
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide exchange studies of novel Pd(II) NNN-pincer complexes†
Seher Kuyuldar ab,
Clemens Burda*b and
William B. Connick‡
a
ab,
Clemens Burda*b and
William B. Connick‡
a
aDepartment of Chemistry, University of Cincinnati, 2600 Clifton Ave., Cincinnati, OH 44221, USA
bDepartment of Chemistry, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106, USA. E-mail: burda@case.edu
First published on 15th August 2019
Abstract
Palladium(II) complexes with an NNN type pincer ligand (pip2NNN = 2,6-bis(piperdyl-methyl)pyridine) are synthesized and characterized. Electronic and 1H NMR spectra point to decreasing filled/filled repulsions between the dπ(Pd) orbitals and the halide lone pair orbitals along the Cl < Br < I series. For all complexes, the most downfield α-piperidyl resonance of the pip2NNN ligand is sensitive to changes in the coordinated halide while the meta-pyridyl and benzylic resonances are sensitive to changes in the counter anion. This sensitivity is utilized to study halide association and exchange at the fourth coordination site. Conductivity and 1H NMR spectroscopy confirm the interaction between the exogenous anion (Cl−, Br−, BF4−) and Pd(pip2NNN)X+ (X = Cl, Br).
Introduction
Pincer metal complexes in which three donor sites of the ligand stabilize a planar metal center have been studied extensively. Variations to the so-called ECE type ligand (Chart 1) where E symbolizes coordination through flanking NR2, PR2, SR, SeR or OR groups afford complexes with applications especially in catalysis1–8 but also in other fields including sensing,6,9–12 materials science13–19 and medicinal chemistry.18,20 Among various metals, palladium is one of the most studied owing mainly to its importance in organic synthesis. While formation of a carbon–palladium bond provides stability, replacing carbon with other elements such as Si, P, N have been shown to lead to significant changes in reactivity and an lead to significant developments in catalysis4,8,21–27 and cytotoxicity studies.18,20,28–32 | ||
| Chart 1 General depiction of palladium pincer complexes. | ||
A key factor in reactivity is the general lability of the fourth coordination site. For instance, the nature of the monodentate ligand (L) at the fourth coordination site has been shown to provide remarkable control over the electronic structures of palladium and platinum complexes with NCN pincer ligands.33–37
This article presents palladium(II) complexes with the NNN analog of the pip2NCN ligand. The neutral pip2NNN pincer ligand (2,6-bis(piperdyl-methyl)pyridine) shown in Scheme 1 was used to prepare a series of [Pd(pip2NNN)X]+ complexes with various anions (Cl−, Br−, I−, BF4−) and study the effect of the coordinating halide and the counter-anion by 1H NMR spectroscopy. We will able to investigate halide exchange at the fourth coordination site because pip2NNN is sensitive to the kind of halide and mode of interaction. Understanding the role of coordinated halide versus the anion has the potential to shed light into competing metal site interactions in catalytic systems with square planar geometries, especially platinum and palladium catalysts.
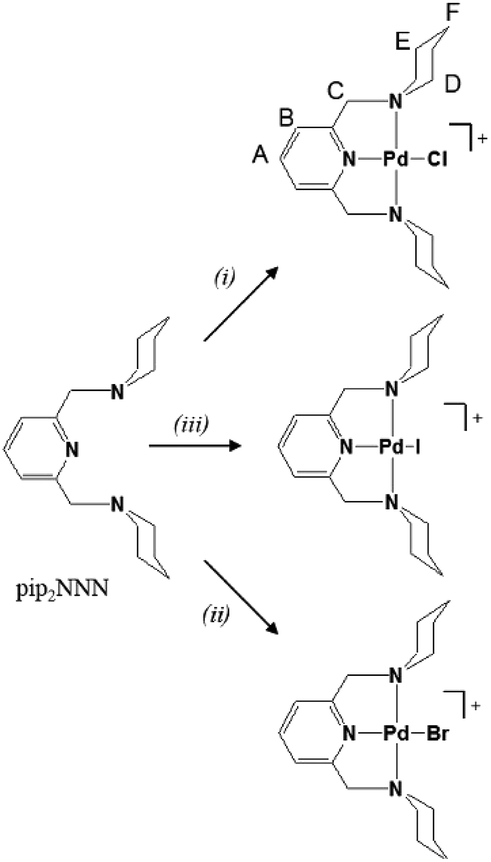 | ||
| Scheme 1 Synthesis of Pd(pip2NNN)X+ (X = Cl, Br, I) salts. (i) Pd(COD)Cl2 or PdCl2, acetonitrile, 40 °C. (ii) Pd(COD)Br2 or PdBr2, acetonitrile, RT. (iii) PdI2, acetonitrile, 50 °C. The unequal protons are labelled (A–F) on Pd(pip2NNN)Cl+. | ||
Results and discussion
Synthesis of Pd(pip2NNN)X+ salts
Salts of Pd(pip2NNN)X+ (X = Cl, Br, I), hereafter referred to as [X]+, are readily prepared by stirring either PdX2 or Pd(COD)X2 (COD = 1,5-cyclooctadiene) in acetonitrile solution of pip2NNN (Scheme 1). Depending on the reaction conditions, salts with X−, PdX42− and/or Pd2X62− counter anions are obtained. More information about the synthesis, isolation, elemental analyses, UV-visible absorption spectroscopy, and positive and negative mass spectroscopy and of the PdX42− and/or Pd2X62− salts are provided in the ESI.† Briefly, the X−, PdX42− and/or Pd2X62− salts have nearly identical 1H NMR spectra in acetonitrile, but are readily distinguished by their colors, solubilities, negative anion mass spectra, electronic absorption spectra, and elemental analyses. The Cl−, Br− and I− salts are obtained as pure compounds, whereas samples containing PdX42− or Pd2X62− are usually mixed with one or both of the other two anions. The halide salts form yellow to orange solutions, whereas salts containing PdnX2n+22− anions (n = 1, 2) are only weakly soluble in acetonitrile and give yellow solutions in the case of X = Cl, Br and red-brown solutions in the case of X = I. 1H NMR and mass spectra, as well as elemental analyses, confirm that the yellow/orange major products isolated from the filtrates are the halide salts of [X]+. In contrast, for each reaction, the elemental analysis of the less soluble minor product is consistent with having PdX42− and/or Pd2X62− as counter anions.Electronic spectroscopy
To better understand the electronic structures of the complexes with the pip2NNN ligand, absorption spectra of acetonitrile solutions of the halide salts of [X]+ were recorded. The data are collected in Table 1 and the spectra are shown in Fig. 1.| Compound | λmax/nm (ε/cm−1 M−1) |
|---|---|
| [Pd(pip2NNN)Cl]Cl | 209 (34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200), 252 (13150), 277sh (4300), 362 (1000) 200), 252 (13150), 277sh (4300), 362 (1000) |
| [Pd(pip2NNN)Br]Br | 218 (29300), 266sh (10200), 277sh (6700), 376 (850) |
| [Pd(pip2NNN)I]I | 205 (32700), 249 (29600), 277sh (11400), 297sh (5600), 421 (800) |
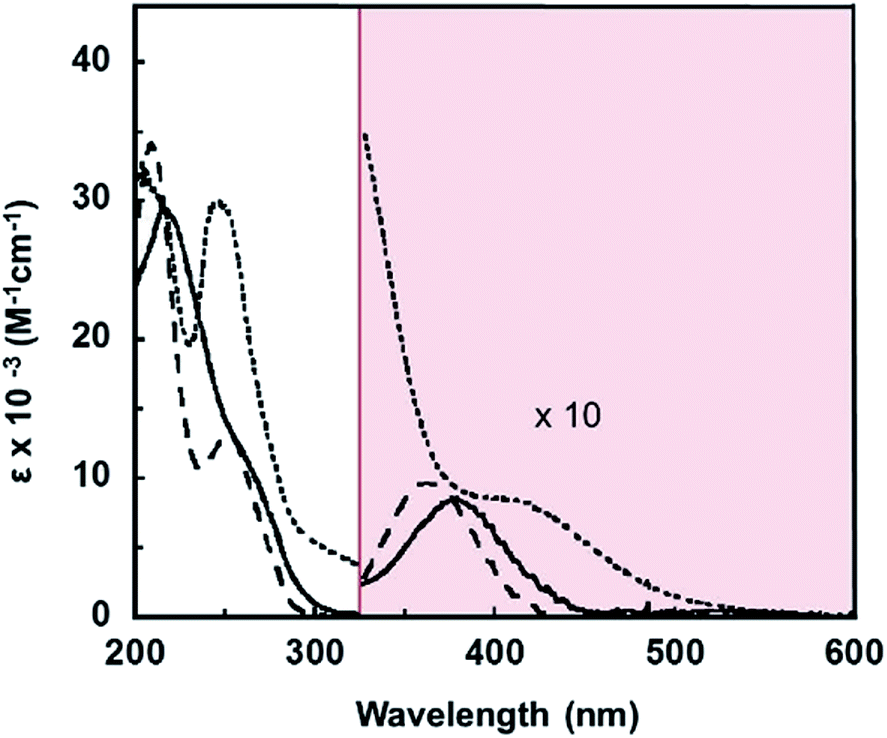 | ||
| Fig. 1 UV-visible absorption spectra of [Pd(pip2NNN)Cl]Cl (- - -), [Pd(pip2NNN)Br]Br (—) and [Pd(pip2NNN)I]I (⋯) in acetonitrile. The shaded area depicts tenfold magnification of the measured spectra. | ||
For [X]X, the UV region is dominated by intense absorptions between 200 and 280 nm (Fig. 1). Since the free ligand absorbs moderately in this region (265 nm, 3800 cm−1 M−1), these bands most likely have some contribution from ligand-based transitions. There is a moderately intense shoulder at 297 nm (5600 cm−1 M−1) in the [I]I spectrum. Pd(pip2NCN)X (X = Cl, Br, I) complexes exhibit slightly weaker absorption bands at shorter wavelengths (265–285 nm, 2000–4000 cm−1 M−1) that have been assigned as having significant MLCT character involving the pip2NCN− ligand.38 The MLCT transitions of the pip2NNN complexes are expected to occur at longer wavelengths because the greater π-acceptor capability of pip2NNN. Therefore, this band is tentatively assigned as having significant MLCT character involving pip2NNN. The transition is anticipated to be shifted to shorter wavelengths in the spectra of the [Cl]Cl and [Br]Br complexes, and hence obscured by other transitions. In support of this assignment, it is noteworthy that the lowest spin-allowed MLCT transition of Pd(4-mbpy)I2 (4-mbpy = 4,4′-dimethyl-2,2′-bipyridine) occurs near 306 nm (18400 cm−1 M−1, in DMF).39
A broad charge transfer feature appears at longer wavelengths in the spectrum of each halide complex, [Cl]Cl (362 nm, 1000 cm−1 M−1, FWHM = 2100 cm−1); [Br]Br (376 nm, 850 cm−1 M−1, FWHM = 2250 cm−1) and [I]I (421sh nm, 800 cm−1 M−1, FWHM = 2300 cm−1). A comparison to related complexes suggests that this band is unlikely to have MLCT character. For example, the lowest spin-allowed metal-to-ligand(pyridyl) charge-transfer band of Pt(2,6-bis(aminomethyl)pyridine) (OH)+ in aqueous solution is shifted to the blue of 320 nm,40 and the corresponding transition for the palladium(II) analog is expected to occur at even shorter wavelengths.
Similarly, the lowest spin-allowed metal-to-ligand(bpy) charge-transfer transition of Pd(bpy)Cl2 (bpy = 2,2′-bipyridine) occurs near 320 nm in aqueous solution;41 the MLCT transition of [X]+ is expected to occur at shorter wavelengths because of the stabilization of the unoccupied π*(bpy) level relative to the π*(pip2NNN) level. On the other hand, there are several examples of palladium(II) complexes that are believed to exhibit LMCT transitions in the >300 nm region,42–44 including trans-Pd(PPh3)2Cl2 (CH2Cl2: 345 nm, 20135 cm−1 M−1, FWHM ∼ 3000 cm−1),45,46 cis-Pd(dbcpe)Br2 (CH3CN: 354 nm, 14600 cm−1 M−1), cis-Pd(dbcpe)I2 (CH3CN: 396 nm, 6300 cm−1 M−1)47 and Pd(TPA)Cl+ (DMSO: 338 nm, 485 cm−1 M−1; 380 nm, 416 cm−1 M−1)48 (dbcpe = 1,2-bis[di(benzo-15-crown-5)phosphino]ethane ligand; TPA = tris(2-pyridylmethyl)amine). By analogy, the long wavelength band in the spectra of [X]+ is tentatively assigned to a transition having significant pπ(X−) → dx2−y2(Pd) charge-transfer character. There is considerable variability in the intensities of reported long wavelength LMCT transitions of palladium(II) complexes. The comparatively low intensities of the bands in the spectra of [X]+ may be a consequence of spin-forbidden character. An alternative explanation is that these bands arise from a ligand field transition. The red shift of this band along the halide series Cl < Br < I is in accord with either assignment.
The ∼1000 cm−1 red shift from Cl to Br and the ∼2800 cm−1 red shift from Br to I fall within the range of shifts in LMCT bands reported for square planar d8 complexes such as Pd(dbcpe)X2 (800, 3000 cm−1),47 trans-[Pd2(P(Et)2CH2P(Et)2)2X4] (Cl–Br, 1690 cm−1)49 and Ni(Cynp3)X+ (700, 3000 cm−1, Cynp3 = tris(2-dicyclohexylphosphinoethyl)amine).50
1H NMR spectroscopy
A general labeling scheme for inequivalent protons is shown in Scheme 1 for [Pd(pip2NNN)Cl]+. The 1H NMR spectra of [X]+ (X = Cl, Br, I) in acetonitrile exhibit patterns consistent with C2 symmetry and are qualitatively similar to those of their pip2NCN− palladium and platinum analogs.33,37 A triplet and a doublet due to the para and meta protons of the pyridyl ring (A, B) occur between 7.4 and 8.15 ppm (Fig. 2). The benzylic protons (C) give rise to a singlet near 4.6 ppm, suggesting a relatively low barrier to move the benzylic groups in and out of the coordination plane. For [Cl]+, the α-piperidyl proton resonance at 3.29 ppm (D′′) has the appearance of a doublet and is assigned accordingly to the equatorial proton; the resonance at 4.02 ppm (D′) has the appearance of a triplet and is assigned to the axial proton. These assignments reflect the expectation of strong coupling between the axial α- and β-protons.51 The remaining aliphatic proton resonances (E, F) appear as complex multiplets further up field (1.4–1.9 ppm). As expected, the spectra of the complexes with X− and PdnX2n+22− counter-anions are essentially identical in CD3CN.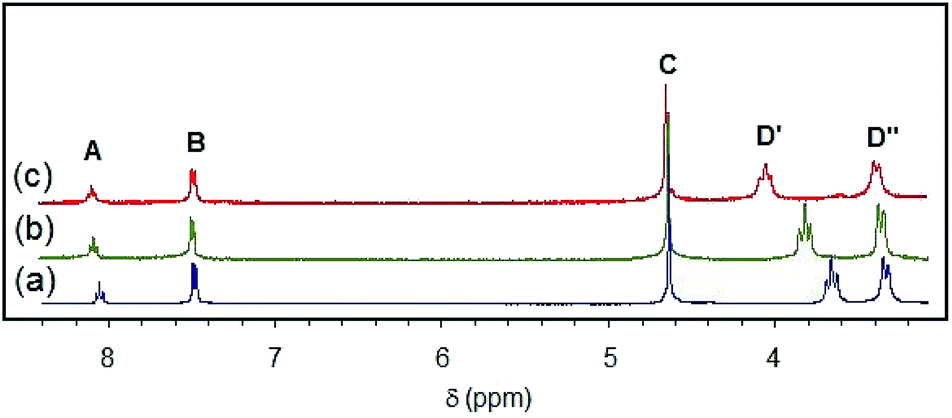 | ||
| Fig. 2 1H NMR spectra of (a) [Pd(pip2NNN)Cl]Cl, (b) [Pd(pip2NNN)Br]Br, (c) [Pd(pip2NNN)I]I in CD3CN. | ||
With the exception of the axial α-piperidyl proton resonances (D′) each of the pip2NNN resonances is shifted downfield from that observed for the corresponding Pd(pip2NCN)X complex. For example, in CD3CN the para- and meta-pyridyl proton resonances (A/B) are shifted downfield by ∼1 ppm ([Cl]+, 1.12/0.74; [Br]+, 1.13/0.75; [I]+, 1.1/0.72 ppm) from those of Pd(pip2NCN)X. The shifts are smaller for the benzylic protons, C ([Cl]+, 0.37 ppm; [Br]+, 0.37 ppm; [I]+, 0.36 ppm) and the equatorial α-piperidyl proton, D′′ ([Cl]+, 0.15 ppm; [Br]+, 0.12 ppm; [I]+, 0.12 ppm). By contrast, the axial α-piperidyl proton resonance, D′, is shifted upfield by 0.19 and 0.14 ppm in the spectra of [Cl]+ and [Br]+, respectively, and downfield by 0.05 ppm in the spectrum of [I]+. Thus, the gap between D′ and D′′ resonances (D′–D′′: [Cl]Cl, 0.36; [Br]Br, 0.47; [I]I, 0.67 ppm) is smaller than observed for Pd(pip2NCN)X (X = Cl, 0.67; Br, 0.74; I, 0.76 ppm) and increases along the series Cl < Br < I (Cl, 0.33; Br, 0.48; I, 0.69 ppm). When [X]+ is treated with AgBF4 to give Pd(pip2NNN) (solvent)+, the α-piperidyl protons appear as a singlet at 3.37 ppm, indicating that Pd–N(piperidyl) bond cleavage and ring inversion are fast on the NMR timescale.
As noted for the M(pip2NCN)X (M = Pd, Pt; X = Cl, Br, I) series,33,37 the 1H NMR resonances for [X]+ undergo a slight downfield shift along the Cl < Br < I series (Fig. 2). The deshielding effect going from the chloro to iodo complex is greatest (0.40 ppm) for the piperidyl axial α-proton resonance (D′), exceeding shifts observed for the analogous M(pip2NCN)X complexes (Pt, 0.30; Pd, 0.16 ppm).52 The sensitivity of the axial proton resonances is consistent with crystal structure data for [Cl]Cl and [Br]BF4 showing that the axial protons are 0.8–1.0 Å closer to the halide ligand than the equatorial protons when the Pd center is at the equatorial position of the piperidyl N atom. The sensitivity of the remaining resonances to halide ligand substitution decreases along the A > D′′ > C > B series, which can be rationalized in terms of through-bond and through–space interactions. The trend along the Cl < Br < I series opposes the relative electronegativities of the halogen groups, as well as patterns in 195Pt NMR experimental and computational results.53 However, this behavior has been noted for related compounds54 and is consistent with structural, spectroscopic and reactivity patterns of many transition metal complexes.55 Antipin, Grushin and coworkers have argued that similar trends in crystallographic and NMR data for trans-Pd(PPh3)2(Ph)X (X = F, Cl, Br, I) can be understood in terms of filled/filled repulsions between the dπ orbitals of the metal center and the lone pair orbitals of the halide ligands. Infrared studies of five-coordinate RuHX(CO)(P(CMe3)2Me)2 (X = F, Cl, Br, I) have established that the carbonyl stretching frequency increases along the F < Cl < Br < I series, indicating that filled/filled repulsions decrease along this series.54,56 The deshielding of the pip2NNN ligand resonances along the Cl < Br < I series is likewise consistent with decreasing filled/filled repulsions and electron releasing properties of the Pd–X unit.
Anion dependence of the chemical shift
The 1H NMR spectra of samples of [X]+ salts dissolved in CDCl3 are qualitatively similar to those obtained for samples dissolved in CD3CN with the surprising difference that the chemical shifts of the cation are strongly dependent on the nature of the anion. The influence of the counter anion is strongest for the meta-pyridyl and benzylic resonances which is the reverse of the sensitivity of these resonances to changes in the coordinated halide ligand. As shown in Fig. 3(a) and (b), the spectra of [Cl]BF4 and [Cl]Cl in CDCl3 are distinctly different. The meta-pyridyl (B) and benzylic (C) proton resonances of the BF4− salt are shifted upfield by 0.27 and 0.23 ppm, respectively. By contrast, the chemical shifts of the remaining resonances are nearly identical to those of the chloride salt. When slightly more than one equivalent of tetrabutylammonium chloride, TBACl, is added to a chloroform solution of [Cl]BF4, the benzylic and meta-pyridyl resonances shift back to where they appeared for the [Cl]Cl complex. However, when TBABF4 is added to a sample of [Cl]BF4, no appreciable change (<0.05 ppm) is observed in the chemical shift of any resonance. Likewise, addition of TBACl to the [Cl]Cl sample does not shift any of the [Cl]Cl resonances significantly.57 The strong influence of one equivalent of chloride ion on the NMR spectrum is indicative of an interaction between the cation and exogenous chloride anion.58–60 These effects are attenuated in CD3CN. The 1H NMR spectra of [Cl]Cl and [Cl]BF4 in CD3CN are very similar (<0.07 ppm difference), and addition of TBACl does not alter either spectrum. It is reasonable to expect that Cl−, as well as [Cl]+ and BF4−, is better solvated in the higher dielectric solvent (ε: CH3CN, 37.5; CHCl3, 5.5). In keeping with this interpretation, addition of one equivalent of TBABF4 to a CDCl3 solution of the [Cl]Cl modestly shifts the resonances toward their positions in the [Cl]BF4 spectrum; for example, the benzylic resonance C shifts upfield by 0.09 ppm. These observations are consistent with decreasing cation/halide interaction with increasing ionic strength.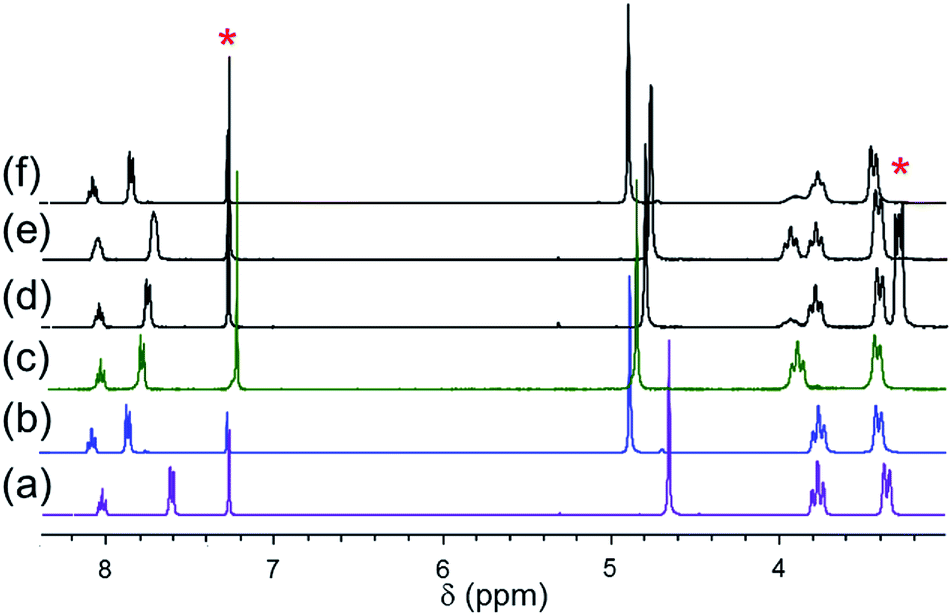 | ||
| Fig. 3 1H NMR spectra of (a) [Cl][BF4], (b) [Cl]Cl, (c) [Br]Br, (d) 1:1 [Cl][BF4]/TBABr, (e) 1:1 [Cl][BF4]/[Br]Br, (f) 1:1 [Cl]Cl/[Br]Br in CDCl3. * denotes solvent residual resonance and TBABr resonance. | ||
Conductivity measurements in acetonitrile confirm that [Cl]BF4 and [Cl]Cl are essentially 1:1 electrolytes (0.12 mM [Cl]Cl, 138 S cm2 mol−1; 0.12 mM [Cl]BF4, 155 S cm2 mol−1). Interestingly, even in dilute chloroform solution, both salts are essentially non-electrolytes (0.12 mM [Cl]Cl, <2 S cm2 mol−1; 0.12 mM [Cl]BF4, <2 S cm2 mol−1). This observation and the aforementioned 1H NMR data indicate that the cation–anion interaction for [Cl]Cl in CDCl3 is stronger than for [Cl]BF4 and places the metal complex in a significantly different chemical environment than for [Cl]BF4. The similarity between the spectra of [Cl]BF4 in CDCl3 and CD3CN, as well as the weak Lewis basicity of BF4−, are consistent with conventional ion-pairing. On the other hand, the cation–anion interaction for [Cl]Cl in CDCl3 causes a more significant perturbation of the NMR spectrum, possibly because the chloride counterion is engaged in an inner-sphere interaction with the metal to form a five-coordinate complex. Such a structure must be fluxional or symmetric such that the equivalency of the piperidyl groups is preserved on the NMR timescale.
The sensitivity of certain resonances to substitution of the halide ligand or the counter-anion is convenient for investigations of the influence of halide anion in mixtures. For example, when one equivalent of TBABr was added to a CDCl3 solution of [Cl]BF4 (Fig. 3(d)), each resonance (except D′) appears at chemical shifts that are close to the average values for pure [Cl]BF4 and [Br]Br solutions (e.g., C: 4.79 ppm (4.65 + 4.89)/2 = 4.77 ppm; B: 7.75 ppm (7.83 + 7.61)/2 = 7.72 ppm). Although the solution is a mixture of several species (i.e., [X]BF4 and [X]X where X = Cl, Br), the coordinated halide has little effect on the chemical shifts and consequently they are close to the averaged values. The D′ protons give rise to two distinct resonances in a 3:1 intensity ratio (Fig. 3(d)). The less intense resonance (3.92 ppm) is nearly coincident with that of the equatorial α-piperidyl proton resonance of [Br]Br (3.93 ppm), whereas the more intense resonance (3.75 ppm) is coincident with the α-piperidyl proton resonance of [Cl]Cl (3.75 ppm). Thus, only about 25% of the chloride ligand is replaced by bromide confirming the preference for chloride over bromide discussed previously. Assuming that the coordinated halide does not influence the chemical shifts of B and C (i.e., δ[Cl]BF4) = δ([Br]BF4); δ([Cl]Br = δ([Br]Br); δ([Br]Cl) = δ([Cl]Cl)) and that the [Cl]X:[Br]X ratio is 3:1 (i.e., the [X]Cl:[X]Br ratio is 1:3; X = Cl, Br), we estimate from the observed chemical shift that in a 1:1 [Cl]BF4:TBABr mixture, the BF4− ion pair and the halide adduct [X]X are in a 9:10 ratio. This implies that the interaction strengths of BF4− and Br− are similar. Furthermore, making similar assumptions about a 1:1 [Cl]BF4:TBACl mixture leads to the conclusion that the BF4− ion pair and the Cl− adduct [Cl]Cl are in a 2:5 ratio, confirming that association with Cl− is stronger than with BF4−.
When [Cl]BF4 is mixed with one equivalent of [Br]Br at room temperature (22 °C), the aromatic and benzylic resonances are broad and appear at average chemical shifts of the corresponding resonances of the pure solutions (Fig. 4(e)). The two D′ piperidyl resonances appear in a 1:1 intensity ratio and are coincident with the D′ resonances in the spectra of [Cl]Cl and [Br]Br, respectively. As the [Cl]BF4/[Br]Br mixture is cooled, the aromatic and benzylic resonances split, revealing two sets of nearly overlapping resonances (Fig. 4(c–d)). By contrast, the D′ and other piperidyl resonances sharpen (Fig. 4). The coalescence temperatures (Tc) of A, B and C are ∼25 °C. The two D′ resonances coalesce at ∼60 °C but no coalescence is observed for the diastereotopic α-piperidyl resonances, D′ and D′′, at ≤60 °C (Fig. 4(f)). The Eyring plot of D′ resonance is slightly nonlinear, becoming more shallow at low temperature. This behavior suggests underlying complexity, such as a change in rate-limiting step. Under the assumption of linearity, ΔH‡ and ΔS‡ are estimated to be approximately 11 kcal mol−1 and −0.01 kcal mol−1 K−1, respectively. These values are in good agreement with those for halide exchange reactions of square planar Pt(II)61,62 and Pd(II)63 complexes with amine ligands (ΔH‡, 10 to 22 kcal mol−1; ΔS‡, −30 to −16 cal mol−1 K−1).
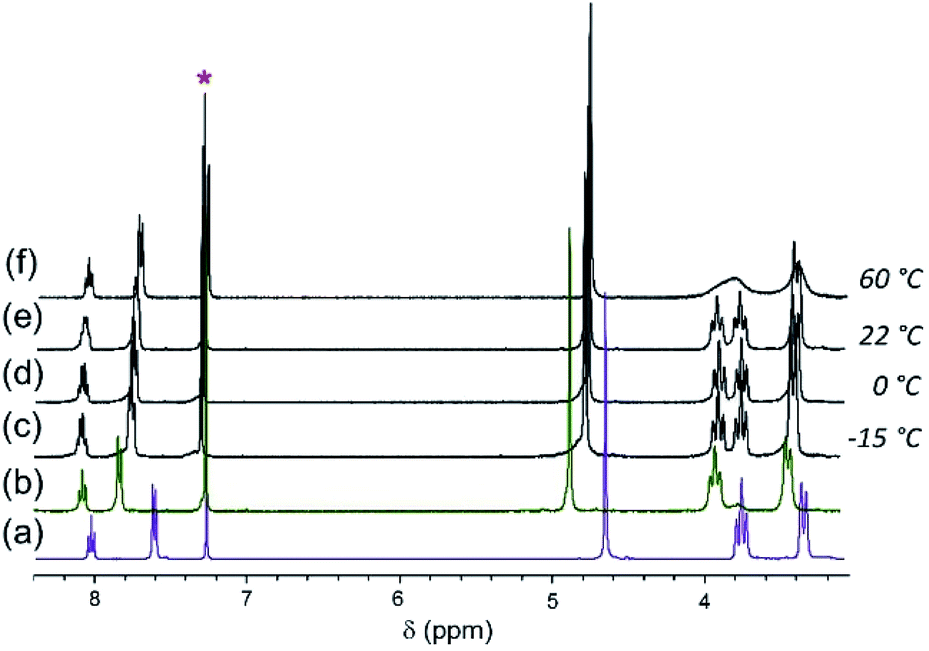 | ||
| Fig. 4 1H NMR spectra of (a) [Cl][BF4] and (b) [Br]Br in CDCl3. Spectra of a 1:1 mixture of [Cl]BF4/[Br]Br at different temperatures (c)–(f) as shown in the picture. * denotes solvent residual resonance. | ||
Because of the similarities in the spectra of [Cl]Cl and [Br]Br in CDCl3, the changes in chemical shifts are comparatively modest when [Cl]Cl is mixed with one equivalent of [Br]Br (Fig. 5(e)). As expected, [Cl]+ is favored over [Br]+, as indicated by the 3:1 intensity ratio of the D′ resonances. As the solution is cooled, the aromatic and benzylic resonances, A, B and C, broaden but no splitting is observed at ≥−15 °C (Fig. 5(c–d)). On the other hand, the piperidyl resonances, D′ and D′′ (and also E and F, not shown here), are sharp at <0 °C. The two D′ resonances coalesce at approximately 40 °C. At 60 °C, the diastereotopic α-piperidyl resonances, D′ and D′′, are slightly closer to coalescence than in the spectrum of 1:1 [Cl]BF4/[Br]Br (Fig. 4(f)). The estimation of thermodynamic parameters for the halide ligand exchange is complicated due to the inequality in the intensity ratio of the two D′ resonances.
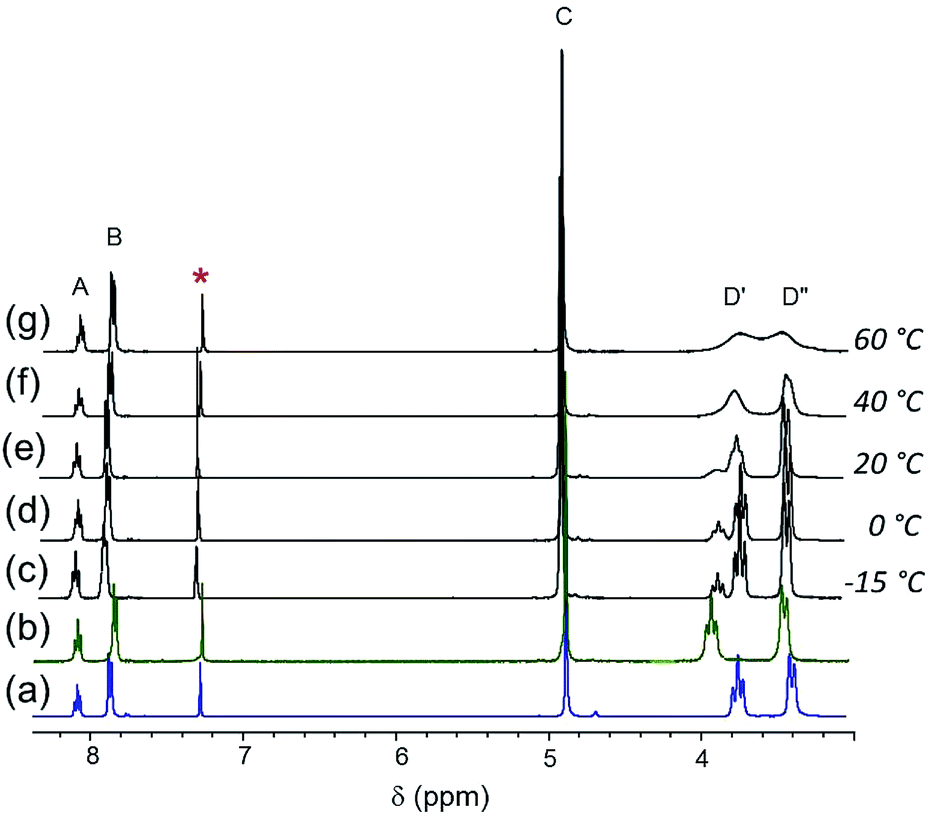 | ||
| Fig. 5 1H NMR spectra of (a) [Cl]Cl and (b) [Br]Br in CDCl3. Spectra of 1:1 mixture of [Cl]Cl/[Br]Br at different temperatures (c)–(g) as shown in the picture. * denotes solvent residual resonance. | ||
Additionally, as the D′ resonances approach coalescence, the D′ and D′′ resonances begin to move toward each other (Fig. 5(g)). Consequently, because of the exchange process between D′ and D′′, the system can no longer be analyzed as two resonances coalescing. Treating the system as an unequally populated two-site exchange system,64,65 with a coalescence temperature between 40–45 °C gives the barrier to halide ligand exchange (ΔG‡) between 15.7–15.9 kcal mol−1. Assuming the same ΔS‡ as the 1:1 [Cl]BF4/[Br]Br mixture (−0.01 kcal mol−1), ΔH‡ is calculated to be in the 12.5–12.7 kcal mol−1 range. On the other hand, the barrier to exchange for the [Cl]BF4/[Br]Br mixture is calculated to be between 16.2–16.4 kcal mol−1 and ΔH‡ at a coalescence temperature between 55–60 °C is in the 12.9–13.1 kcal mol−1 range.
For a rate-determining intra-molecular rearrangement (e.g., [Br]Cl ↔ [Cl]Br), the dynamic process responsible for the coalescence of two different D′ resonances, assigned to [Cl]+ and [Br]+, is proposed to involve a 5-coordinate transition state. In other words, the barrier to halide ligand exchange likely reflects the instability of a 5-coordinate transition state species relative to [X]+ or the [X]+/anion adduct. Therefore, assuming that the BF4− is not directly involved, for both [Cl]BF4/[Br]Br and [Cl]Cl/[Br]Br mixtures, the transition state species is expected be essentially the same. Apart from the error introduced by the estimation methods, the variation in the barrier to halide ligand exchange values can be attributed to the presence of BF4− in one of the mixtures. The facts that the conductivity measurements suggest a strong interaction between the cation and BF4− and the mixing experiments show that [X]BF4 forms even if there is enough halide to coordinate to all [X]+ in a solution (X = Cl, Br), indicate that displacement of BF4− is required before halide ligand exchange can occur. If the displacement of BF4− is involved in the rate determining step, the overall reaction pathway is anticipated to have a higher activation barrier. Another possibility is that the rate determining step is bimolecular, and the rate of exchange and the coalescence temperature depend on halide ion concentration. Since the halide ion concentration is higher for the [Cl]Cl/[Br]Br mixture, a lower barrier is expected, which is qualitatively consistent with what we have observed. It is noteworthy that the ionic strength of the mixture is not expected to have a significant influence since according to conductivity measurements the solutions do not contain many free ions. Although the water resonance shifts upfield as the temperature is raised in both mixtures, this does not influence the chemical shifts of the complexes' resonances measurably.
There is precedent for the interaction of four-coordinate palladium(II) complexes with exogenous halide anions.66,67 For example, the conductivity measurements and NMR spectroscopy show that five coordinate Pd(N^N^N)(CH3)Cl (N^N^N = 2-(2-((2′-pyridylmethylene)amino)ethyl)pyridine) is favored at low temperatures in chlorinated solvents, whereas a square planar geometry with Cl− as the counter anion is observed in acetonitrile. At higher temperatures, the neutral species with bidentate N^N^N ligand forms regardless of the solvent.66 Not surprisingly, the flexibility of the N^N^N ligand stabilizes the five coordinate species at low temperatures in non-coordinating solvents. A similar Pd(II) complex with a phosphorus-bis(nitrogen) ligand, [Pd(η3-PNN)CH3]Cl (PNN = N-(2-(diphenylphosphino)benzylidene)(2-((2-pyridyl)ethyl)amine)) is reported to be ionic in acetonitrile and molecular in chloroform as indicated by conductivity measurements.67 Compared to the N^N^N and PNN ligands, the pip2NNN ligand is more rigid, and regardless of solvent or temperature no evidence of asymmetric or bi-dentate coordination is observed by 1H NMR spectroscopy. The preservation of symmetry in the 1H NMR spectra could be due to fast exchange, ion pairing or formation of an adduct that preserves the mirror plane symmetry. In the latter case, one possibility is to position the pip2NNN and the two halide ligands in the same coordination plane. A more likely possibility is the preservation of the C2v symmetry by positioning the halide ligands above and below the plane defined by pip2NNN.
To assess the strength of the association between the halide counterion and the palladium cation, 1H NMR spectra were recorded of a chloroform solution of [Cl]Cl at different concentrations. The effective association constant was estimated68 to be ∼104 M−1 from variations in the meta-pyridyl resonance shifts, indicating strong binding of the counter halide (Fig. 6). The fact that [Cl]Cl solutions are nonconductive even at low concentrations supports the notion that the interaction between the [Cl]+ and Cl− is strong. The red curve represents the fitting carried out with the assumption that there are only two species in solution, namely one with a specific interaction between the cation and the anion and the other fully dissociated. The species with a specific interaction is represented by the [Cl]Cl chemical shift and the fully dissociated species, [Cl]+Cl−, is represented by the [Cl]BF4 chemical shift. Two factors are considered for the poor fit of the red curve. First is the presence of a third form of the complex, [Cl]+/Cl−, in which the cation and the anion are paired in a non-specific manner. The second reason is that the [Cl]BF4 chemical shift is not a good representative for the chemical shift of the fully dissociated [Cl]+Cl− species. On the contrary, the lack of conductivity and 1H NMR spectroscopy suggest an interaction between the [Cl]+ cation and the BF4− counter anion. Thus, the [Cl]BF4 spectrum may resemble more to the spectrum of the ion paired species. However, it is not expected to be the same since the interaction of Cl− with the cation is stronger than BF4.
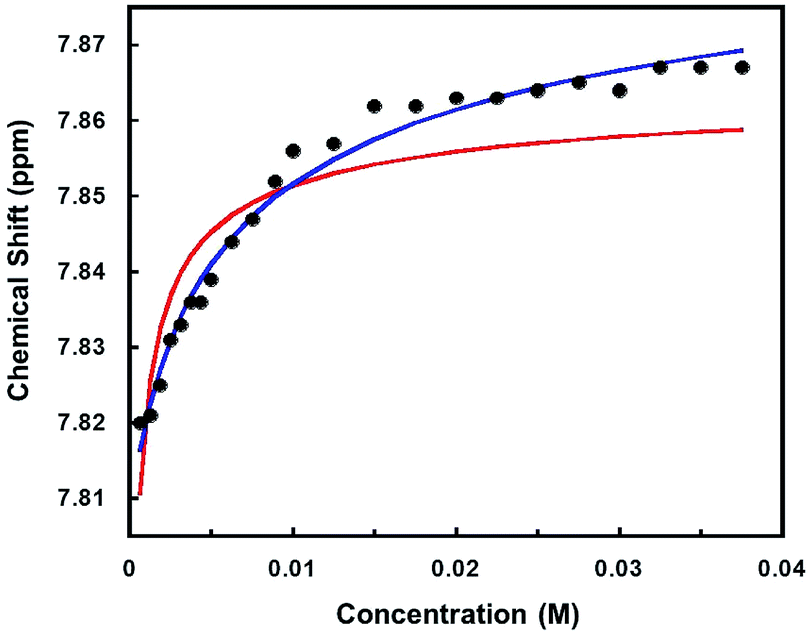 | ||
| Fig. 6 The chemical shifts of meta CH protons vs. concentration of [Cl]Cl (0.0006–0.04 M) in CDCl3. The red and blue curves illustrate the best-fit curves according to:δ = δ[Cl]+X− + ((δ[Cl]Cl − δ[Cl]+X−)/2C)(Kd + 2C − [(Kd + 2C)2 − 4C2]1/2), where C is the concentration of [Cl]+. For the red curve, X = BF4, Ka = 1/Kd, = 2.4 × 104 M−1, the chemical shift at infinite dilution (δ[Cl]+X−) is assumed to be that of a solution of [Cl]BF4 (7.618 ppm) and the chemical shift of the fully associated species (δ[Cl]Cl) is assumed to be that of a solution of [Cl]Cl (7.867 ppm). For the blue curve, the chemical shifts at infinite dilution and full association are optimized to obtain the best fit, X = Cl, Ka = 194 M−1, δ[Cl]+X− = 7.808 ppm, δ[Cl]Cl = 7.897 ppm. | ||
When the chemical shifts of the two limiting species are assumed to be different than the ones of [Cl]Cl and [Cl]BF4 and allowed to be varied along with Ka, a better fit is obtained (Fig. 6, blue curve). In this case, the calculated chemical shifts of the species at full association (7.897 ppm) and infinite dilution (7.808 ppm) are higher than the chemical shifts of [Cl]Cl (7.867 ppm) and [Cl]BF4 (7.618 ppm), respectively, whereas the Ka (∼102) is two orders of magnitudes lower. Since conductometry does not support a high degree of dissociation at low concentrations, it is probable that the chemical shift calculated for the species at infinite dilution is actually that of the ion paired species ([Cl]+/Cl−). It should be added that unlike other Pd(II) complexes,58 significant formation of charged aggregates at high concentrations is not supported by conductometry. No appreciable change in the conductivity with concentration increase was recorded when the concentration of the [Cl]Cl solution was raised to 0.01 M.
An associative mechanism is favored for ligand exchange reactions of related Pd(II) pincer ligand complexes,69,70 as in the case of insertion of CO into the Pd–C bond of Pd(Me4NNN)R+ type cations (Me4NNN = 2,6-bis(dimethylamine-methyl)-pyridine; R = methyl, phenyl, naphthyl). In that case, the transient species were modeled as Pd being coordinated by the Me4NNN ligand in a bidentate fashion along with the R and CO groups. Interestingly, although a five-coordinate structure was not energetically favored; the transition state was found to be stabilized by an interaction between the non-coordinated amine and the Pd center. In general, a five-coordinate intermediate(s) and/or a transition state(s) with either square pyramidal or trigonal bipyramidal geometries are proposed for square planar Pd(II) complexes. As illustrated in Scheme 2, the accumulated data are consistent with association of the cation and halide anion prior to halide exchange, which is suggested to involve a five-coordinated intermediate.
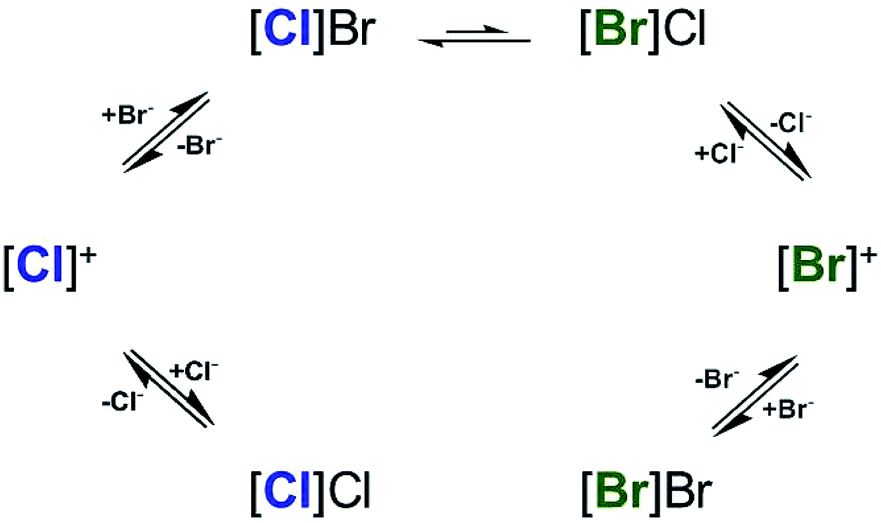 | ||
| Scheme 2 Suggested mechanism of halide exchange in CDCl3 based on the 1HNMR data (Fig. 5). [X]X represents [Pd(pip2NNN)(X)]+ associated with X− (X = Cl, Br). | ||
There is considerable evidence that the equilibrium lies toward the Cl− coordination. A reaction profile for the top portion of the scheme describing Cl/Br exchange is shown in Scheme 3. The reaction profile takes shape depending on the nature of the leaving and the entering ligands. When the leaving group, Cl−, is bonded more strongly to Pd than the entering group, Br−, the transition state is anticipated to have more Pd⋯Cl bond dissociation character. It is noteworthy that, since in most cases Br− bonds more tightly to Pd(II) than Cl−, a reaction profile in which the transition state with more Pd⋯Br bond dissociation character is more commonly encountered. In the present case, the size of the Cl− ligand and the filled/filled repulsions between the dπ orbitals of the metal center and the lone pair orbitals of the halide ligand are anticipated to contribute to the preference for Cl− over Br− coordination.
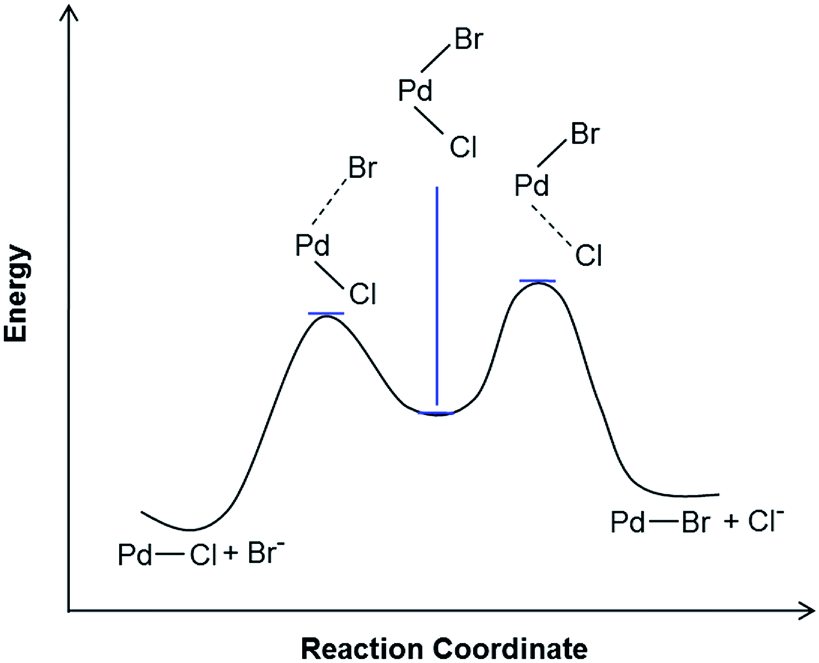 | ||
| Scheme 3 Proposed reaction profile for the halide exchange reaction; [Cl]+ + Br− → [Br]+ + Cl−. The transition state is proposed to have more Pd⋯Cl bond dissociation character. | ||
Conclusions
Although palladium complexes with ECE type pincer ligands have been studied extensively, neutral NNN pincer ligands with pyridine central moieties have not been explored as much. New square planar palladium(II) complexes with pip2NNN pincer ligand have been prepared: [Pd(pip2NNN)X]X (X = Br, I). Unlike the NCN pincer analogue, formation of bridged Pd2X62− or single PdX42− anions (X = Cl, Br, I) from the same reaction set up is observed.1H NMR spectra of [Pd(pip2NNN)X]X show that the presence of the halide anion has the strongest influence on the benzylic and meta-CH resonances, whereas variations in the halide ligand have the strongest influence on the furthest downfield α-piperidyl resonance. This difference in sensitivity is used to study halide association. The difficulty in determination of the chemical shifts of the fully associated and fully dissociated species in a mixed anion solution lead to a somewhat broad estimation of the effective association constant (∼104 M−1 to 10−2 M−1). An associative mechanism where the transition state has more Pd⋯Cl dissociation character is anticipated which indicates preference of Cl versus Br coordination due to the smaller size of Cl and the stronger filled/filled repulsions between Pd dπ orbitals the Cl lone pair orbitals. The importance of exogenous halide anions on the kinetics of outer-sphere two-electron transfer has been noted for 5-coordinate palladium(II) complexes.71,72 The solvent and anion dependence of the interaction between the exogenous anion (Cl−, Br−, BF4−) and the palladium cation (Pd(pip2NNN)X+, X = Cl, Br) open possibilities for further studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to Dr Janette Krause for help with characterization. We thank the National Science Foundation (Grant CHE0134975) and the Arnold and Mabel Beckman Foundation for support. We also thank CWRU for support of the Center for Chemical Dynamics.References
- K. J. Szabó, Synlett, 2006, 811–824 CrossRef.
- L. González-Sebastián and D. Morales-Morales, J. Organomet. Chem., 2019, 893, 39–51 CrossRef.
- M. R. Eberhard, Org. Lett., 2004, 6, 2125–2128 CrossRef CAS PubMed.
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- M. Q. Slagt, D. A. P. van Zwieten, A. J. C. M. Moerkerk, R. J. M. K. Gebbink and G. van Koten, Coord. Chem. Rev., 2004, 248, 2275–2282 CrossRef CAS.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS PubMed.
- D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, 1st edn, 2007 Search PubMed.
- D. Morales-Morales, Pincer Compounds, Elsevier, 2018 Search PubMed.
- P. O'Leary, C. A. van Walree, N. C. Mehendale, J. Sumerel, D. E. Morse, W. C. Kaska, G. van Koten and R. J. M. K. Gebbink, Dalton Trans., 2009, 4289–4291 RSC.
- M. Albrecht, R. A. Gossage, G. van Koten and A. L. Spek, Chem. Commun., 1998, 1003–1004 RSC.
- M. Albrecht, N. J. Hovestad, J. Boersma and G. van Koten, Chem.–Eur. J., 2001, 7, 1289–1294 CrossRef CAS.
- M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nature, 2000, 406, 970–974 CrossRef CAS PubMed.
- W. W. Gerhardt, A. J. Zucchero, J. N. Wilson, C. R. South, U. H. F. Bunz and M. Weck, Chem. Commun., 2006, 2141–2143 RSC.
- J. He, A. M. Bohnsack, N. W. Waggoner, S. G. Dunning, V. M. Lynch, W. C. Kaska and S. M. Humphrey, Polyhedron, 2018, 143, 149–156 CrossRef CAS.
- G. Rodríguez, M. Albrecht, J. Schoenmaker, A. Ford, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2002, 124, 5127–5138 CrossRef PubMed.
- M. T. Johnson, Z. Džolić, M. Cetina, M. Lahtinen, M. S. G. Ahlquist, K. Rissanen, L. Öhrström and O. F. Wendt, Dalton Trans., 2013, 42, 8484–8491 RSC.
- I. Davidi, D. Hermida-Merino, K. Keinan-Adamsky, G. Portale, G. Goobes and R. Shenhar, Chem.–Eur. J., 2014, 20, 6951–6959 CrossRef CAS PubMed.
- S. G. Churusova, D. V. Aleksanyan, A. A. Vasil'ev, E. Y. Rybalkina, O. Y. Susova, Z. S. Klemenkova, R. R. Aysin, Y. V. Nelyubina and V. A. Kozlov, Appl. Organomet. Chem., 2018, 32, e4360 CrossRef.
- S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CrossRef CAS PubMed.
- N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodríguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784–2797 CrossRef CAS.
- M. Basauri-Molina, S. Hernández-Ortega and D. Morales-Morales, Eur. J. Inorg. Chem., 2014, 4619–4625 CrossRef CAS.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254–15255 CrossRef CAS PubMed.
- J. Takaya and N. Iwasawa, J. Synth. Org. Chem., 2013, 71, 417–424 CrossRef CAS.
- M. Mazzeo, M. Lamberti, A. Massa, A. Scettri, C. Pellecchia and J. C. Peters, Organometallics, 2008, 27, 5741–5743 CrossRef CAS.
- N. Solin, J. Kjellgren and K. J. Szabó, J. Am. Chem. Soc., 2004, 126, 7026–7033 CrossRef CAS PubMed.
- V. Subramaniyan, B. Dutta, A. Govindaraj and G. Mani, Dalton Trans., 2019, 48, 7203–7210 RSC.
- E. Peris, J. A. Loch, J. Mata and R. H. Crabtree, Chem. Commun., 2001, 201–202 RSC.
- S. G. Churusova, D. V. Aleksanyan, E. Yu. Rybalkina, O. Yu. Susova, V. V. Brunova, R. R. Aysin, Y. V. Nelyubina, A. S. Peregudov, E. I. Gutsul, Z. S. Klemenkova and V. A. Kozlov, Inorg. Chem., 2017, 56, 9834–9850 CrossRef CAS PubMed.
- S. G. Churusova, D. V. Aleksanyan, E. Yu. Rybalkina, Y. V. Nelyubina, A. S. Peregudov, Z. S. Klemenkova and V. A. Kozlov, Polyhedron, 2018, 143, 70–82 CrossRef CAS.
- F.-F. Hung, S.-X. Wu, W.-P. To, W.-L. Kwong, X. Guan, W. Lu, K.-H. Low and C.-M. Che, Chem.–Asian J., 2017, 12, 145–158 CrossRef CAS PubMed.
- T. Thirunavukkarasu, H. A. Sparkes, K. Natarajan and V. G. Gnanasoundari, Appl. Organomet. Chem., 2018, 32, e4403 CrossRef.
- J.-Y. Lee, J.-Y. Lee, Y.-Y. Chang, C.-H. Hu, N. M. Wang and H. M. Lee, Organometallics, 2015, 34, 4359–4368 CrossRef CAS.
- H. Jude, J. A. Krause Bauer and W. B. Connick, Inorg. Chem., 2002, 41, 2275–2281 CrossRef CAS PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- H. Jude, J. A. Krause Bauer and W. B. Connick, Inorg. Chem., 2005, 44, 1211–1220 CrossRef CAS PubMed.
- L.-L. Shi, Y. Liao, G.-C. Yang, Z.-M. Su and S.-S. Zhao, Inorg. Chem., 2008, 47, 2347–2355 CrossRef CAS PubMed.
- S. Tastan, J. A. Krause and W. B. Connick, Inorg. Chim. Acta, 2006, 359, 1889–1898 CrossRef CAS.
- L. F. Olsson, Inorg. Chem., 1986, 25, 1697–1704 CrossRef CAS.
- V. Anbalagan, R. Srinivasan and K. S. Pallavi, Transition Met. Chem., 2001, 26, 603–607 CrossRef CAS.
- A. Hofmann, D. Jaganyi, O. Q. Munro, G. Liehr and R. van Eldik, Inorg. Chem., 2003, 42, 1688–1700 CrossRef CAS PubMed.
- P. M. Gidney, R. D. Gillard and B. T. Heaton, J. Chem. Soc., Dalton Trans., 1973, 132–134 RSC.
- F. D. Lewis, G. D. Salvi, D. R. Kanis and M. A. Ratner, Inorg. Chem., 1993, 32, 1251–1258 CrossRef CAS.
- W. Zhang, C. Bensimon and R. J. Crutchley, Inorg. Chem., 1993, 32, 5808–5812 CrossRef CAS.
- H. Kunkely and A. Vogler, J. Organomet. Chem., 1998, 559, 215–217 CrossRef CAS.
- C. L. Choi and D. Phillips, Mol. Phys., 1998, 94, 547–554 CrossRef CAS.
- K. H. Leung, W. Szulbinski and D. L. Phillips, Mol. Phys., 2000, 98, 1323–1330 CrossRef CAS.
- X.-X. Lu, E. C.-C. Cheng, N. Zhu and V. W.-W. Yam, Dalton Trans., 2006, 1803–1808 RSC.
- Z. Hui Zhang, X. He Bu, Z. Ang Zhu and Y. Ti Chen, Polyhedron, 1996, 15, 2787–2792 CrossRef.
- C. B. Pamplin, S. J. Rettig, B. O. Patrick and B. R. James, Inorg. Chem., 2003, 42, 4117–4126 CrossRef CAS PubMed.
- P. Stoppioni, R. Morassi and F. Zanobini, Inorg. Chim. Acta, 1981, 52, 101–106 CrossRef CAS.
- S. M. Ansari, W. Robien, M. Schlederer and P. Wolschann, Monatshefte Chem., 1989, 120, 1003–1014 CrossRef CAS.
- The chemical shifts for Pt(pip2NCN)X were determined in CDCl3.
- T. M. Gilbert and T. Ziegler, J. Phys. Chem. A, 1999, 103, 7535–7543 CrossRef CAS.
- J. P. Flemming, M. C. Pilon, O. Y. Borbulevitch, M. Y. Antipin and V. V. Grushin, Inorg. Chim. Acta, 1998, 280, 87–98 CrossRef CAS.
- K. G. Caulton, New J. Chem., 1994, 18, 25–41 CAS.
- J. T. Poulton, M. P. Sigalas, K. Folting, W. E. Streib, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1994, 33, 1476 CrossRef CAS.
- As expected from the spectrum of TBACl, addition of TBACl shifts the water resonance in the [Cl]Cl and [Cl]BF4 spectra. Nevertheless, when a few equivalents of water are added to either [Cl]Cl or [Cl]BF4, no substantial shift is observed for the [Cl]+ resonances.
- B. Crociani, F. di Bianca, A. Giovenco and T. Boschi, Inorg. Chim. Acta, 1987, 127, 169–182 CrossRef CAS.
- A. Irving, K. R. Koch and M. Matoetoe, Inorg. Chim. Acta, 1993, 206, 193–199 CrossRef CAS.
- R. Romeo, N. Nastasi, L. M. Scolaro, M. R. Plutino, A. Albinati and A. Macchioni, Inorg. Chem., 1998, 37, 5460–5466 CrossRef CAS PubMed.
- U. Belluco, R. Ettorre, F. Basolo, R. G. Pearson and A. Turco, Inorg. Chem., 1966, 5, 591–593 CrossRef CAS.
- M. A. Tucker, C. B. Colvin and D. S. Martin, Inorg. Chem., 1964, 3, 1373–1383 CrossRef CAS.
- R. Roulet and H. B. Gray, Inorg. Chem., 1972, 11, 2101–2104 CrossRef CAS.
- H. Shanan-Atidi and K. H. Bar-Eli, J. Phys. Chem., 1970, 74, 961–963 CrossRef CAS.
- W. Egan, PhD thesis, Princeton University, 1971.
- R. E. Rulke, J. M. Ernsting, A. L. Spek, C. J. Elsevier, P. W. N. M. van Leeuwen and K. Vrieze, Inorg. Chem., 1993, 32, 5769–5778 CrossRef CAS.
- R. E. Ruelke, V. E. Kaasjager, P. Wehman, C. J. Elsevier, P. W. N. M. van Leeuwen, K. Vrieze, J. Fraanje, K. Goubitz and A. L. Spek, Organometallics, 1996, 15, 3022–3031 CrossRef CAS.
- C. S. Wilcox, Frontiers in Supramolecular Organic Chemistry and Photochemistry, 1991, pp. 123–143 Search PubMed.
- C. H. Langford and H. B. Gray, Ligand Substitution Processes, W. A. Benjamin, Inc., Reading, 1966 Search PubMed.
- R. J. Cross, in Advances in Inorganic Chemistry, ed. A. G. Sykes, Academic Press, 1989, vol. 34, pp. 219–292 Search PubMed.
- S. Chatterjee, Cooperative Two-Electron Reagents of Lower Transition Metals of Group 10, University of Cincinnati, 2009 Search PubMed.
- D. E. Janzen, D. G. VanDerveer, L. F. Mehne, D. A. d. S. Filho, J.-L. Brédas and G. J. Grant, Dalton Trans., 2008, 1872–1882 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section. Synthesis, UV visible absorption spectra and positive and negative ion spectra for the PdX42− and/or Pd2X62− salts. See DOI: 10.1039/c9ra05423e |
| ‡ William B. Connick passed away in 2018. |
| This journal is © The Royal Society of Chemistry 2019 |