
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into mineral carbonation and utilization in cement-based materials at solid–liquid interfaces
Shu-Yuan
Pan
 *a,
Barry
Lai
b and
Yang
Ren
b
*a,
Barry
Lai
b and
Yang
Ren
b
aDepartment of Bioenvironmental Systems Engineering, National Taiwan University, Taipei City 10617, Taiwan. E-mail: sypan@ntu.edu.tw
bX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 2nd October 2019
Abstract
In order to ensure the viability of CO2 mineralization and utilization using alkaline solid waste, a mechanistic understanding of reactions at mineral–water interfaces was required to control the reaction pathways and kinetics. In this study, we provided new information for understanding the reactions of CO2 mineralization and utilization at mineral–water interfaces. Here we have carried out high-energy synchrotron X-ray analyses to characterize the changes of mineral phases in petroleum coke fly ash during CO2 mineralization and their subsequent utilization as supplementary cementitious materials in cement mortars. The 2-D synchrotron patterns were converted to 1-D diffraction patterns and the results were then interpreted via the Rietveld refinement. The results indicated that there was a continuous source of calcium ions mainly due to the dissolution of CaO and Ca(OH)2 in fly ash. This would actually enhance the driving force of saturation index at the solid–fluid interfacial layer, and then could eventually result in the nucleation and growth of calcium carbonate (calcite) at the interface. A small quantity of CaSO4 (anhydrite) in fly ash was also dissolved and simultaneously converted into calcite. In addition, the calcium sulfate in fly ash would effectively prevent the early hydration of tricalcium aluminate in blended cement, and thus could avoid the negative impact on its strength development. The proposed reaction mechanisms were also qualitatively verified by X-ray fluorescence mapping and electron microscopy. These results would help to design efficient reactors and cost-effective processes for CO2 mineralization and utilization in the future.
Introduction
CO2 mineralization and utilization have been considered as one of the most promising practices to mitigate the global anthropogenic CO2 emissions while achieving a circular economy.1,2 The NASEM report in 2019 conveyed that mineral carbonation would be one of three main utilization pathways for gaseous carbon waste stream utilization.3 Kirchofer et al.4 reported that the CO2 sequestration potential via mineralization using natural alkaline sources in the US would range between 1.8% and 23.7% of the total CO2 emissions. In the case of using industrial alkaline solid waste, the annual mitigation potential via mineralization in the US would be approximately 7.6 million tonnes of CO2.5 Mineralization technologies could also offer great potential for CO2 utilization in the short to medium term as they could produce construction materials that possess tremendous market value around the world,3i.e., over 48 billion metric tons of construction materials are needed annually.6Extensive studies on the performance of CO2 mineralization using alkaline solid wastes have been reported.7–9 Industrial alkaline solid wastes, such as petroleum coke fly ash (PCFA) and steel slag, are abundant and considered as the low-cost feedstocks for the advancement of CO2 mineralization and utilization technologies. In our previous study,10 we have evaluated the performance of CO2 mineralization using PCFA and its subsequent utilization of reaction products as supplementary cementitious materials (SCMs) in blended cement mortars. The results indicated that up to 95.6% of the CO2 removal ratio was achieved in the flue gas by CO2 mineralization using PCFA. The analysis performed for the product utilization suggested that cement blended with a 5% substitution of carbonated PCFA could exhibit superior compressive strength at 28 days, when compared to that of fresh PCFA.10
CO2 mineralization and utilization using alkaline solid wastes are a heterogeneous (CO2–fluid–solid) reaction system, which could typically involve complex, inorganic constituents in the aqueous solution such as calcium, aluminum, sodium, potassium, sulfate, carbonate, and chloride ions. In the literature, only few fundamental studies on CO2 geological sequestration have been carried out to elucidate the reaction mechanism of supercritical CO2–saline water–rock interfaces,11,12 although their reaction conditions were not similar to CO2 mineralization (e.g., under ambient pressures). For cementation with alkaline solid wastes, similar approaches to carbonating cementitious materials under the CO2 atmosphere during the curing stage, so-called CO2 curing, have been developed and evaluated. For instance, Zhang et al.13 found that CO2 curing could effectively enhance the hydration of Portland cement and thus promote its compressive strength at 28 days. Lippiatt et al.14 also developed a combined hydration–carbonation system using supersaturated aqueous CO2 solution to overcome the process huddles because the rate of typical hydration reactions of cement (cementation) was much higher than that of carbonation reactions in atmosphere.
Despite recent progress on performance evaluation, mechanistic understanding is still needed to unravel the CO2 mineralization and utilization at interface levels for further controlling the rate of reaction pathways. Also, the role of the carbonated product as SCMs in cement mortars was unclear due to its complex chemistry of cementation during hydration and strength development of cementitious materials. The strength development of cement-based materials involves a number of reactions, such as the hydration of Ca3SiO5 (cement chemistry notation, C3S), Ca2SiO4 (C2S) and Ca3Al2O6 (C3A) in the presence of minor components such as sulfates and carbonates. In order to elucidate the hydration reactions of different types of Portland cements, great efforts have been made on applying the advanced analytical techniques, such as in situ X-ray diffraction,15 high-pressure X-ray diffraction,16,17 X-ray absorption near edge structure spectrometry18 and nanoscale tomography.19 These studies could provide significant impacts on modeling the kinetics and morphological evolution of cement hydration. However, when utilizing the solid waste products from mineralization as SCMs in cement mortars or concrete, it was difficult to understand the different mechanisms of cement hydration reactions due to the complex compositions of mineralized solid wastes. High-energy synchrotron X-ray analyses provide great opportunities for determining mineralogical and structural characteristics efficiently and accurately in complex sample environments. An ordinary XRD may require more than 10 hours, in the case of alkaline solid wastes,20 to get qualified diffraction patterns (e.g., signal-to-noise ratios) for mineral phase quantification using Rietveld refinement.
To the best of our knowledge, little-to-no research has been reported on the fundamental mechanisms of CO2 mineralization and utilization using PCFA due to its chemical and mineralogical complexity. In this study, we provided a new information for understanding the relevant reactions at mineral–water interfaces. Here, we performed high-energy synchrotron X-ray analyses, including quantitative X-ray diffraction and micro-probe X-ray fluorescence, to characterize the changes of mineral phases in PCFA for CO2 mineralization and utilization. We have also examined the morphology and elemental distributions of PCFA before and after CO2 mineralization via electron microscope to verify the findings obtained from the synchrotron X-ray analyses. Thus, this study could provide an insight into the elucidation of the mechanisms and reaction pathways at interface levels for CO2 mineralization and utilization using PCFA via high-energy synchrotron X-ray technique.
Results and discussion
Reaction pathways of mineral carbonation using PCFA
In order to elucidate the reaction mechanisms, the PCFA-based mineralization was carefully examined using high-energy synchrotron X-ray technique. Table 1 presents the physico-chemical compositions of fresh and carbonated PCFA determined by the XRF. Compared to iron and steel slags, the chemical compositions of the fresh PCFA was relatively simple where the main components were found to be CaO (∼63%) and SO3 (∼31%). The results indicated that the specific surface area of PCFA increased from 3720 to 4890 cm2 g−1 after CO2 mineralization, whereas, the mean diameter of PCFA decreased from 16.8 to 13.2 μm. The decrease in the mean diameter could probably be due to the formation of small particles of carbonate precipitates on the surface of the PCFA.| Items | Constituents | Units | F-PCFA | C-PCFA |
|---|---|---|---|---|
| a The chemical compositions were analyzed by the XRF. b The mineralogy was quantified by the Rietveld method according to the high-energy synchrotron XRD. In this study, only a slight difference in the intensity of some major peaks was observed. | ||||
| Physical properties | Density | g cm−3 | 2.50 | 2.44 |
| Specific surface area | cm2 g−1 | 3720 | 4890 | |
| Fineness | cm2 g−1 | 3810 | 8770 | |
| Mean diameter | μm | 16.8 | 13.2 | |
| D [4,3] | μm | 22.7 | 18.3 | |
| D [3,2] | μm | 6.45 | 5.03 | |
| Chemical compositions (XRF)a | CaO | % | 62.8 | 60.1 |
| SiO2 | % | 3.08 | 2.89 | |
| Al2O3 | % | 1.01 | 0.98 | |
| MgO | % | 0.83 | 1.24 | |
| Fe2O3 | % | 0.70 | 0.60 | |
| SO3 | % | 31.0 | 33.0 | |
| Na2O | % | 0.06 | 0.04 | |
| K2O | % | 0.43 | 0.33 | |
| Total | % | 99.9 | 99.2 | |
| Mineralogy (quantitative XRD)b | Anhydrite (CaSO4) | % | 91.9 | 87.0 |
| Lime (CaO) | % | 4.2 | — | |
| Quartz (SiO2) | % | 1.9 | 2.1 | |
| Calcite (CaCO3) | % | 1.5 | 11.0 | |
| Portlandite (Ca(OH)2) | % | 0.5 | — | |
Fig. 1 shows the mineralogical characteristics of the fresh and carbonated PCFA obtained by synchrotron X-ray diffraction (XRD). According to the XRD results, calcium species in the PCFA were found to be the major components that could react with flue gas CO2 to form carbonate participates. Before carbonation, that the anhydrite (CaSO4) peak was found to be the major mineral phases of the fresh PCFA (Fig. 1a and c), and the peaks due to lime (CaO), portlandite (Ca(OH)2) and quartz (SiO2) were present as the minor phases. After carbonation, no presence of lime and portlandite was observed, while the intensity of calcite significantly increased in the carbonated PCFA (Fig. 1b and d). For both the fresh and carbonated PCFA, no amorphous phase was found as there was no diffuse peak or broad hump that could be clearly observed from the XRD patterns. This observation was consistent with the findings reported in the literature.21,22
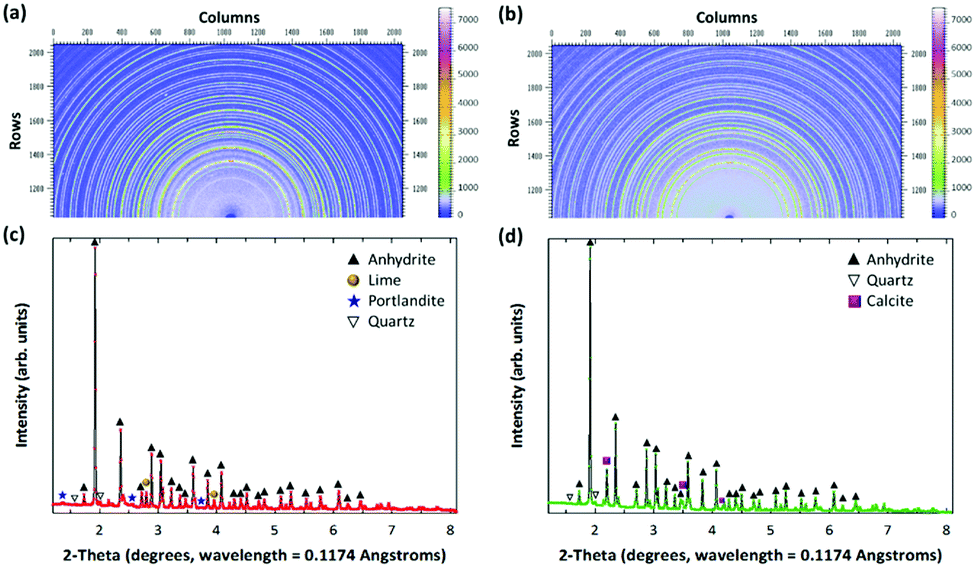 | ||
| Fig. 1 Elucidation of CO2 mineralization mechanism through high-energy synchrotron 2-D patterns of (a) fresh and (b) carbonated PCFA, and their associated 1-D XRD patterns of (c) fresh and (d) carbonated PCFA. The 2-D patterns (a and b) were calibrated using a standard CeO2 sample and then converted to 1-D patterns (c and d) using Fit2D program. The wavelength of X-ray at Argonne National Laboratory is 0.1174 Angstroms. | ||
The mechanisms and pathways of CO2 mineralization using PCFA were proposed as follows; the lime (CaO) and portlandite (Ca(OH)2) in the fresh PCFA were first dissolved and leached into the water as shown in eqn (1) and (2), respectively. Wei et al.23 suggested that, upon contact with water, the reactive minerals (e.g., lime and portlandite) of calcium-bearing components in fly ash would readily dissolve within the first few minutes. These processes could enhance the alkalinity of the solution by releasing calcium ions, which was actually beneficial to CO2 mineralization via accelerated carbonation.24 It was also noted that, as the pH of the slurry increased, other alkaline compounds such as sodium and potassium would gradually release from the glassy phases in solid wastes,23,25 thereby resulting in the formation of metal-depleted silica rims within the ash particles.
Lime:
| CaO(s) + H2O(aq) → Ca2+(aq) + 2OH−(aq) | (1) |
Portlandite:
| Ca(OH)2(s) + H2O(aq) → Ca2+(aq) + 2OH−(aq) + H2O(aq) | (2) |
The high alkalinity of the solution due to the formed hydroxide ions (OH−) would accelerate the dissolution of gaseous CO2 into carbonate ions (when the pH of solution is above 10.3), as described by eqn (3). It was also noted that this process will gradually acidify the solution due to the formation of protons, which is disadvantageous to further dissolution of CO2 into solution and thus the carbonation reaction.
| CO2(g) + OH−(aq) → CO32−(aq) + H+(aq) | (3) |
The leached calcium ions (eqn (1) and/or (2)) would simultaneously react with carbonate ions (eqn (3)) to form calcium carbonate precipitates, as shown in eqn (4). It was noted that, depending on the operating condition such as temperatures and the presence of liquid water, metastable CaCO3 phases (e.g., amorphous, vaterite and aragonite) might exist prior to the formation of stable calcite.26 The overall accelerated carbonation was an exothermic reaction, where the heat of formation 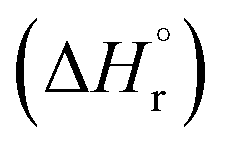 and Gibbs free energy
and Gibbs free energy 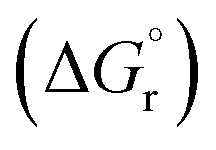 of the reaction (eqn (2)–(4)) at 298.15 K and 1 atm were −113.1 and −73.1 kJ mol−1, respectively.20
of the reaction (eqn (2)–(4)) at 298.15 K and 1 atm were −113.1 and −73.1 kJ mol−1, respectively.20
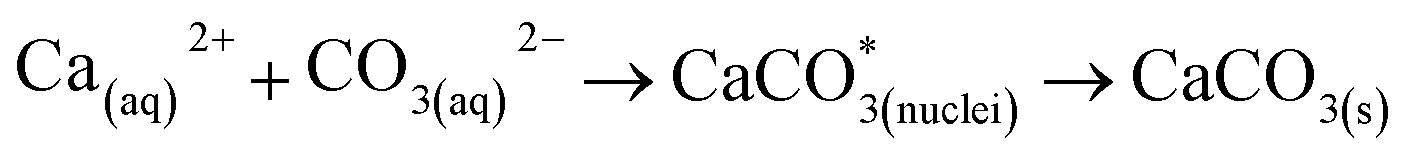 | (4) |
In addition, according to the results in Table 1, the content of anhydrite (CaSO4) in PCFA was slightly reduced from 91.9% to 87.0% after CO2 mineralization. This might be attributed to the dissolution of anhydrite (into calcium and carbonate ions) and the simultaneous growth of calcite, as described by eqn (5). The Gibbs free energy of the overall reaction at 298.15 K and 1 atm were −24.9 kJ mol−1. Roncal-Herrero et al.27 also found that the direct carbonation of anhydrite would result in a negative volume change with the generation of microporosity, thereby increasing the specific surface area of materials. This observation was consistent with the findings in this study that the specific surface area of PCFA increased after CO2 mineralization.
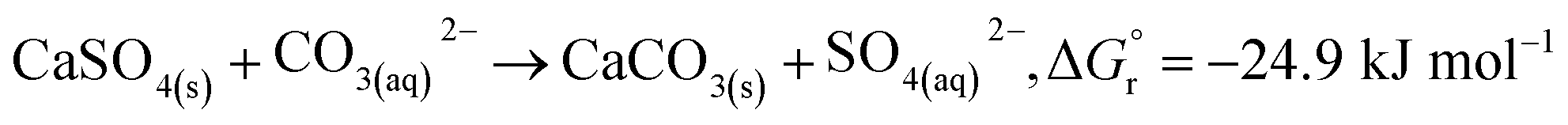 | (5) |
The mass quantity of calcite in PCFA increased from 1.5% to 11.0% after CO2 mineralization, as presented in Table 1. The increase of calcite would be contributed from the complete conversion of lime (4.2%) and portlandite (0.5%) by eqn (1) and (2), respectively, along with the partial conversion of anhydrite by eqn (5). According to the stoichiometric ratio and mass balance, about 2.4% (out of 91.9%) of anhydrite in fresh PCFA was reacted with carbonate ions to form calcite.
The aforementioned reaction pathways of CO2 mineralization using PCFA were verified by scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDS). As shown in Fig. 2a, the fresh PCFA exhibited relatively smooth surfaces with irregular shapes of particles. According to the EDS analyses (Fig. 2b), the ratios of Ca, C, S, Si and Al atoms on the spot of red cross symbol in the SEM image were found to be 8.3 ± 0.6%, 55.1 ± 3.9%, 4.1 ± 0.3%, 0.8 ± 0.1% and 0.4 ± 0.1%, respectively. The EDS results were in good agreement with the results of the XRF (Table 1). The elemental distribution also showed that the fresh PCFA was rich in sulfur (Fig. 2c) and calcium (Fig. 2d) elements, with minor distribution of silicon (Fig. 2e) and aluminum (Fig. 2f) elements. For some PCFA particles, the elements of Al, Si, and Ca were found with very high signals. It was presumed that Ca coexisted together with Al and Si as calcium aluminosilicates, which might be with poor crystallinity and thus could not be well detected by the XRD.
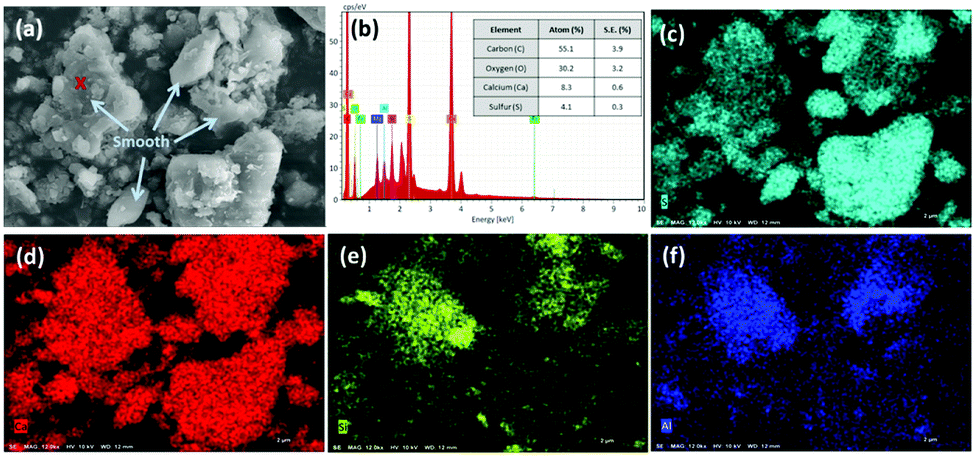 | ||
| Fig. 2 (a) Scanning electron microscopy (SEM) image of fresh PCFA, and (b) energy dispersive X-ray spectroscopy (EDS) for the spot with the red cross symbol on SEM image. Elemental mapping of (c) sulfur, (d) calcium, (e) silicon, and (f) aluminium. | ||
In contrast, after accelerated carbonation, the surface of the carbonated PCFA was found to be covered with rhombohedral (calcite) crystals, with a size of 0.1–0.5 μm (Fig. 3a). The carbonated PCFA was rich is sulfur (Fig. 3c) and calcium (Fig. 3d) elements with minor amounts of silicon (Fig. 3e) and aluminum (Fig. 3f). According to the EDS results (Fig. 3b), the ratio of carbon atom after carbonation (57.3 ± 4.0%) was found to increase, appreciably confirming the formation of carbonate precipitates in PCFA. There was a slight decrease in the ratios of other atoms such as Ca (7.9 ± 0.6%), S (3.7 ± 0.3%), Si (0.8 ± 0.1%) and Al (0.2 ± 0.1%) due to the presence of relatively more carbon atoms in carbonated PCFA. It was thus suggested that the carbonate precipitates would be physically beneficial to the strength development of blended cement as they could provide favorable sites for the nucleation as well as the growth of hydration products.28
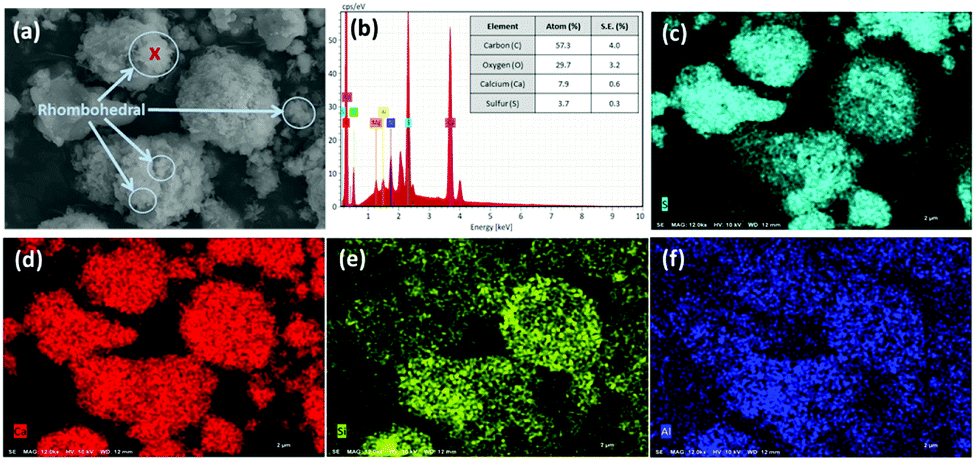 | ||
| Fig. 3 (a) Scanning electron microscopy (SEM) image of carbonated PCFA, and (b) energy dispersive X-ray spectroscopy (EDS) for the spot with the red cross symbol on SEM image. Elemental mapping of (c) sulfur, (d) calcium, (e) silicon, and (f) aluminum. | ||
Mechanism of product utilization as SCMs in cement mortars: hydration chemistry
After successful CO2 mineralization, the obtained products (i.e., carbonated PCFA) could be utilized as SCMs in blended cement mortars. This could reduce the use of Portland cements for construction works, thereby indirectly avoiding the CO2 emissions during cement manufacturing.29 In addition, the elimination of lime and portlandite in PCFA after CO2 mineralization would be beneficial to the subsequent utilization as SCMs in cement mortars. Also, the SO3 composition (i.e., the CaSO4 phase) in the PCFA would exhibit a remarkable influence on the cement chemistry and initial strength development. Here, we performed high-energy synchrotron XRD (Fig. 4) to elucidate the mechanisms of strength development for cement mortars blended with fresh and carbonated PCFA. The major peaks in the synchrotron XRD patterns were identified and then their quantities were simulated by the Rietveld refinement. Table 2 presents the mineralogy of blended cement mortars with fresh and carbonated PCFA.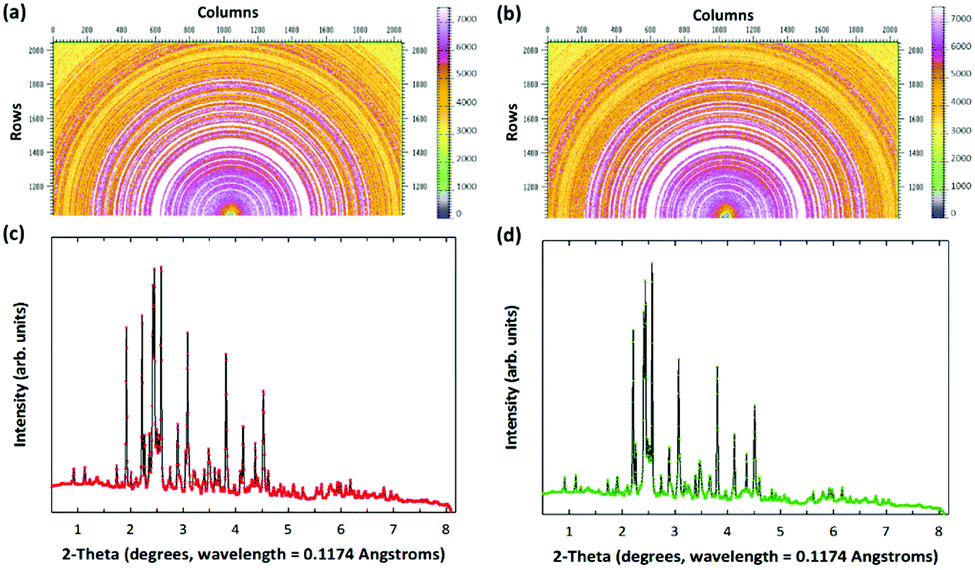 | ||
| Fig. 4 High-energy synchrotron 2-D patterns of blended cement mortars with (a) fresh and (b) carbonated PCFA, and their associated 1-D XRD patterns of (c) fresh and (d) carbonated PCFA. The wavelength of X-ray at Argonne National Laboratory is 0.1174 Angstroms. | ||
| Mineralogy | Formula | Units | Blended cement mortars | |
|---|---|---|---|---|
| With F-PCFA | With C-PCFA | |||
| a The mineralogy of PCFA was quantified by the Rietveld method according to the high-energy synchrotron X-ray diffraction patterns. | ||||
| Hatrurite | Ca27Si9O45 | % | 27.1 | 38.7 |
| Wollastonite | CaSiO3 | % | — | 18.3 |
| Sillimanite | Al2SiO5 | % | 5.1 | — |
| Anhydrite | CaSO4 | % | 15.4 | — |
| Hatrurite | Ca9Si3O15 | % | 14.2 | 22.2 |
| Dmitryivanovite | CaAl2O4 | % | 14.6 | — |
| Ettringite | Ca6Al2S3O49.496H60.602 | % | 0.7 | 7.1 |
| Cancrinite | Ca0.04C0.5H6O14.25Si3 | % | — | 4.1 |
| Na3.58K0.15 | ||||
| Ca2Al2O5 | % | 7.6 | — | |
| Foshagite | Ca4Si3O11H2 | % | 3.5 | — |
| Afwillite | Ca3Si2O10H6 | % | 11.7 | 9.6 |
| Portlandite | Ca(OH)2 | % | — | — |
In cement chemistry, the contents of available C3S (tricalcium silicate) and C2S (dicalcium silicate) are considered as the key constituents for strength development of cement-based materials. C3S is the most important, abundant (usually >70%)30 component of Portland cement. The rapid hydration of C3S (eqn (6)) is responsible for the initial strength (<28 days).31 C2S is the second most abundant component, comprising 15–30% of clinker. The hydration of C2S is much slow and contributes to the ultimate strength after 28 days (up to two years). It was observed that ∼70% of the total C3S as well as ∼30% of the total C2S were reacted in a cement paste at 28 days.32
| 2Ca3SiO5 + 6H2O → 3CaO·2SiO2·3H2O + 3Ca(OH)2, ΔH = 114.16 kJ mol−1 | (6) |
| 2Ca2SiO4 + 4H2O → 3CaO·2SiO2·3H2O + Ca(OH)2, ΔH = 38.75 kJ mol−1 | (7) |
The hydration of C3S and C2S would result in the formation of the calcium–silicate–hydrate (C–S–H) phases. As shown in eqn (6) and (7), the hydration product (CaO·2SiO2·3H2O) was one type of the C–S–H phases. For cement mortars blended with the fresh PCFA, other C–S–H phases including foshagite (3.5%) and afwillite (11.7%) were also observed, as presented in Table 2. In the case of mortars blended with the carbonated PCFA, only the C–S–H phase of afwillite (9.6%) was found as the hydration products.
The C3A phase in Portland cement also plays a crucial role in strength development, as its hydration will result in a large amount of hardening heat (eqn (8)). A rapid C3A hydration might cause an adverse impact on the strength after 28 days due to the formation of microcracks, thereby weakening the bond in cement matrix.33 However, the C3A phase would react with sulfates (e.g., the anhydrite in PCFA) as described by eqn (9) to form ettringite (hexacalcium aluminate trisulfate hydrate) during the early hydration. Thus, this reaction could prevent the early hydration of C3A and avoid inducing a stiffening of the hydrating paste.34 In the case of cement mortars blended with the fresh PCFA, other C3A hydration products such as CaO·Al2O3 (dmitryivanovite, CA) and (CaO)2·Al2O3 (cancrinite, C2A) were also formed.
| C3A + 6H2O → C3A·6H2O, ΔH = −245 kJ mol−1 | (8) |
| C3A + 3CaSO4 (in PCFA) + 32H2O → C3A·3CaSO4·32H2O (ettringite), ΔH = −452 kJ mol−1 | (9) |
With a substantial quantity of CaSO4 in PCFA, the ettringite (eqn (9)) could be readily formed and could also cover the C3A grains to avoid further hydration of C3A and thus improve the ultimate strength of blended cement. Meantime, the deposited ettringite would react with C3A at the contact surface, as shown in eqn (10), to form calcium-aluminate-monosulfate (s-AFm). Geng et al.35 applied nanotomographic and spectromicroscopic techniques to evaluate the C3A hydration in the presence of gypsum (CaSO4). They found that, as soon as being in contact with sulfur-containing solution, C3A would undergo rapid dissolution owing to the quick growth of ettringite as well as the formation of AFm phases (but quickly vanish), and then followed by slow dissolution of C3A, which might probably be due to ion-complexation.35 Similar observation was found in the case of C3A–gypsum system suggesting both the nucleation and growth of the s-AFm phases as the rate controlling mechanism.15
| 2C3A + C3A·3CaSO4·32H2O (ettringite) + 4H2O → 3C3A·CaSO4·12H2O (s-AFm), ΔH = −238 kJ mol−1 | (10) |
In addition to CaSO4, the presence of CaCO3 would chemically influence the hydration of C3A phases in blended cement. For instance, the CaCO3 would delay and slow down the reaction of sulfate with C3A in blended cement by forming the monocarboaluminate phases (eqn (11)). The existing carboaluminates were found to be quantitatively more and potentially stiffer than that of sulfoaluminates (e.g., ettringite). In the case of mortars blended with carbonate PCFA, the reaction of ettringite with carbonates was found to result in other complex carbonate-silicate products of sodium, calcium and aluminium such as cancrinite (∼4.1%).
| C3A·3CaSO4·32H2O (ettringite) + CaCO3 → C3A·CaCO3·xH2O (carboaluminate) + … | (11) |
It was noteworthy to mention that due to the occurrence of CaCO3 in blended cement, the formation of ettringite would be promoted during the early hydrations (<30 min) by suppressing the formation of AFm, as shown in eqn (12). In this study, the amount of ettringite formed in the blended cement with carbonated PCFA was significantly higher than that of fresh PCFA (see Table 2). The contents of ettringite in cement mortars blended with fresh and carbonated PCFA at 90 days were found to be 0.7% and 7.1%, respectively.
| 3C3A·CaSO4·12H2O (AFm) + 2CaCO3 + 18H → 2C3A·CaCO3·11H + C3A·3CaSO4·32H2O (ettringite) | (12) |
The CaCO3 could also directly react with the C3A phase, leading to the formation of the hexagonal monocarbonate phase (C3A·CaCO3·11H2O), as shown in eqn (13). This reaction is exothermic and faster than that of C3S hydration.36 The unstable phase in eqn (13) after one day would gradually convert to the monocarbonate phase and C3AH6, as shown in eqn (14). These reactions could enhance hydration heat and offer mechanical strength to the cement (especially for the initial strength development).37,38 Therefore, the blended cement with carbonated PCFA could generally exhibit superior compressive strength compared to that of fresh PCFA.
| 2C3A + 1.5CaCO3 + 0.5CH + 22.5H → C3A·CaCO3·11H + C3A·0.5CaCO3·0.5CH·11.5H (unstable phase) | (13) |
| 2C3A·0.5CaCO3·0.5Ca(OH)2·11.5H → C3A·CaCO3·11H + C3AH6 | (14) |
Apart from these, in some cases, the partial decomposition of monocarbonate phases to C3AH6 and CaCO3 might also occur after seven days, as shown in eqn (15), depending upon the temperature and humidity of the surrounding environment. This was usually considered as one of the main reasons leading to the decrease in compressive strength after three days.38
| C3A·CaCO3·11H → C3AH6 + CaCO3 + 5H2O | (15) |
In this study, no presence of CaCO3 in blended cements with either fresh or carbonated PCFA was observed (see Table 2), indicating the complete consumption of CaCO3 during cement hydrations. Apart from the chemical enhancement by CaCO3 in carbonated PCFA, the fine particle size and high surface area of the carbonated PCFA would provide more active sites for hydration reactions.
It was worthy noted that we applied the synchrotron XRD technique to identify the hydration products of blended cement due to the fact of complex reactions of hydration. Regarding the amorphous content in cement-based materials, extensive studies on quantitative phase analysis for Portland cements (without any additions) have been conducted. Snellings et al.39 found that, based on the results of both external and internal standard methods, the fresh Portland cements did not contain amorphous materials. However, depending upon the sample preparation and comminution procedure, a small portion of amorphous content might be observed in Portland cements.40 Apart from the fresh Portland cements, for blended cements with either fresh or carbonated PCFA, no significant quantity of amorphous phases was observed from the XRD patterns in this study. In the future, the amorphous (or unknown) phases should be included in the Rietveld refinement to increase the accuracy of the estimated amounts of the crystalline phases in cement-based materials.41
Qualitative evidences from elemental mapping using X-ray fluorescence
Here we applied the XRF mapping technique for identifying the elemental distributions of PCFA and PCFA-blended cements to provide qualitative evidences to the proposed pathways of CO2 mineralization and utilization. Fig. 5 shows the XRF elemental distribution of fresh and carbonated PCFA. The results indicated that a relative low content of silicon was observed both in fresh (Fig. 5a) and carbonated (Fig. 5e) PCFA. In both cases, the sulfur element (Fig. 5b and f) was mostly combined with the calcium element (Fig. 5c and g), indicating the presence of CaSO4 compound in PCFA. These observations were consistent with the findings presented in Table 1. They also provided the evidence of the calcium-bearing phases such as CaO and Ca(OH)2 would be the main reaction component in PCFA.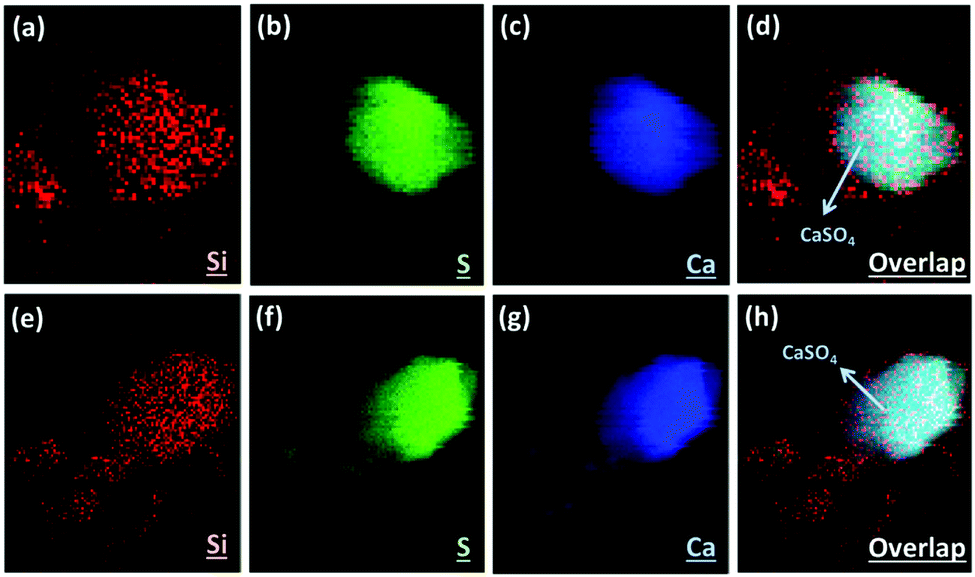 | ||
| Fig. 5 Imaging of elemental distribution by X-ray fluorescence: (a–d) fresh and (e–h) carbonated PCFA. Colocalization of silicon (red), sulfur (green) and calcium (blue) signals is presented as the overlap of these three colors (d and h). | ||
Fig. 6 shows the XRF elemental distribution of cement mortars blended with either fresh or carbonated PCFA. In the case of blended cement with fresh PCFA, the abundant silicon element (Fig. 6a) from Portland cements was adhesively bonded with the calcium-bearing (Fig. 6c) phases, including the CaSO4 (Fig. 6b), from the fresh PCFA. This observation indicated that the formation of C–S–H phases might involve the CaSO4 phase (Fig. 6d) in the fresh PCFA. With the presence of CaCO3 in carbonated PCFA, in contrast, most of the silicon element (Fig. 6e) was bonded with the calcium element (Fig. 6g) rather than the sulfur element (Fig. 6f). This observation could provide the evidences for the CaCO3 component in carbonated PCFA to reduce or slow down the reaction of CaSO4 with Portland cement due to the formation of monocarboaluminate. In the blended cement with carbonated PCFA, most of the C–S–H phases was not incorporated with the composition of sulfur element (Fig. 6h).
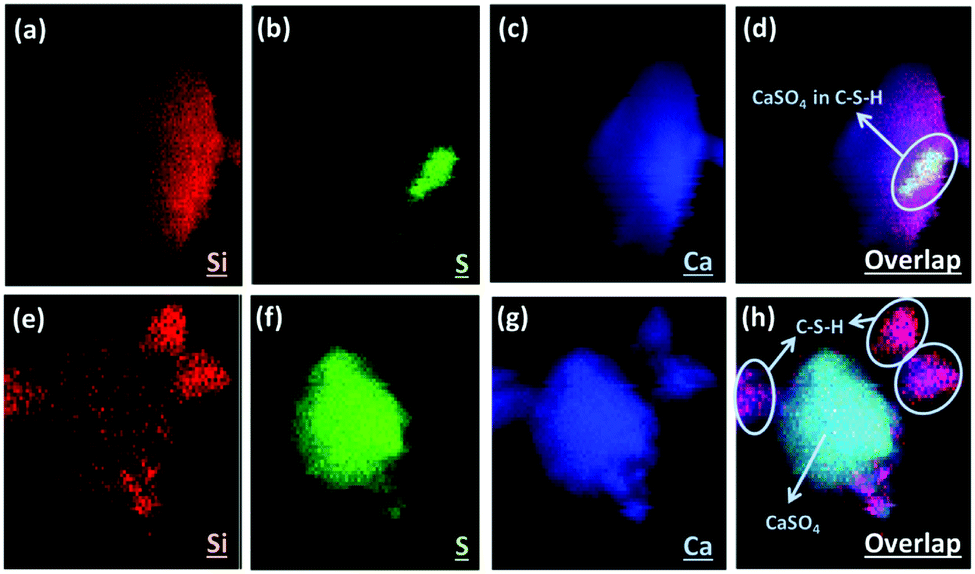 | ||
| Fig. 6 Imaging of elemental distribution by X-ray fluorescence for blended cement with (a–d) fresh and (e–h) carbonated PCFA. Colocalization of silicon (red), sulfur (green) and calcium (blue) signals is presented as the overlap of these three colors (d and h). | ||
Mechanistic elucidation of CO2 mineralization and utilization at interface levels
In this study, the synchrotron techniques including XRD and XRF were conducted to determine the reaction mechanisms of CO2 mineralization and utilization using PCFA. Based on the aforementioned findings, the reaction pathways at the mineral–water interface for CO2 mineralization and its subsequent product utilization were proposed, as shown in Fig. 7a and b, respectively. For CO2 mineralization, the gaseous CO2 molecule was first dissolved in the aqueous solution via Route and then converted to bicarbonate and/or carbonate ions via Route
and then converted to bicarbonate and/or carbonate ions via Route  , depending upon the pH value of the solution. The main reactive components in fresh PCFA were found to be calcium oxide (CaO) and calcium hydroxide (Ca(OH)2). They would gradually hydrate via Route
, depending upon the pH value of the solution. The main reactive components in fresh PCFA were found to be calcium oxide (CaO) and calcium hydroxide (Ca(OH)2). They would gradually hydrate via Route  and then release calcium (by Route
and then release calcium (by Route  ) and hydroxide (by Route
) and hydroxide (by Route  ) ions from the inside particle into the solution. The dissolution of CaO and Ca(OH)2 could provide a continuous source of Ca2+ ions, which in turn could increase the driving force of saturation index at the solid–fluid interfacial layer. Thus, the generated carbonate and calcium ions would eventually result in the nucleation and growth of the calcium carbonate (i.e., calcite) phase at the interface (Route
) ions from the inside particle into the solution. The dissolution of CaO and Ca(OH)2 could provide a continuous source of Ca2+ ions, which in turn could increase the driving force of saturation index at the solid–fluid interfacial layer. Thus, the generated carbonate and calcium ions would eventually result in the nucleation and growth of the calcium carbonate (i.e., calcite) phase at the interface (Route  ) and/or in the bulk solution (Route
) and/or in the bulk solution (Route  ). The comprehensive analysis indicated that most of the calcite products would deposit and cover the surface of the PCFA particle, forming a protective (product) layer outside the particle. Wang et al.42 revealed that the rates of nucleation of the newly formed phases would normally depend on the solid–fluid interfacial energy and kinetic barriers regarding desolvation, attachment, detachment, and diffusion.
). The comprehensive analysis indicated that most of the calcite products would deposit and cover the surface of the PCFA particle, forming a protective (product) layer outside the particle. Wang et al.42 revealed that the rates of nucleation of the newly formed phases would normally depend on the solid–fluid interfacial energy and kinetic barriers regarding desolvation, attachment, detachment, and diffusion.
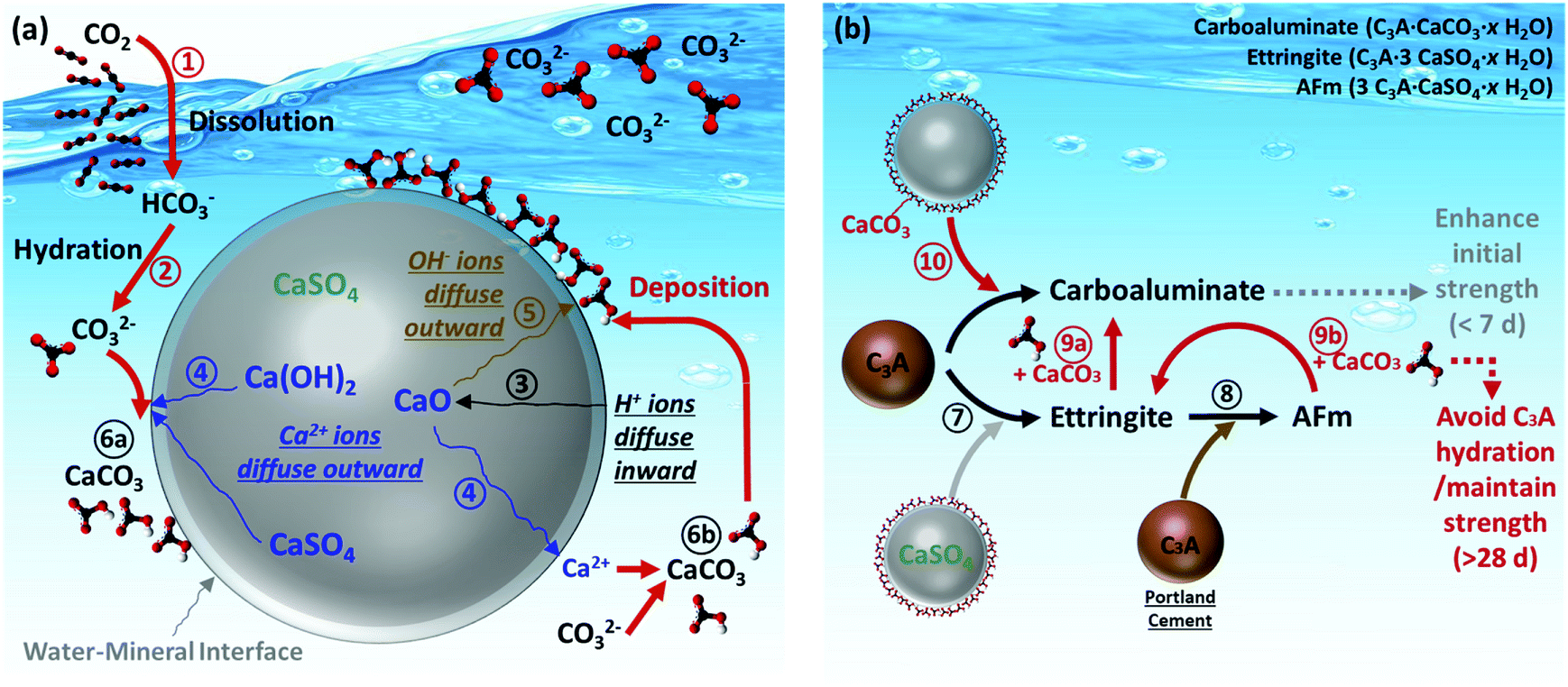 | ||
| Fig. 7 Reaction pathways of (a) CO2 mineralization using PCFA at water–mineral interface, and (b) utilization of carbonated PCFA products in blended cement mortars. | ||
In order to reduce the use of Portland cement, the carbonated PCFA could be partially blended with Portland cement for the production of cement mortars or concretes. The significant quantity of calcium sulfate (CaSO4) in PCFA would easily react with the C3A phase in Portland cement to form ettringite phase via Route  and then further convert to the AFm phase via Route
and then further convert to the AFm phase via Route  . These reactions could effectively prevent the early hydration of C3A, and thus avoid the negative impact on its strength development after 28 days. With the presence of carbonated products (i.e., CaCO3) in PCFA, part of the formed ettringite and AFm phases would convert to carboaluminate via Route
. These reactions could effectively prevent the early hydration of C3A, and thus avoid the negative impact on its strength development after 28 days. With the presence of carbonated products (i.e., CaCO3) in PCFA, part of the formed ettringite and AFm phases would convert to carboaluminate via Route  . The carbonated product would also directly react with C3A to form carboaluminate (e.g., C3A·CaCO3·11H) via Route
. The carbonated product would also directly react with C3A to form carboaluminate (e.g., C3A·CaCO3·11H) via Route  . It was noted that the formation of carboaluminate phases at early stage would enhance the initial strength of cement mortars, especially for 3 days. Therefore, it suggested that the use of carbonated PCFA in cement materials would potentially offer great benefits in terms of strength development.
. It was noted that the formation of carboaluminate phases at early stage would enhance the initial strength of cement mortars, especially for 3 days. Therefore, it suggested that the use of carbonated PCFA in cement materials would potentially offer great benefits in terms of strength development.
Implications to interfacial chemistry of waste-based cementitious materials
This study applied high-energy synchrotron techniques to elucidate the reaction mechanisms of CO2 mineralization using PCFA and its product utilization as SCM in cement mortars. Several important information and findings were obtained as the reference of future research. For instance, CO2 was successfully mineralized as calcite (CaCO3) through accelerated carbonation of PCFA. In the case of either fresh or carbonated PCFA, there was no presence of CaCO3 in blended cement mortars after 90 days, suggesting the complete consumption (reaction) of CaCO3 in the course of cement hydrations. In general, the addition of CaCO3 in cement mortars could provide both physical (e.g., more active sites) and chemical (e.g., the formation of hexagonal monocarbonate in eqn (13)) enhancement for hydration reactions.The findings from this investigation could provide an insight into the kinetics of hydration reactions for PCFA-blended cement mortars. However, the products of hydration reactions at different curing ages (e.g., 1, 3, 7, 28, 56 and 90 days) were not yet studied in details. As the aforementioned, the amounts of the reaction products, such as carboaluminate, ettringite and the AFm phase, would vary in the course of the hydration, thereby affecting the strength development at the early and late stages. Our future research will be focused on the hydration products at different ages, as well as the kinetics of hydration reactions for PCFA-blended cement mortars using the calorimetry and synchrotron techniques (such as in situ XRD, nanoscale tomography and X-ray absorption near edge structure spectrometry).
Conclusions
This study could provide an insight into the mechanistic understanding of reactions at mineral–water interfaces for CO2 mineralization and utilization exemplified by PCFA. We found that, for CO2 mineralization using PCFA, the formation of calcite (CaCO3) products would be attributed to the complete conversion of lime and portlandite, along with the partial conversion of anhydrite in the fresh PCFA. The surface of the carbonated PCFA was covered with rhombohedral calcite crystals, with a size of 0.1–0.5 μm. For the subsequent utilization of mineralized products, the amounts of carboaluminate, ettringite and the AFm phase varied in the course of the hydration, thereby affecting the strength development at the early and late stages. Also, no presence of calcite in blended cements with either fresh or carbonated PCFA was observed, indicating the complete consumption of CaCO3 during cement hydrations. Despite the findings could provide the basis for controlling pathways and kinetics, the exact reactions in fact, especially for hydration of blended cement, were much more complex. Future research directions should focus on the identification of dynamic speciation by in situ high-resolution equipment, measuring local mass transfer and determining the phase characteristics at the molecular levels. This could enable the broad-spectrum collection of thermodynamic and kinetic data that specially emphasize on mass transfer and reaction rate at the solid–fluid interfaces.Experimental section
Mineral carbonation and utilization using PCFA
In this study, PCFA generated from the unit process of a circular fluidized bed boiler in a petrochemical industry (Taiwan) was used. Prior to CO2 mineralization experiments, the PCFA was dried overnight in an oven at 105 °C to eliminate the moisture. Additional grinding process was not required as the particle size of the PCFA was fine enough (e.g., <20 μm) for CO2 mineralization. De-ionized (DI) water with 18.2 MΩ cm−1 was used. High-pressure CO2 gas of 99.5% and N2 gas of 99.5% were purchased from Ching-Fung Gas Corporation (Taipei, Taiwan). For the mineralization experiments, the dried PCFA was mixed with DI water to form a PCFA slurry at a liquid-to-solid ratio of 10. The formed PCFA slurry was highly alkaline with a pH value of >13.2, thereby rapidly resulting in the formation of carbonate ions once the CO2 gas was introduced into the reactor. The details of experimental procedure for CO2 mineralization could be referred to our previous study.10For the PCFA utilization experiments, the PCFA was blended with clinker at a substitution ratio of 5%. In order to prepare cement mortar specimens, graded standard sand (Ottawa sand, compliance with ASTM C778) and ordinary Portland cement (type I with a mean diameter of 21.6 μm) were used. The water-to-powder and sand-to-powder ratios were designated at 0.49 and 2.75, respectively, for all cement mortars. The blended cement mortars were cured in lime-saturated water at 23 ± 1 °C for 90 days.
High-energy synchrotron X-ray techniques
The high-energy synchrotron X-ray diffraction (XRD) patterns of PCFA before and after CO2 mineralization were collected at beamline 11-ID-C of the Advanced Photon Source at Argonne National Laboratory. The wavelength of the X-ray beam was 0.11742 Å (photon energy of 105.59 keV). A PerkinElmer area detector was used to collect 2D XRD data. The obtained 2D diffraction patterns were calibrated using a standard CeO2 sample and then converted to 1D patterns (the diffraction intensity versus 2θ angle) using Fit2D program.43In order to quantify the changes of mineral phases in PCFA, the Rietveld refinement was performed using the Match software (Crystal Impact, Bonn, Germany). The parameters of crystal structures were taken from the American Mineralogist Crystal Structure Database in the Crystallography Open Database to interpret the synchrotron XRD patterns in Match. The collection codes for the major peaks included anhydrite (CaSO4, code 96-900-4097), lime (CaO, code 96-900-6713), quartz (SiO2, code 96-900-6302), calcite (CaCO3, code 96-900-9668), portlandite (Ca(OH)2, code 96-900-0114), hatrurite (Ca27Si9O45, code 96-900-8367), hatrurite (Ca9Si3O15, code 96-900-9266), wollastonite (CaSiO3, code 96-900-5778), sillimanite (Al2SiO5, code 96-900-3990), dmitryivanovite (CaAl2O4, code 96-901-3918), ettringite (Ca6Al2S3O49.496H60.602, code 96-901-2923), cancrinite (Ca0.04C0.5H6O14.25Si3Na3.58K0.15, code 96-901-3989), foshagite (Ca4Si3O11H2, code 96-901-1044), and afwillite (Ca3Si2O10H6, code 96-900-7612).
Micro-probe X-ray fluorescence (XRF)
Scanning micro-probe XRF microscopy was carried out at beamline 2-ID-D of the Advanced Photon Source at the Argonne National Laboratory.44 Fresnel zone plates were used as focusing optics in X-ray microprobes at energies between 1 and 10 keV, corresponding to atomic numbers Z between 13 (Zn) and 30 (Zn). Highly focused X-rays (Kα-line fluorescence) were used to excite and map the elements in heterogeneous PCFA with low detection limits at the sub-micron spatial resolution. Areas of interest were raster scanned with 0.8 μm pixel size and a focus diameter of ∼0.5 μm. XRF signals from the PCFA specimen were captured with an energy dispersive Ge detector placed perpendicular to the incident beam direction in the plane of polarization.Morphology and elemental distribution analysis of PCFA
The qualitative analysis of PCFA was carried out with a field-emission scanning electron microscope (FE-SEM) equipped with X-ray energy dispersive spectrometer (EDS). The morphology and elemental distribution analysis (e.g., sulfur, calcium, silicon, and aluminum) of fresh and carbonated PCFA were examined by a FE-SEM (S-4800, Hitachi) and EDS (XFlash Quad 5040, Bruker), respectively. The operating voltage was kept at 10.0 kV, with the electron beam directed at 90° to the specimen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
High appreciation goes to the Ministry of Science and Technology (MOST), Taiwan (R. O. C.) for their financial support under Grant No. 107-2917-I-564-043 and 108-2218-E-002-053-MY3. Dr Ronghui Kou at the X-ray Science Division, Advanced Photon Source, Argonne National Laboratory was highly appreciated for his technical assistance on high-energy synchrotron X-ray techniques. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Shu-Yuan Pan also thanks the financial support from National Taiwan University under the Grant Number 108L7309. We also thank Dr Mengyao Gao at Energy Technology Area, Lawrence Berkeley National Laboratory for her technical assistance on mineralogical analyses.References
- K. S. Lackner, Science, 2003, 300, 1677–1678 CrossRef CAS.
- S. Y. Pan, T. C. Chung, C. C. Ho, C. J. Hou, Y. H. Chen and P. C. Chiang, Sci. Rep., 2017, 7, 17227 CrossRef.
- NASEM, Gaseous Carbon Waste Streams Utilization: Status and Research Needs, Report 978-0-309-48336-0, National Academies of Sciences, Engineering, and Medicine, Washington, DC, 2019 Search PubMed.
- A. Kirchofer, A. Brandt, S. Krevor, V. Prigiobbe and J. Wilcox, Energy Environ. Sci., 2012, 5, 8631 RSC.
- A. Kirchofer, A. Becker, A. Brandt and J. Wilcox, Environ. Sci. Technol., 2013, 47, 7548–7554 CrossRef CAS.
- The Freedonia Group, Global Demand For Construction Aggregates To Exceed 48 Billion Metric Tons In 2015, https://www.concreteconstruction.net/business/global-demand-for-construction-aggregates-to-exceed-48-billion-metric-tons-in-2015_o Search PubMed.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- S. Tian and J. Jiang, Environ. Sci. Technol., 2012, 46, 13545–13551 CrossRef CAS.
- H. Xie, H. Yue, J. Zhu, B. Liang, C. Li, Y. Wang, L. Xie and X. Zhou, Engineering, 2015, 1, 150–157 CrossRef CAS.
- S. L. Pei, S. Y. Pan, Y. M. Li and P. C. Chiang, Environ. Sci. Technol., 2017, 51, 10674–10681 CrossRef CAS PubMed.
- H. Shao, J. R. Ray and Y. S. Jun, Environ. Sci. Technol., 2011, 45, 1737–1743 CrossRef CAS.
- Y. Min, J. D. Kubicki and Y. S. Jun, Environ. Sci. Technol., 2015, 49, 1946–1954 CrossRef CAS.
- D. Zhang, V. C. Li and B. R. Ellis, ACS Sustainable Chem. Eng., 2018, 6, 15976–15981 CrossRef CAS.
- N. Lippiatt, T.-C. Ling and S. Eggermont, Constr. Build. Mater., 2019, 229, 116825 CrossRef CAS.
- A. Quennoz and K. L. Scrivener, Cem. Concr. Res., 2012, 42, 1032–1041 CrossRef CAS.
- G. Geng, J. Li, Y. Zhou, L. Liu, J. Yan, M. Kunz and P. J. M. Monteiro, Cem. Concr. Res., 2018, 108, 38–45 CrossRef CAS.
- G. Geng, R. J. Myers, J. Li, R. Maboudian, C. Carraro, D. A. Shapiro and P. J. Monteiro, Sci. Rep., 2017, 7, 44032 CrossRef.
- P. J. M. Monteiro, L. Clodic, F. Battocchio, W. Kanitpanyacharoen, S. R. Chae, J. Ha and H.-R. Wenk, Cem. Concr. Compos., 2013, 40, 14–20 CrossRef CAS.
- M. D. Jackson, J. Moon, E. Gotti, R. Taylor, S. R. Chae, M. Kunz, A.-H. Emwas, C. Meral, P. Guttmann, P. Levitz, H.-R. Wenk, P. J. M. Monteiro and G. Scherer, J. Am. Ceram. Soc., 2013, 96, 2598–2606 CrossRef CAS.
- S. Y. Pan, P. C. Chiang, Y. H. Chen, C. D. Chen, H. Y. Lin and E. E. Chang, Environ. Sci. Technol., 2013, 47, 13677–13685 CrossRef CAS.
- S. Wansom, P. Chintasongkro and W. Srijampan, Cem. Concr. Compos., 2019, 103, 134–148 CrossRef CAS.
- D.-Y. Lei, L.-P. Guo, W. Sun, J.-p. Liu and C.-w. Miao, Constr. Build. Mater., 2017, 153, 765–773 CrossRef CAS.
- Z. Wei, B. Wang, G. Falzone, E. C. La Plante, M. U. Okoronkwo, Z. She, T. Oey, M. Balonis, N. Neithalath, L. Pilon and G. Sant, J. CO2 Util., 2018, 23, 117–127 CrossRef CAS.
- E. E. Chang, S. Y. Pan, Y. H. Chen, C. S. Tan and P. C. Chiang, J. Hazard. Mater., 2012, 227–228, 97–106 CrossRef CAS.
- S. Y. Pan, P. C. Chiang, Y. H. Chen, C. S. Tan and E. E. Chang, Appl. Energy, 2014, 113, 267–276 CrossRef CAS.
- K. Vance, G. Falzone, I. Pignatelli, M. Bauchy, M. Balonis and G. Sant, Ind. Eng. Chem. Res., 2015, 54, 8908–8918 CrossRef CAS.
- T. Roncal-Herrero, J. M. Astilleros, P. Bots, J. D. Rodríguez-Blanco, M. Prieto, L. G. Benning and L. Fernández-Díaz, Am. Mineral., 2017, 102, 1270–1278 CrossRef.
- D. P. Bentz, A. Ardani, T. Barrett, S. Z. Jones, D. Lootens, M. A. Peltz, T. Sato, P. E. Stutzman, J. Tanesi and W. J. Weiss, Constr. Build. Mater., 2015, 75, 1–10 CrossRef.
- S.-Y. Pan, R. Adhikari, Y.-H. Chen, P. Li and P.-C. Chiang, J. Cleaner Prod., 2016, 137, 617–631 CrossRef CAS.
- H. Manzano, E. Durgun, M. J. Abdolhosseine Qomi, F.-J. Ulm, R. J. M. Pellenq and J. C. Grossman, Cryst. Growth Des., 2011, 11, 2964–2972 CrossRef CAS.
- X. Li, X. Shen, M. Tang and X. Li, Ind. Eng. Chem. Res., 2014, 53, 1954–1964 CrossRef CAS.
- V. K. Peterson, D. A. Neumann and R. A. Livingston, J. Phys. Chem. B, 2005, 109, 14449–14453 CrossRef CAS PubMed.
- W. Kurdiwski, Cement and Concrete Chemistry, Springer, New York London, 2014 Search PubMed.
- S. Pourchet, L. Regnaud, J. P. Perez and A. Nonat, Cem. Concr. Res., 2009, 39, 989–996 CrossRef CAS.
- G. Geng, R. J. Myers, Y.-S. Yu, D. A. Shapiro, R. Winarski, P. E. Levitz, D. A. L. Kilcoyne and P. J. M. Monteiro, Cem. Concr. Res., 2018, 111, 130–137 CrossRef CAS.
- S. Y. Pan, Y. H. Chen, C. D. Chen, A. L. Shen, M. Lin and P. C. Chiang, Environ. Sci. Technol., 2015, 49, 12380–12387 CrossRef CAS.
- S.-Y. Pan, C.-H. Hung, Y.-W. Chan, H. Kim, P. Li and P.-C. Chiang, ACS Sustainable Chem. Eng., 2016, 4, 3045–3052 CrossRef CAS.
- A. P. Luz and V. C. Pandolfelli, Ceram. Int., 2012, 38, 1417–1425 CrossRef CAS.
- R. Snellings, A. Bazzoni and K. Scrivener, Cem. Concr. Res., 2014, 59, 139–146 CrossRef CAS.
- J. Tang, S. Wei, W. Li, S. Ma, P. Ji and X. Shen, Constr. Build. Mater., 2019, 223, 177–184 CrossRef CAS.
- K. L. Scrivener, T. Füllmann, E. Gallucci, G. Walenta and E. Bermejo, Cem. Concr. Res., 2004, 34, 1541–1547 CrossRef CAS.
- L. Wang, C. V. Putnis, E. Ruiz-Agudo, J. Hovelmann and A. Putnis, Environ. Sci. Technol., 2015, 49, 4184–4192 CrossRef CAS.
- A. P. Hammersley, S. O. Svenssoon, M. Hanfland and A. N. Fitch, High Pressure Res., 1996, 14, 235 CrossRef.
- Z. Cai, B. Lai, W. Yun, P. Ilinski, D. Legnini, J. Maser and W. Rodrigues, AIP Conf. Proc., 2000, 507, 472–477 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |