
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinear PVA–DTPA–Gd conjugate for magnetic resonance imaging†
Weibing Xu *a,
Zhiyan Lin*b,
Guichen Lic,
Haitao Longa,
Mingyuan Dua,
Guorui Fua and
Lumei Pu*a
*a,
Zhiyan Lin*b,
Guichen Lic,
Haitao Longa,
Mingyuan Dua,
Guorui Fua and
Lumei Pu*a
aCollege of Science, Gansu Agricultural University, Lanzhou 730000, Gansu Province, P. R. China. E-mail: xuwb@gsau.edu.cn; Tel: +86 931 7631212
bClinical Medical College, Gansu University of Chinese Medicine, Lanzhou 730000, China. E-mail: 915290692@qq.com
cGansu Provincial Key Laboratory of Aridland Crop Science, Gansu Agricultural University, Lanzhou 730070, China
First published on 12th November 2019
Abstract
In this study, we report the preparation and characterization of the PVA–DTPA–Gd conjugate as a potential MRI contrast agent (CA). The r1 value and the r2/r1 ratio were about 5.6 mM−1 s−1 and 1.31, respectively. In vitro toxicity studies not only demonstrated that the polymeric system possessed good biocompatibility, but also proved that the conjugate could be an attractive candidate for CA.
Magnetic resonance imaging (MRI) is the most frequently used tool for the detection and diagnosis of cancer in clinical settings, due to its high-resolution, noninvasive monitoring manner and excellent tissue penetration depth.1–3 However, it is still hard to differentiate tumors from healthy tissues by MRI, due to its unsatisfactory detection sensitivity.4 Many paramagnetic contrast agents (CAs) have been demonstrated and applied to enhance the image contrast and to ultimately highlight the pathological areas. The positive CAs, so-called T1-weighted CAs, generate bright signals,5,6 whereas, the negative CAs with dark signals are called T2-weighted CAs.7 Gd3+-based T1 CAs are famous positive CAs with great safety, which have been widely applied clinically. Unfortunately, some Gd3+-based T1 CAs may trigger the development of nephrogenic systemic fibrosis with renal failure.8 Thus, the signal intensity, which is related to the concentration of CAs in the region of interest, should be improved to decrease its injected doses. Many studies have been focused on the development of various Gd-based T1-weighted contrast agents with high r1 value and low r2/r1 ratio; both parameters are used to characterize the performance of MRI.12 High relaxivity and low r2/r1 ratio could be achieved by a high payload of active magnetic centers according to the SBM theory.9–11 Forming covalent conjugations with macromolecules is an effective way to control the tumbling motion and ensure optimal water residency times.13 Various polymers, such as polyethylene glycol (PEG), poly(N-2-hydroxypropyl methacrylamide) (PHPMA), poly(lactic-co-glycolic acid (PLGA) and chitosan have emerged as promising carriers for delivering the Gd chelates. Typically, Lim et al. conjugated gadolinium chelates to a serial of polyamidoamine dendrimers, and the resultant CAs exhibited high r1 value and long circulation in the blood.14 Telechelic PEG-polymers end-capped with diphenylalanine motifs containing a DOTA–Gd complex bound on a lysine side chain at the centre of the peptide moiety were synthesized with relaxivity of around 11 mM−1 s−1 (B0 = 3.0 T).15 A dendronized heparin–gadolinium polymer CAs was reported and its r1 value was 16.3 mM−1 s−1 (B0 = 3.0 T).16 Guo et al. conjugated Gd–DOTA onto a biodegradable poly[N-(1,3-di-hydroxypropyl)methacrylamide] copolymer backbone through a GSH-sensitive cleavable disulfide bond to produce novel Gd-CAs; its relaxivity was 10.24 mM−1 s−1 (B0 = 3.0 T).17 In another report, multiple chelated Gd(III) ions were attached to the hydrazide modified dextran–poly(glycidyl methacrylate), and the prepared MRI CAs exhibited a satisfactory r1 value of 44.4 mM−1 s−1 (B0 = 3.0 T).18 As a kind of synthetic polymer, polyvinyl alcohol (PVA) is an interesting commercial polymer with the highest annual production of over 1.2 billion kg.19 PVA is non-toxic, biodegradable, biocompatible and low-cost, making it a promising potential component of sustainable and degradable commodity plastics.20–23 A few studies on PVA-based CAs have been published and the majority of them involved complicated methods that are not conducive to the large-scale production of CAs, with increased security risks and cost. For example, Cilliers et al. reported the synthesis and characterization of PVA embolic particles modified with DTPA–Gd, with the help of dangerous n-butyl lithium.24 Tachibana et al. fabricated poly(vinyl alcohol)–gadolinium CA via two complex and costly steps. (i) PVA was stirred with carbonyl diimidazole, then 1,3-propanediamine was added to the mixture and the product was obtained by freeze-drying. (ii) The aminated PVA was reacted with DOTA–NHS–ester to form the PVA–DOTA.25
In order to simplify the reaction process of PVA-based CAs, a novel preparation strategy for PVA–DTPA–Gd conjugate was proposed and realized in this study. As Scheme 1 shows, PVA–DTPA was synthesized by grafting DTPA onto PVA via the reaction between the anhydrides of DTAP and –OH groups in PVA. The biodegradable PVA–DTPA–Gd conjugate was successfully constructed by the chelating reaction between DTPA and Gd3+. By varying the feed ratio, the loaded content of DTPA could be facilely and securely regulated. The macromolecular contrast agents could efficiently retard rotational motion, increase rotational correlation time and prevent rapid renal clearance.
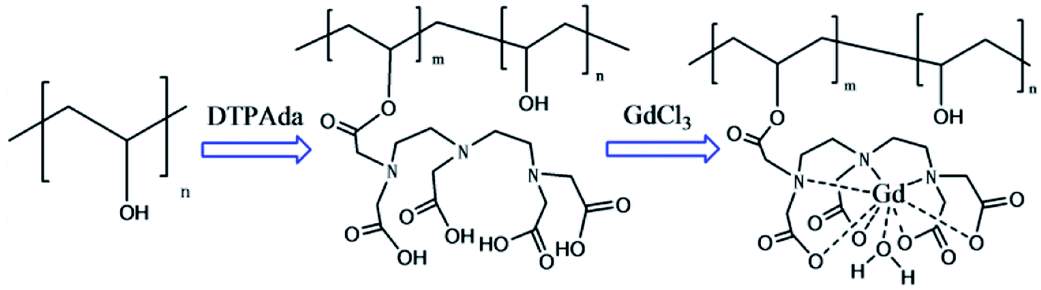 | ||
| Scheme 1 The synthesis of the PVA–DTPA–Gd conjugate. | ||
A series of characterizations was applied to confirm the successful preparation of the polymer. Firstly, the free PVA, PVA–DTPA and PVA–DTPA–Gd were all characterized by FT-IR, and the results are given in Fig. S1.† Compared with the spectrum of PVA, a pair of new vibration peaks appeared in the spectrum of PVA–DTPA–Gd at 1724 and 1639 cm−1, which could be ascribed to the stretching vibration absorption peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the ester bond and free carboxyl.26 Furthermore, a stretching vibration absorption peak of C–N appeared at 1225 cm−1.27 All the characteristic peaks demonstrated the successful synthesis of PVA–DTPA. For PVA–DTPA–Gd, the peak at 1639 cm−1 disappeared and a new vibration peak appeared at 1591 cm−1, suggesting that the coordination interaction between the CO group in the PVA–DTPA and metal Gd3+ ions may take place.28 Additionally, a weak peak at 520 cm−1 was found in the PVA–DTPA–Gd conjugate, which could be assigned to the stretching vibration of the Gd–O bond.28
O in the ester bond and free carboxyl.26 Furthermore, a stretching vibration absorption peak of C–N appeared at 1225 cm−1.27 All the characteristic peaks demonstrated the successful synthesis of PVA–DTPA. For PVA–DTPA–Gd, the peak at 1639 cm−1 disappeared and a new vibration peak appeared at 1591 cm−1, suggesting that the coordination interaction between the CO group in the PVA–DTPA and metal Gd3+ ions may take place.28 Additionally, a weak peak at 520 cm−1 was found in the PVA–DTPA–Gd conjugate, which could be assigned to the stretching vibration of the Gd–O bond.28
The polymer composition was also studied by 1H NMR spectroscopy, and the NMR spectra of the PVA and PVA–DTPA are shown in Fig. S2.† The spectra show a broad singlet signal observed at 4.70 ppm, which corresponds to the chemical shifts of protons in the –OH group from the PVA repeating units (Fig. S2a†). The broad singlet signal at δ = 3.86 ppm could be attributed to the methane proton from the –CH–(OH) group. The spectrum also showed a characteristic signal of methylene protons from the main polymer chain of the PVA backbone at δ = 1.40–1.90 ppm.29 In Fig. S2b,† the resonance signals corresponding to methylene protons (–N–(CH2)2–N–) and –CH2COOH from the DTPA group were located at δ = 3.78 and 3.02 ppm, respectively (marked as k and l),30,31 suggesting that the DTPA group was successfully bonded to the PVA.
The thermal stability curves of the PVA and PVA–DTPA–Gd conjugate are presented in Fig. S3.† The low-temperature weight loss from 25 °C to 200 °C in both samples corresponds to the evaporation of the physically adsorbed water.32 The PVA showed a dramatic mass loss of ∼60% from around 250 °C to 350 °C, which was due to the oxidation of –OH groups in the polymer.33 The slow mass loss that occurred between 350 °C and 500 °C could be assigned to the combustion of the polymer. The weight loss of the PVA–DTPA–Gd conjugate also included two parts. The smooth and sharp weight losses were observed when the temperature ranged from 100 °C to 260 °C and 260 °C to 500 °C, respectively. By assuming that pure Gd2O3 with a weight percentage of 12.8% in PVA–DTPA–Gd conjugate, was the main product of the TGA measurement, it could be estimated that the Gd3+ content in the initial PVA–DTPA–Gd conjugate was about 11.2%, and the rest could be the PVA–DTPA.34
The molecular weight and polydispersity index of PVA and the PVA–DTPA–Gd conjugate were determined via GPC (Fig. S4†). The results clearly indicated that the obtained curves are monomodal and symmetrical with a relatively narrow molecular weight distribution for both samples.35 Tailing peaks were observed at neither the lower nor higher molecular weight sides, indicating the complete and successful synthesis of the target polymer.36 The main molecular characteristics are listed in Fig. S4.† The Mw and Đ values of PVA were determined to be 5.73 × 104 g mol−1 and 1.139, respectively, and the Mw and Đ values of the PVA–DTPA–Gd were 6.36 × 104 g mol−1 and 1.223, respectively. Previous studies demonstrated that the longitudinal relaxivities were molecular weight dependent: the higher the molecular weight, the greater the longitudinal relaxivity.37 It was also found that the molecular weights of the polymer samples were greater than 5 × 104 g mol−1, which may result in the high r1 value of CAs.
The morphology of the PVA–DTPA–Gd conjugate was analyzed by TEM (Fig. S5†). It was found that the PVA–DTPA–Gd exhibited an irregular size and looked like flakes, and the size was almost hundreds of nanometers. Furthermore, the EDX analysis of the PVA–DTPA–Gd shown in Fig. S5b† successfully demonstrated the distribution of Gd, O, N and C elements in the polymer conjugate. The result further implied that the Gd3+ ions were fully confined within the polymer during the process of coordination, and the two components of Gd3+ and PVA–DTPA were strongly coupled together in the conjugate. Furthermore, the signal of the Cu element was also found in Fig. S5b,† which might have been due to the copper mesh for testing. Additionally, it can be clearly observed that the PVA–DTPA–Gd conjugate had an irregular spherical structure with the diameter varying from 120 to 150 nm (Fig. S6†). The particle sizes obtained from TEM were smaller as compared to those obtained from DLS, which may be ascribed to the DLS samples being thoroughly dissolved in water, while the TEM samples were initially dispersed in buffer and then dried before measurements. Therefore, the DLS data represented the real hydrodynamic diameters.
In Fig. S7a.† The C, O, N and Gd elements are clearly shown in the XPS survey spectrum of the PVA–DTPA–Gd conjugate, also indicating that the Gd3+ ions were successfully intercalated into the polymer. Fig. S7b–d† shows the C 1s, O 1s and Gd 4d XPS spectra, respectively. The deconvoluted C 1s XPS spectra showed six peaks with energies of 284.4 eV, 284.8 eV, 285.2 eV, 285.6 eV, 287.5 eV and 288.4 eV, which could be assigned to the C atoms coming from C–C, C–O, C–O–C, C–N, O–CO and COOH, respectively.38,39 Furthermore, two distinct peaks could be identified in the high-resolution O 1s spectrum (Fig. S7c†): the peak located at 532.3 eV for the C–O groups and the peak at 533.6 eV from the CO group.39 In order to confirm that Gd3+ was successfully seeded in the PVA–DTPA–Gd polymer, high-resolution XPS spectra of Gd 4d were obtained (Fig. S7d†). Two strong binding peaks were observed; the binding energy of 142.8 eV could correspond to the Gd 4d5/2 energy level, and 153.6 eV could correspond to the Gd 4d3/2 energy level.40 The appearance of the Gd 4d binding peaks indicated the presence of the Gd3+ oxidation state in the PVA–DTPA–Gd. Regarding the aforementioned results, the existence of Gd–N and Gd–O bonds demonstrated the successful formation of the PVA–DTPA–Gd conjugate.
The stability of the PVA–DTPA–Gd was also tested, and the results are listed in Fig. S8.† In Fig. S8a,† the amount of Gd3+ released by the PVA–DTPA–Gd conjugate was carefully determined by UV-Vis. A calibration curve was obtained according to the functional relation between the Gd3+ concentration and absorption intensity. It was found that the absorption curves showed similar shapes, even under different concentrations (Fig. S8a† inset). The maximum absorption wavelength was located at around 656 nm. The intensity of the UV absorption peak increased along with the increasing of the concentration of PVA–DTPA–Gd and a good linear correlation was established between them with a correlation coefficient of R2 = 0.997. Thus, the amount of Gd3+ at each time interval could be calculated according to this linear equation. Fig. S7b† shows the release kinetics curve and the related UV spectrum is exhibited in the inset. Remarkably, it was found that the negligible Gd3+ was released in 75 h, indicating that the PVA–DTPA–Gd conjugate has excellent stability.
The cellular toxicity of CAs is an important aspect in biological applications. In this work, the cytotoxicity of the free DTPA–Gd and PVA–DTPA–Gd conjugate on a normal HUVEC cell line was evaluated via MTT assay. The viability of the cells was tested after incubation with free DTPA–Gd and the PVA–DTPA–Gd conjugate for 48 h, and both of the concentrations of the tested targets were selected in the range of 10 to 100 μg Gd3+ mL−1. According to Fig. 1, the viability of the HUVEC cells was still higher than 90% after incubation with the PVA–DTPA–Gd conjugate and free DTPA–Gd, respectively, even at a concentration of 100 μg Gd3+ mL−1 for a long incubation time (48 h). These results indicate that both the free DTPA–Gd and the PVA–DTPA–Gd conjugate demonstrate no serious toxic effects at high doses. In addition, it was found that the cytotoxicity of PVA–DTPA–Gd was lower than that of the free DTPA–Gd, demonstrating that the conjugated Gd ions are not easily released due to the excellent binding strength of DTPA to Gd3+ ions and the minimized cell interaction with Gd3+. Therefore, the nephrotoxicity risk induced by the conjugate is potentially lower than that of free DTPA–Gd at the same Gd dosages.41
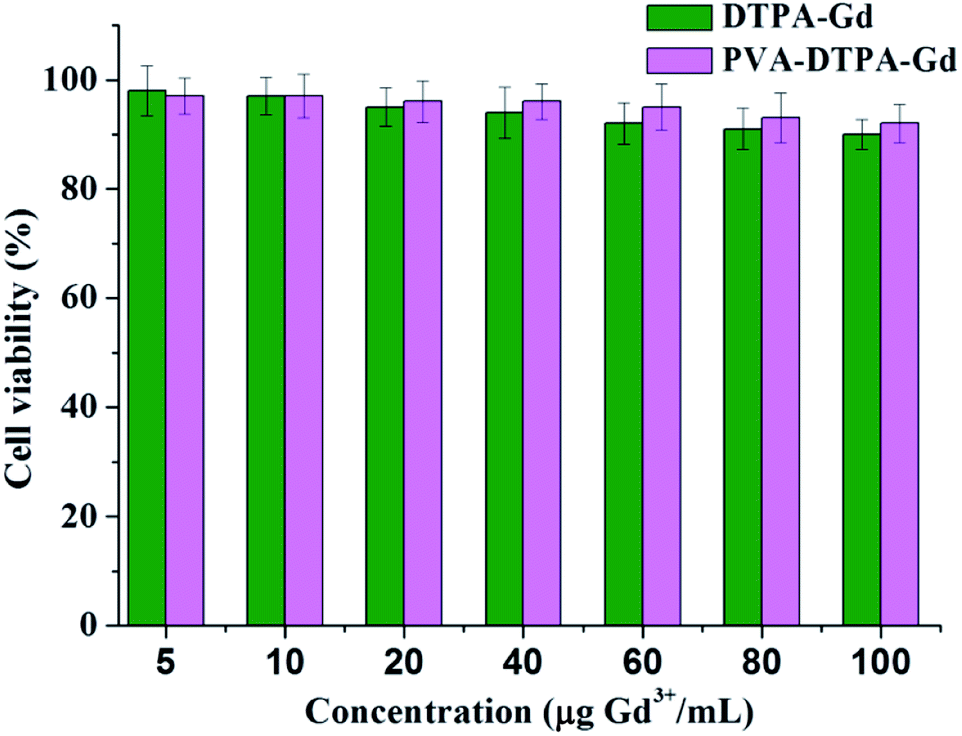 | ||
| Fig. 1 Cytotoxicity studies of PVA–DTPA–Gd in HUVEC cells after incubation for 48 h. | ||
Hemolysis is the destruction of the erythrocytes (red blood cells, RBCs) in the blood, while hemocompatibility is a necessary component of safety. Due to the interactions of the cell-polymer with the cell membrane, the membrane integrity is often measured as a proxy for hemocompatibility. When the cell membrane integrity of the RBCs is destroyed, hemoglobin is released and thus, the results can be used in quantitative analysis. The materials can then be defined as non-hemolytic (0–2% of hemolysis), slightly hemolytic (2–5%), and hemolytic (5–100%).40 As such, the blood compatibility of the PVA–DTPA–Gd conjugate was determined and the results are given in Fig. 2 and Fig. S9;† no hemolysis was observed, even at 1100 μg mL−1 of the experimental agents. When the concentration of the conjugate was 730 μg mL−1, the hemolysis was about 2.53 ± 0.31%, meaning that the conjugate is non-hemolytic. When the concentration of the conjugate increased to 1100 μg mL−1, the conjugate showed 4.67 ± 0.28% hemolysis, which is slightly hemolytic. These results indicate that that the prepared conjugate has no destructive effect on red blood cells, and it has good blood compatibility. This may be due to the fact that the acidic groups contained in the polymer hindered its interaction with red blood cells.
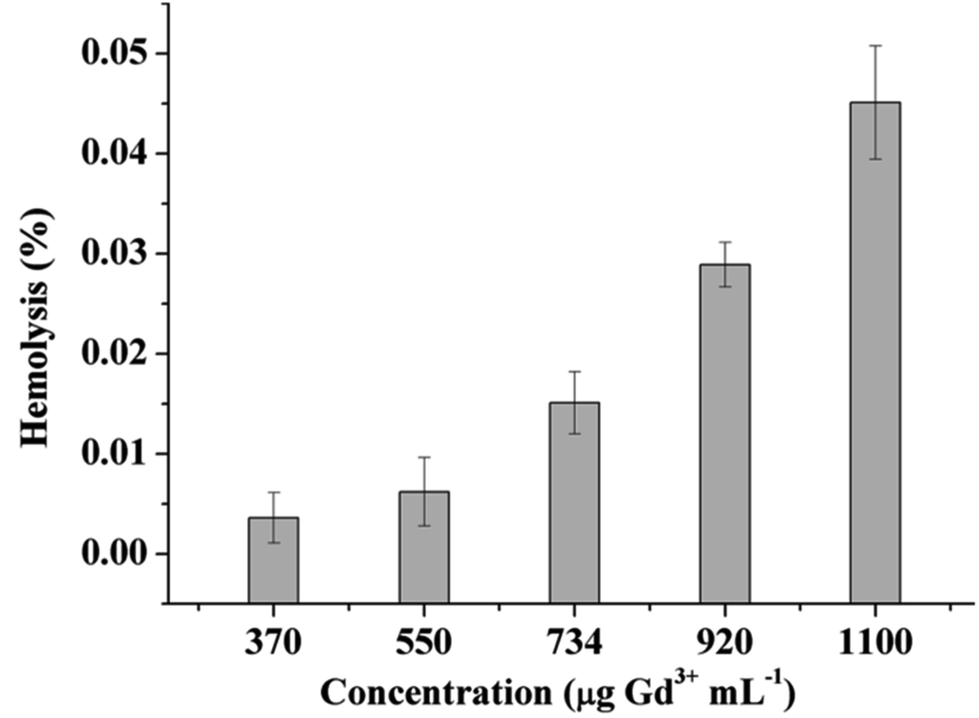 | ||
| Fig. 2 The hemolysis rate of PVA–DTPA–Gd. | ||
To evaluate the capability of the as-prepared PVA–DTPA–Gd conjugate in MRI application, longitudinal proton relaxation times (T1) and spin–spin relaxation times (T2) of the conjugate were determined. Based on this, the r1 and r2 relaxivity was calculated to be 5.6 and 7.3 mM−1 s−1 (Fig. 3a and b), respectively, by the linear fit of Gd3+ concentration vs. 1/T1 and 1/T2. Additionally, the r2/r1 ratio, another important parameter for characterizing contrast agents, was about 1.31, indicating that this conjugate is a kind of standard T1 CAs. Based on this, the r1 value of the PVA–DTPA–Gd conjugate was about 1.24 times higher than that of the DTPA–Gd (r1 = 4.5 mM−1 s−1). The r2/r1 ratio was lower than that of the corresponding small-molecule contrast agent, and the improved relaxivity can be explained by the complexation leading to slower molecular tumbling, thus reducing the relaxation time of water.42,43 These results successfully demonstrated the excellent contrast effect of the conjugate prepared in this work. The T1-weighted MR images of the PVA–DTPA–Gd conjugate and free DTPA–Gd solutions are exhibited in Fig. 3c and S9,† respectively. It is clear that the brightness of the MR images was enhanced with the increase in the concentration of Gd3+, as shown in Fig. 3c. It was also found from the black and white images that the conjugate exhibited better MRI efficiency as compared to the free DTPA–Gd (Fig. S10†). These results have demonstrated the great potential of the polymer conjugate as an effective contrast agent for T1-weighted MRI.
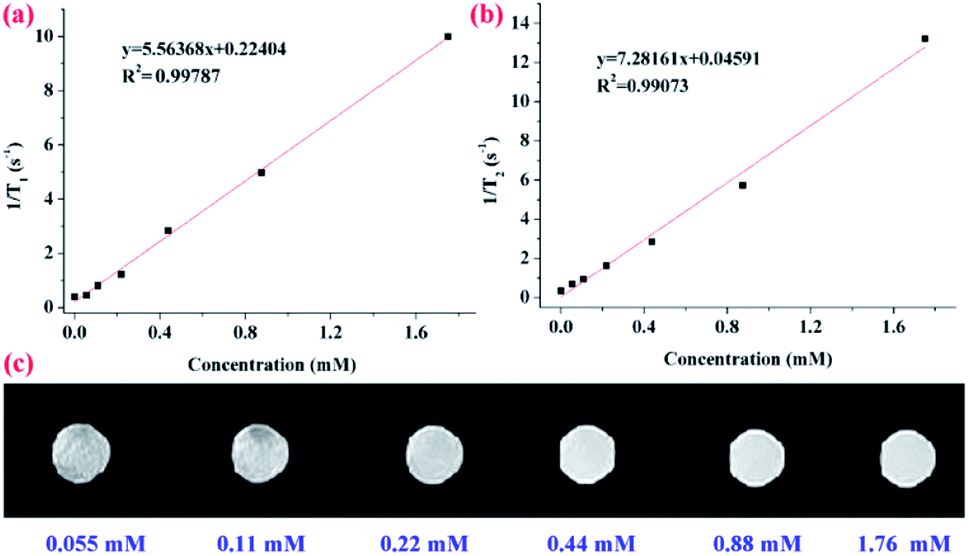 | ||
| Fig. 3 (a) T1, (b) T2-relaxation rate and (c) T1-weighted MRI images of PVA–DTPA–Gd. | ||
Conclusions
In this work, the small molecule chelating agent DTPA was covalently attached to PVA via a simple reaction. The loaded content of DTPA could be facilely regulated by adjusting the feed ratio. The MRI CAs of PVA–DTPA–Gd were successfully prepared via the reaction between the PVA–DTPA and Gd3+, and it showed satisfactory contrast effect. The mechanism of the contrast effect of the CAs was discussed, and the experimental results demonstrated that the polymer CAs had good compatibility.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the support of scientific research start-up funds for openly-recruited doctors (No: 2017RCZX-42) and special funds for discipline construction of Gansu Agricultural University (No: GAU-XKJS-2018-131). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Gansu Agricultural University and approved by the Animal Ethics Committee of Gansu Agricultural University.Notes and references
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi and J. G. Park, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- P. Mi, D. Kokuryo, H. Cabral, H. Wu, Y. Terada, T. Saga, I. Aoki, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2016, 11, 724 CrossRef CAS.
- W. Shang, C. Zeng, Y. Du, H. Hui, X. Liang, C. Chi, K. Wang, Z. Wang and J. Tian, Adv. Mater., 2017, 29, 1604381 CrossRef.
- T. Courant, V. G. Roullin, C. Cadiou, M. Callewaert, M. C. Andry, C. Portefaix and L. V. Elst, Angew. Chem., Int. Ed., 2012, 51, 9119–9122 CrossRef CAS.
- L. Sun, X. Li, X. Wei, Q. Luo, P. Guan, M. Wu and Q. Gong, ACS Appl. Mater. Interfaces, 2016, 8, 10499–10512 CrossRef CAS.
- Z. Zhou, Z. Zhao, H. Zhang, Z. Wang, X. Chen, R. Wang and J. Gao, ACS Nano, 2014, 8, 7976–7985 CrossRef CAS.
- F. Su, S. Agarwal, T. Pan, Y. Qiao, L. Zhang, Z. Shi and V. D. Kodibagkar, ACS Appl. Mater. Interfaces, 2018, 10, 1556–1565 CrossRef CAS.
- A. T. M. A. Rahman, P. Majewski and K. Vasilev, Contrast Media Mol. Imaging, 2013, 3, 92 CrossRef PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- E. Terreno, D. Delli Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019–3042 CrossRef CAS.
- N. Bloembergen and L. O. Morgan, J. Chem. Phys., 1961, 34, 842–850 CrossRef CAS.
- Z. Shen, T. Chen, X. Ma, W. Ren, Z. Zhou, G. Zhu, A. Zhang, Y. Liu, J. Song, Z. Li, H. Ruan, W. Fan, L. Lin, J. Munasinghe, X. Chen and A. Wu, ACS Nano, 2017, 11, 10992 CrossRef CAS.
- Z. Zhou, Y. Mondjinou, S. H. Hyun, A. Kulkarni, Z. R. Lu and D. H. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 22272–22276 CrossRef CAS.
- J. Lim, B. Turkbey, M. Bernardo, L. H. Bryant Jr, M. Garzoni, G. M. Pavan, T. Nakajima, P. L. Choyke, E. E. Simanek and H. Kobayashi, Bioconjugate Chem., 2012, 23, 2291–2299 CrossRef CAS.
- C. Diaferia, E. Gianolio, A. Accardo and G. Morelli, J. Pept. Sci., 2017, 23, 122–130 CrossRef CAS.
- C. Guo, L. Sun, W. She, N. Li, L. Jiang, K. Luo and Z. Gu, Polym. Chem., 2016, 7, 2531–2541 RSC.
- S. Guo, X. Xiao, X. Wang, Q. Luo, H. Zhu, H. Zhang and K. Luo, Biomater. Sci., 2019, 7, 1919–1932 RSC.
- H. Wang, T. T. Dai, B. L. Lu, S. L. Li, Q. Lu, V. Mukwaya and H. J. Dou, Chin. J. Polym. Sci., 2018, 36, 391–398 CrossRef CAS.
- M. Rostagno, S. Shen, I. Ghiviriga and S. A. Miller, Polym. Chem., 2017, 8, 5049–5059 RSC.
- C. Kelly, C. DeMerlis, D. Schoneker and J. Borzelleca, Food Chem. Toxicol., 2003, 41, 719 CrossRef CAS.
- C. DeMerlis and D. Schoneker, Food Chem. Toxicol., 2003, 41, 319 CrossRef CAS.
- J. Pajak, M. Ziemski and B. Nowak, Chemik, 2010, 64, 523 CAS.
- W. Wan, A. D. Bannerman, L. Yang and H. Mak, Poly (vinyl alcohol) cryogels for biomedical applications, Springer International Publishing, Cham, 2014, pp. 283–321 Search PubMed.
- R. Cilliers, Y. Song, E. K. Kohlmeir, A. C. Larson, R. A. Omary and T. J. Meade, Magn. Reson. Med., 2008, 59, 898–902 CrossRef CAS.
- Y. Tachibana, J. I. Enmi, C. A. Agudelo, H. Iida and T. Yamaoka, Bioconjugate Chem., 2014, 25, 1243–1251 CrossRef CAS.
- A. O. Ba-Salem, M. N. Shaikh, N. Ullah and M. Faiz, Inorg. Chem. Commun., 2015, 56, 5–7 CrossRef CAS.
- J. Roosen and K. Binnemans, J. Mater. Chem. A, 2014, 2, 1530–1540 RSC.
- P. Doeblin, R. Schilling, M. Wagner, R. Luhur, A. Huppertz, B. Hamm and T. Durmus, Eur. J. Radiol., 2014, 83, 660–664 CrossRef.
- Y. Ou, K. Chen, H. Cai, H. Zhang, Q. Gong, J. Wang and K. Luo, Biomater. Sci., 2018, 6, 1177–1188 RSC.
- J. C. Yuan, X. L. Xie, X. W. Zeng, H. Y. Guo and C. P. Miao, Chin. Chem. Lett., 2012, 23, 875–878 CrossRef CAS.
- L. Sun, X. Li, X. Wei, Q. Luo, P. Guan, M. Wu and Q. Gong, ACS Appl. Mater. Interfaces, 2016, 8, 10499–10512 CrossRef CAS.
- I. V. Pylypchuk, D. Kołodyńska, M. Kozioł and P. P Gorbyk, Nanoscale Res. Lett., 2016, 11, 168 CrossRef.
- W. Xu, B. Mu, W. Zhang and A. Wang, RSC Adv., 2015, 5, 64065–64075 RSC.
- S. Ding, J. S. Chen and X. W. D. Lou, Chem.–Eur. J., 2011, 17, 13142–13145 CrossRef CAS.
- S. G. Krimmer, H. Pan, J. Liu, J. Yang and J. Kopeček, Macromol. Biosci., 2011, 11, 1041–1051 CrossRef CAS.
- K. A. Günay and H. A. Klok, Polym. Chem., 2016, 7, 970–978 RSC.
- A. David, P. Kopeckova, A. Rubinstein and J. Kopecek, Bioconjugate Chem., 2001, 12, 890–899 CrossRef CAS.
- F. H. Wang, K. Bae, Z. W. Huang and J. M. Xue, Nanoscale, 2018, 10, 5642–5649 RSC.
- W. Xu, B. Mu and A. Wang, Electrochim. Acta, 2017, 224, 113–124 CrossRef CAS.
- Y. Pan, J. Yang, Y. Fang, J. Zheng, R. Song and C. Yi, J. Mater. Chem. B, 2017, 5, 92–101 RSC.
- Y. Cao, L. Xu, Y. Kuang, D. Xiong and R. Pei, J. Mater. Chem. B, 2017, 5, 3431–3461 RSC.
- Y. Y. Cheng, L. B. Zhao, Y. W. Li and T. W. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC.
- A. K. Duncan, P. J. Klemm, K. N. Raymond and C. C. Landry, J. Am. Chem. Soc., 2012, 134, 8046–8049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental part, the IR spectra, NMR spectra, TGA curve, GPC trace, TEM image, EDS curves, DLS curve, XPS spectra, stability and hemolysis studies of PVA–DTPA–Gd, MRI images of free DTPA–Gd. See DOI: 10.1039/c9ra05607f |
| This journal is © The Royal Society of Chemistry 2019 |