
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew red-emitting phosphor RbxK3−xSiF7:Mn4+ (x = 0, 1, 2, 3): DFT predictions and synthesis†
Seunghun Jang‡
a,
June Kyu Park‡a,
Minseuk Kimb,
Kee-Sun Sohn b,
Chang Hae Kim*a and
Hyunju Chang*a
b,
Chang Hae Kim*a and
Hyunju Chang*a
aKorea Research Institute of Chemical Technology, 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, Republic of Korea
bFaculty of Nanotechnology and Advanced Materials Engineering, Sejong University, Seoul 05006, Republic of Korea. E-mail: changhae@krict.re.kr; hjchang@krict.re.kr; Fax: +82 42 860 7508; Tel: +82 42 860 7364
First published on 2nd December 2019
Abstract
Finding new phosphors through an efficient method is important in terms of saving time and cost related to the development of phosphor materials. The ability to identify new phosphors through preliminary simulations by calculations prior to the actual synthesis of the materials can maximize the efficiency of novel phosphor development. In this paper, we demonstrate the use of density functional theory (DFT) calculations to guide the development of a new red phosphor. We performed first-principles calculations based on DFT for pristine and Mn-doped RbxK3−xSiF7 (x = 0, 1, 2, 3) and predicted their stability, electronic structure, and luminescence properties. On the basis of the results, we then synthesized the stable Rb2KSiF7:Mn4+ red conversion phosphor and investigated its luminescence, structure, and stability. As a result, we confirmed that Rb2KSiF7:Mn4+ emitted red light with a longer wavelength than that emitted by K3SiF7:Mn4+ and a wavelength similar to that of K2SiF6:Mn4+. These results show that DFT calculations can provide rational insights into the design of a phosphor material before it is synthesized, thereby reducing the time and cost required to develop new red conversion phosphors.
1. Introduction
White-light-emitting diodes (WLEDs), which have been highlighted as next-generation light sources, have been widely studied because of their outstanding features, which include high durability, high energy conversion efficiency, low power consumption, and environmental friendliness.1–3 To obtain white light, the strong blue-light emission from an InGaN light-emitting diode (LED) chip must be converted by color-conversion phosphor materials. Among numerous available conversion phosphors, an excellent red phosphor that exhibits a good color-rendering index is needed for WLEDs. Conventionally, CaAlSiN3:Eu2+ (CASN:Eu2+) and K2SiF6:Mn4+ are the representative red conversion phosphors with superior performance.4–7 Because of their extensive color gamut and better color-rendering index, these phosphors have been widely used. In addition, a novel red narrow-band phosphor, K3SiF7:Mn4+, with fluorescence characteristics similar to those of the K2SiF6:Mn4+ phosphor has recently been introduced.8 Although some progress has been made in terms of enhancing the feasibility of its synthesis and improving its decay time, the K3SiF7:Mn4+ phosphor has a shortcoming in that the wavelength of its main photoluminescence (PL) peak is shorter than that of K2SiF6:Mn4+, which inhibits its color-rendering index or reduces its color gamut.To address this problem, we considered substituting the K+ ions in K3SiF7:Mn4+ with Rb+ ions. When Rb+, which is a member of the same periodic group as K+ but has a larger atomic radius, is substituted for K+ in the K3SiF7:Mn4+ system, expansion of the lattice is expected. Such a change in the lattice is reasonably presumed to affect the luminescent properties of the system.
Meanwhile, theoretical approaches for the design of new phosphors can offer more effective ways to explore new conversion phosphor materials.5,9 In our previous work, on the basis of first-principles studies based on density functional theory (DFT), we proposed that Sn- or Bi-doped CASN would be an effective alternative to Eu-doped CASN phosphors. Following our theoretical study, Sn- and Bi-doped CASN red phosphors were successfully prepared and their phosphor properties were experimentally demonstrated.5,10
In the present work, we performed first-principles calculations based on DFT for pristine and Mn-doped RbxK3−xSiF7 (x = 0, 1, 2, 3) and predicted their stability, electronic structure, and luminescence properties. In addition, on the basis of the knowledge gained from first-principles calculations, we synthesized Rb2KSiF7:Mn4+ as a stable red conversion phosphor and verified its PL properties, structure, and stability. The Rb2KSiF7:Mn4+ emitted red light with a wavelength longer than that of the red light emitted by K3SiF7:Mn4+ and similar to that of the light emitted by K2SiF6:Mn4+. These results also show good agreement with the theoretical calculation results that indicated that the simulated emission wavelength of Rb2KSiF7:Mn4+ is the longest among the investigated phosphors. These results demonstrate that DFT calculations can provide rational insights into the properties of a phosphor material before it is synthesized, thereby reducing the time required to develop new red conversion phosphors.
2. Computational and experimental details
2.1. DFT calculations
The atomic and electronic structures of the RbxK3−xSiF7 (x = 0, 1, 2, 3) host materials were examined using the Vienna ab initio simulation package (VASP).11,12 The exchange–correlation functional was approximated using the Perdew–Burke–Ernzerhof (PBE) expression.13 The electron–ion interactions were modeled using the projector-augmented wave (PAW) method.14 The electronic wave functions were expanded in a basis set of plane waves using a kinetic energy cutoff of 500 eV. Geometry relaxation steps were performed under the criterion that ionic forces were reduced to less than 0.01 eV Å−1.Mn-doped RbxK3−xSiF7 systems with Mn atoms substituted at Si sites were analyzed using a 1 × 1 × 2 supercell structure. To optimize the geometry of each modeled structure, the k-space integration steps for pristine and Mn-doped RbxK3−xSiF7 structures were performed with finite sampling of the k-points on 7 × 7 × 9 and 7 × 7 × 5 meshes in the Brillouin zone, respectively. For the calculations of electronic structures, such as density of states (DOS) and band-structure calculations, the k-space integration steps for the pure and Mn-doped RbxK3−xSiF7 structures were performed with finite sampling of the k-points on 9 × 9 × 11 and 9 × 9 × 7 meshes in the Brillouin zone, respectively.
2.2. Synthesis
The novel red phosphor Rb2KSiF7:Mn4+ was successfully synthesized at room temperature via a two-step procedure, as shown in Fig. 1(a) and (b). Step 1 is the preparation of Rb2SiF6:Mn4+; step 2 is sintering and washing to obtain the novel red phosphor Rb2KSiF7:Mn4+.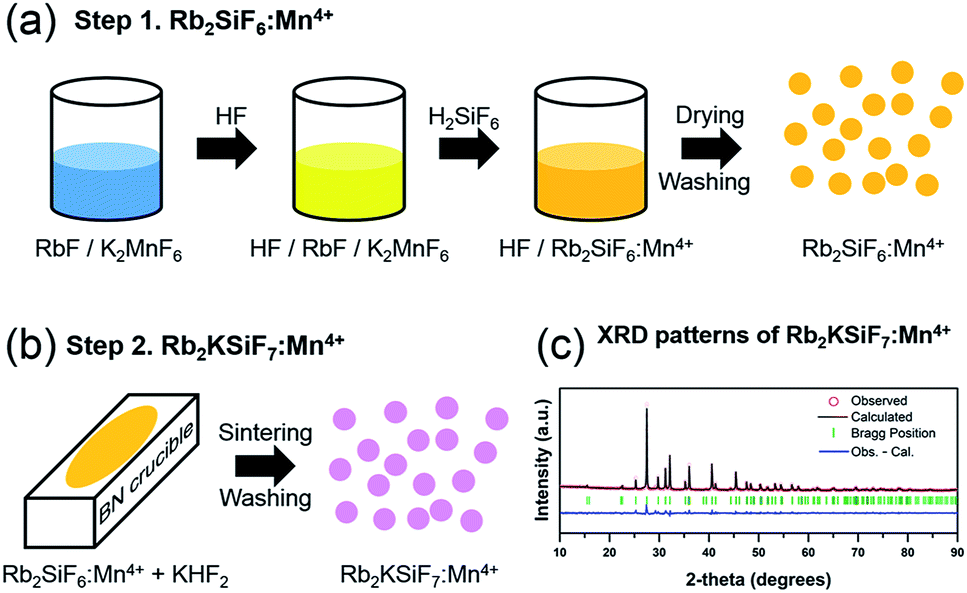 | ||
| Fig. 1 Overall experimental procedure for the synthesis of the novel red phosphor Rb2KSiF7:Mn4+: (a) procedure for the synthesis of Rb2SiF6:Mn4+ and (b) procedure for conversion from Rb2SiF6:Mn4+ to Rb2KSiF7:Mn4+. (c) XRD pattern of the Rb2KSiF7:Mn4+ with its corresponding Rietveld refinement (solid line) and residual (observed–calculated). | ||
A schematic of the liquid-state reaction (LSR) method is shown in Fig. 1(a). The raw materials used in the synthesis were Rb2SiF6 (Fluka, 99.0%), KF (Acros, 99%), KMnO4 (Junsei Chemical Co., Ltd., 99.3%), KHF2 (Sigma-Aldrich, 99%), HF (Avantor, 48%), H2SiF6 (SAMCHUN, 40%), and H2O2 (SAMCHUN, 34.5%). All chemical reagents were used as obtained without further purification. The overall synthesis process was conducted in a safe environment.
| 2KMnO4(s) + 2KF(s) + 10HF(aq) + 3H2O2 → 2K2MnF6(s) + 8H2O(l) + 3O2(g) |
2.3. Characterization
The crystal structures of the phosphors (Rb2KSiF7:Mn4+) were determined by X-ray diffraction (XRD, Rigaku D/Max 220V) using Cu Kα radiation (Ni filter, voltage: 40 kV, current: 40 mA), as shown in Fig. 1(c). To investigate the morphology and the size of the prepared phosphors, we used scanning electron microscopy (SEM; JEOL JSM-6360). The composition of the samples was analyzed by energy-dispersive X-ray spectrometry (EDS) using a spectrometer (Bruker Quantax 200) with an energy resolution <127 eV (HV: 20.0 kV). The optical characteristics of the phosphors were examined at room temperature using a photoluminescence spectrometer (DARSA PRO-5000) and a Xe light source (500 W). Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) of the phosphors were conducted on a Thermo Plus EVO II (TG8120 series, DSC 8230) at a heating rate of 5 °C min−1 under flowing N2 gas.3. Results and discussion
Fig. 2(a) shows the atomic geometries of the RbxK3−xSiF7 structures. With increasing x in RbxK3−xSiF7, the lattice parameters and cell volume increase, as shown in Table 1, because the atomic radius of Rb (2.35 Å) is slightly larger than that of K (2.20 Å).15 The trend of the calculated lattice parameters with increasing Rb content is consistent with the trend of the experimental lattice parameters.16,17 In particular, in the cases of binary cations (Rb+ and K+), various structural configurations in which Rb+ or K+ ions occupy positions in the unit cell are possible (see Fig. S1 in ESI†). The RbK2SiF7 and Rb2KSiF7 atomic geometries shown in Fig. 2(a) were chosen as the structures with the most stable total energy among the various investigated configurations. These two energetically preferred structures exhibit interesting structural features. The RbK2SiF7 and Rb2KSiF7 atomic geometries include K-2D-layer (green trapezium) and K-1D-chain (red arrow) substructures within their atomic structures, respectively (see Fig. S2 in ESI†). We speculated that such substructures energetically stabilize their associated atomic structures.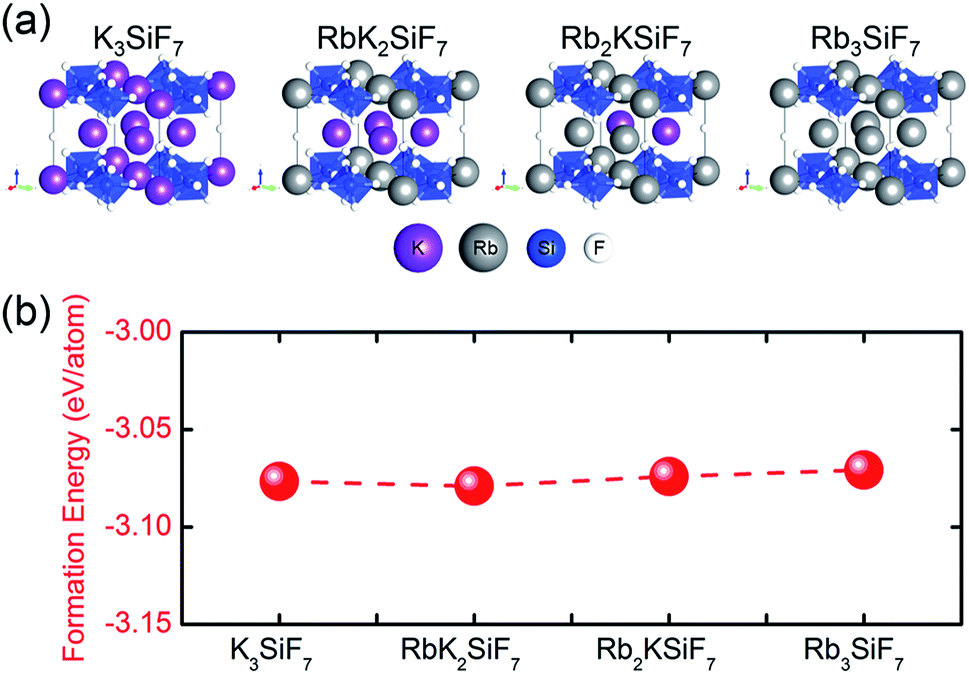 | ||
| Fig. 2 (a) Atomic geometries of RbxK3−xSiF7. The violet, pink, blue, and white balls represent K, Rb, Si, and F atoms, respectively. (b) Bulk formation energies of the RbxK3−xSiF7 structures. | ||
| Tetragonal (P4/mbm) | Experiment | Theory (this work) | ||||
|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | a (Å) | b (Å) | c (Å) | |
| K3SiF7 | 7.74 | 7.74 | 5.56 | 7.87 | 7.87 | 5.64 |
| RbK2SiF7 | 7.94 | 7.94 | 5.72 | |||
| Rb2KSiF7 | 7.88 | 7.88 | 5.72 | 8.04 | 8.04 | 5.82 |
| Rb3SiF7 | 7.95 | 7.95 | 5.82 | 8.14 | 8.14 | 5.92 |
In addition, we calculated the bulk formation energies of the RbxK3−xSiF7 structures. The formation energy, ΔHf, for RbxK3−xSiF7 is given by 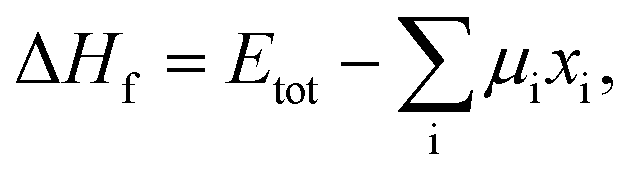 where Etot is the total energy of the structure, μi is the chemical potential of element i, and xi is the number of elements i in the structure.18 In terms of the design of a new host material for conversion phosphors, the DFT formation energy can be an important indicator for determining whether a phosphor material can be synthesized. As shown in Fig. 2(b), all of the RbxK3−xSiF7 structures have negative formation energies, which indicates that all of the structures we considered can be synthesized. Among these compounds, all except Rb2KSiF7 have been reported experimentally.16,17
where Etot is the total energy of the structure, μi is the chemical potential of element i, and xi is the number of elements i in the structure.18 In terms of the design of a new host material for conversion phosphors, the DFT formation energy can be an important indicator for determining whether a phosphor material can be synthesized. As shown in Fig. 2(b), all of the RbxK3−xSiF7 structures have negative formation energies, which indicates that all of the structures we considered can be synthesized. Among these compounds, all except Rb2KSiF7 have been reported experimentally.16,17
Furthermore, we calculated the energy band structures and DOSs of the RbxK3−xSiF7 system (see Fig. S3 in ESI†). With increasing Rb content, the bandgap of the host material system decreases from 5.59 eV to 5.33 eV. This decrease of the bandgap energy is related to the increase of the lattice parameters with increasing Rb content. Because of the large radius of Rb atoms, an increase of the Rb content (with Rb substitution at the K sites) leads to an expansion of the RbxK3−xSiF7 unit-cell volume. Accordingly, the increase of the cell volume leads to weakening of the binding forces of the atoms' valence electrons. Consequently, the valence electrons become freer with increasing interatomic distance, which eventually reduces the bandgap which corresponds to the energy required for valence electrons to be promoted to the conduction band (CB).
From the atomic projected DOSs, we find that, although the CB is derived mainly from the Rb d state, the F p state contributes to the upper valence band (VB) (see Fig. S4 in ESI†). An enlarged image of the CB region from 5 eV to 10 eV is shown in the inset of each DOS diagram. As the Rb content increases, the Rb d state (in the CB) and Rb p state (near −8 eV, marked with a blue star) are enhanced. These Rb-related DOSs do not appear to strongly affect the F- or Si-related states.
One of the important factors in the development of conversion red phosphors is their emission wavelength. If the emission wavelength of the candidate material can be predicted before the material is actually synthesized, the phosphor development time would be dramatically reduced, leading to the efficient design of effective phosphor materials. Consequently, the ability to predict the emission energy of the doping system is important. For Mn-doped phosphor systems, the emission light is known to originate from the spin-flip transition of Mn4+, such as the transition from the 2Eg excited state to the 4A2g ground state. As shown in Fig. 3(a), low-spin (1 μB) and high-spin (3 μB) states correspond to the 2Eg excited state and the 4A2g ground state, respectively.19 Therefore, the emission energy of Mn4+ can be predicted by calculating the total energy difference between the low-spin state and the high-spin state. The emission energy was calculated according to the equation Emission Energy = Elow-spin − Ehigh-spin, where Elow-spin and Ehigh-spin are the total energies of Mn4+ doped systems with relaxed low-spin (1 μB) and high-spin (3 μB) states, respectively.
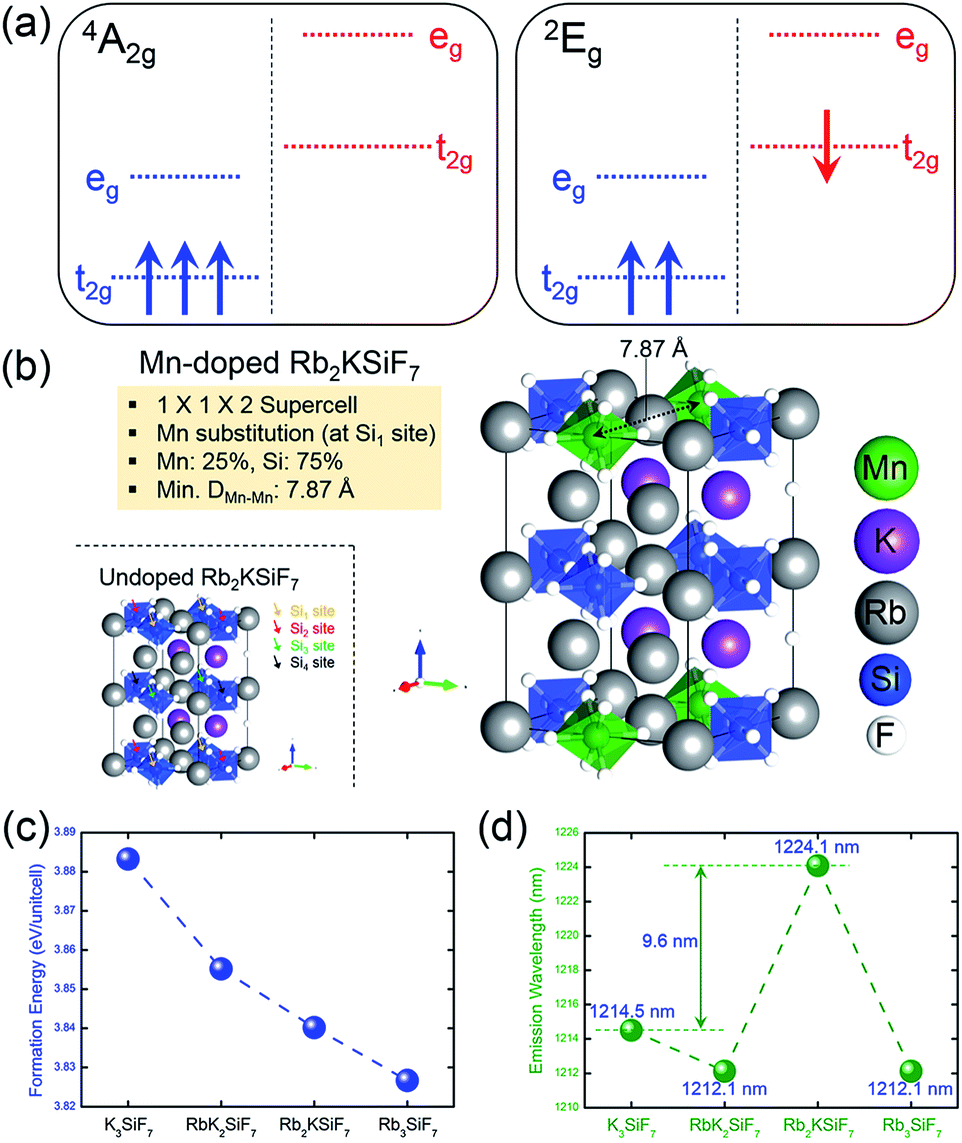 | ||
| Fig. 3 (a) Schematic energy diagrams of the 4A2g ground state and 2Eg excited state of Mn4+. (b) Atomic geometries of Rb2KSiF7 (undoped) and Rb2KSi0.75Mn0.25F7 (Mn-doped) structures. The green, violet, pink, blue, and white balls represent Mn, K, Rb, Si, and F atoms, respectively. (c) Formation energies of RbxK3−xSiF7:Mn4+ with Mn substituted at Si sites and (d) the simulated emission wavelengths of the Mn-substituted products. | ||
To calculate emission energies that account for the effects of Mn doping on the four RbxK3−xSiF7 (x = 0, 1, 2, 3) host materials, we first calculated the atomic and electronic structures of Mn-doped RbxK3−xSiF7. Fig. 3(b) shows the atomic geometry of the Rb2KSi0.75Mn0.25F7 structure. To minimize the effect of Mn–Mn interaction on the electronic structure, we introduced a 1 × 1 × 2 super cell structure. Among four Si sites (Si1, Si2, Si3, and Si4 sites), a Si atom was replaced with a Mn atom and the minimum Mn–Mn interatomic distance was 7.8 Å. Fig. S5 in ESI† shows the atomic projected DOSs on the 4A2g ground (left side) and 2Eg excited (right side) states of the RbxK3−xSiF7:Mn4+ structures. In all of the DOSs, both the t2g and eg states of Mn atoms are hybridized with the F p states. That is, the t2g and eg states between the bandgap of the host are donated by the Mn 3d and the F 2p orbitals. In addition, the 4A2g ground and 2Eg excited states have high-spin (3 μB) and low-spin (1 μB) configurations, respectively. In particular, in the 2Eg excited state, a small Jahn–Teller distortion will eventually manifest as splitting of the t2g and eg states, as shown in the right side of Fig. S5 in ESI†.19 These DOS results are similar to those reported in a previous study.19 No particular trend of the mid-gap states (t2g and eg states) with respect to the Rb content was found.
From the total energies of the ground states, we also calculated the formation energies for substitution of Mn at the Si site in RbxK3−xSiF7. The formation energy for Mn substitution, EformMn-sub, was calculated to assess the ease with which a Mn atom can replace a Si atom in a certain host material. It was calculated according to the equation EformMn-sub = EMn:RKSF + μSi − (ERKSF + μMn), where EMn:RKSF is the total energy of the ground state of RbxK3−xSiF7:Mn4+, ERKSF is the total energy of undoped RbxK3−xSiF7, μSi is the total energy per atom of bulk Si, and μMn is the total energy per atom of bulk Mn. As shown in Fig. 3(c), as the Rb content increases, the formation energy for Mn substitution decreases. This trend is attributed to the lattice expansion induced by the Rb substitution promoting Mn doping (at Si sites).
The results in Fig. 3(d) confirm that the simulated emission energy value fluctuates irrespective of the Rb content. Notably, the Rb2KSiF7:Mn4+ exhibits the longest simulated emission wavelength among the RbxK3−xSiF7:Mn4+ phosphors. On the basis of this result, we expected the light emission from Rb2KSiF7:Mn4+ to shift toward longer emission wavelengths compared with the wavelengths of the emissions from K3SiF7:Mn4+. In terms of material design of a new conversion phosphor, the aforementioned results provide an important guideline for the synthesis of host materials.
On the basis of the material insights gained thus far through theoretical investigations, we attempted to synthesize a new red conversion phosphor corresponding to Mn-doped RbxK3−xSiF7 (x > 1) using various methods. As a result, we obtained three phosphors: K3SiF7:Mn4+, Rb3SiF7:Mn4+ and Rb2KSiF7:Mn4+. The K3SiF7:Mn4+ phosphor has already been reported.8 Thus, we successfully synthesized two new conversion red phosphors, Rb3SiF7:Mn4+ and Rb2KSiF7:Mn4+, with a host material containing Rb. For reference, RbK2SiF7:Mn4+, which has not been reported in the ICSD database, was also not obtained in the present study. In addition, Rb3SiF7:Mn4+, which we successfully synthesized, was very unstable, exhibiting sensitivity to moisture; it structure fragmented soon after it was synthesized. However, the Rb2KSiF7:Mn4+ red phosphor exhibited very stable structural characteristics.
The procedure for the synthesis of Rb2KSiF7:Mn4+ is detailed in Fig. 1(a) and (b) and in Section 2.2. The crystallographic data (see Table S1 in ESI†) determined from Rietveld refinement with XRD patterns of the synthesized Rb2KSiF7:Mn4+, as the first conclusive evidence of the successful synthesis of Rb2KSiF7:Mn4+, shows good agreement with the reported experimental lattice parameters for Rb2KSiF7 in Table 1.16,17
Fig. 4(a) shows the normalized PL (λex = 460 nm) and photoluminescence excitation (PLE) (λem = 630 nm) spectra of the K2SiF6:Mn4+, K3SiF7:Mn4+ and Rb2KSiF7:Mn4+ phosphors. As shown in the right side of the Fig. 4(a), all three phosphors not only show very strong and narrow red luminescence in the wavelength region near 630 nm but also show very stable excitation from 330 nm to 480 nm. To examine the PL results more closely, the spectral region from 600 nm to 650 nm was enlarged (Fig. 4(b)). Here, Rb2KSiF7:Mn4+ clearly emits longer-wavelength light than K3SiF7:Mn4+ and has an emission wavelength almost equal to that of K2SiF6:Mn4+. Furthermore, the PL efficiency of Rb2KSiF7:Mn4+ sample (having the highest PL) corresponds to 85% of that of K2SiF6:Mn4+ (93% of that of K3SiF7:Mn4+), as shown in Fig. S6 in ESI.† These results indicate that luminescence with an emission wavelength similar to that of the conventional K2SiF6:Mn4+ phosphor can be induced from a Rb-containing quaternary fluoride phosphor. It also shows good agreement with the theoretical calculation results indicating that the simulated emission wavelength of Rb2KSiF7:Mn4+ is the longest among the investigated phosphors (Fig. 3(d)); thus, such calculation results can provide rational insights into the properties of a phosphor material before it is synthesized.
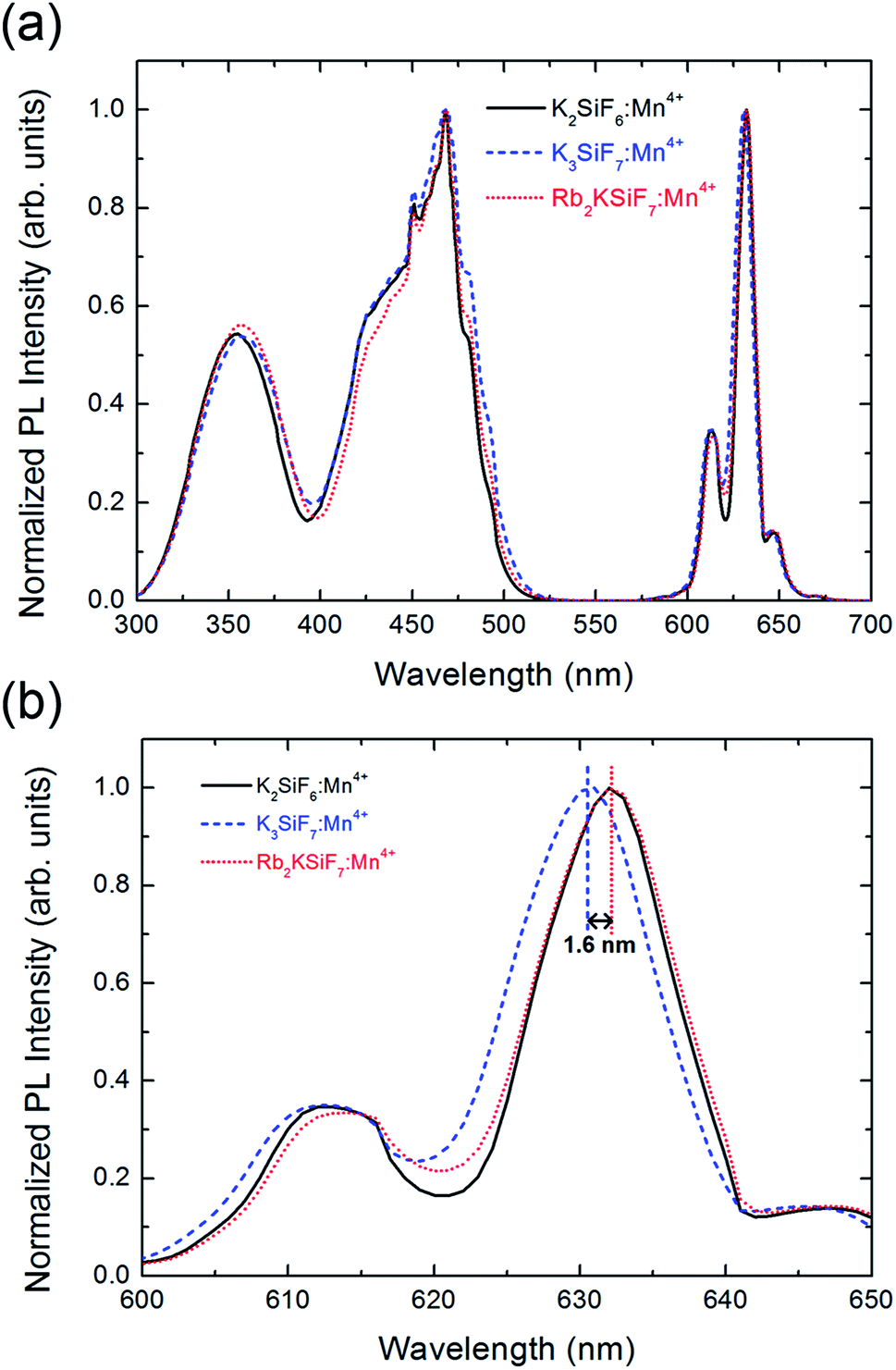 | ||
| Fig. 4 (a) Normalized PL (λex = 460 nm) and PLE (λem = 630 nm) spectra of K2SiF6:Mn4+, K3SiF7:Mn4+, and Rb2KSiF7:Mn4+ phosphors. (b) An enlargement of the PL spectra in the wavelength range from 600 nm to 650 nm. | ||
Particle morphology is a key factor for phosphor materials used in white LEDs. It is influenced by various physicochemical parameters such as sintering temperature, holding time, precursor materials, chemical composition, crystal structure, and homogeneity, especially when the powders are produced by the solution process via amorphous precursor particles. The morphologies of the particles of the synthesized phosphor materials were observed by SEM. Fig. 5(a) and (b) show the morphology of the Mn4+-doped Rb2KSiF7 powder. Particles with various sizes (approximately 50–100 μm) and shape distributions are observed to be dispersed as shown in Fig. 5(a). In assessing the suitability of the red conversion phosphor for use in white LEDs, not only its morphological properties but also its thermal characteristics should be considered. Fig. 5(d) displays the TGA and DTA curves of the Rb2KSiF7:Mn4+ phosphor. The TGA result shows over two the steps weight drops at around 700 °C and 800 °C. Moreover, DTA exhibits two DTA peaks at 710 °C and 790 °C. These results indicate that the Rb2KSiF7:Mn4+ phosphor is not only decomposed over the two steps into SiF4 and alkali metal fluorides (RbF or KF) but also thermally stable to 750 °C.20 Apart from thermal stability of phosphor structure, we checked the temperature-dependent PL properties of Rb2KSiF7:Mn4+ (see Fig. S7 in ESI†). As a results, the luminescent property of Rb2KSiF7:Mn4+ phosphor exhibits thermally stable properties up to 125 °C, but, above 150 °C, PL quenching occurs rapidly. Even if the temperature was lowered again, PL intensity degraded by high temperature was not recovered.
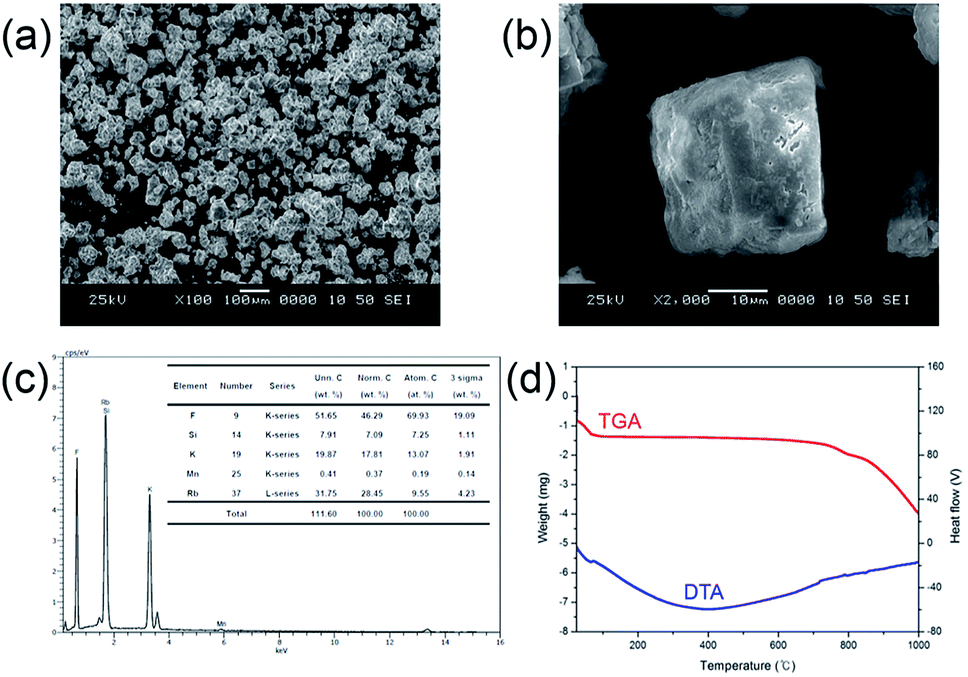 | ||
| Fig. 5 (a) and (b) SEM images, (c) EDS diagram, and (d) TGA and DTA curves of the Rb2KSiF7:Mn4+ phosphor. | ||
4. Conclusions
We performed first-principles calculations based on DFT for pristine and Mn-doped RbxK3−xSiF7 (x = 0, 1, 2, 3) and predicted their stability, electronic structure, and luminescence properties. According to the results of calculations for the RbxK3−xSiF7 (x = 0, 1, 2, 3) host structures performed prior to the calculation for the Mn-doped system, all of the compounds exhibit a large negative bulk formation energy, which indicates that all of the structures considered in this study could be synthesized. Moreover, as the content of Rb increases in the RbxK3−xSiF7 host system, their bandgap energy decreases from 5.59 eV to 5.33 eV. From the atomic projected DOSs, we found that, while the CB is derived mainly form the Rb d state, the upper VB arises from the F p state. For the Mn-doped RbxK3−xSiF7 phosphor system, we calculated the formation energy for Mn substitution and the simulated emission energy of each structure. As a result, we found that, with increasing Rb content, the formation energy for Mn substitution decreases. This trend was attributed to the Rb substitution expanding the lattice because of larger size of Rb ion than K ion. The lattice expanding could promote Mn doping at Si sites.On the basis of the knowledge gained through the simulations, we next synthesized red conversion phosphors and investigated their luminescence, structure, and stability. As a result, we successfully prepared the stable new red conversion phosphor Rb2KSiF7:Mn4+. In addition, the Rb2KSiF7:Mn4+ emitted at a longer wavelength than the K3SiF7:Mn4+ and exhibited an emission wavelength similar to that of K2SiF6:Mn4+. This similarity indicated that a red phosphor with a luminescence wavelength similar to that of the conventional K2SiF6:Mn4+ phosphor was prepared as a quaternary fluoride phosphor containing Rb. It also showed good agreement with our theoretical calculation results, which shows that Rb2KSiF7:Mn4+ exhibited the longest simulated emission wavelength among the four Mn-doped structures.
Collectively, these results demonstrate that DFT calculations can provide rational insights into the properties of a red phosphor material before it is synthesized, thereby reducing the time and cost required to develop new red conversion phosphors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Nano Material Technology Development Program through the National Research Foundation of Korea funded by the Ministry of Science and ICT (NRF-2016M3A7B4025408).Notes and references
- Y. H. Kim, P. Arunkumar, B. Y. Kim, S. Unithrattil, E. Kim, S.-H. Moon, J. Y. Hyun, K. H. Kim, D. Lee, J.-S. Lee and W. B. Im, Nature, 2017, 16, 543–550 CrossRef CAS PubMed.
- M. Zhao, H. Liao, L. Ning, Q. Zhang, Q. Liu and Z. Xia, Adv. Mater., 2018, 30, 1802489 CrossRef PubMed.
- S. Liang, M. Shang, H. Lian, K. Li, Y. Zhang and J. Lin, J. Mater. Chem. C, 2017, 5, 2927–2935 RSC.
- X. Piao, K.-I. Machida, T. Horikawa, H. Hanzawa, Y. Shimomura and N. Kijima, Chem. Mater., 2007, 19, 4592–4599 CrossRef CAS.
- S. Jang, J. Im, B. K. Bang, C. H. Kim, H. Chang and K.-J. Kong, RSC Adv., 2015, 5, 39319–39323 RSC.
- T. Takahashi and S. Adachi, J. Electrochem. Soc., 2008, 155, E183–E188 CrossRef CAS.
- H. F. Sijbom, R. Verstraete, J. J. Joos, D. Poelman and P. F. Smet, Opt. Mater. Express, 2017, 7, 3332–3365 CrossRef CAS.
- M. Kim, W. B. Park, B. Bang, C. H. Kim and K.-S. Sohn, J. Am. Ceram. Soc., 2017, 100, 1044–1050 CrossRef CAS.
- Y. Zhuo, A. M. Tehrani, A. O. Oliynyk, A. C. Duke and J. Brgoch, Nat. Commun., 2018, 9, 4377 CrossRef PubMed.
- C. H. Kim, B. K. Bang, J. K. Park, K.-J. Kong, H. Chang, J. Im, S. Jang and K. S. Choi, KR 101790541, 2017.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199 CrossRef CAS.
- D. L. Deadmore and W. F. Bradley, Acta Crystallogr., 1962, 15, 186–189 CrossRef CAS.
- B. Hofmann and R. Hoppe, Z. Anorg. Allg. Chem., 1979, 458, 151–162 CrossRef CAS.
- S. Kirklin, J. E Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 15010 CrossRef CAS.
- M. H. Du, J. Mater. Chem. C, 2014, 2, 2475–2481 RSC.
- R. Verstraete, H. F. Sijbom, J. J. Joos, K. Korthout, D. Poelman, C. Detavernier and P. F. Smet, ACS Appl. Mater. Interfaces, 2018, 10, 18845–18856 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Atomic geometries for RbK2SiF7 and Rb2KSiF7, band structures and density of states (DOS) for RbxK3−xSiF7 structures, atomic and orbital projected DOSs of Rb3SiF7, atomic projected DOS on the ground and excited states of RbxK3−xSiF7:Mn4+ structures. See DOI: 10.1039/c9ra05929f |
| ‡ S. J. and J. K. P. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |