
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal unfolding and refolding of a lytic polysaccharide monooxygenase from Thermoascus aurantiacus†
Raushan K. Singh a,
Benedikt M. Blossomb,
D. A. Russo‡
c,
B. van Oortd,
R. Croced,
P. E. Jensenc,
C. Felbyb and
M. J. Bjerrum*a
a,
Benedikt M. Blossomb,
D. A. Russo‡
c,
B. van Oortd,
R. Croced,
P. E. Jensenc,
C. Felbyb and
M. J. Bjerrum*a
aDepartment of Chemistry, University of Copenhagen, Copenhagen, Denmark. E-mail: mobj@chem.ku.dk
bDepartment of Geosciences and Natural Resource Management, University of Copenhagen, Frederiksberg C, Denmark
cDepartment of Plant and Environmental Sciences, University of Copenhagen, Frederiksberg C, Denmark
dBiophysics of Photosynthesis, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
First published on 19th September 2019
Abstract
Lytic polysaccharide monooxygenases (LPMOs) are copper-containing enzymes which promote the degradation of recalcitrant polysaccharides like cellulose or chitin. Here, we have investigated the thermostability of an LPMO from Thermoascus aurantiacus (TaLPMO9A). TaLPMO9A was found to retain most of its initial activity after incubating at 100 °C while its apparent melting temperature (Tm) is 69 °C at neutral pH. Interestingly, our studies show that holoTaLPMO9A, apoTaLPMO9A and deglycosylated TaLPMO9A can fold back to their original conformation upon lowering the temperature. In the presence of β-mercaptoethanol the protein does not refold. Activity of TaLPMO9A and refolded TaLPMO9A was studied by an Amplex® Red assay as well as by TaLPMO9A catalysed oxidation of phosphoric acid swollen cellulose (PASC). These studies confirm the functional regain of TaLPMO9A activity upon going through one cycle of unfolding and refolding. The thermal unfolding and refolding of TaLPMO9A was measured spectroscopically. Utilizing the two-state model, detailed thermodynamic parameters were obtained for holoTaLPMO. Furthermore, we have investigated the kinetics of TaLPMO9A unfolding and refolding. Our results have implications in understanding LPMO stability, which is crucial for the efficient application of LPMOs as biocatalysts during biomass degradation.
Introduction
Lignocellulosic biomass, predominantly consisting of recalcitrant plant cell wall polymers, constitutes the largest source of biomass of the biosphere.1,2 Therefore, the development of strategies to harness lignocellulosic biomass may hold the key to the production of green renewable energy and its bioproducts and has huge potential to reduce the environmental pollution caused by petroleum-based energy sources.3,4 One potential exploitation route is enzymatic degradation, by utilizing a variety of hydrolytic enzymes. One notable development of this field was the discovery of a group of copper-dependent enzymes called lytic polysaccharide monooxygenases (LPMOs).5,6 LPMOs cleave β-(1→4) glycosidic bonds by oxidising the C1 and/or C4 carbon in polysaccharides.6–10 LPMOs have been identified in fungi, bacteria, viruses, invertebrates, and algae.11–13 They are currently classified into seven auxiliary activities families (AA9, AA10, AA11, AA13, AA14, AA15 and AA16) following the carbohydrate-active enzymes (CAZy) database.14–16 All LPMOs have a characteristic flat, solvent exposed binding surface and an essential, highly conserved, mononuclear active site type-2 copper coordinating, t-shaped histidine-brace.5,10,17 The crystal structure and EPR spectra of an LPMO from Thermoascus aurantiacus (TaLPMO9A) provided valuable structural insights into the LPMO active-site and histidine brace coordination sphere of the copper ion.5 In presence of a soluble redox active agent such as ascorbic acid, TaLPMO9A has been reported to oxidize substrate at both C1 and C4 carbons. However, TaLPMO9A predominantly releases C4-oxidized oligosaccharides.18 Addition of TaLPMO9A to the cocktail of cellulose-degrading enzymes had a boosting effect on cellulose saccharification. Here, the operational stability of LPMOs is crucial due to the long incubation period of cellulose saccharification.18Although LPMOs have been studied intensively in recent years, the focus of the work has been predominantly on elucidating the reaction mechanism, substrate specificity and characterization of new family members.5,19–28 To date, there are very few studies dealing with LPMO stability, a crucial aspect for commercial success as a biocatalyst. In particular, thermal stability is of great interest due to the fact that catalytic enzymes start to unfold and get inactivated with increasing temperature.29 Unfolding and thermal stability in LPMOs has been explored in a recent study showing that MtLPMO9B from Myceliophthora thermophila C1 show irreversible unfolding at temperature higher than 70 °C.30 In another study, site-directed mutagenesis was applied to improve the Tm of the Streptomyces coelicolor LPMO10C (ScLPMO10C) up to 12 °C by introducing two additional disulphide bridges.31 The apparent melting temperatures for TaLPMO9A (with and without the methylated N-terminal His1 residue) was found to be 77 °C using differential scanning calorimetry.18 However, the potential reversibility of thermal unfolding in LPMOs and the thermodynamics of unfolding and refolding have not been studied till now. Thermodynamic studies of LPMOs can provide valuable insights into the structural changes they undergo at higher temperature. Additionally, information on the reversibility of thermal unfolding in LPMOs can contribute to the application of LPMOs as biocatalysts for thermal sterilization and on–off control in bioreactors.32
In the present work, we show that TaLPMO9A is highly thermostable with a characteristic transition midpoint (Tm) for the unfolding of the backbone of 69 °C at neutral pH. Furthermore, it is demonstrated that thermal unfolding of TaLPMO9A is reversible. We performed a spectroscopic study of the thermal unfolding/refolding of TaLPMO9A, utilizing a simple two-state model, thus allowing the calculation of thermodynamic parameters of unfolding/refolding. The results of this study provide insight into the thermal unfolding mechanism and stability at higher temperature.
Materials and methods
Materials
All chemicals were of the highest purity grade available and were purchased from Sigma-Aldrich unless stated otherwise. Ampliflu™ Red (10-acetyl-3,7-dihydroxyphenoxazine) was purchased from Cayman chemical company. TaLPMO9A (Thermoascus aurantiacus) expressed in Aspergillus oryzae was donated by Novozymes A/S (Bagsværd, Denmark). PNGase F (peptide-N-glycosidase F) from Elizabethkingia miricola and horseradish peroxidase (HRP) were purchased from Sigma-Aldrich (St. Louis, USA).Purification of TaLPMO9A
Copper(I) chloride was used to saturate TaLPMO9A (Thermoascus aurantiacus) under anaerobic environment on ice for 2 h. The TaLPMO9A preparation was then filtered and injected into an ÄKTA chromatography system (Amersham Pharmacia Biotech, Sweden) equipped with a Hiload 26/60 Superdex 75 prep grade column (Pharmacia Biotech) equilibrated with 20 mM MOPS buffer (pH 7.0). HoloTaLPMO9A (i.e. copper loaded TaLPMO9A, hereafter TaLPMO9A) was eluted in a single peak using 20 mM MOPS buffer (pH 7.0) with a flow rate of 1 mL min−1. The fractions corresponding to the single peak were pooled, concentrated using an Amicon Ultra-15 centrifugal filter (3 kDa, Merck Millipore Ltd. Ireland) and stored at 4 °C for further use. The purified TaLPMO9A was analysed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a Mini-PROTEAN TGX precast gel from Bio-Rad Laboratories with a gradient of 4–15% polyacrylamide. Protein bands were visualized by staining with Coomassie blue. Precision Plus Protein Standard was used for apparent mass determination.To obtain apoTaLPMO9A (i.e. copper free TaLPMO9A), purified holoTaLPMO9A was dialyzed using a D-tube Dialyzer Maxi (6–8 kDa) from Millipore (Billerica, MA, USA). A D-tube loaded with purified holoTaLPMO9A was suspended in a beaker containing 20 mM phosphate buffer (pH 7.0) supplemented with 10 mM EDTA. The dialysis was performed at 4 °C for 12 h. During the course of dialysis, the buffer was continuously stirred using a magnetic stirrer and was replaced twice. After 12 h of dialysis, the apoTaLPMO9A was buffer exchanged and concentrated using Amicon Ultra-15 centrifugal filter (3 kDa, Merck Millipore Ltd. Ireland).
Copper and protein estimation
The copper content of the TaLPMO9A was analysed using high resolution inductively coupled plasma-MS (ICP-MS) using an Aurora M90 ICP-MS system (Bruker, Germany). Data were collected using Bruker Quantum software. Optimization parameters for plasma flow, auxiliary flow, sheath gas nebulizer flow were 16.5, 2.0, 0.2, and 1.0 L min−1, respectively. The metal content was determined by comparison with standard curve generated from known concentrations of internal standard solution. Standard curve was plotted from 0 to 50 ppb using IV-ICPMS-71A solution (Inorganic ventures, Christiansburg, VA, USA). Instrument was calibrated and 2% nitric acid was run as control before samples. TaLPMO9A (1–2 mg mL−1) was appropriately diluted in 2% nitric acid. An average of three analyses of each sample was taken.Protein concentrations of purified TaLPMO9A were estimated based on its absorbance at 280 nm using a NanoDrop spectrophotometer (Fisher Scientific, USA). The TaLPMO9A sequence was used to calculate the extinction coefficient of the TaLPMO9A (ε280 = 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 630 M−1 cm−1) with an online tool available at https://web.expasy.org/protparam/.
630 M−1 cm−1) with an online tool available at https://web.expasy.org/protparam/.
Deglycosylation and mass spectroscopy (MS)
For deglycosylation, TaLPMO9A (1 mg mL−1, 50 μL) was treated with PNGase F (500 units mL−1, 2 μL) in 20 mM ammonium bicarbonate (pH 8.0). The reaction mixture was incubated at 37 °C for 12 h and then heated to 100 °C for 5 minutes to stop the reaction. The reaction mixture containing deglycosylated TaLPMO9A was frozen for subsequent MS analysis.33The intact molecular weight analyses were performed using a Bruker microTOF focus electrospray mass spectrometer (Bruker Daltonik GmbH, Germany). Samples were washed on a MassPREP On-Line Desalting column (2.1 × 10 mm; Waters, USA) and introduced to the electrospray source with a flow rate of 200 μL h−1 using an Agilent LC system. Data analysis was performed with DataAnalysis version 3.3 (Bruker Daltonik GmbH, Germany). The molecular weight of the samples was calculated by maximum entropy deconvolution.
Circular dichroism (CD)
CD spectra were recorded on a Jasco J-815 circular dichroism spectrometer equipped with a temperature-controlled cell holder. The instrument was flushed with nitrogen, with a flow rate of 10 L min−1 throughout the experiment. The transition from the folded state to the unfolded state of the TaLPMO9A (0.66 mg mL−1) was recorded at 222 nm using a 1 mm cuvette. Temperature ranged from 30 to 90 °C (unfolding) and 90 to 30 °C (refolding) with four different heating and cooling rates, 1.0 °C, 0.5 °C, 0.2 °C and 0.1 °C min−1. The fraction of the folded protein (fN) at each temperature is calculated using the expression fN = (ΘU − ΘT)/(ΘU − ΘN), where ΘN and ΘU are the measured ellipticity of the folded and the unfolded protein respectively.34 ΘT is the ellipticity at each temperature. For the “far-UV” spectral region (260–180 nm) CD spectra was recorded at a bandwidth of 2 nm using 0.1 mm round cuvette at 25 °C. All measurements were performed in 20 mM MOPS buffer (pH 7.0). Data were analysed with SpectraManager 2 (Jasco, Germany). To calculate the content of secondary structural elements of unfolded and refolded TaLPMO9A online tool available at http://bestsel.elte.hu/index.php was used.35 CD spectra was used from 190 to 250 nm. For the thermodynamic functions of the TaLPMO9A unfolding/refolding we used the transition curve, van't Hoff's plot and van't Hoff's equation. ΔH and ΔS were derived from the slope and intercept of van't Hoff's plot, respectively.Thermal unfolding based on intrinsic tryptophan fluorescence
For each condition, 10 μL of sample per capillary were prepared. The protein samples were loaded into UV capillaries (NanoTemper Technologies, Germany) and experiments were carried out using the NanoTemper Tycho NT.6 (NanoTemper Technologies, Germany). The temperature gradient was set to an increase of 20 °C min−1 in a range from 35 °C to 95 °C. Protein unfolding was measured by detecting the temperature-dependent change in tryptophan fluorescence at emission wavelengths of 330 and 350 nm. Inflection temperatures (Ti) were determined by detecting the maximum of the first derivative of the fluorescence ratios (F350/F330). For this, an 8th order polynomial fit was calculated for the transition region. Next, the first derivative of the fit was formed and the peak position (at Ti) was determined.Unfolding and refolding kinetics
The kinetics of thermal unfolding was monitored using time course measurement on a circular dichroism spectrometer. The transition from the folded state to the unfolded state of TaLPMO9A (0.5 mg mL−1) was recorded by following change in ellipticity (222 nm) upon heating the protein at 72, 74, 76, 78, and 80 °C for 1 hour. Spectra were recorded in pre-equilibrated buffer at each temperature. Single exponential decay equation, fN = yf + ae−kut was applied to fit the folded protein fraction (fN), where ku and t are the rate of unfolding and time, respectively.34 The activation energy (Eua) of unfolding was determined from the slope of an Arrhenius plot.The refolding kinetics of TaLPMO9A were performed by monitoring the change in ellipticity (222 nm) of the unfolded TaLPMO9A. TaLPMO9A was unfolded by heating at 80 °C for 10 min immediately before the refolding kinetic experiment. The refolding kinetics study was carried out by allowing the unfolded TaLPMO9A to refold at 60, 62, 64, 66, and 68 °C for 3 hours. As described above, the rate of refolding (kf) and the activation energy of refolding (Efa) were estimated by fitting the fN using single an exponential decay equation and from the slope of an Arrhenius plot, respectively.
Amplex® Red assay
In the absence of substrate, LPMOs undergo a futile side reaction thereby producing H2O2. The produced H2O2 is directly proportional to the amount of LPMO added to the assay mixture. H2O2 produced as the result of side reaction is then determined by HRP coupled conversion of Amplex® Red to resurofin.36 Horseradish peroxidase (HRP)-coupled Amplex® Red (10-acetyl-3,7-dihydroxyphenoxazine) assay was performed on SpectraMax M2 multi-detection microplate reader (Molecular Devices, USA) using a black 96-well plate with clear bottom from Nunc (Thermo Fisher, USA).36 All the reactions were performed in 20 mM sodium phosphate buffer (pH 6.0) at 37 °C. 200 μL of reaction mixture contained 100 μM EDTA, 80 μM ascorbate, 50 μM Ampliflu™ Red, 20 U mL−1 HRP and 2.0 μg mL−1 TaLPMO9A. Resorufin fluorescence was recorded with an excitation and emission wavelength of 557 nm and 583 nm, respectively. Fluorescence was measured from the bottom for 10 min with 5 s shaking before measurements began.Enzymatic oxidation of phosphoric acid swollen cellulose (PASC)
PASC was prepared by treating microcrystalline cellulose (avicel) with phosphoric acid as described by Wood et al. (1988).37 Avicel (4 g) was suspended into phosphoric acid, 100 mL (86% w/v) at 60 °C and stirred for an hour. Mili-Q water (1900 mL) was slowly dripped into the solution. The prepared suspension was then moved to 4 °C for sedimentation. The supernatant was then discarded and the remaining suspension was washed with Mili-Q water at least four times. The pH of the suspension was neutralized by Na2CO3 and stored at 4 °C in the dark until further use.PASC oxidation was performed using both the purified TaLPMO9A and the refolded TaLPMO9A in the presence of ascorbate (2 mM) in 100 mM citrate buffer (pH 6.2) at 50 °C for 19 h. Reaction was terminated by the addition of 0.1 M NaOH and samples were centrifuged immediately. The separated supernatant was then filtered (0.22 μM) and analysed for the presence of oligosaccharides using High-Performance Anion Exchange Chromatography (HPAEC).
High-performance anion exchange chromatography
Oligosaccharides released from TaLPMO9A catalysed oxidation of PASC were detected using an ICS-5000+ system with a CarboPac PA1 column (two 2 × 50 mm guard columns followed by a 2 × 250 mm analytical column) and a PAD detector (Thermo Scientific, USA) as described by Möllers et al. (2017).38 The following gradients were used for the elution (eluent A, 0.1 M NaOH; eluent B, 1 M NaOAc in 0.1 M NaOH): 100% A:0% B to 90% A:10% B (10 min), then to 70% A:30% B (25 min) and lastly 0% A:100% B (30 min). 100% A:0% B was used for the column reconditioning. The peaks for cello-oligosaccharides were interpreted following the elution profiles of commercially available non-oxidized oligosaccharides (DP2–DP6). Based on previous publications the peaks appearing after 25 min correspond to the C4-oxidized products.21,38
Results and discussion
The TaLPMO9A is a widely studied LPMO from Thermoascus aurantiacus due to its industrial relevance in decomposing lignocellulosic biomass.5,39 However, very little is known regarding the thermal stability and structural properties at elevated temperatures of this enzyme. Therefore, TaLPMO9A was copper loaded and purified until it was free from any visible impurities (Fig. S1†). We then proceeded to undertake a detailed study of the thermal stability and thermodynamics of folding/unfolding of the TaLPMO9A.Thermal stability of TaLPMO9A
Initially, the thermal stability of the TaLPMO9A was investigated by measuring the activity of the enzyme after incubation at elevated temperature. Surprisingly, only a gradual decrease in activity was observed with increasing incubation time at 100 °C (Fig. 1A). The TaLPMO9A showed only 19% decrease in activity after 30 min of incubation at 100 °C. The activity was reduced to 72% after 45 min of incubation at 100 °C. In order to analyse changes in the secondary structure of the protein, we proceeded with a CD spectroscopy based analysis. The far-UV spectral region showed two peaks corresponding to 222 nm and 195 nm (Fig. 1B). It is clear that the secondary structure changes gradually with an increase of incubation time at 100 °C. The 222 nm minima start to shift to 200 nm and, simultaneously, the 195 nm peak gradually disappeared with increasing incubation time. After 120 min, the 222 nm minima disappeared and distinctly appeared at 200 nm. This shift towards shorter wavelength clearly indicates that the TaLPMO9A is unfolding but not into a fully random structure, as described before for LPMOs 9B and 9D from Myceliophthora thermophila C1.30 Furthermore, CD spectra in far-UV spectral region for TaLPMO9A passed through an isosbestic point at 215 nm at all temperatures from 40 to 90 °C (Fig. S2†). This suggests that in the temperature range of 40 to 90 °C, unfolding of TaLPMO9A is a two-state process without formation of significant amounts of an intermediate.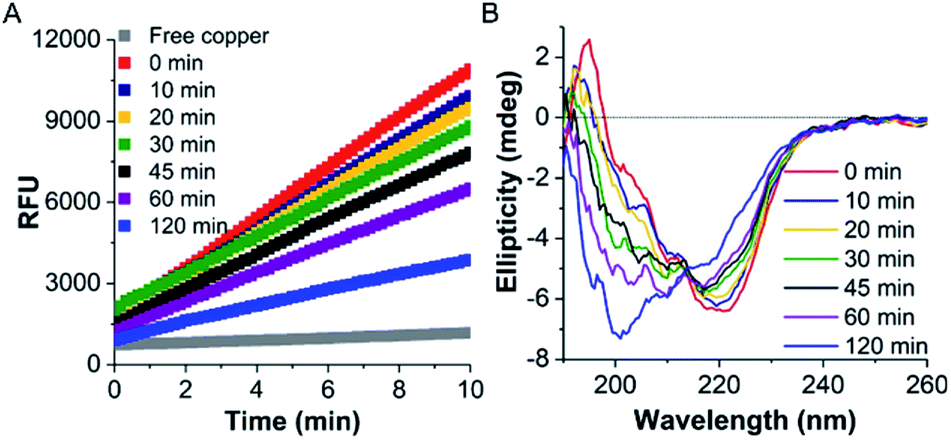 | ||
| Fig. 1 Thermal stability of TaLPMO9A. (A) TaLPMO9A activity measured by the Amplex® Red assay at 37 °C after exposure to 100 °C for different time intervals. TaLPMO9A was kept on ice for 10 min before performing the Amplex® Red assay. Amplex® Red assay mixture contained 100 μM EDTA, 80 μM ascorbate, 50 μM Ampliflu™ Red, 20 U mL−1 HRP and 2.0 μg mL−1 TaLPMO9A. (B) CD spectra of TaLPMO9A (1 mg mL−1, in 20 mM MOPS buffer, pH 7.0) at 25 °C after exposure to 100 °C for different time intervals. An average buffer baseline spectrum was subtracted from average of three scans at each condition. | ||
Next we monitored the stability of TaLPMO9A by intrinsic protein fluorescence. TaLPMO9A contains ten tryptophan residues, therefore, changes in the tryptophan fluorescence intensity upon protein unfolding allow for the determination of a defined transition point (Fig. 2). From these experiments, apparent midpoint transition temperatures, in the following referred to as Ti were determined from the first derivative of the fluorescence traces (Fig. S3A†). The effect of TaLPMO9A concentration on thermal stability was monitored for concentrations ranging from 0.1 mg mL−1 to 5 mg mL−1. Ti decreased with increased TaLPMO9A concentration (Fig. S3A†). The decrease in Ti at high TaLPMO9A concentrations may be attributed to a reversible co-operative interaction of denatured monomeric protein during the unfolding.40 Additionally, Ti determined in different buffer systems and pH environments (pH 4.0 to pH 9.0) revealed pronounced effects of both (Fig. 2B and S3B†). The Ti value of TaLPMO9A decreased from acidic pH to alkaline condition. The highest Ti (89.5 °C) was found to be at pH 5.0 in citrate buffer (20 mM). The decrease in Ti at pH values lower than 5.0, suggests that the protonation has destabilising effect on TaLPMO9A.41 On the other hand, if we compare the effect of different buffer systems (citrate, phosphate and trizma), TaLPMO9A seems to be more stable in Trizma buffer.
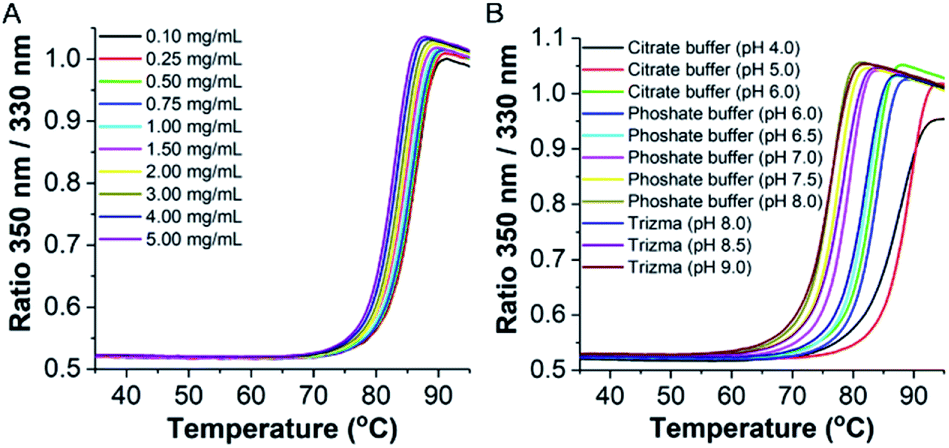 | ||
| Fig. 2 Thermostability at various conditions. Stability of TaLPMO9A at different protein concentrations (20 mM MOPS buffer, pH 7.0) (A) and at various pH (1 mg mL−1 of TaLPMO9A) (B) determined by temperature-dependent change in tryptophan fluorescence at emission wavelengths of 330 and 350 nm using the NanoTemper Tycho NT.6. | ||
Unfolding and refolding
Following the observation that the TaLPMO9A was still active after incubation at 100 °C we proceeded to probe the unfolding reversibility of TaLPMO9A by following the CD signal at 222 nm during gradual heating to 90 °C followed by cooling down to 30 °C. TaLPMO9A was subjected to unfolding by increasing the temperature from 30 °C to 90 °C with 1 °C min−1 temperature ramp in 20 mM MOPS buffer (pH 7.0) (Fig. 3A). The TaLPMO9A showed almost no change in ellipticity with the increase in temperature up to 50 °C. A steep decrease in ellipticity was observed after 70 °C indicating the onset of unfolding. The apparent Tm was observed to be 74.3 °C for the TaLPMO9A. Following similar experimental conditions, the Tm for apoTaLPMO9A and deglycosylated TaLPMO9A were determined to be 65.2 and 67.9 °C, respectively (Fig. 3B and C). The lower Tm for the apoTaLPMO9A is due to the absence of copper from the active site leading to a less stable structure. This has also been reported for LPMOs from Neurospora crassa and Bacillus amyloliquefaciens.42 Additionally, in regards to the deglycosylated TaLPMO9A, the decreased Tm value confirms that the sugar groups linked to the surface of the TaLPMO9A may be acting as a protectant against harsh physical conditions, including high temperature. To investigate the refolding of thermally unfolded TaLPMO9A, the protein was cooled down from 90 to 30 °C by gradually decreasing the temperature at 1 °C min−1. During the refolding cycle, the TaLPMO9A was found to be in an unfolded state down to 60 °C upon which it started to refold, and it regained the initial minimal ellipticity at 222 nm of the completely folded state at 50 °C. It is interesting to note that TaLPMO9A thermal unfolding is reversible in all three forms i.e. holoTaLPMO9A, apoTaLPMO9A and deglycosylated TaLPMO9A. This indicates strongly that neither the coordinated copper-ion at the active site nor the glycosylation have an impact on the reversibility of LPMO refolding.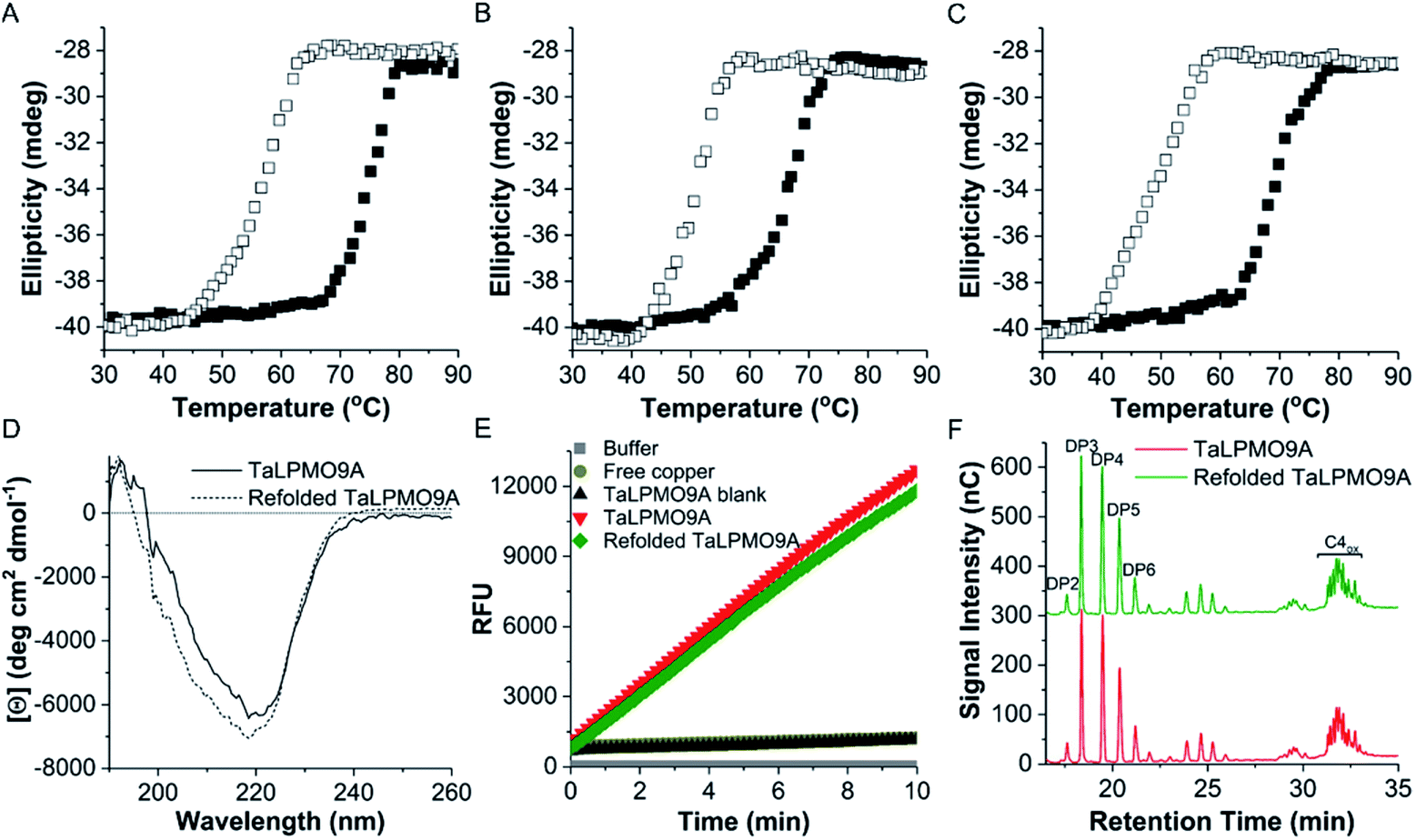 | ||
| Fig. 3 Thermal unfolding of TaLPMO9A. The molar ellipticity measured at 222 nm was monitored continuously in the temperature range from 30 to 90 °C for TaLPMO9A (A), apoTaLPMO9A (B) and deglycosylated TaLPMO9A (C). The transition from folded state to unfolded state of TaLPMO9A was recorded using 1 mm cuvette, temperature ranging from 30 to 90 °C and 90 to 30 °C with a constant increment of 1.0 °C min−1 for unfolding. Similarly, 1.0 °C min−1 cooling rates was used for refolding of TaLPMO9A. Solid and hollow squares represent the unfolding and refolding, respectively. (D) shows the far-UV CD spectra measured at the beginning of unfolding experiment and after the refolding step of TaLPMO9A and refolded TaLPMO9A. Far-UV spectral region (260–190 nm) was recorded using 0.1 mm round cuvette at 25 °C. The mean residue ellipticity is calculated on the basis of mean residue weight (MRW). (E) Activity of TaLPMO9A and refolded TaLPMO9A measured by Amplex® Red assay. TaLPMO9A was subjected to unfolding and refolding by heating it from 30 to 90 °C and then cooling it from 90 to 30 °C at the rate of 1.0 °C min−1. Amplex® Red assay was performed in 20 mM sodium phosphate buffer (pH 6.0) at 37 °C. (F) TaLPMO9A and refolded TaLPMO9A catalysed oxidation of cellulose analysed by HPAEC. Refolded TaLPMO9A was prepared by heating TaLPMO9A from 30 to 90 °C and then cooling it from 90 to 30 °C at the rate of 1.0 °C min−1. PASC oxidation was performed for 19 hours with 2 mM ascorbate as electron donor and in citrate buffer (pH 6.2, 100 mM). The peaks were assigned according to the elution profile of commercially available non-oxidized cello-oligosaccharides. DP2, cellobiose; DP3, cellotriose; DP4, cellotetraose; DP5, cellopentaose; DP6, cellohexaose. The C4-oxidized glucose (4-keto aldose) peaks appear after a retention time of 30 min. | ||
The CD spectra in the far-UV spectral region (190–260 nm) of the native TaLPMO9A were found to be nearly identical before unfolding (at 25 °C) and after going through the unfolding and refolding cycle (25 °C) (Fig. 3D). The calculated secondary structures derived from the far-UV CD spectrum for TaLPMO9A before unfolding (at 25 °C) and after going through the unfolding/refolding cycle (25 °C) suggests a regaining of the secondary structure (Fig. 3D). The purified TaLPMO9A before unfolding contains 12.4% α helices, 31.5% β sheets, 13.5% turn and 42.6% others. Upon going through unfolding/refolding cycle the secondary structure components for TaLPMO9A were calculated to be 11.7% α helices, 36.0% β sheets, 11.8% turns and 40.5% others. The crystal structure of TaLPMO9A (PDB: 2YET) shows 11.6%, 30.7% and 57.7% of α helices, β sheets, and others, respectively. The increase in β sheet might be an actual effect but could be due to experimental variation. Nonetheless, the refolded TaLPMO9A almost entirely regains its native helical content.
To compare the catalytic activity of TaLPMO9A and refolded TaLPMO9A (after unfolding/refolding cycle), Amplex® Red and PASC oxidation assays were performed under standard experimental conditions. As shown in Fig. 3E, the Amplex® Red assay indicated no significant difference in the activities for TaLPMO9A and refolded TaLPMO9A. Similarly, no measureable differences were seen in the oxidized products for TaLPMO9A and refolded TaLPMO9A catalysed oxidation of PASC (Fig. 3F). Thus, it is evident from the results that the refolded TaLPMO9A regained its oxidative activity after going through an unfolding/refolding cycle.
To further investigate the reversibility of thermal unfolding, the TaLPMO9A unfolding and refolding was also performed in the presence of the cysteine disulphide bond reducing reagent β-mercaptoethanol (β-ME, added before heating). The data shown in Fig. 4 clearly indicates that, with increasing temperatures from 30 to 90 °C the TaLPMO9A unfolds. However, when the unfolded TaLPMO9A is cooled down from 90 to 30 °C in the presence of β-ME the refolding process is not observed. The lack of reversibility in the unfolded TaLPMO9A is ascribed to the prevention of reformation of the disulphide bonds. The TaLPMO9A crystal structure (2YET) contains two disulfide bonds in each subunit (Cys97–Cys101 and Cys56–Cys178) (Fig. S4†).5 When the TaLPMO9A unfolds, the disulphide bonds are reduced by β-ME. From the above results it can be concluded that during the unfolding of TaLPMO9A, in absence of β-ME, the protein retains its native disulphide bonds intact.43 The two disulphide bonds are thus critical for the structural stability of the TaLPMO9A at high temperatures.31
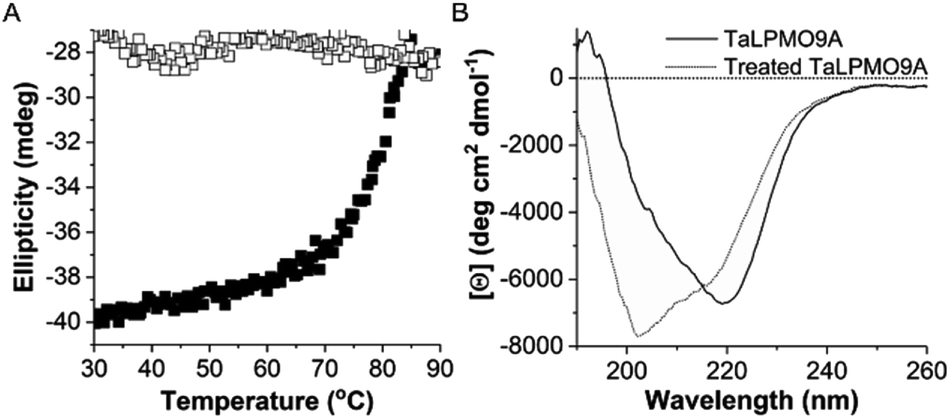 | ||
| Fig. 4 Thermal unfolding of TaLPMO9A in presence of β-ME. (A) The molar ellipticity measured at 222 nm was monitored continuously in the temperature range from 30 to 90 °C for the TaLPMO9A (0.66 mg mL−1) in the presence of β-ME (10 mM). The transition from folded state to unfolded state of TaLPMO9A was recorded using a 1 mm cuvette, with temperatures ranging from 30 to 90 °C and 90 to 30 °C with an increment of 1.0 °C min−1 for unfolding and a 1.0 °C min−1 cooling rate for refolding. Solid and hollow squares represent the unfolding and refolding, respectively. (B) The far-UV CD spectra measured for TaLPMO9A and β-ME (10 mM) treated TaLPMO9A. β-ME (10 mM) was mixed with TaLPMO9A (1 mg mL−1) and incubated at 100 °C for 10 minutes. The far-UV spectral region (260–190 nm) was recorded using a 0.1 mm round cuvette at 25 °C. The mean residue ellipticity is calculated on the basis of mean residue weight (MRW). | ||
Thermodynamic parameters
To study the thermodynamic parameters of unfolding and refolding of TaLPMO9A with intact disulphide bonds, we recorded the ellipticity at 222 nm with different temperature ramps (1.0, 0.5, 0.2 and 0.1 °C min−1). The ellipticity (222 nm) was measured from 25 °C to 90 °C and from 90 °C to 25 °C for TaLPMO9A unfolding and refolding, respectively (Fig. 5). Unfolding of TaLPMO9A started at 68, 66, 64 and 62 °C for a 1.0, 0.5, 0.2, and 0.1 °C min−1 temperature ramp, respectively (Fig. 5A). Accordingly, a decrease in melting temperature (Tm) with slower heating rates was also observed (Fig. 5A and Table S1†). This indicates that the unfolding is a process that, at high heating rates is limited by the heating rate. After achieving the unfolding at 90 °C, where ellipticity (222 nm) became constant, we also monitored the refolding of TaLPMO9A by decreasing the temperature from 90 °C to 25 °C applying various cooling rates (1.0, 0.5, 0.2 and 0.1 °C min−1) (Fig. 5B). The transition curve for refolding at various cooling rates (Fig. S5B†) showed differences in the onset of refolding (see Methods section for the calculation of the folded fraction). Consequently, a lower half refolding temperature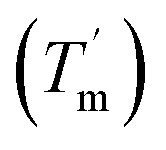 was observed with faster cooling rate (Fig. 5B and Table S1†).
was observed with faster cooling rate (Fig. 5B and Table S1†).
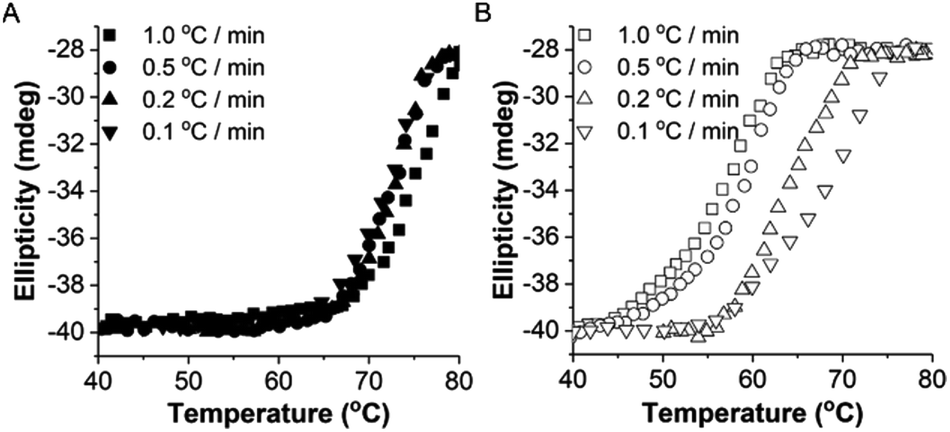 | ||
| Fig. 5 Unfolding and refolding of TaLPMO9A. Reversible thermal unfolding of TaLPMO9A. The ellipticity at 222 nm was monitored continuously in the temperature range from (A) 40 to 80 °C and (B) 80 to 40 °C at different temperature ramps. Thermal unfolding and refolding were performed at 0.1, 0.2, 0.5, and 1.0 °C min−1 temperature ramps. The scans at each condition were smoothed using Savitzky–Golay filter with a 5 point and polynomial order 2. | ||
For a specific heating and cooling rate, such as 1 °C min−1, the melting temperature (Tm) is much higher than the half refolding temperature 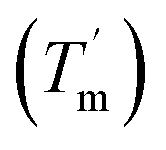 which indicates hysteresis in the unfolding and refolding process of TaLPMO9A.44 From this difference in Tm and
which indicates hysteresis in the unfolding and refolding process of TaLPMO9A.44 From this difference in Tm and 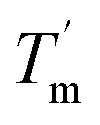 it can be inferred that during a fast temperature ramp, there is not sufficient time for TaLPMO9A to reach an equilibrium at a specific temperature, thereby, causing hysteresis. However, this hysteresis decreases with a decreasing temperature ramp for the unfolding and refolding cycle. Table S1† shows that the difference between Tm and
it can be inferred that during a fast temperature ramp, there is not sufficient time for TaLPMO9A to reach an equilibrium at a specific temperature, thereby, causing hysteresis. However, this hysteresis decreases with a decreasing temperature ramp for the unfolding and refolding cycle. Table S1† shows that the difference between Tm and 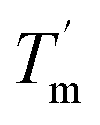 decreased with a decrease in temperature ramp (from 1.0 °C min−1 to 0.1 °C min−1). Also, the decrease in
decreased with a decrease in temperature ramp (from 1.0 °C min−1 to 0.1 °C min−1). Also, the decrease in 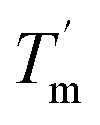 is more prominent than that of Tm. This implies that TaLPMO9A refolding process is much slower than that of unfolding. The unfolding and refolding process was analyzed by applying the simplest model (N↔U). Here, the TaLPMO9A is assumed to exist in the native (N) or in the unfolded (U) state. Fig. 6 and S5† show a plot of the apparent transition temperature and the apparent van't Hoff enthalpy change (ΔH). Both the apparent Tm and the apparent ΔH were found to be dependent on the scanning rate. ΔH of TaLPMO9A unfolding (0.1 °C min−1 heating rate) and refolding (0.1 °C min−1 cooling rate) was observed to be 245 and 247 kJ mol−1, respectively (Fig. 6 and Table S2†). We have extrapolated the unfolding and refolding data from Fig. 6A by considering the average of unfolding/refolding to obtain values that should correspond to values close to the equilibrium. At equilibrium, the CD spectra were very similar before and after a heating cycle and unfolding and refolding transitions were super-imposable. The reversibility and equilibrium condition found in the temperature-induced unfolding of TaLPMO9A allowed the estimation of ΔH, ΔS, and Tm. ΔH and ΔS values for TaLPMO9A unfolding/refolding were found to be 246 kJ mol−1 and 0.72 kJ mol−1, respectively (Fig. S6 and Table S2†). The native state of TaLPMO9A is stabilized at 25 °C and 37 °C by a free energy change (ΔG) of 13 and 4 kJ mol−1, respectively. The apparent Tm of TaLPMO9A was estimated to be 69.3 °C under conditions close to the equilibrium. ΔH for glutamate dehydrogenase, a hyperthermostable enzyme from Thermotoga maritima is reported to be 218 kJ mol−1 (at pH 7.15).45
is more prominent than that of Tm. This implies that TaLPMO9A refolding process is much slower than that of unfolding. The unfolding and refolding process was analyzed by applying the simplest model (N↔U). Here, the TaLPMO9A is assumed to exist in the native (N) or in the unfolded (U) state. Fig. 6 and S5† show a plot of the apparent transition temperature and the apparent van't Hoff enthalpy change (ΔH). Both the apparent Tm and the apparent ΔH were found to be dependent on the scanning rate. ΔH of TaLPMO9A unfolding (0.1 °C min−1 heating rate) and refolding (0.1 °C min−1 cooling rate) was observed to be 245 and 247 kJ mol−1, respectively (Fig. 6 and Table S2†). We have extrapolated the unfolding and refolding data from Fig. 6A by considering the average of unfolding/refolding to obtain values that should correspond to values close to the equilibrium. At equilibrium, the CD spectra were very similar before and after a heating cycle and unfolding and refolding transitions were super-imposable. The reversibility and equilibrium condition found in the temperature-induced unfolding of TaLPMO9A allowed the estimation of ΔH, ΔS, and Tm. ΔH and ΔS values for TaLPMO9A unfolding/refolding were found to be 246 kJ mol−1 and 0.72 kJ mol−1, respectively (Fig. S6 and Table S2†). The native state of TaLPMO9A is stabilized at 25 °C and 37 °C by a free energy change (ΔG) of 13 and 4 kJ mol−1, respectively. The apparent Tm of TaLPMO9A was estimated to be 69.3 °C under conditions close to the equilibrium. ΔH for glutamate dehydrogenase, a hyperthermostable enzyme from Thermotoga maritima is reported to be 218 kJ mol−1 (at pH 7.15).45
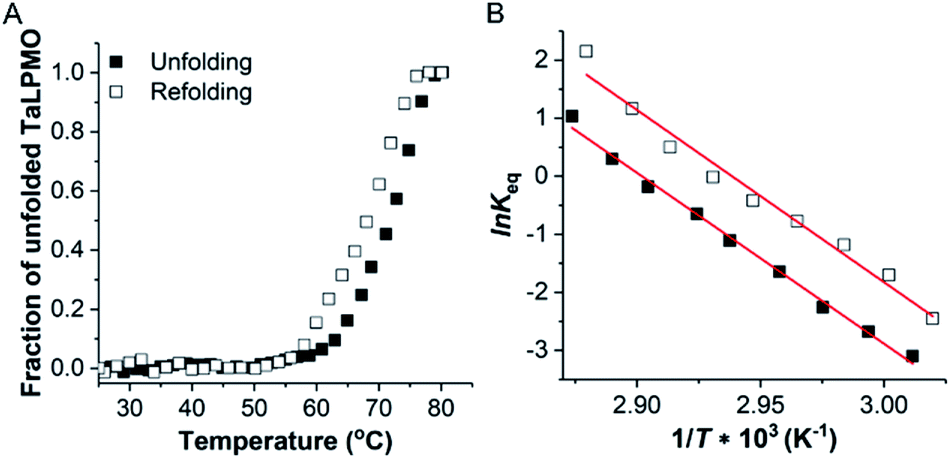 | ||
| Fig. 6 Thermodynamic calculation using van't Hoff plot. Reversible thermal unfolding of TaLPMO9A. (A) Transition curve for the ellipticity measured at 222 nm in the temperature range from 25 to 80 °C and 80 to 25 °C, at a temperature ramp of 0.1 °C min−1. (B) van't Hoff plot for the calculation of thermodynamic parameters of the TaLPMO9A unfolding and refolding. Solid and hollow squares represent the unfolding and refolding, respectively. | ||
Unfolding and refolding kinetics
To investigate the kinetics of TaLPMO9A unfolding, we recorded the ellipticity at 222 nm at different temperatures (from 72 to 80 °C) for 1 h using a CD spectrophotometer (Fig. 7). The protein was unable to unfold, or the unfolding was very slow, at temperatures below 72 °C. At higher temperatures (80, 78 and 76 °C) TaLPMO9A starts to unfold rapidly and unfolded completely within 15 min at 80 and 78 °C. Unfolding at 72 and 74 °C was comparably slower than the unfolding at 76, 78 and 80 °C. The folded protein fraction (fN) was reduced to less than 0.3 within 30 min at all tested temperatures from 72 to 80 °C (Fig. 7A). The rate of unfolding (ku) was derived from the exponential fit of fN overtime (Fig. S7A†). The activation energy of unfolding (Eua), derived from the slope of ku in the Arrhenius plot (Fig. S7B†), was calculated to be ∼171 kJ mol−1.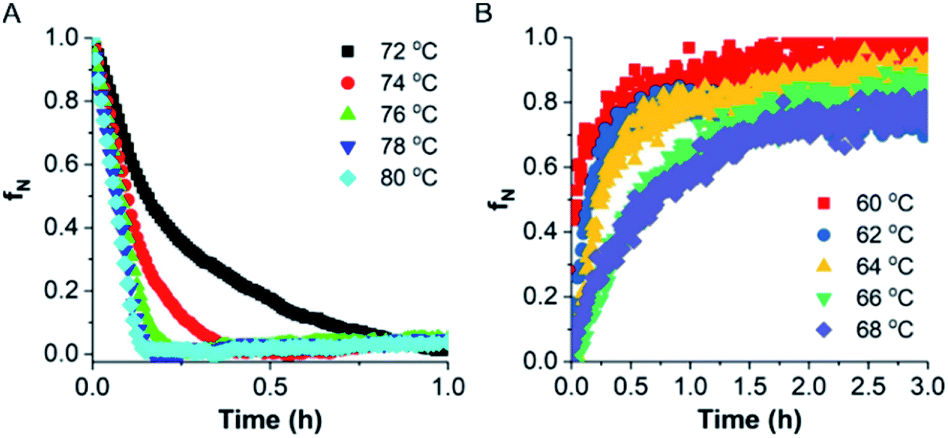 | ||
| Fig. 7 Unfolding and refolding kinetics. (A) Variation in folded fraction during TaLPMO9A (0.5 mg mL−1, in 20 mM MOPS buffer, pH 7.0) unfolding as a function of time at different temperatures. The transition from the folded state to the unfolded state was recorded by following change in ellipticity (222 nm) using a 1 mm cuvette. (B) Variation in folded fraction during TaLPMO9A (0.5 mg mL−1, in 20 mM MOPS buffer, pH 7.0) refolding as a function of time by cooling it down at different temperatures. The transition from the unfolded state to the folded state was recorded by following change in ellipticity (222 nm) using a 1 mm cuvette. The folded protein fraction is presented as fN. | ||
Finally, the refolding kinetics of TaLPMO9A were monitored by recording the change in ellipticity at 222 nm for 3 h. TaLPMO9A was first subjected to unfolding at 80 °C for 10 min then the refolding kinetics were determined by cooling it down within 2 min to different temperatures (from 60 to 68 °C). At 60 °C the unfolded TaLPMO9A regained 50% of its folded conformation within 3 min of incubation (Fig. 7B). Despite the fast initial rate, it took more than 60 min to reach a 90% folded state. At higher temperatures, such as 68 and 66 °C, it does not return to its fully folded state after 3 h of incubation. This shows that the refolding process of TaLPMO9A is much slower at temperatures close to Tm. In case of lower temperatures such as 60, 62 and 64 °C, the refolding process tends to be faster. The rate of TaLPMO9A refolding (kf) was calculated from single exponential fit (Fig. S7C†) of data shown in Fig. 6B. The activation energy of refolding (Efa) was derived from the slope of the Arrhenius plot for the rate of refolding (Fig. S7D†) and was estimated to be ∼−181 kJ mol−1.
Conclusions
An extensive study of the thermal unfolding and refolding of a fully copper loaded TaLPMO9A was conducted using CD spectroscopy. Our resuslts reveal high structural stability at elevated temperatures and reversible thermal unfolding. TaLPMO9A retains more than 80% of its activity after 30 min of incubation at 100 °C. The onset of unfolding and refolding is strongly dependent on the heating rate. The catalytic activity of TaLPMO9A, measured by Amplex® Red and PASC oxidation assays, was fully restored after an unfolding/refolding cycle. Finally, the Tm of the deglycosylated as well as the apoTaLPMO9A are lower than for the native, copper loaded, TaLPMO9A, yet both also fully refold upon cooling. The higher thermostability of native TaLPMO9A may be ascribed to its glycosylation. Additionally, experiments with β-ME show that disulphide bonds are retained during thermal unfolding and are essential for the refolding of TaLPMO9A. CD provides structural information on average for all the states in solution while allowing us to understand the functional stability and thermodynamic parameters of this unique protein during an unfolding/refolding cycle. The knowledge of unfolding/refolding kinetic (Tm, rates) and thermodynamic parameters (ΔH, ΔS) will be especially valuable when using TaLPMO9A independently or as part of the enzyme cocktail in enzymatic biomass conversion. This work contributes to the fast-growing field of enzyme deconstruction of lignocellulose and, in particular, to the industrial application of LPMOs below their melting temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Novo Nordisk Foundation project “Harnessing the Energy of the Sun for Biomass Conversion” (NNF16OC0021832). We also acknowledge the donation of enzymes from Novozymes A/S. We also want to thank our lab technician Thanh H. Madsen for her support. All the data relevant to this study are included in the main paper or the ESI.†References
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- D. Loque, H. V. Scheller and M. Pauly, Curr. Opin. Plant Biol., 2015, 25, 151–161 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J. C. Poulsen, K. S. Johansen, K. B. Krogh, C. I. Jorgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sorlie and V. G. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- J. W. Agger, T. Isaksen, A. Varnai, S. Vidal-Melgosa, W. G. Willats, R. Ludwig, S. J. Horn, V. G. Eijsink and B. Westereng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6287–6292 CrossRef CAS PubMed.
- W. T. Beeson, C. M. Phillips, J. H. Cate and M. A. Marletta, J. Am. Chem. Soc., 2012, 134, 890–892 CrossRef CAS PubMed.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. G. Eijsink, Biotechnol. Biofuels, 2012, 5, 45 CrossRef CAS PubMed.
- X. Li, W. T. Beeson, C. M. Phillips, M. A. Marletta and J. H. Cate, Structure, 2012, 20, 1051–1061 CrossRef CAS PubMed.
- F. Sabbadin, G. R. Hemsworth, L. Ciano, B. Henrissat, P. Dupree, T. Tryfona, R. D. S. Marques, S. T. Sweeney, K. Besser, L. Elias, G. Pesante, Y. Li, A. A. Dowle, R. Bates, L. D. Gomez, R. Simister, G. J. Davies, P. H. Walton, N. C. Bruce and S. J. McQueen-Mason, Nat. Commun., 2018, 9, 756 CrossRef PubMed.
- J. S. Loose, Z. Forsberg, D. Kracher, S. Scheiblbrandner, R. Ludwig, V. G. Eijsink and G. Vaaje-Kolstad, Protein Sci., 2016, 25, 2175–2186 CrossRef CAS PubMed.
- G. R. Hemsworth, E. M. Johnston, G. J. Davies and P. H. Walton, Trends Biotechnol., 2015, 33, 747–761 CrossRef CAS PubMed.
- A. Levasseur, E. Drula, V. Lombard, P. M. Coutinho and B. Henrissat, Biotechnol. Biofuels, 2013, 6, 41 CrossRef CAS PubMed.
- G. R. Hemsworth, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Chem. Biol., 2014, 10, 122–126 CrossRef CAS PubMed.
- C. Filiatrault-Chastel, D. Navarro, M. Haon, S. Grisel, I. Herpoel-Gimbert, D. Chevret, M. Fanuel, B. Henrissat, S. Heiss-Blanquet, A. Margeot and J. G. Berrin, Biotechnol. Biofuels, 2019, 12, 55 CrossRef PubMed.
- F. L. Aachmann, M. Sorlie, G. Skjak-Braek, V. G. Eijsink and G. Vaaje-Kolstad, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18779–18784 CrossRef CAS PubMed.
- D. M. Petrovic, B. Bissaro, P. Chylenski, M. Skaugen, M. Sorlie, M. S. Jensen, F. L. Aachmann, G. Courtade, A. Varnai and V. G. H. Eijsink, Protein Sci., 2018, 27, 1636–1650 CrossRef CAS PubMed.
- M. Gudmundsson, S. Kim, M. Wu, T. Ishida, M. H. Momeni, G. Vaaje-Kolstad, D. Lundberg, A. Royant, J. Stahlberg, V. G. Eijsink, G. T. Beckham and M. Sandgren, J. Biol. Chem., 2014, 289, 18782–18792 CrossRef CAS PubMed.
- S. Kim, J. Stahlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 149–154 CrossRef CAS PubMed.
- D. Cannella, K. B. Möllers, N. U. Frigaard, P. E. Jensen, M. J. Bjerrum, K. S. Johansen and C. Felby, Nat. Commun., 2016, 7, 11134 CrossRef CAS PubMed.
- C. Sygmund, D. Kracher, S. Scheiblbrandner, K. Zahma, A. K. Felice, W. Harreither, R. Kittl and R. Ludwig, Appl. Environ. Microbiol., 2012, 78, 6161–6171 CrossRef CAS PubMed.
- L. Nekiunaite, M. O. Arntzen, B. Svensson, G. Vaaje-Kolstad and M. Abou Hachem, Biotechnol. Biofuels, 2016, 9, 187 CrossRef PubMed.
- I. J. Kim, H. J. Youn and K. H. Kim, Process Biochem., 2016, 51, 1445–1451 CrossRef CAS.
- B. Bissaro, A. K. Rohr, G. Muller, P. Chylenski, M. Skaugen, Z. Forsberg, S. J. Horn, G. Vaaje-Kolstad and V. G. H. Eijsink, Nat. Chem. Biol., 2017, 13, 1123–1128 CrossRef CAS PubMed.
- J. A. Hangasky, T. C. Detomasi and M. A. Marletta, Trends in Chemistry, 2019, 1, 198–209 CrossRef.
- K. E. Frandsen, T. J. Simmons, P. Dupree, J. C. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- E. D. Hedegard and U. Ryde, Chem. Sci., 2018, 9, 3866–3880 RSC.
- R. K. Singh, M. K. Tiwari, R. Singh and J. K. Lee, Int. J. Mol. Sci., 2013, 14, 1232–1277 CrossRef CAS PubMed.
- M. Frommhagen, A. H. Westphal, R. Hilgers, M. J. Koetsier, S. W. A. Hinz, J. Visser, H. Gruppen, W. J. H. van Berkel and M. A. Kabel, Appl. Microbiol. Biotechnol., 2018, 102, 1281–1295 CrossRef CAS PubMed.
- M. Tanghe, B. Danneels, M. Last, K. Beerens, I. Stals and T. Desmet, Protein Eng., Des. Sel., 2017, 30, 401–408 CrossRef CAS PubMed.
- M. Iwakura, D. Nakamura, T. Takenawa and Y. Mitsuishi, Protein Eng., 2001, 14, 583–589 CrossRef CAS PubMed.
- P. H. Jensen, N. G. Karlsson, D. Kolarich and N. H. Packer, Nat. Protoc., 2012, 7, 1299–1310 CrossRef CAS PubMed.
- P. Saha, C. Manna, J. Chakrabarti and M. Ghosh, Sci. Rep., 2016, 6, 29541 CrossRef PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- R. Kittl, D. Kracher, D. Burgstaller, D. Haltrich and R. Ludwig, Biotechnol. Biofuels, 2012, 5, 79 CrossRef CAS PubMed.
- T. M. Wood, Methods Enzymol., 1988, 160, 19–25 CAS.
- K. B. Möllers, H. Mikkelsen, T. I. Simonsen, D. Cannella, K. S. Johansen, M. J. Bjerrum and C. Felby, Carbohydr. Res., 2017, 448, 182–186 CrossRef PubMed.
- K. S. Johansen, F. Xu, P. H. Walton, B. McBrayer, H. Lund and C. L. Soong, European Pat., EP3339442, 2012.
- P. D. Ross and A. Shrake, J. Biol. Chem., 1988, 263, 11196–11202 CAS.
- R. Kazlauskas, Chem. Soc. Rev., 2018, 47, 9026–9045 RSC.
- D. Kracher, M. Andlar, P. G. Furtmuller and R. Ludwig, J. Biol. Chem., 2018, 293, 1676–1687 CrossRef CAS PubMed.
- J. Y. Chang and L. Li, FEBS Lett., 2002, 511, 73–78 CrossRef CAS PubMed.
- B. T. Andrews, D. T. Capraro, J. I. Sukowska, J. N. Onuchic and P. A. Jennings, J. Phys. Chem. Lett., 2013, 4, 180–188 CrossRef CAS PubMed.
- V. Consalvi, R. Chiaraluce, L. Giangiacomo, R. Scandurra, P. Christova, A. Karshikoff, S. Knapp and R. Ladenstein, Protein Eng., 2000, 13, 501–507 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05920b |
| ‡ Current address: Institute for Inorganic and Analytical Chemistry, Bioorganic Analytics, Friedrich Schiller University Jena, Jena, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |