DOI:
10.1039/C9RA05883D
(Paper)
RSC Adv., 2019,
9, 32453-32461
Inhibitory effect of a natural phenolic compound, 3-p-trans-coumaroyl-2-hydroxyquinic acid against the attachment phase of biofilm formation of Staphylococcus aureus through targeting sortase A
Received
29th July 2019
, Accepted 17th September 2019
First published on 11th October 2019
Abstract
The antibiofilm activity and molecular mechanism of a natural phenolic compound, 3-p-trans-coumaroyl-2-hydroxyquinic acid (CHQA) against Staphylococcus aureus were investigated in this study. Crystal violet staining and XTT reduction assay demonstrated that CHQA could prominently prevent the biofilm formation of S. aureus accompanied with decrease in metabolic activity of biofilm cells. Meanwhile, microscopic observations revealed that CHQA caused a huge collapse on the architecture of S. aureus biofilm. Moreover, CHQA specifically inhibited the initial attachment phase of biofilm development and reduced S. aureus adhesion to fibrinogen. Fluorescence resonance energy transfer assay and molecular simulation showed that CHQA inhibited the activity of S. aureus sortase A (SrtA) through binding to the active region via non-covalent interactions. Additionally, CHQA efficiently reduced S. aureus attachment to stainless steel. Hence, these results suggested CHQA as a potential bacterial biofilm inhibitor which achieved antibiofilm activity through affecting the attachment phase of biofilm formation by targeting SrtA.
1. Introduction
Staphylococcus aureus is a significant foodborne pathogen which causes various diseases of high morbidity and mortality worldwide.1,2S. aureus can adapt and survive in a wide variety of stress conditions, such as high temperature, high salt concentration, low pH, and dessication.1 The presence of S. aureus in food processing environments poses a serious risk of food contamination, leading to food poisoning among consumers.3 Foodborne diseases provoked by S. aureus are still a pressing and challenging issue facing the food industry.
Biofilm formation is important for the survival of S. aureus in food processing environments, and is a main cause of the occurrence of foodborne outbreaks.4,5 Biofilms are complex microbial aggregates adhering to biotic or abiotic surfaces embedded by a self-secreted extracellular polymeric matrix.6,7 A particular characteristic of biofilms is that they confer favorable growth environments to pathogens and are highly resistant to antimicrobials and disinfectants.3,8 Undesirable bacterial biofilms formed on food processing contact surfaces caused many problems, including production of virulence factor, persistence of bacterial pathogens, and recurrent cross contamination, resulting in substantial health risks for consumers.5,9 Control of S. aureus biofilm remains tenuous with commercial available disinfectants, preservatives, and even single antibiotic7,10 Moreover, the potential negative effects of synthetic sanitizers on human health are receiving growing attention. Therefore, it is imperative to search for safe and efficient biofilm inhibitors with novel targets to combat foodborne pathogens.
Adhesion to host surface is a crucial initial step for biofilm formation. S. aureus secreted a formidable array of cell wall-anchored proteins that associated with binding host matrix components to initiate bacterial adherence and biofilm development.11 Part of surface proteins share a typical sorting signal with a conserved C-terminal LPXTG motif and have a direct role in biofilm formation.12,13 Importantly, the anchoring of LPXTG proteins to the bacterial cell wall is reliant on a membrane transpeptidase sortase A (SrtA) which recognizes the LPXTG motif and catalyzes the covalent attachment of these proteins to cell wall peptidoglycan.14 A previous study has indicated that srtA knockout mutants of S. aureus failed to display surface proteins and were defective in the establishment of infections.15 Further research revealed that loss of SrtA reduced biofilm phenotype in S. aureus,16 and inhibition of SrtA activity attenuated the biofilm formation of S. aureus.17 In addition, it was demonstrated that overexpression of SrtA resulted in increased levels of biofilm formation in some staphylococcal strains.18 Therefore, SrtA appears to be a promising target for identifying biofilm inhibitors due to its important role in S. aureus biofilm formation.12
Plant secondary metabolites are main sources of antibacterial and antibiofilm agents. Numerous polyphenols have been demonstrated to significantly inhibit the biofilm formation of pathogens,19,20 and many of them showed a potent inhibitory activity against SrtA of S. aureus, such as morin, myricetin, quercetin,21 chlorogenic acid,22 kaempferol,10 and isovitexin.23 Previously, we have reported a novel phenolic compound, 3-p-trans-coumaroyl-2-hydroxyquinic acid (CHQA, Fig. 1A) from pine needles of Cedrus deodara, which exhibited strong antibacterial activity against S. aureus through damaging bacterial cell membrane and disturbing cellular functions.24,25 In the present study, we evaluated the inhibitory effect of CHQA on the biofilm formation by S. aureus, and further elucidated that the primary antibiofilm mechanism of CHQA might be due to the inhibition of the attachment phase of biofilm formation by targeting S. aureus SrtA.
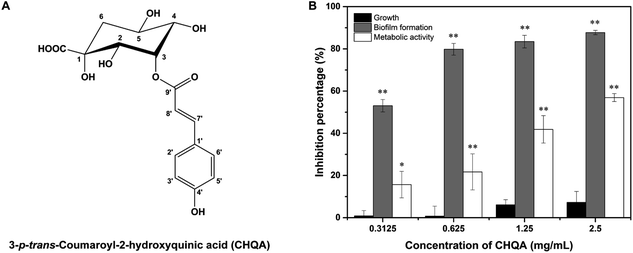 |
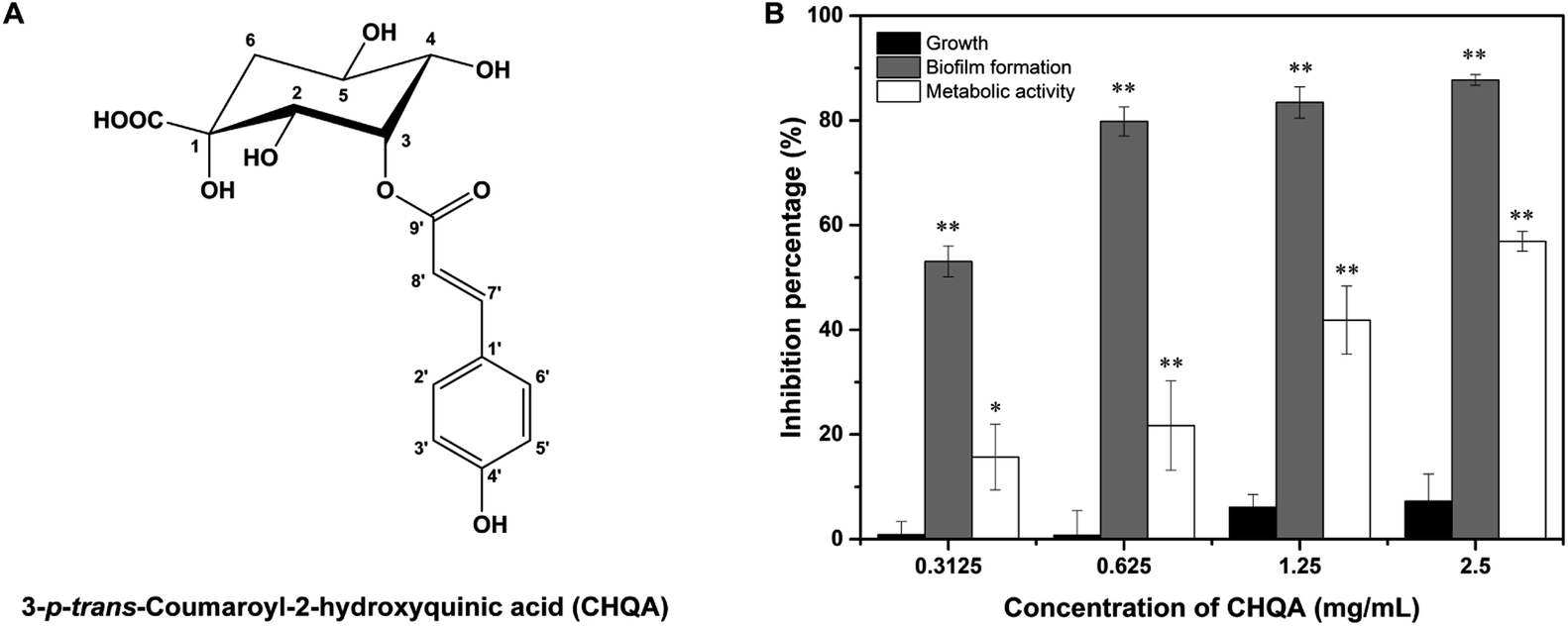
| | Fig. 1 (A) Chemical structure of 3-p-trans-coumaroyl-2-hydroxyquinic acid (CHQA). (B) Effect of CHQA on the cell growth, biofilm formation and biofilm metabolic activity of S. aureus. *p < 0.05, **p < 0.01. | |
2. Materials and methods
2.1. Materials, bacterial strain and culture conditions
CHQA (HPLC ≥ 98%) was purified from pine needles of C. deodara according to our previous reported method.26 A stock solution of CHQA in Milli-Q water was prepared for use in all experiments. S. aureus ATCC 29213 was obtained from the China Medical Culture Collection Center (Beijing, China). S. aureus was cultured in tryptone soy broth (TSB) supplemented with 1% glucose at 37 °C overnight to obtain the logarithmic phase cells for biofilm assay.
2.2. Measurement of biofilm biomass and bacterial growth
The prevention efficacy of CHQA on the bacterial growth and biofilm formation of S. aureus was assessed as a previous reported method.6 In brief, logarithmic phase S. aureus bacterial suspension was prepared in TSB with 1% glucose at a density of 1 × 106 CFU mL−1. Aliquots of 100 μL of serial twofold dilutions of CHQA in TSB with 1% glucose were added into a sterile 96-well microplate, followed by addition of 100 μL of the bacterial suspension. The plate was incubated at 37 °C for 24 h to allow biofilm formation. After incubation, the optical density at 600 nm was measured to determine the inhibitory percentage of CHQA on the bacterial growth of S. aureus with a microplate reader (Spectra MAX-190; Molecular Devices Co., Sunnyvale, USA).
To evaluate the prevention efficacy on biofilm formation, the bacterial suspension was decanted and the planktonic S. aureus was removed by washing the wells twice with PBS (0.01 M, pH 7.2). The biofilm formed on the well bottom was stained with 200 μL of 0.4% crystal violet solution for 5 min, and washed twice with distilled water. Then, 200 μL of 20% glacial acetic acid was added to each well to dissolve the stained biofilm for 30 min and the biofilm biomass was quantified by recording the absorbance at 570 nm. The inhibition percentages of growth and biofilm were calculated as the following formulas, respectively.
| Growth inhibition percentage (%) = [1 − (treated OD600 nm/control OD600 nm)] × 100 |
| Biofilm inhibition percentage (%) = [1 − (treated A570 nm/control A570 nm)] × 100 |
2.3. Assessment of metabolic activity of biofilm cells
The 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide sodium salt (XTT) reduction assay was performed to evaluate the metabolic activity of S. aureus biofilm cells as previously reported.3 Biofilm of S. aureus was formed in the absence or presence of CHQA by the procedure described in the biofilm biomass measurement assay. After removal of planktonic cells, 120 μL of a fresh mixed solution of XTT (Sigma Aldrich Co., St. Louis, USA) and phenazine methosulfate (Sigma Aldrich Co., St. Louis, USA) prepared in PBS was added to each well to keep the final concentrations at 100 μg mL−1 and 10 μg mL−1, respectively. The plate was incubated in the dark at 37 °C for 3 h. The absorbance at 492 nm was measured by the microplate reader referred above. The inhibition percentage of metabolic activity was calculated by the following formula.
| Inhibition percentage (%) = [1 − (treated A490 nm/control A490 nm)] × 100. |
2.4. Observation of S. aureus biofilm
Biofilm of S. aureus was grown on glass slides for the light microscopic observation.27 Briefly, sterile round glass sides (ϕ 14 mm) were placed in each well of a 24-well microplate. Then, 500 μL of different concentrations of CHQA in TSB with 1% glucose and 500 μL of logarithmic phase S. aureus inoculum were added to the microplate, and incubated at 37 °C for 24 h. Subsequently, the glass slides were washed thrice with PBS and stained with 0.4% crystal violet. After 5 min, the slides were washed again with distilled water to remove the excess stain and air dried. The stained biofilms were observed with a light microscope (Nikon Eclipse E200, Tokyo, Japan).
The effect of CHQA on the biofilm architecture was further analyzed by a scanning electronic microscope (SEM) with a slightly modified method.3 In brief, the biofilms of S. aureus were developed on glass slides as described above. Then, the glass slides were placed in 24-well microplate, submerged with 2.5% glutaraldehyde at 4 °C for 3 h, and gently rinsed with PBS. Subsequently, the biofilms on the slides were dehydrated in a graded ethanol series of 25%, 50%, 70%, 90% and 100% at 4 °C for 10 min each. After a further critical-point drying, the specimen was sputter-coated with gold and examined under a SEM (JSM-7500, JEOL, Tokyo, Japan).
2.5. Preformed biofilm disruption assay
The disruption ability of CHQA on the preformed biofilm of S. aureus was evaluated according to a previous reported method.28 In brief, aliquots of 100 μL of logarithmic phase S. aureus suspension (1 × 106 CFU mL−1) were inoculated to each well of a 96-well microplate and incubated at 37 °C. After 24 h, the planktonic S. aureus was discarded and the established one-day-old biofilms were washed with PBS. Then, different concentrations of CHQA prepared in TSB with 1% glucose were added to the wells and further incubated for 24 h. The biomass of biofilm formed on the well bottom was assessed by crystal violet staining method according to the procedure described above.
2.6. Analysis of inhibition phase of biofilm formation
The inhibitory effect of CHQA on the different phase of biofilm formation by S. aureus was explored according to a previous reported method with minor exceptions.10 Briefly, 100 μL of S. aureus suspension was prepared in TSB supplemented with 1% glucose at a cell density of 1 × 106 CFU mL−1 and added to each well of a 96-well microplate. Then, CHQA was added to the wells at 0, 1, 2, 3, 4, 8 and 12 h respectively to keep the final concentration of 2.5 mg mL−1. After incubation at 37 °C for a total of 24 h, the biofilm biomass was quantified by crystal violet staining assay as described above.
2.7. Fibrinogen binding assay
Adhesion of S. aureus to immobilized fibrinogen was measured by fibrinogen binding assay as previously reported with few modifications.10 In brief, logarithmic phase S. aureus (1 × 106 CFU mL−1) was cultured in the presence or absence of CHQA with shaking at 37 °C for 5 h. The bacteria were collected by centrifugation at 3000 rpm for 5 min, washed and resuspended in PBS to an OD600 nm of 0.35. Each well of polystyrene 96-well plate was coated with 200 μL of 100 μg mL−1 bovine fibrinogen at 4 °C overnight. The plate was washed with PBS, and 100 μL of the bacterial suspension was added and incubated for 1 h at 37 °C. Then, the supernatant was removed, and the cells were washed with PBS and fixed with 25% formaldehyde for 30 min. The biomass of the adherent S. aureus cells was determined by the crystal violet staining method as already described. The inhibition percentage was calculated according to the adherence rate compared to the control by the following formula. Inhibition percentage (%) = 100 × (C − T)/C, where C and T were the absorbance values of the control and CHQA treated group, respectively.
2.8. Measurement of sortase A activity
Sortase A activity was measured by a fluorescence resonance energy transfer method as reported previously with some modifications.10 The assay was performed in the wells of a 96-well black plate. The synthetic fluorescent peptide Dabcyl-QALPETGEE-Edan (GL Biochem Ltd., Shanghai, China) was used as a model substrate. Recombined SrtAΔN59 was purified from E. coli and the enzyme purity was examined by SDS-PAGE. Briefly, different concentrations of CHQA were added to the reaction buffer (50 mM Tris·HCl, 150 mM NaCl, 5 mM CaCl2, pH 7.5) containing 10 μM SrtAΔN59 in a final volume of 300 μL. The plate was incubated at 37 °C for 30 min. Subsequently, 10 μM of substrate peptide was added and keep a further incubation at 37 °C for 1 h. The fluorescence intensity was measured at 495 nm with an excitation wavelength of 350 nm using a microplate reader (PE envision, PerkinElmer Co., Waltham, USA), and the fluorescence changes were used to calculate the inhibitory rates.
2.9. Molecular modeling of CHQA and sortase A
The potential binding mode of CHQA to S. aureus sortase A was explored by a molecular docking method performed on AutoDock 4.0.2 The starting crystal structure of S. aureus SrtA was obtained from the Protein Data Bank (PDB code: 1T2P). The co-crystallized water molecules and counterions were removed from SrtA structure and polar hydrogen atoms were added using the AutoDock Tools. The structure of CHQA for docking was prepared by defining rotatable bonds and merging non-polar hydrogen atoms. Standard docking procedure for a rigid protein and a flexible ligand was performed. Subsequently, the complex structure obtained from molecular docking was used as the original structure to perform the molecular dynamics simulation with Gromacs 2018.4 package29 using the Charmm2730 all-atom force field combining the TIP3P water model.31 The SrtA-CHQA system was run for 20 ns of molecular dynamics simulation with a time step of 2 fs. Energy decomposition was calculated by Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method.32 Analysis of the trajectories was performed on VMD and Gromacs tools.
2.10. Stainless steel model assay
Stainless steel 304 coupons (2 cm × 2 cm × 1 mm thickness) were used for biofilm development as previously described method.5 After cleaning and sterilization, coupons were immersed in S. aureus bacterial suspension and incubated at 37 °C for 2 h. Each coupon with adherent cells was gently washed with 0.1% peptone water and submerged in different concentrations of CHQA for 10 min. Following the treatment, coupons were washed again to remove excess CHQA, immersed in 10 mL of 0.1% peptone water and sonicated at 55 kHz for 10 min. Coupons treated with 0.1% peptone water were served as the negative control. S. aureus in the sonicated samples were counted as the adherent cells by serial dilution and plating method.
2.11. Statistical analysis
All experiments were conducted in triplicate. One-way analysis of variance was performed on SPSS 19.0 software (IBM Co., Armonk, USA) using LSD or Games–Howell test according to the homogeneity of variances test. Data differences were considered statistically significant at p < 0.05.
3. Results and discussion
3.1. CHQA prevented the biofilm formation of S. aureus without affecting bacterial growth
Biofilm biomass was determined by crystal violet staining assay to evaluate the effect of CHQA on S. aureus biofilm formation. As shown in Fig. 1B, CHQA significantly (p < 0.01) prevented the biofilm formation of S. aureus in a dose dependent manner. The reduction in biofilm biomass ranged from 53% to 88% when the concentrations of CHQA were increased from 0.3125 to 2.5 mg mL−1. Since an ideal antibiofilm inhibitor is expected to exert no pressure on bacterial growth, the antibacterial activities of all tested concentrations of CHQA were assessed. The results showed that all tested concentrations of CHQA hardly affected the growth of planktonic S. aureus, causing a minor (<10%) and no significant (p > 0.05) decrease when compared to the control. It was consistent with our previous report that the minimum inhibitory concentration of CHQA against S. aureus ATCC 29213 was 5 mg mL−1.24 These results demonstrated that the inhibitory effect of CHQA on the biofilm formation of S. aureus was mainly attributed to the antibiofilm activity rather than dependent on the inhibition of planktonic bacterial growth.
3.2. CHQA decreased the metabolic activity of S. aureus biofilm cells
The inhibitory efficacy of CHQA on the biofilm formation of S. aureus was further verified by measuring the metabolic activity of S. aureus biofilm cell. The results indicated that CHQA significantly (p < 0.01) reduced the cellular metabolic activity of S. aureus biofilm by 22%, 42% and 57% at concentrations of 0.625, 1.25 and 2.5 mg mL−1, respectively (Fig. 1B). Numerous phytochemicals have been reported to prevent biofilm formation of pathogens accompanied with reduction in metabolic activity of biofilm, such as gallic acid, caffeic acid, chlorogenic acid, and morin.3,6 It is noteworthy that the reduction percentage of metabolic activity was lower than the inhibition rate of biofilm formation at same concentration of CHQA, suggesting that CHQA might inhibit S. aureus biofilm formation through other mechanism besides decreasing the metabolic activity of bacterial cells in biofilm.
3.3. Light microscope and SEM images of S. aureus biofilm
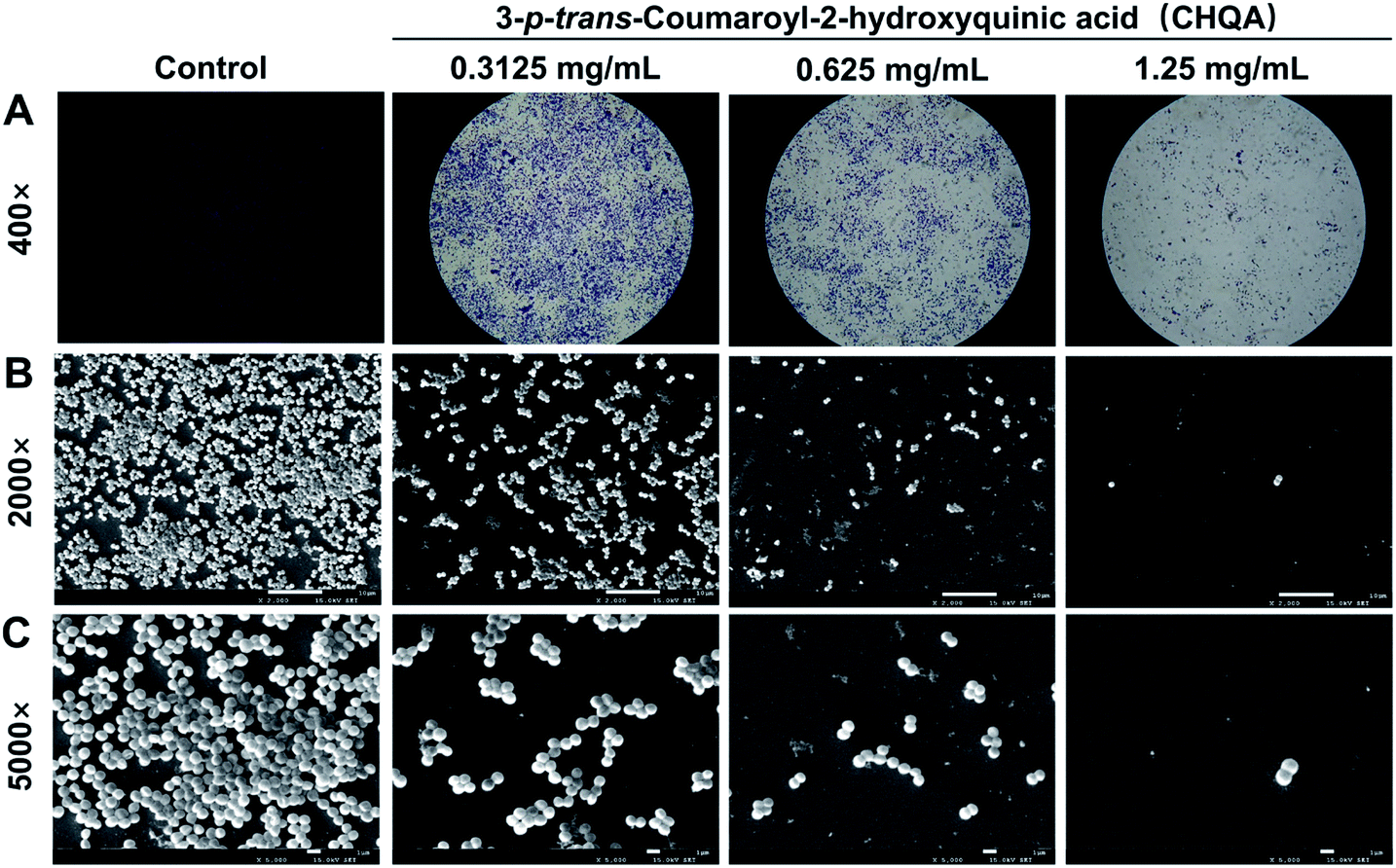
To visually disclose the impact of CHQA on S. aureus biofilm, the changes in morphology and architecture of biofilm were observed by light microscope and SEM (Fig. 2). The light microscope images captured after crystal violet staining displayed the overall morphology of S. aureus biofilm, which revealed that CHQA caused a huge collapse on the biofilm and a remarkable decrease in the number of adherent S. aureus cells. Furthermore, SEM analysis with higher magnification clearly showed that CHQA dramatically disrupted the architecture of S. aureus biofilm. Biofilms in the control group exhibited typical complex structure with multilayered cell clusters embedded in an extracellular polymeric substance. In contrast, S. aureus exposed to CHQA produced scant biofilms organized by small bacterial clumps or even single cell. Additionally, part of S. aureus cells were damaged and some debris was loosely attached to the glass surface. Formation of characteristic biofilm architecture is an important step in development of complicated biofilm with extracellular polymeric matrix that prevented antibiotics from reaching the bacteria and enhanced pathogenicity and resistance of S. aureus.33 As a consequence, the suppressed production and loosed architecture of S. aureus biofilm caused by CHQA would ultimately attenuate the resistance of bacteria.
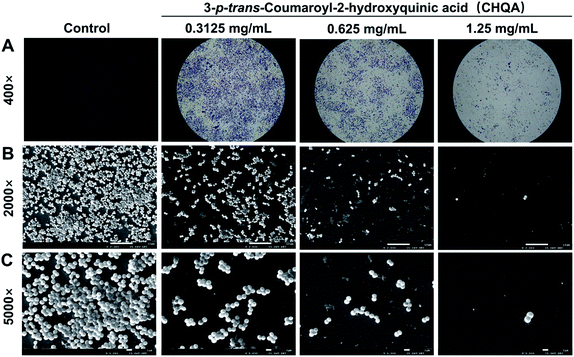 |
| | Fig. 2 Light microscope images (A) and scanning electron micrographs (B and C) of S. aureus biofilm formed on glass slides in the presence and absence of CHQA. | |
3.4. CHQA had no disruption effect on the preformed biofilm of S. aureus
The elimination efficacy of CHQA on the preformed one-day-old S. aureus biofilm was further investigated. Unfortunately, removal of S. aureus established biofilm was not achieved by CHQA even at high concentration of 10 mg mL−1 (Table 1). This is probably because bacteria that reside in biofilm were more resistant to antimicrobial agents through robust extracellular matrix, metabolic dormancy, and molecular persistence program.34 It could be speculated that CHQA exhibited inhibitory effect on S. aureus biofilm production by disturbing cell–surface interaction and cell–cell interaction, thereby impacting the adherence of planktonic S. aureus cell. These results were coincident with a previous study which reported that morin failed to disrupt the mature biofilms of Listeria monocytogenes although it prominently inhibited the biofilm formation.6 Bacterial adhesion to the surface changed from reversible to irreversible and consequently formed persistent biofilm.35 So, these findings suggested that prevention of bacterial adhesion is critical for combating biofilm as it was difficult to eradicate once biofilm formed.
Table 1 Effect of CHQA on the one-day-old preformed biofilm of S. aureusa
| Groups |
Control |
3-p-trans-Coumaroyl-2-hydroxyquinic acid (mg mL−1) |
| 10 |
5 |
2.5 |
1.25 |
0.625 |
|
Each value was expressed as a mean ± standard deviation (n = 3). There was no significance (p > 0.05) between the A570 nm values of control and CHQA treated group at all tested concentrations.
|
|
A
570 nm
|
1.28 ± 0.15 |
1.33 ± 0.12 |
1.24 ± 0.06 |
1.39 ± 0.09 |
1.47 ± 0.31 |
1.46 ± 0.20 |
3.5. CHQA inhibited the attachment phase of S. aureus biofilm formation
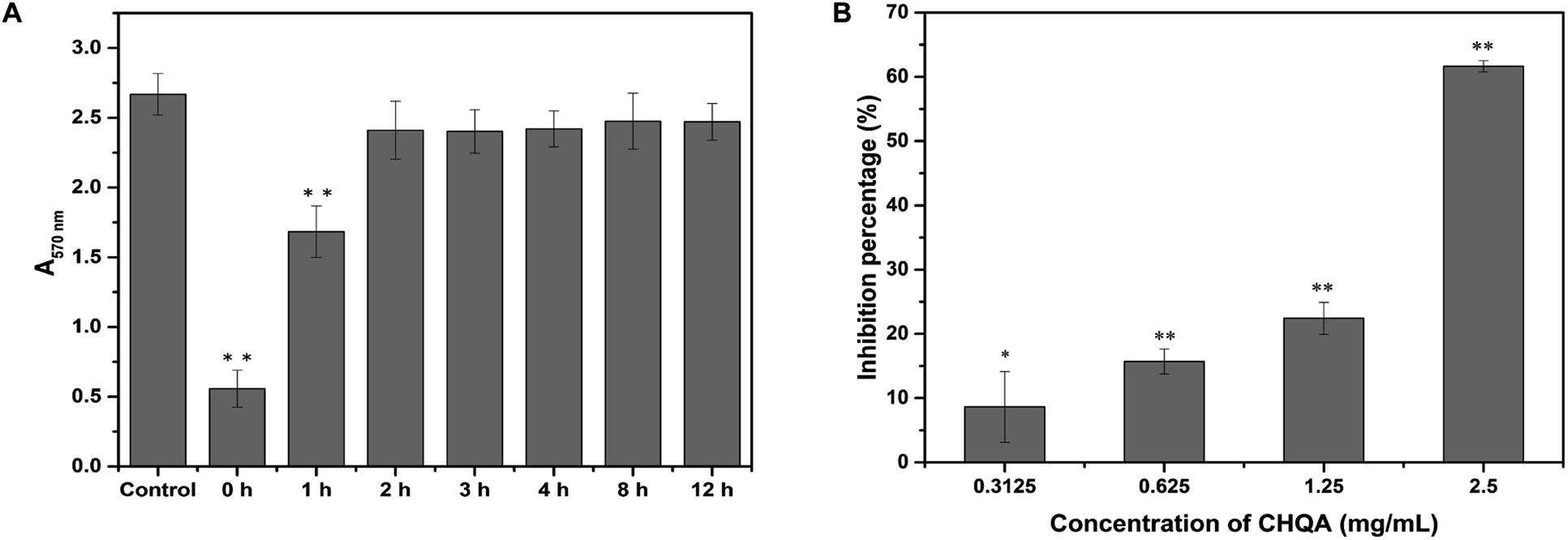
It was recognized that bacterial biofilm development consists of three sequential phases: attachment of cells to a surface, accumulation of cells to form microcolonies and differentiation of biofilm into a mature structure.11,35 The preceding results showed that CHQA markedly prevented S. aureus biofilm formation when co-cultured with bacteria, whereas it could not destroy the established biofilm. So, addition of CHQA at different time points during biofilm formation was performed to clarify which phase of biofilm development was affected by CHQA. As illustrated in Fig. 3A, compared with the control group, CHQA significantly (p < 0.01) reduced the biofilm biomass of S. aureus when added immediately and after incubation for 1 h. However, after S. aureus was incubated for 2, 3, 4, 8, and 12 h, respectively, the addition of CHQA had no effect and the biofilms were completely resistant to CHQA. Similar observations were reported for kaempferol that specifically inhibited the attachment phase of biofilm formation of S. aureus.10 After attached to surfaces, bacterial would proliferate, aggregate and recruit cells from the surrounding to form and differentiate into biofilm,36 thus the preventive effect of CHQA on the attachment phase of S. aureus would undoubtedly hinder the subsequent biofilm development. These findings were in accordance with the observations obtained in the preformed biofilm disruption assay, which further revealed that CHQA prevented S. aureus biofilm formation by acting at the initial attachment stage of biofilm development.
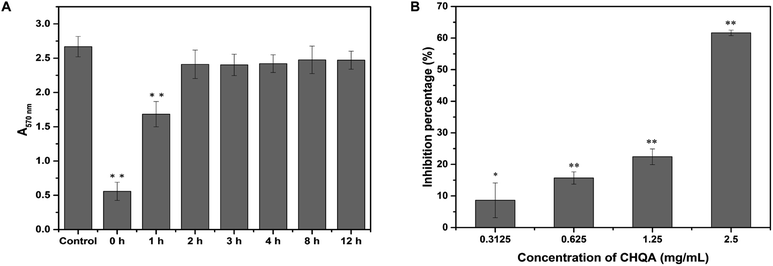 |
| | Fig. 3 (A) Effect of CHQA on the different phases of S. aureus biofilm development. (B) Effect of CHQA on the adhesion of S. aureus to fibrinogen. *p < 0.05, **p < 0.01. | |
3.6. CHQA reduced S. aureus adhesion to fibrinogen
The binding of S. aureus surface anchored proteins to the host matrix proteins initiated cell adherence, which mediated the attachment phase of biofilm formation.11,37 Some of the LPXTG surface proteins of S. aureus can recognize different host matrix components, such as fibrinogen, fibronectin, collagen and cytokeratin.11 Accordingly, we used fibrinogen as a substrate for bacterial adhesion to explore the influence of CHQA on the adhesion capacity of S. aureus. As expected, CHQA significantly (p < 0.05) inhibited the adhesion of S. aureus to fibrinogen (Fig. 3B). The inhibition percentage of S. aureus adhesion was approximately 62% with treatment of CHQA at 2.5 mg mL−1. These results were consistent with previous studies that reported many natural products exerted inhibitory effect on the adhesion of S. aureus to fibrinogen.2,23,38,39 Hence, it can be inferred that CHQA might inhibit the attachment phase of biofilm formation by affecting the adhesion of S. aureus to host surface, eventually lead to weak biofilm formation.
3.7. CHQA inhibited the activity of S. aureus SrtA
Since sortase A plays a vital role in the adhesion of S. aureus to host matrix components and subsequent biofilm formation, we further evaluated the effect of CHQA on the activity of S. aureus SrtA. As shown in Fig. 4, inhibition of SrtA activity was caused by CHQA in a dose dependent manner. The activity of S. aureus SrtA was inhibited by 74% after treated with 156.25 μg mL−1 of CHQA, and the IC50 value was detected to be 57.05 μg mL−1. Chlorogenic acid, a structural analogue of CHQA, has also been demonstrated to exhibit potent inhibitory activity against S. aureus SrtA.2 Notably, lower concentrations of CHQA were required to achieve comparable inhibitory efficacy on SrtA activity compared with that used in the fibrinogen binding assay or biofilm inhibition assay. The discrepant results are possibly because the purified SrtA enzyme was more susceptible to CHQA in vitro. These findings indicated that CHQA interfered the anchoring of surface proteins and adhesion of S. aureus by inhibiting SrtA activity, so as to prevent the bacterial biofilm formation. However, further research was required to explore the molecular interaction mode between CHQA and S. aureus SrtA.
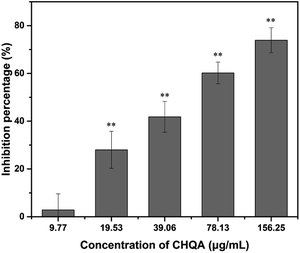 |
| | Fig. 4 Inhibitory effect of CHQA on the activity of S. aureus SrtA in vitro. **p < 0.01. | |
3.8. Binding mode of CHQA with S. aureus SrtA
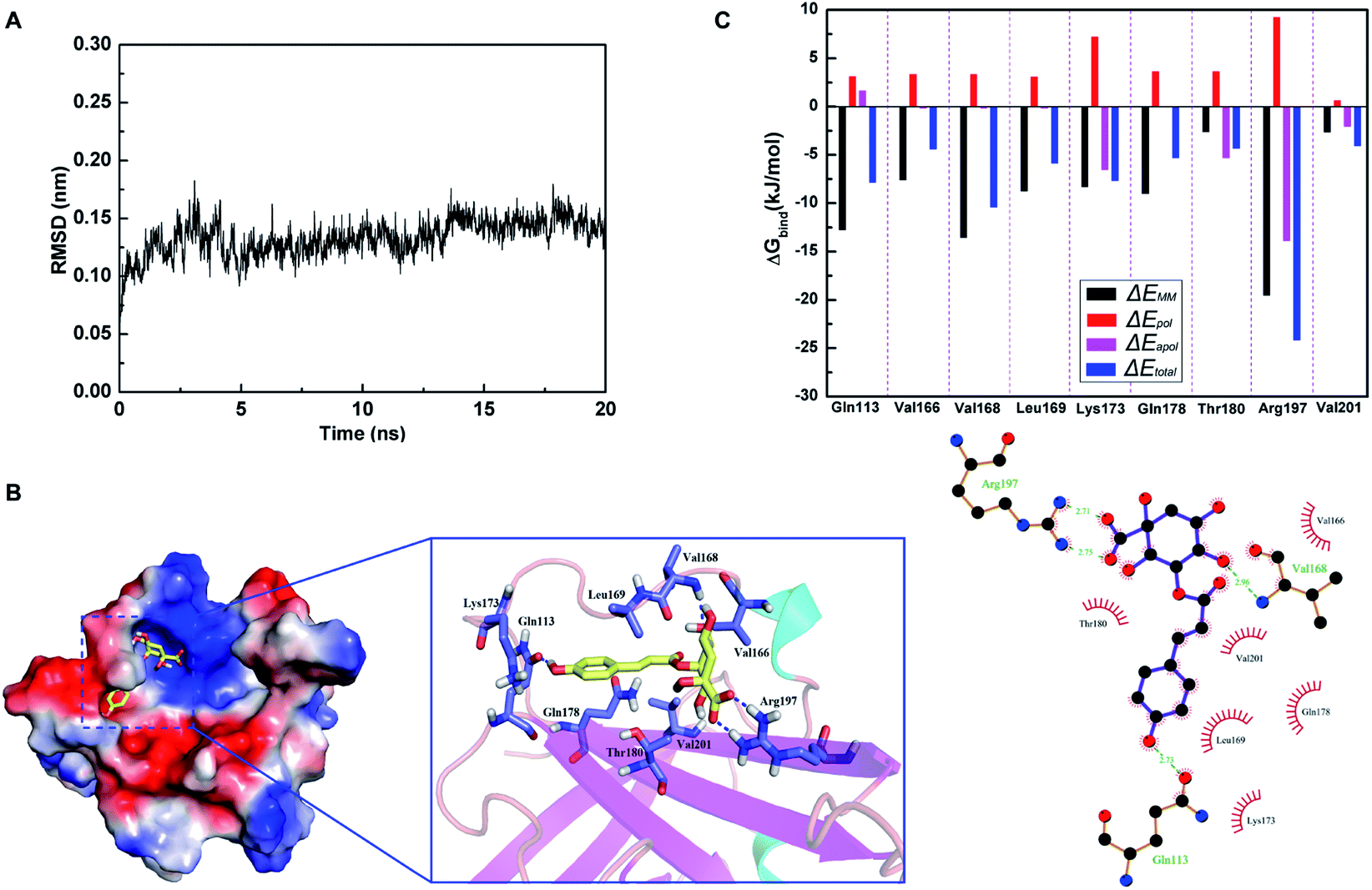
Based on the results of SrtA activity inhibitory assay, molecular docking and molecular dynamics simulation were performed to investigate whether CHQA could bind directly to S. aureus SrtA and clarify the potential binding mechanism. Root mean square deviation (RMSD) values of the backbone atoms of the SrtA-CHQA system were calculated to determine the dynamic stability of the model (Fig. 5A), which indicated that the complex achieved equilibrium at 10 ns and the structure was stabilized during the simulation. As shown in Fig. 5B, CHQA localized to the catalytic pocket of SrtA via hydrogen bonds and hydrophobic interactions as important driving forces. In detail, the negatively charged carboxyl group on the cyclohexane of CHQA was oriented to the positively charged region of SrtA, and formed two hydrogen bonds with Arg197 residue. Moreover, a hydrogen bond between the C-4 hydroxyl group on the cyclohexane of CHQA and Val168 was also present, while the aromatic ring of CHQA extended into the groove of SrtA and formed a hydrogen bond with the backbone of Gln113 residue. The surrounding residues of Val166, Leu169, Lys173, Gln178, Thr180 and Val201 were involved in hydrophobic interactions with CHQA. In addition, the total binding free energy for the SrtA-CHQA complex was calculated to be −47.2 kJ mol−1, implying that CHQA had a strong ability to bind to S. aureus SrtA.
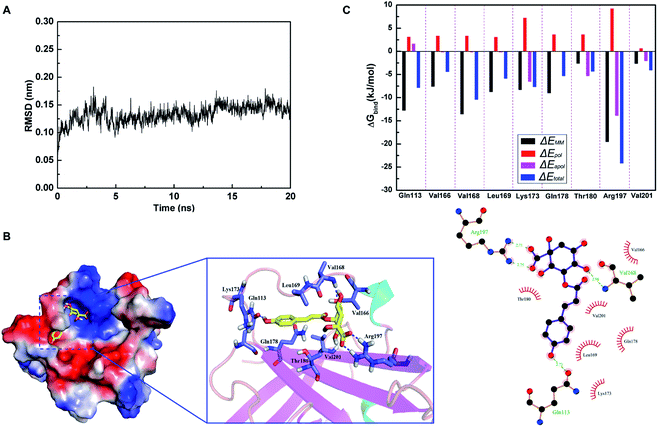 |
| | Fig. 5 The binding mode of CHQA to S. aureus SrtA based on molecular dynamics simulation. (A) The root mean square deviation (RMSD) of the backbone atoms of the SrtA-CHQA system as a function of time. (B) The 3D structure of SrtA-CHQA complex and the interaction of CHQA with the key residues of SrtA. (C) Decomposition of the binding energy on a per-residue basis at the binding sites of SrtA-CHQA complex. | |
To better understand the contribution of each residue to the binding system, the ΔEMM (bond, angle and dihedral interactions, van der Waals and electrostatic interactions energy), ΔEpol (polar solvation energy), ΔEapol (nonpolar solvation energy), and ΔEtotal (total energy) were calculated. As shown in Fig. 5C, residues Gln113, Val168 and Arg197 that were involved in hydrogen bonding contributed more to the binding energy with ΔEtotal values of −7.9, −10.5, and −24.3 kJ mol−1, respectively. The results indicated that these three residues might be critical for the binding of CHQA to SrtA. Previous studies have demonstrated that many SrtA inhibitors could interact with Arg197 residue which was recognized as one of the residues of active site triad of S. aureus SrtA.22,38,40,41 Substitution of Arg197 residue results in complete loss of catalytic activity of SrtA,42 thus the two strong hydrogen bonds between Arg197 and CHQA might affect the activity of SrtA. Therefore, it was suggested that CHQA inhibited the enzymatic activity of S. aureus SrtA through binding to the active region via non-covalent interactions.
3.9. CHQA reduced S. aureus attachment to stainless steel
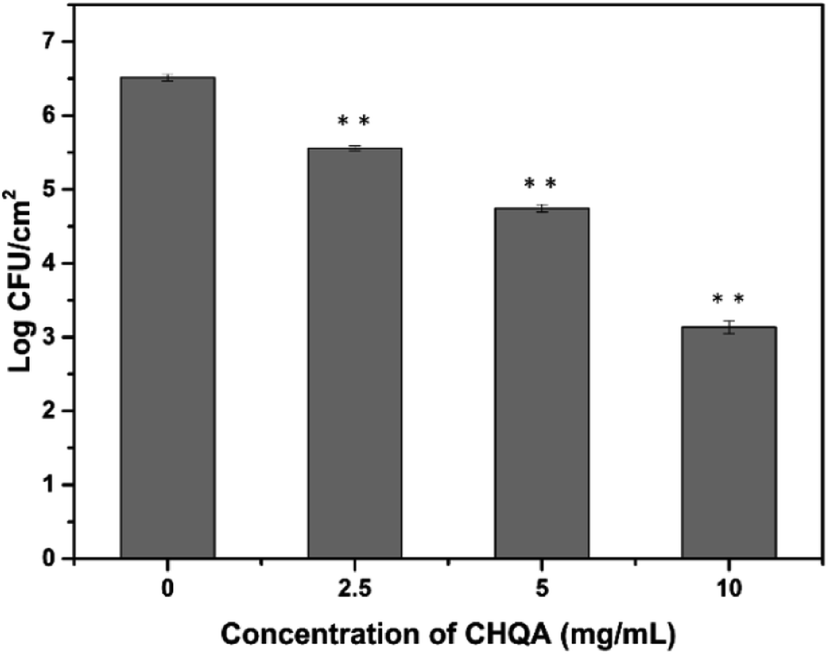
Stainless steel is widely used throughout the food industry, whereas its hydrophilic property usually favors bacterial attachment and biofilm formation.5 Considering the promising antibiofilm activity of CHQA against biofilms in food industry, adhesion of S. aureus to food-grade stainless steel was conducted to simulate food processing environments. As shown in Fig. 6, cell density of adhesive S. aureus on the stainless steel was 6.51 log CFU mL−1 after incubation for 2 h. However, the populations of adherent cells were significantly (p < 0.01) reduced to 5.56, 4.75, and 3.14 log CFU mL−1 after treatment with CHQA at concentrations of 2.5, 5, and 10 mg mL−1, respectively. The results revealed that CHQA could inhibit S. aureus attachment to stainless steel after a short contact time of 10 min, suggesting a potential of CHQA to control of bacterial biofilm in food industry. Nevertheless, in food processing plants, food contact surfaces are complex with the presence of food residues which lead to changes in surface properties and bacterial attachment. Therefore, underestimated results might be obtained in the stainless steel model assay, and the inhibitory efficacy of CHQA against biofilms formed by pathogens in real food premises need to be further investigated.
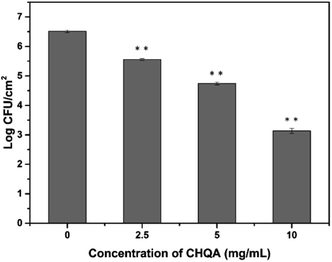 |
| | Fig. 6 Effect of CHQA on the population of S. aureus cells adhered to stainless steel. **p < 0.01. | |
4. Conclusions
In the present study, the antibiofilm activity of CHQA against S. aureus was evaluated and the potential molecular mechanism was further elucidated. The results revealed that CHQA efficiently prevented the biofilm formation of S. aureus without affecting the bacterial growth, leading to a reduction in the metabolic activity of biofilm cells and a huge collapse on the biofilm architecture. Moreover, it was found that CHQA inhibited the biofilm formation by affecting the initial attachment phase of biofilm development, whereas it had no disruption effect on the established one-day-old S. aureus biofilm. Further results showed that CHQA significantly inhibited the activity of S. aureus SrtA through binding to the active region via hydrogen bonds and hydrophobic interactions, so as to cause weak bacterial adhesion and subsequent inhibition of biofilm formation. In addition, CHQA exhibited a potent ability to reduce the number of sessile S. aureus cells adhered on stainless steel after a short contact time. All these findings suggested that CHQA might be a promising candidate to combat bacterial biofilm in the food processing environments.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31571936) and the Project funded by China Postdoctoral Science Foundation (2019M653411).
Notes and references
- J. Kadariya, T. C. Smith and D. Thapaliya, BioMed Res. Int., 2014, 2014, 827965 CrossRef.
- L. Wang, C. Bi, H. Cai, B. Liu, X. Zhong, X. Deng, T. Wang, H. Xiang, X. Niu and D. Wang, Front. Microbiol., 2015, 6, 1031 Search PubMed.
- Â. Luís, F. Silva, S. Sousa, A. P. Duarte and F. Domingues, Biofouling, 2014, 30, 69–79 CrossRef.
- A. I. Doulgeraki, C. P. Di, A. Ianieri and G. E. Nychas, Res. Microbiol., 2017, 168, 1–15 CrossRef CAS.
- J. B. Engel, C. Heckler, E. C. Tondo, D. J. Daroit and S. M. P. Da, Int. J. Food Microbiol., 2017, 252, 18–23 CrossRef CAS.
- M. Sivaranjani, S. Gowrishankar, A. Kamaladevi, S. K. Pandian, K. Balamurugan and A. V. Ravi, Int. J. Food Microbiol., 2016, 237, 73–82 CrossRef CAS.
- F. Cappitelli, A. Polo and F. Villa, Food Eng. Rev., 2014, 6, 29–42 CrossRef.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS.
- N. A. Al-Shabib, F. M. Husain, I. Ahmad, M. S. Khan, R. A. Khan and J. M. Khan, Food Control, 2017, 79, 325–332 CrossRef CAS.
- D. Ming, D. Wang, F. Cao, H. Xiang, D. Mu, J. Cao, B. Li, L. Zhong, X. Dong and X. Zhong, Front. Microbiol., 2017, 8, 2263 CrossRef.
- D. E. Moormeier and K. W. Bayles, Mol. Microbiol., 2017, 104, 365–376 CrossRef CAS.
- S. Cascioferro, M. Totsika and D. Schillaci, Microb. Pathog., 2014, 77, 105–112 CrossRef CAS.
- S. D. Oniga, C. Araniciu, M. D. Palage, M. Popa, M. C. Chifiriuc, G. Marc, A. Pirnau, C. I. Stoica, I. Lagoudis, T. Dragoumis and O. Oniga, Molecules, 2017, 22, 1827 CrossRef.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS.
- S. K. Mazmanian, G. Liu, E. R. Jensen, E. Lenoy and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5510–5515 CrossRef CAS.
- E. O'Neill, C. Pozzi, P. Houston, H. Humphreys, D. A. Robinson, A. Loughman, T. J. Foster and J. P. O'Gara, J. Bacteriol., 2008, 190, 3835–3850 CrossRef.
- X. Hou, M. Wang, Y. Wen, T. Ni, X. Guan, L. Lan, N. Zhang, A. Zhang and C.-G. Yang, Bioorg. Med. Chem. Lett., 2018, 28, 1864–1869 CrossRef CAS.
- M. J. Sibbald, X. M. Yang, E. Tsompanidou, D. Qu, M. Hecker, D. Becher, G. Buist and J. M. van Dijl, Proteomics, 2012, 12, 3049–3062 CrossRef CAS.
- L. Slobodníková, S. Fialová, K. Rendeková, J. Kováč and P. Mučaji, Molecules, 2016, 21, 1717 CrossRef.
- F. Blando, R. Russo, C. Negro, L. De Bellis and S. Frassinetti, Antioxidants, 2019, 8, 117 CrossRef CAS.
- S. S. Kang, J. G. Kjm, T. H. Lee and K.-B. Oh, Biol. Pharm. Bull., 2006, 29, 1751–1755 CrossRef CAS.
- C. Bi, L. Wang, X. Niu, H. Cai, X. Zhong, X. Deng, T. Wang and D. Wang, Biotechnol. Lett., 2016, 38, 1341–1347 CrossRef CAS.
- D. Mu, H. Xiang, H. Dong, D. Wang and T. Wang, J. Microbiol. Biotechnol., 2018, 28, 1426–1432 CrossRef CAS.
- Y. Wu, J. Bai, K. Zhong, Y. Huang, H. Qi, Y. Jiang and H. Gao, Molecules, 2016, 21, 1084 CrossRef.
- Y. Wu, J. Bai, X. Liu, L. Liu, K. Zhong, Y. Huang and H. Gao, RSC Adv., 2018, 8, 4969–4975 RSC.
- Y.-P. Wu, X. Liang, X.-Y. Liu, K. Zhong, B. Gao, Y.-N. Huang and H. Gao, J. Funct. Foods, 2015, 14, 605–612 CrossRef CAS.
- I. A. S. V. Packiavathy, S. Priya, S. K. Pandian and A. V. Ravi, Food Chem., 2014, 148, 453–460 CrossRef CAS.
- E. M. Costa, S. Silva, F. K. Tavaria and M. M. Pintado, J. Appl. Microbiol., 2017, 122, 1547–1557 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS.
- A. D. Mackerell Jr, M. Feig and C. L. Brooks III, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS.
- A. Kannappan, S. Gowrishankar, R. Srinivasan, S. K. Pandian and A. V. Ravi, Microb. Pathog., 2017, 110, 313–324 CrossRef CAS.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS.
- R. Roy, M. Tiwari, G. Donelli and V. Tiwari, Virulence, 2018, 9, 522–554 CrossRef CAS.
- M. Chen, Q. Yu and H. Sun, Int. J. Mol. Sci., 2013, 14, 18488–18501 CrossRef.
- M. Otto, Annu. Rev. Med., 2013, 64, 175–188 CrossRef CAS.
- B. Zhang, Z. Teng, X. Li, G. Lu, X. Deng, X. Niu and J. Wang, Front. Microbiol., 2017, 8, 1715 CrossRef.
- C. Bi, X. Dong, X. Zhong, H. Cai, D. Wang and L. Wang, Molecules, 2016, 21, 1285 CrossRef.
- P. Ouyang, X. He, Z.-W. Yuan, Z.-Q. Yin, H. Fu, J. Lin, C. He, X. Liang, C. Lv and G. Shu, Toxins, 2018, 10, 385 CrossRef CAS PubMed.
- A. K. Kahlon, A. S. Negi, R. Kumari, K. K. Srivastava, S. Kumar, M. P. Darokar and A. Sharma, Appl. Microbiol. Biotechnol., 2014, 98, 2041–2051 CrossRef CAS PubMed.
- C. Araniciu, O. Oniga, G. Marc, M. D. Palage, L. Marutescu, M. C. Chifiriuc, C. I. Stoica, I. Ioana and S. D. Oniga, Farmacia, 2018, 66, 627–634 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiao-Yan
Liu
a,
Jin-Rong
Bai
a,
Xiao-Yan
Liu
a,
Jin-Rong
Bai