17 mg g−1 to 65.81 mg g−1 with an increase in the reuse cycles and the decline efficiency was 6.21% after four cycles of regeneration experiments. In addition, the adsorption capacity of α-Fe2O3/g-C3N4-0.5 slightly declined by 9.80%, from 27.36 mg g−1 to 24.68 mg g−1 for MB after four cycles. Also, the XRD peaks of MO/MB after photocatalysis and adsorption of the materials did not change (Fig. S9†). These phenomena indicate that α-Fe2O3/g-C3N4-0.5 has good reusability and chemical stability.3.7 Adsorption mechanisms
The α-Fe2O3/g-C3N4-0.5 sample was measured by EDS after the adsorption of MO and MB. As shown in Fig. S2b and c,† a new peak at approximately 2.3 keV appeared, respectively. This result indicates the successful adsorption of MO and MB on α-Fe2O3/g-C3N4-0.5. Thus, to further explore the potential adsorption mechanism of dyes on the as-prepared sample, α-Fe2O3/g-C3N4-0.5 was measured by surface sensitive XPS spectra before and after the adsorption of MO and MB. Besides, the high-resolution XPS scans of C 1s and N 1s were also obtained. As can be seen in Fig. 6a, there were four peaks in the XPS survey spectrum of α-Fe2O3/g-C3N4-0.5, including N 1s, O 1s, Fe 2p, and C 1s. After adsorption, the new peak S 2p was found on α-Fe2O3/g-C3N4-0.5, which is attributed to the S element of MO and MB. These results also demonstrate that MO and MB were successfully adsorbed on the as-prepared sample, which are consistent with the EDS results. The O 1s spectrum of α-Fe2O3/g-C3N4-0.5 showed a characteristic peak at 529.02 eV, which is ascribed to the lattice oxygen in α-Fe2O3. However, the peak position of lattice oxygen in pure α-Fe2O3 shifted by ∼0.63 eV (529.65 eV) (Fig. S10†). This result suggests that α-Fe2O3 and g-C3N4 are bound by chemical bonds rather than a simple physical mixture. Before and after adsorption, the C 1s and N 1s spectra of α-Fe2O3/g-C3N4-0.5 provide some evidence of the interaction mechanism between the as-prepared sample and adsorbates. The N 1s spectrum of α-Fe2O3/g-C3N4-0.5 showed four peaks at 398.41, 399.70, 400.90 and 404.17 eV (Fig. 6c). The peak at 398.41 eV is the prime binding energy, which can be ascribed to the sp2 N of the triazine ring (C3N3) or heptazine units (C6N7), while the peak at 399.70 is ascribed to the tertiary nitrogen N–(C)3 and that at 400.90 eV is attributed the amine functions (–NHχ).42 Furthermore, the peak at 404.17 eV may be related to charging effects. In the C 1s spectrum of α-Fe2O3/g-C3N4-0.5, the four peaks observed at 285.04, 286.70, 287.94 and 289.27 eV are ascribed to the sp2 C–N, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups, and N
N groups, and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N2 of the triazine ring (C3N3) or heptazine (C6N7) units,43 and sp2 hybridized C atoms in CO, respectively (Fig. 6b).44 After adsorption, the peak positions at 287.94 eV (C 1s) and 398.41 eV (N 1s) for α-Fe2O3/g-C3N4-0.5 shifted for MO by ∼0.36 eV (288.30 eV) and ∼0.38 eV (398.79 eV) and for MB by ∼0.29 eV (288.23 eV) and ∼0.26 eV (398.67 eV), respectively (Fig. 6d–g). The binding energies of the various elements on α-Fe2O3/g-C3N4-0.5 are listed in Table 2. The peak area ratio at 297.94 eV decreased from 64.70% to 57.12% for MB and 55.36% for MO. Similarly, the peak area ratio at 398.41 eV also decreased from 54.20% to 43.13% for MB and to 41.96% for MO, which indicates the reduced electron density of the N and C atoms after adsorption.45 This can be due to the π–π electron donor–acceptor (EDA) interaction between the π electron system of the heptazine (C6N7)/triazine ring (C3N3) units and the electronic density in the aromatic rings of MO/MB.46 It is worth noting that the changes in the binding energy (EB) of 297.94 eV (C 1s) and 398.41 eV (N 1s) and peak area ratio of α-Fe2O3/g-C3N4-0.5 + MO were slightly more than that of α-Fe2O3/g-C3N4-0.5 + MB, suggesting that the π–π EDA interaction between the as-prepared sample and MO was more powerful than that for MB.47 This also explains why the maximum adsorption capacity of α-Fe2O3/g-C3N4-0.5 for MO is higher than that for MB. Hence, the results of this series of experiments and characterization effectively demonstrate that the adsorption of MO/MB was mainly determined by π–π EDA interaction and electrostatic interaction, respectively.
C–N2 of the triazine ring (C3N3) or heptazine (C6N7) units,43 and sp2 hybridized C atoms in CO, respectively (Fig. 6b).44 After adsorption, the peak positions at 287.94 eV (C 1s) and 398.41 eV (N 1s) for α-Fe2O3/g-C3N4-0.5 shifted for MO by ∼0.36 eV (288.30 eV) and ∼0.38 eV (398.79 eV) and for MB by ∼0.29 eV (288.23 eV) and ∼0.26 eV (398.67 eV), respectively (Fig. 6d–g). The binding energies of the various elements on α-Fe2O3/g-C3N4-0.5 are listed in Table 2. The peak area ratio at 297.94 eV decreased from 64.70% to 57.12% for MB and 55.36% for MO. Similarly, the peak area ratio at 398.41 eV also decreased from 54.20% to 43.13% for MB and to 41.96% for MO, which indicates the reduced electron density of the N and C atoms after adsorption.45 This can be due to the π–π electron donor–acceptor (EDA) interaction between the π electron system of the heptazine (C6N7)/triazine ring (C3N3) units and the electronic density in the aromatic rings of MO/MB.46 It is worth noting that the changes in the binding energy (EB) of 297.94 eV (C 1s) and 398.41 eV (N 1s) and peak area ratio of α-Fe2O3/g-C3N4-0.5 + MO were slightly more than that of α-Fe2O3/g-C3N4-0.5 + MB, suggesting that the π–π EDA interaction between the as-prepared sample and MO was more powerful than that for MB.47 This also explains why the maximum adsorption capacity of α-Fe2O3/g-C3N4-0.5 for MO is higher than that for MB. Hence, the results of this series of experiments and characterization effectively demonstrate that the adsorption of MO/MB was mainly determined by π–π EDA interaction and electrostatic interaction, respectively.
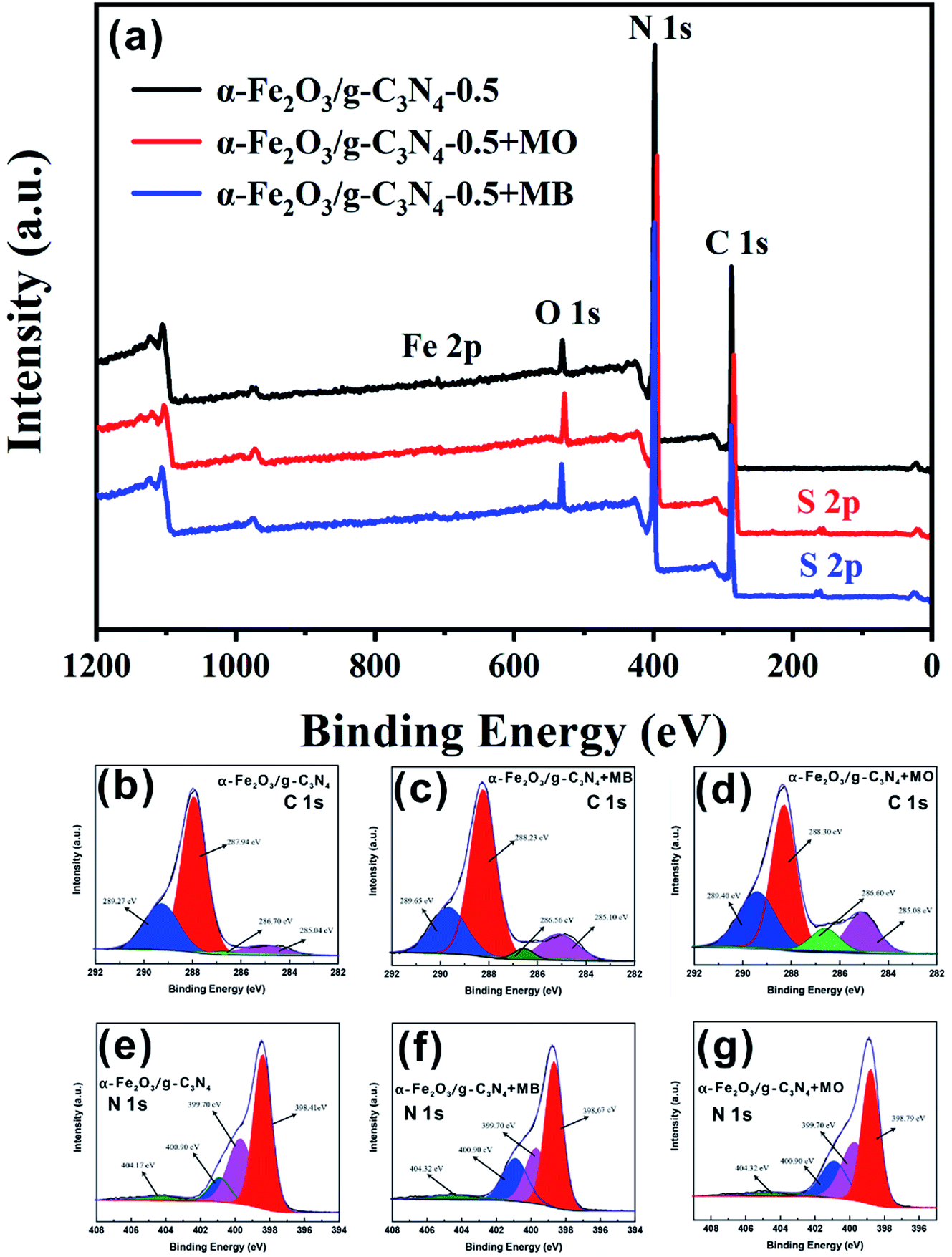 | ||
| Fig. 6 XPS spectra (a) and corresponding O 1s (b–d) and N 1s (e–g) spectra of α-Fe2O3/g-C3N4-0.5 before adsorption (b and e), after MB adsorption (c and f), and after MO adsorption (d and g). | ||
| Peak | α-Fe2O3/g-C3N4-0.5 | α-Fe2O3/g-C3N4-0.5 + MO | α-Fe2O3/g-C3N4-0.5 + MB | |||
|---|---|---|---|---|---|---|
| Peak position (eV) | Area (%) | Peak position (eV) | Area (%) | Peak position (eV) | Area (%) | |
| C 1s | 287.94 | 64.70 | 288.30 | 55.36 | 288.23 | 57.12 |
| N 1s | 398.41 | 54.20 | 398.79 | 41.96 | 398.67 | 43.13 |
3.8 Photocatalytic activity of α-Fe2O3/g-C3N4-0.5
To further investigate the maximum removal efficiency of the target dyes by the as-prepared materials, photocatalytic experiments of MO and MB on α-Fe2O3/g-C3N4-0.5 were performed. As shown in Fig. 7a, the as-prepared sample showed excellent photocatalytic capacity for MO with 79.49% removal after 150 min. The simulation of the photocatalytic experiments indicates that the photocatalytic of MO fitted the first-order kinetic model (Fig. S11a†). The kinetic constants, k (min−1), for the photodegradation of MO by α-Fe2O3/g-C3N4-0.5 was 3.13-fold higher than that of g-C3N4 (Fig. 7b). In addition to the removal of MO, α-Fe2O3/g-C3N4-0.5 also exhibited excellent photodegradation ability for MB with 80.93% removed after 150 min (Fig. 7d). In addition, the first-order kinetic model was also closer to the photocatalytic experiments of MB (Fig. 7e). The k (min−1) over α-Fe2O3/g-C3N4-0.5 increased 1.14-fold compared to that on g-C3N4 (Fig. S11b†). Overall, the photocatalytic ability of α-Fe2O3/g-C3N4-0.5 to remove dyes improved greatly compared with that of g-C3N4. To study the photocatalytic performance of the composite materials over g-C3N4 for target pollutants, DRS and PL characterization of the two materials were performed. Fig. S12† shows the UV-vis diffuse reflectance spectra of g-C3N4 and α-Fe2O3/g-C3N4-0.5. Pure g-C3N4 and α-Fe2O3/g-C3N4-0.5 have an absorption edge at about 440 nm. It is worth noting that α-Fe2O3/g-C3N4-0.5 exhibited enhanced light absorption in the visible region, and thus can generate more photogenerated charge carriers to improve the catalytic activity of the material.48 Fig. S13† shows the room temperature PL spectra of g-C3N4 and α-Fe2O3/g-C3N4-0.5. The emission peaks of the composite significantly decreased compared with that of g-C3N4, indicating that the introduction of α-Fe2O3 significantly reduced the electron–hole recombination of the composite materials.49 This is consistent with the excellent activity of the composites from the catalytic data. To study the photochemical properties of the materials, the time–current curves of α-Fe2O3, g-C3N4 and α-Fe2O3/g-C3N4 were measured (Fig. S14†). g-C3N4 and α-Fe2O3 showed a low photocurrent density, which is attributed to the fast recombination of photogenerated electrons and holes in g-C3N4 and α-Fe2O3. The α-Fe2O3/g-C3N4-0.5 composite showed the highest photocurrent density, indicating the efficient photogenerated charge transfer between g-C3N4 and α-Fe2O3.50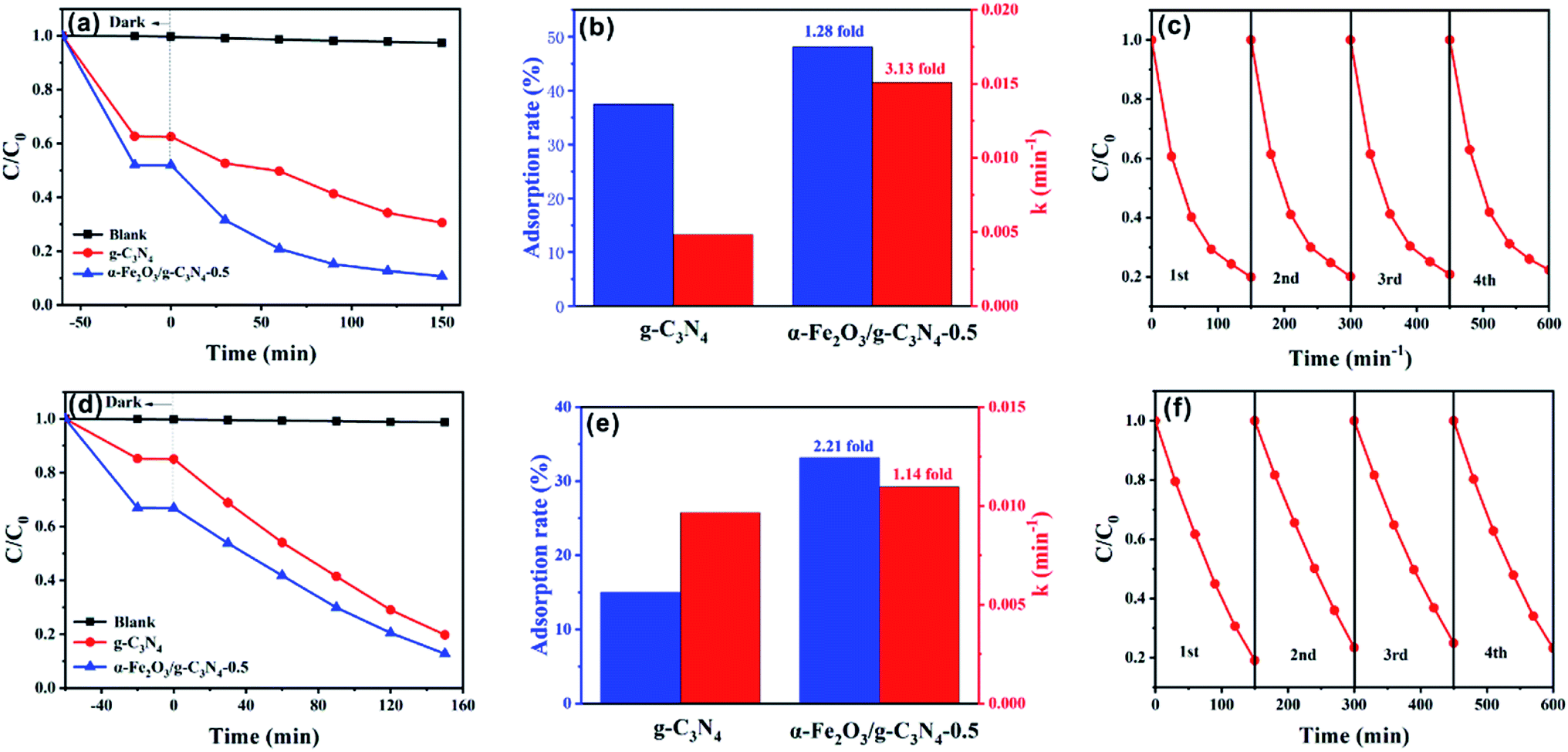 | ||
| Fig. 7 Adsorption and photoreduction of MO (a) and MB (d). Adsorption rate and photoreduction kinetic constant comparisons of MO (b) and MB (e) over g-C3N4, and α-Fe2O3/g-C3N4-0.5 under visible light irradiation (λ ≥ 420 nm). Cycling runs for photoreduction of MO (c) and MB (f) over α-Fe2O3/g-C3N4-0.5. m/v = 0.25 g L−1, and T = 298 ± 1 K. | ||
Furthermore, similar to adsorbents, the stability of catalysts is also a crucial factor for their practical and industrial applications. To evaluate the stability of the catalyst, α-Fe2O3/g-C3N4-0.5 was investigated in a four-cycle test (Fig. 7c and f). After the fourth cycle, the MO removal rate decreased from 79.49% to 71.63%, while the removal rate of MB declined from 80.93% to 72.75%. The results show that the photocatalyst was stable and reusable.
Finally, to investigate the photocatalytic mechanism with respect to MO degradation over α-Fe2O3/g-C3N4-0.5, TEOA, IPA and SOD were added as scavengers of hydroxyl radicals (·OH), holes (h+), and superoxide radicals (·O2−), respectively.51 As shown in Fig. S15,† the photodegradation of MO by α-Fe2O3/g-C3N4-0.5 was significantly suppressed by 71.13% in the presence of TEOA, whereas a slight increase of 1.44% and 6.58% was found after the addition of IPA and SOD, respectively. Hence, the holes (h+) played a significant role in MO photodegradation. Besides MO, the degradation of MB by α-Fe2O3/g-C3N4-0.5 was drastically suppressed by 77.55% and 65.72% in the presence of EDTA and IPA, respectively, whereas a weak inhibition of 3.57% was obtained after the addition of BQ. These results show that hydroxyl radicals (·OH) and holes (h+) were the two most important reactive species for the photodegradation of MB by α-Fe2O3/g-C3N4-0.5.52 If the double charge transfer mechanism is applicable, the photogenerated electrons on the CB (−1.27 eV) of g-C3N4 will transfer to the CB (0.32 eV) of α-Fe2O3, and the photogenerated holes on the VB (2.52 eV) of α-Fe2O3 will also transfer to the VB (1.57 eV) of g-C3N4 because of the low VB and CB positions of α-Fe2O3 relative to that of the original g-C3N4. However, OH− cannot be oxidized by photogenerated holes on the VB of g-C3N4 to generate ·OH because OH−/·OH has a relatively low standard redox potential (2.3 eV) (Fig. S16†).53 Therefore, the Z-scheme mechanism may be more reasonable.
4. Conclusion
Novel α-Fe2O3/g-C3N4 composites were synthesized via a one-step hydrothermal method to remove dyes from wastewater. The results indicated that α-Fe2O3/g-C3N4-0.5 was the best among the series with the maximum adsorption capacities of 69.91 mg g−1 for MO at pH 3.0 and 29.46 mg g−1 for MB at pH 8.0. The adsorption process was affected by ionic strength and solution pH. Electrostatic interaction and π–π EDA interaction were the dominant mechanisms involved in the adsorption of MO/MB on α-Fe2O3/g-C3N4-0.5, respectively. In addition, α-Fe2O3/g-C3N4-0.5 exhibited excellent catalytic properties for the photocatalytic degradation of MO/MB. Hydroxyl radicals (·OH) and holes (h+) were the main reactive species for the photodegradation of MB by α-Fe2O3/g-C3N4-0.5, while holes (h+) played a significant role in the decomposition of MO. Hence, the synthesized α-Fe2O3/g-C3N4 composite provides an efficient method for removing dyes from wastewater and it has the potential for practical application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Major Science and Technology Program for Water Pollution Control and Treatment (2017ZX07603-003) and the National Key R&D Program of China (2018YFD0900801).References
- S. Wong, H. H. Tumari, N. Ngadi, N. B. Mohamed, O. Hassan, R. Mat and N. A. Saidina Amin, J. Cleaner Prod., 2019, 206, 394–406 CrossRef CAS.
- Y. Gao, S.-Q. Deng, X. Jin, S.-L. Cai, S.-R. Zheng and W.-G. Zhang, Chem. Eng. J., 2019, 357, 129–139 CrossRef CAS.
- K. Y. Foo and B. H. Hameed, Desalination, 2011, 275, 302–305 CrossRef CAS.
- X. He, K. B. Male, P. N. Nesterenko, D. Brabazon, B. Paull and J. H. Luong, ACS Appl. Mater. Interfaces, 2013, 5, 8796–8804 CrossRef CAS PubMed.
- Q. Liao, D. Zou, W. Pan, W. Linghu, R. Shen, Y. Jin, G. Feng, X. Li, F. Ye, A. M. Asiri, H. M. Marwani, Y. Zhu, X. Wu and W. Dong, J. Mol. Liq., 2018, 258, 275–284 CrossRef CAS.
- A. Mittal, A. Malviya, D. Kaur, J. Mittal and L. Kurup, J. Hazard. Mater., 2007, 148, 229–240 CrossRef CAS PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- G. Lui, J.-Y. Liao, A. Duan, Z. Zhang, M. Fowler and A. Yu, J. Mater. Chem. A, 2013, 1, 12255 RSC.
- B. Y. Tan, X. H. Zhang, Y. J. Li, H. Chen, X. Z. Ye, Y. Wang and J. F. Ye, Chem.–Eur. J., 2017, 23, 5478–5487 CrossRef CAS PubMed.
- J. Han, Z. Du, W. Zou, H. Li and C. Zhang, Chem. Eng. J., 2015, 262, 571–578 CrossRef CAS.
- P. Xiao, P. Wang, H. Li, Q. Li, Y. Shi, X. L. Wu, H. Lin, J. Chen and X. Wang, J. Hazard. Mater., 2018, 345, 123–130 CrossRef CAS PubMed.
- X. Cai, J. He, L. Chen, K. Chen, Y. Li, K. Zhang, Z. Jin, J. Liu, C. Wang, X. Wang, L. Kong and J. Liu, Chemosphere, 2017, 171, 192–201 CrossRef CAS PubMed.
- L. Zhou, H. Y. Zhang, H. Q. Sun, S. M. Liu, M. O. Tade, S. B. Wang and W. Q. Jin, Catal. Sci. Technol., 2016, 6, 7002–7023 RSC.
- Y. Zou, P. Wang, W. Yao, X. Wang, Y. Liu, D. Yang, L. Wang, J. Hou, A. Alsaedi, T. Hayat and X. Wang, Chem. Eng. J., 2017, 330, 573–584 CrossRef CAS.
- Y. D. Zou, X. X. Wang, Y. J. Ai, Y. H. Liu, Y. F. Ji, H. Q. Wang, T. Hayat, A. Alsaedi, W. P. Hu and X. K. Wang, J. Mater. Chem. A, 2016, 4, 14170–14179 RSC.
- K. Sridharan, T. Kuriakose, R. Philip and T. J. Park, Appl. Surf. Sci., 2014, 308, 139–147 CrossRef CAS.
- L. Ge and C. C. Han, Appl. Catal., B, 2012, 117, 268–274 CrossRef.
- L. Ge, C. C. Han, J. Liu and Y. F. Li, Appl. Catal., A, 2011, 409, 215–222 CrossRef.
- H. P. Li, J. Y. Liu, W. G. Hou, N. Du, R. J. Zhang and X. T. Tao, Appl. Catal., B, 2014, 160, 89–97 CrossRef.
- E.-t. Liu, H. Zhao, H. Li, G. Li, Y. Liu and R. Chen, New J. Chem., 2014, 38, 2911 RSC.
- X. L. Hu, J. C. Yu, J. M. Gong, Q. Li and G. S. Li, Adv. Mater., 2007, 19, 2324–2329 CrossRef CAS.
- Z. Y. Ma, Y. P. Guan and H. Z. Liu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3433–3439 CrossRef CAS.
- L. Ma, H. Fan, K. Fu, S. Lei, Q. Hu, H. Huang and G. He, ACS Sustainable Chem. Eng., 2017, 5, 7093–7103 CrossRef CAS.
- R. Hu, X. Wang, S. Dai, D. Shao, T. Hayat and A. Alsaedi, Chem. Eng. J., 2015, 260, 469–477 CrossRef CAS.
- D. Xiao, K. Dai, Y. Qu, Y. Yin and H. Chen, Appl. Surf. Sci., 2015, 358, 181–187 CrossRef CAS.
- X.-H. Jiang, Q.-J. Xing, X.-B. Luo, F. Li, J.-P. Zou, S.-S. Liu, X. Li and X.-K. Wang, Appl. Catal., B, 2018, 228, 29–38 CrossRef CAS.
- S. Wang, X. Yang, X. Zhang, X. Ding, Z. Yang, K. Dai and H. Chen, Appl. Surf. Sci., 2017, 391, 194–201 CrossRef CAS.
- J. S. Wu, C. H. Liu, K. H. Chu and S. Y. Suen, J. Membr. Sci., 2008, 309, 239–245 CrossRef CAS.
- L. Huang, Q. Shuai and S. Hu, J. Cleaner Prod., 2019, 215, 280–289 CrossRef CAS.
- Y. Li, X. Zhang, R. Yang, G. Li and C. Hu, RSC Adv., 2015, 5, 32626–32636 RSC.
- Y. Sun, Q. Yue, B. Gao, L. Huang, X. Xu and Q. Li, Chem. Eng. J., 2012, 181–182, 790–797 CrossRef CAS.
- Y. Wan, Q. Liang, T. T. Cong, X. Y. Wang, Y. Y. Tao, M. Y. Sun, Z. Y. Li and S. Xu, RSC Adv., 2015, 5, 66286–66293 RSC.
- M. Kara, H. Yuzer, E. Sabah and M. S. Celik, Water Res., 2003, 37, 224–232 CrossRef CAS PubMed.
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- H. Zheng, Z. Y. Wang, J. Zhao, S. Herbert and B. S. Xing, Environ. Pollut., 2013, 181, 60–67 CrossRef CAS PubMed.
- D. Robati, B. Mirza, M. Rajabi, O. Moradi, I. Tyagi, S. Agarwal and V. K. Gupta, Chem. Eng. J., 2016, 284, 687–697 CrossRef CAS.
- L. Y. Yuan, L. Zhu, C. L. Xiao, Q. Y. Wu, N. Zhang, J. P. Yu, Z. F. Chai and W. Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 3774–3784 CrossRef CAS PubMed.
- M. Teixido, J. J. Pignatello, J. L. Beltran, M. Granados and J. Peccia, Environ. Sci. Technol., 2011, 45, 10020–10027 CrossRef CAS PubMed.
- J. Y. Wang, H. M. Wang, Z. J. Song, D. L. Kong, X. M. Chen and Z. M. Yang, Colloids Surf., B, 2010, 80, 155–160 CrossRef CAS PubMed.
- A. B. Albadarin, M. N. Collins, M. Naushad, S. Shirazian, G. Walker and C. Mangwandi, Chem. Eng. J., 2017, 307, 264–272 CrossRef CAS.
- Z. J. Liang, Z. W. Zhao, T. Y. Sun, W. X. Shi and F. Y. Cui, J. Colloid Interface Sci., 2017, 485, 192–200 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. J. Cui, J. S. Zhang, G. G. Zhang, J. H. Huang, P. Liu, M. Antonietti and X. C. Wang, J. Mater. Chem., 2011, 21, 13032–13039 RSC.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- K. Kamiya, R. Kamai, K. Hashimoto and S. Nakanishi, Nat. Commun., 2014, 5, 5040 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- C. Shen, C. Chen, T. Wen, Z. Zhao, X. Wang and A. Xu, J. Colloid Interface Sci., 2015, 456, 7–14 CrossRef CAS PubMed.
- M. Sun, Q. Zeng, X. Zhao, Y. Shao, P. G. Ji, C. Q. Wang, T. Yan and B. Du, J. Hazard. Mater., 2017, 339, 9–21 CrossRef CAS PubMed.
- B. T. Shi, H. Y. Yin, J. Y. Gong and Q. L. Nie, Mater. Lett., 2017, 201, 74–77 CrossRef CAS.
- Y. Hu, K. Li, Y. Li, H. Liu, M. Guo, X. Ye, Z. Wu and K. Lee, ChemistrySelect, 2019, 4, 617–622 CrossRef CAS.
- X. Ding, D. Xiao, L. Ji, D. Jin, K. Dai, Z. Yang, S. Wang and H. Chen, Catal. Sci. Technol., 2018, 8, 3484–3492 RSC.
- Y. Hu, T. Guo, X. Ye, Q. Li, M. Guo, H. Liu and Z. Wu, Chem. Eng. J., 2013, 228, 392–397 CrossRef CAS.
- B. Tan, X. Ye, Y. Li, X. Ma, Y. Wang and J. Ye, Chemistry, 2018, 24, 13311–13321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05100g |
| This journal is © The Royal Society of Chemistry 2019 |