
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical reduction of different Ag(I)-containing solutions in bioelectrochemical systems for recovery of silver and simultaneous power generation†
Ngo Anh Dao
Ho
a and
Sandhya
Babel
 *b
*b
aFaculty of Environment and Labour Safety, Ton Duc Thang University, 19 Nguyen Huu Tho Street, Tan Phong Ward, District 7, Ho Chi Minh City, Vietnam. E-mail: hongoanhdao@tdtu.edu.vn
bSchool of Biochemical Engineering and Technology, Sirindhorn International Institute of Technology, Thammasat University, P. O. Box 22, Pathum Thani 12121, Thailand. E-mail: sandhya@siit.tu.ac.th
First published on 25th September 2019
Abstract
In this study, dual-chamber bioelectrochemical reactors (i.e., R1, R2, R3, and R4) were employed to investigate the Ag recovery and electricity production from different Ag(I)-containing artificial wastewaters (i.e., Ag+ solution, [Ag(NH3)2]+ and [Ag(S2O3)2]3− complexes, and mixed metal solution). Results showed that the electrochemical reductions of Ag(I) ions in all reactors were rapid reactions. The reaction rate in R1 was the fastest. At the same initial conditions (i.e. Ag(I) concentration of 1000–1080 mg L−1), the Ag recovery efficiency was 81.8% for R3 operated with the [Ag(S2O3)2]3− complex. Although high Ag removal efficiency (i.e., >99%) was found in other reactors, some diffusion of positively charged Ag(I) ions through the membrane was also observed along with the electrochemical reduction. In all cases, pure silver electrodeposits, mainly as dendrites and crystals in different morphologies, were observed at the cathode surfaces when characterized by SEM, EDX, and XRD. The performance of electricity production was evaluated by the open circuit voltage (OCV) and maximum power density (Pmax) obtained during the BES operation. Reactor R1 showed better performance (i.e., OCV of 828 mV, Pmax of 8258 mW m−3), due to its high standard reduction potential. The lower performance in other reactors was due to the complexity of solutions, other co-existing metals (mixed metal solution), and lower standard reduction potential. In general, the existing forms of Ag(I) in solutions affect the Ag(I) reduction rate. This further influences the Ag removal efficiency, morphology of electrodeposits, and power generation.
Introduction
A variety of methods have been studied for the recovery of metals (for instance, silver) from aqueous solutions and wastes.1–4 There has been much effort to develop recovery techniques that are cost-effective and sustainable. In recent times, technologies based on microbe-metal interactions, such as bio-sorption, bioremediation, and bio-reduction, have received much attention as biotechnological metal-recovery processes.5 Microbe-based technology is a cost-effective choice for concentrating diffuse metallic ions from effluents for recovery. However, many difficulties still exist with this method, which limit the practical applicability. Toxicity to microorganisms due to the complex physicochemical conditions of waste streams (e.g., too low or too high pH, high metal concentration, other co-existing recalcitrant compounds) is a major concern. In addition, the biomass after recovery needs to be refined and post-treated by further processes that lead to extra costs, to obtain a recoverable product (pure metals). Thus, other bio-based recovery techniques need to be explored to solve these issues.Recently, bio-electrochemical systems (BESs) have been shown as a platform technology for integrated waste treatment along with energy and resource recovery.6–8 A typical BES consists of an anode and a cathode chamber, separated by an ion exchange membrane (IEM). However, BESs differ in configuration, which depends on the target functions to be accomplished. In the anode chamber, biodegradable substrates, such as organic matter, are oxidized by microorganisms via anaerobic metabolism to harvest electrons. These electrons are then transferred to the anode electrode through different extracellular electron transfer (EET) mechanisms. The electron flow is then directly captured through an electrical circuit and is delivered to the cathode for either electricity production, hydrogen generation, or removal of recalcitrant contaminants.8 In principle, any compound with a high redox potential can serve as the catholyte to accept the electrons.9 Metal ions, therefore, can be utilized as terminal electron acceptors (TEAs) to be reduced, forming metallic electrodeposits on the cathode surface. Based on the BES mechanism, organic-rich wastewater and metal-laden solution are fed separately into the anode and cathode chamber, respectively, where the biological oxidation and electrochemical reduction occur simultaneously. Thus, a BES reactor, which can be considered as a fuel cell, serves the functions of a biological wastewater treatment system and a bio-method for the recovery of metals from aqueous solutions. Fundamental mechanisms and engineering feasibilities of BES technology for the removal and recovery of different metals have been reviewed by some researchers, indicating BES as a promising approach.9–11
Due to the valuable features and widespread use of silver in many industrial applications, the recovery of this precious metal from effluents has been emphasized in industrial waste management and treatment. The components of waste are relatively complex, where a variety of organic and inorganic substances (e.g., ammonia, EDTA, and thiosulfate) usually exists.12–14 Thus, the effluents normally contain Ag at high concentration, in which Ag ions exist in the form of either simple silver salts (e.g., silver nitrate AgNO3) or complexes (e.g., silver(I) monothiosulfate [Ag(S2O3)]−, silver(I) dithiosulfate [Ag(S2O3)2]3−, silver(I) trithiosulfate [Ag(S2O3)3],5–15 or silver(I) diammine [Ag(NH3)2]+ (ref. 13)). Since 2012, a few BES studies of silver recovery coupled with power generation have been conducted, in which the effects of operational parameters (e.g., pH, initial Ag(I) concentration) were investigated.16–20 The influence of different IEMs on BES performance has also been examined, recently.21
However, so far there has been no study that compares the BES performance among different Ag(I)-containing solutions. Different existing forms of Ag(I) that are used as catholytes correspond to different standard redox potentials (E0ca). These redox potentials have a distinct influence on Ag(I) reduction and further affect the recovery efficiency. This is because E0ca is directly related to the working cathodic potential (Eca), a decisive factor in the kinetics of electrochemical reactions. In addition, BES performance is also characterized by the formation, growth, and chemical composition of silver deposits that are formed on the cathode surface. These silver deposits are affected by the types of Ag(I) solutions used during BES operation. The main structure of silver atoms is crystalline (i.e., faced-centered cubic silver), which needs to be obtained when recovering silver from aqueous solution. Thus, a comparison of BES performance for different types of Ag(I) solutions is required to understand the reductive capacity of Ag(I) ions serving as TEAs in the cathode chamber.
In this study, four independent BES reactors are employed to investigate the silver recovery and simultaneous electricity production from artificial Ag(I) solutions. These solutions are simulated based on the predominant Ag(I) forms in real industrial effluents (i.e., silver electroplating, photographic processing). They are (i) Ag+ solution from AgNO3, (ii) [Ag(NH3)2]+ complex, (iii) [Ag(S2O3)2]3− complex, and (iv) multi-metal solution containing Ag+, Fe3+ and Cu2+ as nitrate forms. All reactors are operated at the same initial conditions, and the kinetics of the reactions is analysed to compare the Ag(I) reduction rate and removal efficiency. The morphology and composition of the deposits on the cathode surfaces are also analysed to confirm the silver electrodeposits and show the differences in the morphology due to the different types of Ag(I) solution. The electricity production in all cases is performed through the electrochemical parameters measured from polarization tests. These actual electrochemical parameters are then compared with theoretical thermodynamic values to show the differences in performance. The overall goal is to highlight the role of TEAs in the cathode chamber, as Ag(I) ions exist in different forms, and clarify their effects on BES performance. The findings from this work will be useful for further BES studies to investigate the recovery of silver from real Ag(I)-containing wastewaters.
Materials and methods
Fabrication and installation of BES reactors
Four similar cube-shaped BES reactors, made from acrylic plates, are designated reactors R1, R2, R3, R4, corresponding to four different types of Ag(I) solution. Each reactor consisted of an anode and a cathode chamber (i.e., 12 cm long × 6 cm width × 22 cm high, with a working volume of 1.0 L for each chamber) separated by a cation exchange membrane CMI-7000S (Membrane International, Inc. USA). The membrane, with a surface area of 264 cm2, was pre-treated by immersion in 5% NaCl solution for 12 hours to allow for hydration and expansion. Carbon brush (15 cm × 6 cm), prepared by twisting carbon fiber (Thai Carbonfiber Co., Thailand) with a Ti wire of 1.0 mm in diameter (Prolog Titanium Co., Ltd), served as the anode. The cathode was a graphite plate (90 cm2, Fujian, China). Before making electrical connections with the Ti wire, both electrodes were pre-treated to eliminate contaminants and enhance conductivity.The carbon brush was first cleaned by soaking in pure acetone (overnight). After that, it was treated with acid by soaking in a mixture of ammonium peroxydisulfate (200 g L−1) and concentrated sulfuric acid (100 mL L−1) for 15 min. The anode was then heat-treated in a muffle furnace at 450 °C for 30 min and washed three times with distilled water. According to previous studies, the above procedure increased power generation by 25% due to an increase in the specific area, which facilitated bacterial adhesion and enhanced the charge transfer from the bacteria to the electrode.22–24 The graphite plate cathode was immersed for more than 24 h in 2 mM potassium permanganate solution (KMnO4) and washed thoroughly with distilled water before assembling. All reactors were then connected to a computer equipped with a data logger (Grant Instruments, Cambridge Co., Ltd), to record cell voltage production during experiments (Fig. 1).
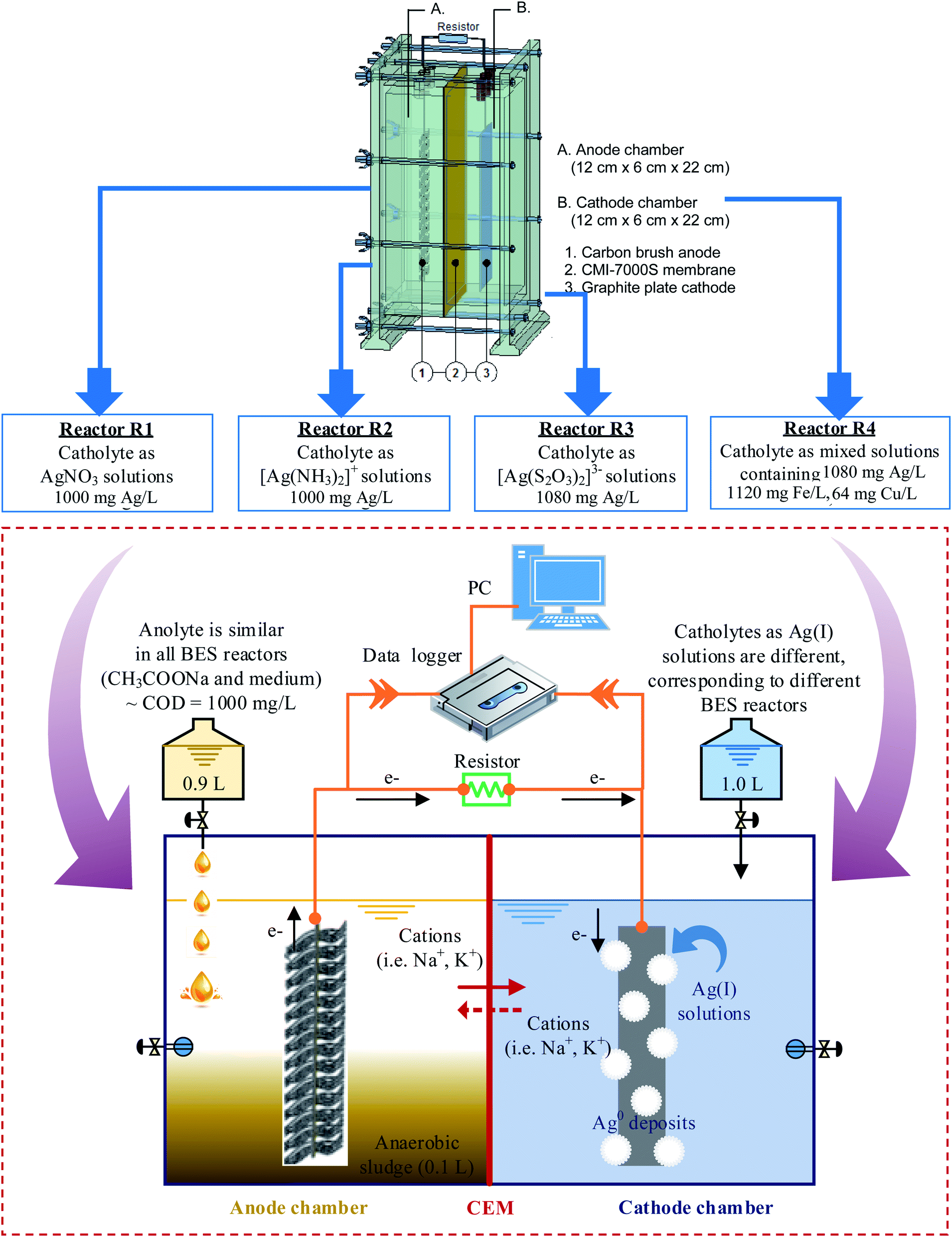 | ||
| Fig. 1 Installation of BES reactors and schematic illustration of operation for Ag recovery. | ||
Inoculation and acclimatization
Mixed-culture inoculum, anaerobic sludge (0.1 L) collected from the digester of a brewery wastewater treatment plant (Pathum Thani, Thailand), was used to activate the biological activities in the anode chambers of all reactors. Compared to pure cultures, the higher diversity of microbial communities in mixed cultures can make bio-electrochemical processes more stable and efficient due to their higher adaptability, stability, and productivity.25,26The anolyte (900 mL), supplied as a nutrient source for the microbial growth, includes (per liter) CH3COONa (1.28 g) as the substrate, Na2HPO4 (3.55 g), KH2PO4 (3.4 g), NH4Cl (0.31 g), and yeast extract (0.2 g). The initial organic loading of the anolyte, as represented by COD concentration, was 1000 mg L−1. It was reported that acetate is a simplest substance that has been extensively used as a carbon source, to induce electrochemical active bacteria (EAB).27 Preliminary results showed that a BES reactor inoculated with acetate provided a faster rate of cell voltage response during the inoculation stage, as compared to glucose.19
The cathode chamber was filled with buffer solution (1.0 L), containing (per liter) NaH2PO4·2H2O (4.77 g), Na2HPO4 (2.75 g), and NaCl (2.93 g). During the inoculation stage, the anode chamber was kept in anaerobic conditions to proceed with the bio-anodic reactions, in which CH3COONa acted as an electron donor. In contrast, the cathode chamber was purged continuously with air (80 mL min−1), to supply oxygen (O2) as an electron acceptor. An electrical circuit made of Ti wire with a 1000 Ω resistor was connected for the electron flow from the anode to the cathode. The system worked as a fuel cell, in which the production of cell voltage (Ecell) was monitored and recorded by a data logger that was connected to a PC (Fig. 1).
The inoculation of all reactors was conducted under batch-fed conditions and room temperature (20–25 °C). The anolytes and catholytes were refreshed at the end of each cycle (i.e., 5–6 days), when the Ecell dropped sharply due to a shortage of substrate in the anode chambers. The system was considered to be successfully acclimated when a stable and repeatable Ecell (i.e., 300–350 mV) was obtained after 6 feeding cycles (5–6 days/each cycle). The airflow was then stopped in the cathode chamber, and BES operation was started simultaneously in all reactors for silver recovery and electricity production.
BES operation for silver recovery and power generation
The BES operation was designed for a batch-feeding mode in 48 hour experiments, in which the anolytes were refreshed to keep the initial organic loading (i.e., COD 1000 mg L−1) similar in all reactors. The catholyte in each reactor was replaced by the corresponding Ag(I) solution, which served as an electron acceptor at this stage. Specifically, Ag+ solution (1000 mg Ag+ per L), [Ag(NH3)2]+ complex (1000 mg Ag(I) per L), [Ag(S2O3)2]3− complex (1080 mg Ag(I) per L), and mixed multi-metal solution (1080 mg Ag+ per L, 1120 mg Fe3+ per L, and 64 mg Cu2+ per L) were fed into the cathode chambers of reactors R1, R2, R3, and R4, respectively. The catholytes were prepared as follows:- For Ag+ and mixed multi-metal solutions: soluble nitrate salts (i.e., AgNO3, Fe(NO3)3, Cu(NO3)2) were dissolved in de-ionized (DI) water. The original pH of the Ag+ solution was adjusted to 2.0 by adding HNO3 acid, while the original mixed solution, as measured, was 2.2.
- For [Ag(S2O3)2]3− complex: AgBr was dissolved in excess 100 mM Na2S2O3 solution. The AgBr was produced by mixing 10 mM AgNO3 (corresponding to 1080 mg Ag/L) with 10 mM KBr. The excess Na2S2O3 solution prevents the formation of the sparingly-soluble silver(I) thiosulfate precipitate Na[Ag(S2O3)]. The soluble complex of [Ag(S2O3)2]3− can be formed which is theoretically the most stable since it has the lowest Gibbs free energy of formation.28 The original pH of the [Ag(S2O3)2]3− complex was 7.39 and was not adjusted during the operation.
- For [Ag(NH3)2]+ complex: Ag2SO4 (Ag+ concentration of 1000 mg L−1) was dissolved in an excess amount of 30% NH4OH solution and de-ionized (DI) water. The mixture was heated gently on a hotplate with occasional stirring until the solid Ag2SO4 dissolved completely. Silver existed in the form of [Ag(NH3)2]+ complex. The original catholyte pH was about 10.2 and was not adjusted during the operation.
The anode and cathode chambers were purged with N2 gas (80 mL min−1) for 15 min to eliminate the dissolved oxygen. The whole system was then kept in anaerobic conditions by sealing with gas-tight septa during the operation. An external resistance (Rext) of 5 Ω was connected to the electrical circuit to maximize the current transferred from the anode to the cathode (Fig. 1).
During the operation for silver recovery and electricity production, acetate (CH3COO−) ions served as electron donors in the anode chamber, whereas Ag(I) ions in different forms replaced O2 to act as electron acceptors in the cathode chamber. The anodic and cathodic reactions are hypothesized as follows:
• The ideal biological reaction of the electron donors in the anode chambers of all reactors is:29
| 2HCO3− + 9H++ 8e− ↔ CH3COO− + 4H2O, Ean = −0.292 V (vs. SHE) | (1) |
• In contrast, the possible reactions of electron acceptors in the cathode chamber of each reactor are shown in the following equations:30
| Reactor R1: Ag(aq)+ + e− → Ag0(s), E0ca = +0.799 V (vs. SHE) | (2) |
| Reactor R2: [Ag(NH3)2](aq)+ + e− → Ag0(s) + 2NH3, E0ca = +0.373 V (vs. SHE) | (3) |
| Reactor R3: [Ag(S2O3)2](aq)3− + e− → Ag0(s) + 2S2O32−, E0ca = +0.016 V (vs. SHE) | (4) |
| Reactor R4: Ag(aq)+ + e− → Ag0(s), E0ca1 = +0.799 V (vs. SHE) | (5a) |
| Cu(aq)2+ + 2e− → Cu0(s), E0ca2 = +0.337 V (vs. SHE) | (5b) |
| Fe(aq)3+ + e− → Fe(aq)2+, E0ca3a = +0.771 V (vs. SHE) | (5c1) |
| Fe(aq)2+ + 2e− → Fe0(s), E0ca3b = −0.44 V (vs. SHE) | (5c2) |
The given hypothesis is that all reactions occur spontaneously and dominantly, except for reactor R4. In reactor R4, the cathodic reactions, shown in eqn (5b) and (5c2), are not energetically favourable. Accordingly, pure silver was expected to be recovered as metallic deposits on the cathode surface, accompanied by power generation as a by-product in all BES reactors.
Polarization curve analysis was also conducted in all BES reactors to characterize them as microbial fuel cell (MFC) systems. The analysis started when the reactors were operated for 10 hours. The reactors were first kept in open circuit to obtain the stable open circuit voltage (OCV), and the Rext was then varied gradually from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Ω to 5 Ω. The Ecell was observed at about 15 minutes, and the temporarily stable values were recorded to avoid changes in the substrate/product during the measurement. The current density (I) and power density (P) were then calculated, based on the stable Ecell obtained at each Rext.
000 Ω to 5 Ω. The Ecell was observed at about 15 minutes, and the temporarily stable values were recorded to avoid changes in the substrate/product during the measurement. The current density (I) and power density (P) were then calculated, based on the stable Ecell obtained at each Rext.
All chemicals used in this study are analytical grade and obtained from Fisher Chemical (Thermo Fisher Scientific, Belgium and Fisher Scientific, UK), Ajax Finechem Pty Ltd, Merck Millipore (Darmstadt, Germany), and Laboratory Reagents and Fine Chemicals (Loba Chemie Pvt. Ltd, India). The solutions and reagents were prepared by using DI water.
Analytical methods and calculation
The anolytes and catholytes in all reactors were sampled every 4 hours to determine the remaining COD and Ag(I) concentration. Before analysis, all samples were filtered through syringe filters (NY 0.45 μm (Allpure)) to eliminate particles or suspended biomass, which may come out when sampling. The Ag(I) concentration in the catholytes was measured by an Inductively Coupled Plasma (ICP) Spectrometer (Optima 8000, PerkinElmer, USA). Soluble COD measurement was based on the Closed Reflux, Titrimetric method.31 All measurements were performed in duplicate and the results reported are the average values.The decrement of COD concentration (ΔCs, mg L−1) at time t was calculated as
| ΔCs = COD0 − CODt | (6) |
The silver removal efficiency (RE%) at time t was calculated as
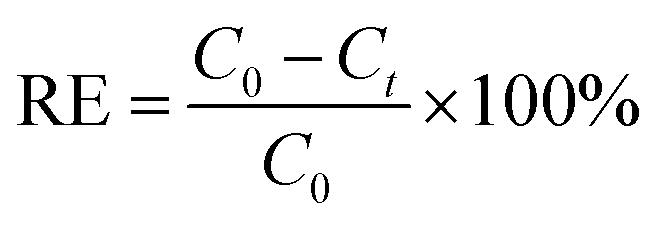 | (7) |
For electrical performance, the Ecell (mV) production was monitored with a data logger (Grant Instruments, Cambridge Co., Ltd) every 10 min during the operation and analyzed by using Squirrel View software 2010. The power density, P (mW m−3), and coulombic efficiency, CE (%), were calculated as follows:
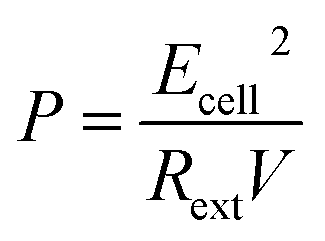 | (8) |
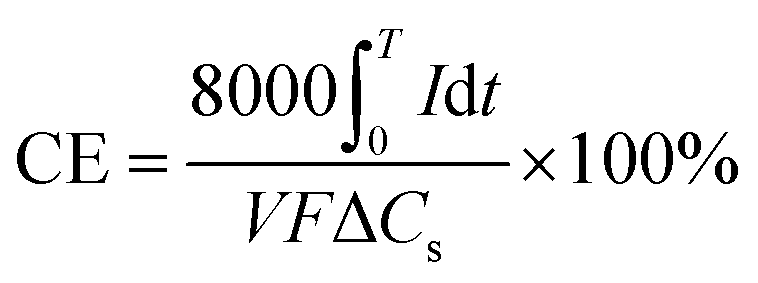 | (9) |
In the above equations, COD0 is the initial COD of anolyte fed into the anode chamber (1000 mg L−1). CODt is the COD at time t (mg L−1). C0 is the initial Ag(I) concentration in the catholyte (mg L−1), and Ct is the remaining Ag(I) concentration in the catholyte at time t (mg L−1). Rext is the external resistance installed in the system. Rext was fixed at (i) 1000 Ω during the inoculation stage, (ii) 5 Ω during operation, and was varied from 10000 to 5 Ω during polarization curve analysis. I (mA) is the current calculated by Ecell/Rext. Then, the current density was obtained as Ecell/(Rext × V). V is the working volume of the anode chamber (1.0 L), and F is the Faraday constant (96485 C mol−1 e−).
After 2 months of BES inoculation and operation, all reactors were emptied. The graphite plate cathodes were taken out of the reactors and air-dried at room temperature. Deposits covering the cathode surfaces were scraped to characterize the morphology and chemical composition by a scanning electron microscope (SEM) (SU8030 or S-3400N, Hitachi, USA and JSM-7800F, JEOL, USA), equipped with an energy dispersive X-ray (EDX) detector (Metek, Apollo XP 2060, USA and X-MaxN, Oxford Instruments, UK).
Results and discussion
Kinetic analysis of Ag(I) reduction and Ag removal efficiency
Four BES reactors (i.e., R1, R2, R3, and R4) were operated at the same conditions (i.e., initial COD loading of 1000 mg L−1, initial Ag(I) concentration of 1000–1080 mg L−1). The decrease of Ag(I) concentration in the catholytes of each reactor during operation is shown in Fig. 2. The remaining Ag(I) concentration in the catholytes of R1, R2, R3, and R4 after 48 hours of operation was 3.45, 3.28, 197.25, and 3.15 mg L−1, respectively. The Ag(I) reduction trends in reactors R1, R2, and R4 were similar (Fig. 2), whereas a slightly sloped curve of Ag(I) reduction was found in reactor R3. This may be due to the existing form of Ag(I) in R3 (i.e., a negatively charged complex), as compared to the common forms used in the other reactors (i.e., positively charged free ions or complexes). The charge state of Ag(I) in the catholyte is also an important factor, as it influences the migration of ions between the two chambers to maintain the electroneutrality. This influences the reduction rate of the anodic and cathodic reactions. Thus, high Ag removal efficiency (RE) (over 99%) was found for R1, R2, and R4, whereas a lower RE of 81.8% was obtained in R3 (Table 1).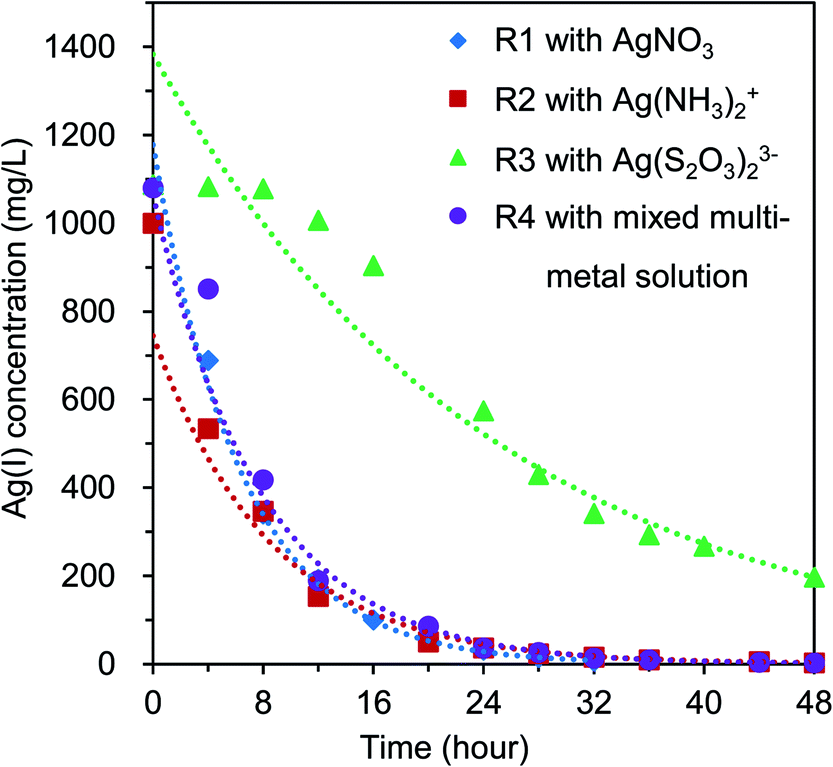 | ||
| Fig. 2 Ag(I) reduction by time in BES reactors (i.e., R1, R2, R3, and R4) containing respective Ag(I) solutions. | ||
| a C 0 and Ct in the equations are interpolated values from the kinetic reactions. R2 is the correlation coefficient. b RE was calculated based on the actual C0 and Ct obtained from experiments, as shown in eqn (7). | ||||
|---|---|---|---|---|
| Types of Ag(I) solutions | Ag+ as AgNO3 (reactor R1) | [Ag(NH3)2]+ complex (reactor R2) | [Ag(S2O3)2]3− complex (reactor R3) | Ag+ in multi-metal solution (reactor R4) |
| Initial Ag(I) concentration, C0 (mg L−1) in the catholyte fed to the reactor | 1000 | 1000 | 1080 | 1080 |
| pH adjustment | YES (pHadjusted = 2.0) | NO (pHoriginal = 10.2) | NO (pHoriginal = 7.39) | YES (pHadjusted = 2.0) |
| Kinetic equation for first-order reaction of Ag(I) removal, dC/dt = −kC or Ct = C0e−kta |
C t = 1177.3e−0.155t (R2 = 0.9969) | C t = 744.71e−0.117t (R2 = 0.9838) | C t = 1382.6e−0.041t (R2 = 0.9582) | C t = 1063.4e−0.129t (R2 = 0.9908) |
| Actual silver removal efficiency after 48 hours, RE (%)b | 99.3% | 99.6% | 81.8% | 99.7% |
| Loss of silver due to Ag(I) diffusion through CMI-7000S membrane | YES | YES | NO | YES |
This can be explained by the low standard reduction potential of [Ag(S2O3)2]3− complex in R3 (i.e., E0([Ag(S2O3)2]3−/Ag0) = + 0.016 V vs. SHE), as compared to that of Ag+ in R1 and R4 (i.e., E0(Ag+/Ag0) = + 0.799 V vs. SHE) and [Ag(NH3)2]+ in R2 (i.e., E0(Ag(NH3)2+/Ag0) = + 0.373 V vs. SHE) (eqn (2)–(5a)). Basically, the reduction potential of cathodic electron acceptors (Eca) directly affects the thermodynamic cell voltage and influences the actual Ecell production, which plays an important role in the reduction rate. A decrease of Ag(I) concentration was hypothesized due to the electrochemical reduction of Ag(I) ions by accepting electrons transferred from the anode, as shown in eqn (2)–(5a).
To compare the Ag(I) removal rate among the 4 reactors, the kinetic reactions of Ag(I) reduction in the system were analyzed. Results showed that the reduction followed and matched the first-order reaction (i.e., Ct = C0e−kt), where k is the reaction constant (h−1) (Table 1). The reduction rate of Ag+ ions in reactor R1 was faster than that of the other reactors, as the reaction constant k was calculated to be 0.155, 0.117, 0.041, and 0.129 (h−1) in reactors R1, R2, R3, and R4, respectively. The linear regression coefficient R2 was >0.95 in all cases (Table 1). This result indicated that the removal rate of Ag+ ion was more rapid than that of Ag(I) complexes. This finding is in line with that found by Tao, et al.,18 in which the reduction of Ag+ ion was more favorable and rapid than the [Ag(S2O3)]− complex.
However, the catholyte pH was adjusted in reactors R1 and R4 to maintain the acidity (i.e., pH of 2.0) to keep silver in a dissolved state (existing as Ag+ ions), which results in a high cost of operation. Furthermore, Ag loss due to the diffusion of positively charged Ag(I) ions from the cathode chamber to the anode chamber through the CMI-7000S membrane was found in reactors R1, R2, and R4 during the experiments and is reported in previous publications.20,21 Although a fast decrease of Ag(I) concentration and high Ag removal efficiency (i.e., >99%) were obtained with reactors R1, R2, and R4, it was ascribed to the electrochemical reduction of Ag(I) to form the recovered product as Ag0 deposits on the cathode surface and by the precipitation on the membrane surface and diffusion to the anode chamber. In reactor R3, [Ag(S2O3)2]3− complex was reduced spontaneously only through the electrochemical reaction in the cathode chamber without diffusion or loss of silver. Thus, the Ag removal efficiency as shown in Table 1 (i.e., 81.8%) could be interpreted as Ag recovery efficiency. Since [Ag(S2O3)2]3− complex is a typical and commonly existing form of silver in real wastewater, it is believed that BES technology is feasible for practical situations.
Performance in electricity production
The effects of Ag(I) forms on power generation are evaluated by considering the electrochemical parameters obtained from the polarization curve test. In this analysis, the external load of the electrical circuit in four reactors was changed by decreasing periodically the Rext from 10000 Ω to 5 Ω. The temporarily stable cell voltages (Establecell) were recorded at pseudo-steady state. The current density (I) and power density (P) were then calculated using Ohm's law. Polarization curves (E1–E4) represented Establecell as a function of I (Fig. 3a), while power curves (P1–P4) described P as a function of I (Fig. 3b).
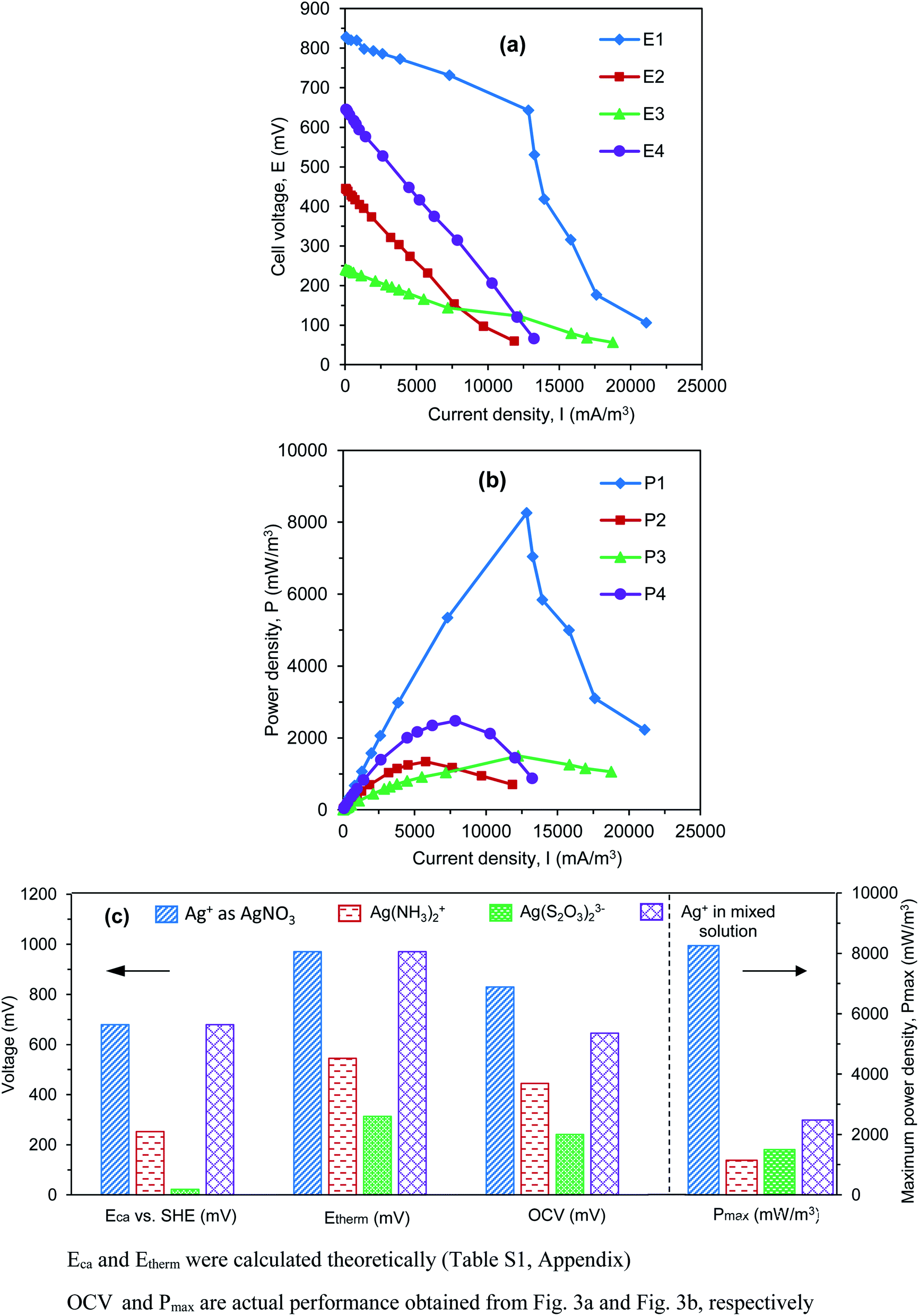 | ||
| Fig. 3 (a) Polarization curves (E1, E2, E3, and E4); (b) power curves (P1, P2, P3, and P4); and (c) actual electrochemical parameters obtained from reactors R1, R2, R3, and R4 in comparison with the thermodynamic values. | ||
In Fig. 3a, when the highest Rext is applied (i.e., 10000 Ω), corresponding to the lowest I produced (i.e., ∼0 mA m−3), the measured Establecell is defined as the open circuit voltage (OCV). The OCV is normally used to evaluate the actual performance of a fuel cell. Fig. 3a shows that there was a significant difference in OCV values among the four reactors. The highest OCV of 828 mV was found in reactor R1, where Ag+ solution served as the catholyte. In reactor R4, although a Ag+ salt was used, OCV decreased dramatically to 645 mV due to the effects of other metallic salts that co-exist in the catholyte (i.e., Cu2+ and Fe3+). When Ag(I) complexes ([Ag(NH3)2]+ and [Ag(S2O3)2]3−) were investigated in reactors R2 and R3, lower OCVs (i.e., 445 and 241 mV, respectively) were found. These results confirm the impacts of the theoretical cathodic reduction potential (i.e., Eca) on the thermodynamic cell voltage (i.e., Etherm = Eca − Ean), which directly influences the actual value (i.e., OCV). When different Ag(I) solutions were examined, the higher the Eca produced by the Ag(I) solution, the higher the OCV obtained in the system. A comparison of the electrochemical values (calculated theoretically, i.e., Eca and Etherm) and the actual performance (i.e., OCV) obtained in each reactor, corresponding to a specific Ag(I) solution, is summarized in Fig. 3c.
In terms of power curves (Fig. 3b), the maximum power density (Pmax) as the peak of the P curve was highest for reactor R1 (i.e., Pmax of 8258 mW m−3), as compared to R2 (i.e., Pmax of 1147 mW m−3), R3 (i.e., Pmax of 1500 mW m−3), and R4 (i.e., Pmax of 2472 mW m−3) (Fig. 3c). This shows that the cathodic reduction of Ag+ ions in a BES reactor is energetically more favorable than other Ag(I) complexes and the mixed solution, which was due to the higher Eca of the catholyte, leading to a higher cell voltage and power density. Although the actual OCV obtained in reactor R2 with [Ag(NH3)2]+ complex (445 mV) was higher than that in reactor R3 with [Ag(S2O3)2]3− complex (241 mV), Pmax2 was lower (1147 mW m−3), as compared to Pmax3 (1500 mW m−3). This may be due to the effects of internal resistance (Rint) resulted from the resistance of the electrolyte and membrane in the system. In BES studies, it was reported that linear polarization curves (E) are most often encountered. The Rint, in which the ohmic resistance (RΩ) is dominant, can be determined from the slope of E curves.29 In this study, the slope of E2 (as shown in Fig. 3a) is sharper than that of E3, indicating a higher Rint in reactor R2. This result was explained by the effects of fouling at the surface of the CMI-7000S membrane. This fouling was due to the formation of inorganic precipitates and the diffusion of positively charged [Ag(NH3)2]+ complex in reactor R2, causing higher Rint of the membrane and lower performance in electricity production.20
Generally, the reactor configurations and electrode materials influence the BES performance due to their effects on internal resistance and cell voltage production. Thus, in order to enhance the performance of electricity production, electrode material that have high redox activity and low-cost are more attractive. For example, ultrathin Ni(OH)2 nanosheets assembled into three-dimensional (3D) interspersed flower-like nickel hydroxide provided a large contact area with the electrolyte, reduced the polarization of the electrochemical reaction, and provided more active sites, which yielded a high capacitance and improved the electrochemical performance.32
Characterization of electrodeposits at the cathode surfaces
Prior to use, the graphite plate cathodes in the four reactors are the color of black-carbon. After 2 months of inoculation and operation, bright deposits were observed on the cathode surfaces in all cases. To confirm the successful BES operation for the recovery of silver, these deposits were scraped from the cathode and characterized by SEM, EDX, and XRD.In SEM analysis, different morphologies of the cathodic deposits were found. Specifically, deposits with polyhedral and small dendritic structures were detected on the cathode of R1 operated with AgNO3 solution (Fig. 4b). This polyhedral form was comparable to the crystal structure of silver atoms (i.e., faced-centered cubic silver). For R2 operated with [Ag(NH3)2]+, larger dendritic structures were found (Fig. 4c). These dendrites have long trunks and short branches which look like a fishbone. A similar dendritic crystal structure was also detected in the cathode surface of R4 when a mixed solution containing Ag+ was used (Fig. 4e). For the [Ag(S2O3)2]3− complex, different-sized clusters of deposits were observed on the cathode surface of R3 (Fig. 4d). This morphology of deposits on R3 was different from the others, where incoherent crystal particles or dendrites were normally formed. The morphology of cathodic deposits in the four reactors was affected by the Ecell output, which is related to the intensity of electron flow for the reduction of Ag(I) in the catholyte, as shown in eqn (2)–(5). It was reported that in an electrochemical reaction, the current density is related to the reaction rate and influences the nucleation and growth of silver particles, as shown in Fig. 4a (i.e., from polyhedral aggregates to long thick rods, followed by thick dendritic structures, and a mixture of dendrites and small cubic particles).33
 | ||
| Fig. 4 (a) Typical formation and growth of silver deposits in an electrochemical system (inferred from the study conducted by Liu et al.);33 (b), (c), (d), and (e) SEM images obtained in this study for the characterization of electrodeposits on the cathode surface of reactors R1, R2, R3, and R4, respectively. | ||
The SEM results were verified by EDX (Fig. 5), in which sharp peaks at an energy level of 2.98 keV were found in all cases, indicating the deposits were comprised of pure silver. The XRD shown in Fig. S1† (Appendix) also confirmed the EDX results. The XRD results matched the standard patterns of silver (i.e., JCPDS card no. 04-0783). All peaks can be indexed to face-centered-cubic silver, where the diffraction peaks occur at 2θ values of about 38°, 44°, 64°, 77°, and 81°, corresponding to the reflection of (111), (200), (220), (311), and (222) planes, respectively. No peaks from other phases (e.g., S-related compounds as Ag2S and Ag2SO3) were detected. This indicated high-purity electrodeposits of silver achieved in this study.
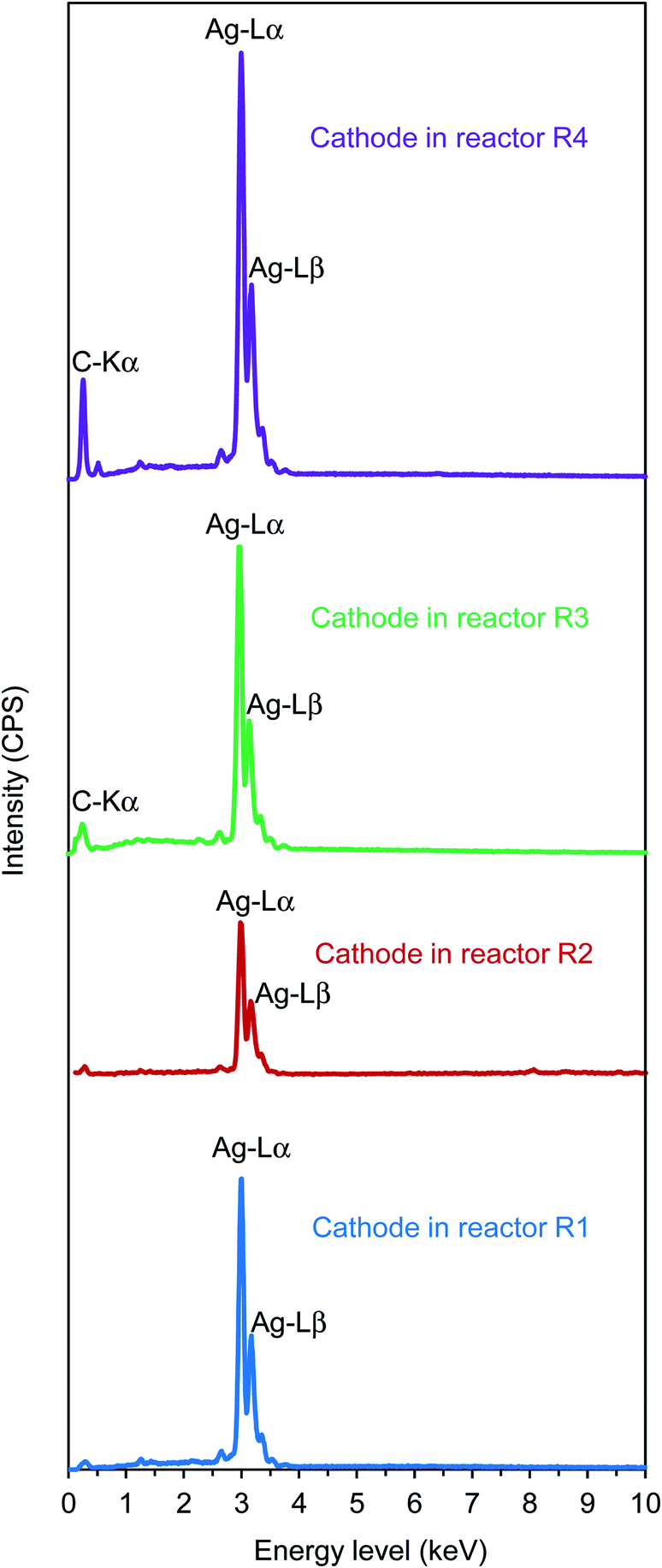 | ||
| Fig. 5 EDX characterization for confirmation of silver deposits on the cathode surfaces. | ||
Conclusions
This study found that different types of Ag(I)-containing solutions (i.e., Ag+ solution, [Ag(NH3)2]+, [Ag(S2O3)2]3− complex, and mixed multi-metal solution containing Ag+, Fe3+, Cu2+) employed in BES reactors resulted in different performance for Ag recovery and simultaneous electricity production. In all four reactors, Ag(I) was reduced electrochemically to form deposits on the cathode surface. The highest reduction rate (i.e., kinetic reaction constant, k of 1.55 h−1) was found with Ag+ solution, as compared to the other Ag(I) solutions (i.e., [Ag(NH3)2]+ complex, k of 0.117 h−1; [Ag(S2O3)2]3− complex, k of 0.041 h−1; and mixed multi-metal solution, k of 0.129 h−1). After BES operation in independent reactors (i.e., 48 hours for each batch of experiments), a recovery efficiency (RE) of 81.8% was obtained with [Ag(S2O3)2]3− complex. High removal efficiencies (i.e., >99%) were found for Ag+, [Ag(NH3)2]+, and the mixed multi-metal solution. This resulted from either cathodic electrochemical reaction or diffusion of positively charged Ag(I) forms through the CMI-7000S membrane, which may cause Ag loss in the system. Different morphologies of electrodeposits formed at the cathode surfaces were found with SEM-EDX-XRD characterization, in which dendrites and crystals were the main structures. In terms of electricity production, Ag+ solution showed the best performance with the highest Pmax (i.e., 8258 mW m−3) and OCV (i.e., 828 mV), which was due to its high standard reduction potential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial grant from the Sirindhorn International Institute of Technology (SIIT), Thammasat University, Thailand for this research.References
- A. D. Bas, E. Y. Yazici and H. Deveci, Recovery of silver from X-ray film processing effluents by hydrogen peroxide treatment, Hydrometallurgy, 2012, 121, 22–27 CrossRef
.
- N. Das, Recovery of precious metals through biosorption—a review, Hydrometallurgy, 2010, 103, 180–189 CrossRef CAS
- O. Kononova, A. Kholmogorov, N. Danilenko, N. Goryaeva, K. Shatnykh and S. Kachin, Recovery of silver from thiosulfate and thiocyanate leach solutions by adsorption on anion exchange resins and activated carbon, Hydrometallurgy, 2007, 88, 189–195 CrossRef CAS
- I. Rivera, A. Roca, M. Cruells, F. Patiño and E. Salinas, Study of silver precipitation in thiosulfate solutions using sodium dithionite. Application to an industrial effluent, Hydrometallurgy, 2007, 89, 89–98 CrossRef CAS
- Y. Nancharaiah, S. V. Mohan and P. Lens, Biological and bioelectrochemical recovery of critical and scarce metals, Trends Biotechnol., 2016, 34, 137–155 CrossRef CAS
- B. E. Logan and K. Rabaey, Conversion of wastes into bioelectricity and chemicals by using microbial electrochemical technologies, Science, 2012, 337, 686–690 CrossRef CAS
- D. Pant, A. Singh, G. Van Bogaert, S. I. Olsen, P. S. Nigam, L. Diels and K. Vanbroekhoven, Bioelectrochemical systems (BES) for sustainable energy production and product recovery from organic wastes and industrial wastewaters, RSC Adv., 2012, 2, 1248–1263 RSC
- H. Wang and Z. J. Ren, A comprehensive review of microbial electrochemical systems as a platform technology, Biotechnol. Adv., 2013, 31, 1796–1807 CrossRef CAS
- A. S. Mathuriya and J. Yakhmi, Microbial fuel cells to recover heavy metals, Environ. Chem. Lett., 2014, 12, 483–494 CrossRef CAS
- Y. Nancharaiah, S. V. Mohan and P. Lens, Metals removal and recovery in bioelectrochemical systems: A review, Bioresour. Technol., 2015, 195, 102–114 CrossRef CAS
- H. Wang and Z. J. Ren, Bioelectrochemical metal recovery from wastewater: a review, Water Res., 2014, 66, 219–232 CrossRef CAS
- W. T. Chen, C. C. Ma, M. H. Lee, Y. C. Chu, L. C. Tsai and C. M. Shu, Silver recovery and chemical oxygen demand (COD) removal from waste fixer solutions, Appl. Energy, 2012, 100, 187–192 CrossRef CAS
- Y. H. Wang, B. S. Wang, B. Pan, Q. Y. Chen and W. Yan, Electricity production from a bio-electrochemical cell for silver recovery in alkaline media, Appl. Energy, 2013, 112, 1337–1341 CrossRef CAS
- E. Y. Yazici, H. Deveci and R. Yazici, Recovery of silver from X-ray film processing effluents using trimercapto-s-triazine (TMT), Sep. Sci. Technol., 2011, 46, 2231–2238 CrossRef CAS
- T. W. Purcell and J. J. Peters, Sources of silver in the environment, Environ. Toxicol. Chem., 1998, 17, 539–546 CrossRef CAS
- C. Choi and Y. Cui, Recovery of silver from wastewater coupled with power generation using a microbial fuel cell, Bioresour. Technol., 2012, 107, 522–525 CrossRef CAS
- B. Lim, H. Lu, C. Choi and Z. Liu, Recovery of silver metal and electric power generation using a microbial fuel cell, Desalin. Water Treat., 2015, 54, 3675–3681 CrossRef CAS
- H. C. Tao, Z. Y. Gao, H. Ding, N. Xu and W. M. Wu, Recovery of silver from silver (I)-containing solutions in bioelectrochemical reactors, Bioresour. Technol., 2012, 111, 92–97 CrossRef CAS
- N. A. D. Ho, S. Babel and K. Sombatmankhong, Factors influencing silver recovery and power generation in bio-electrochemical reactors, Environ. Sci. Pollut. Res., 2017, 24, 21024–21037 CrossRef CAS
- N. A. D. Ho, S. Babel and K. Sombatmankhong, Bio-electrochemical system for recovery of silver coupled with power generation and wastewater treatment from silver (I) diammine complex, Journal of Water Process Engineering, 2018, 23, 186–194 CrossRef
- N. A. D. Ho, S. Babel and F. Kurisu, Bio-electrochemical reactors using AMI-7001S and CMI-7000S membranes as separators for silver recovery and power generation, Bioresour. Technol., 2017, 244, 1006–1014 CrossRef CAS
- Y. Feng, Q. Yang, X. Wang and B. E. Logan, Treatment of carbon fiber brush anodes for improving power generation in air–cathode microbial fuel cells, J. Power Sources, 2010, 195, 1841–1844 CrossRef CAS
- X. Wang, S. Cheng, Y. Feng, M. D. Merrill, T. Saito and B. E. Logan, Use of carbon mesh anodes and the effect of different pretreatment methods on power production in microbial fuel cells, Environ. Sci. Technol., 2009, 43, 6870–6874 CrossRef CAS
- J. Wei, P. Liang and X. Huang, Recent progress in electrodes for microbial fuel cells, Bioresour. Technol., 2011, 102, 9335–9344 CrossRef CAS
- A. S. Mathuriya, Inoculum selection to enhance performance of a microbial fuel cell for electricity generation during wastewater treatment, Environ. Technol., 2013, 34, 1957–1964 CrossRef CAS
- S. A. Patil, V. P. Surakasi, S. Koul, S. Ijmulwar, A. Vivek, Y. Shouche and B. Kapadnis, Electricity generation using chocolate industry wastewater and its treatment in activated sludge based microbial fuel cell and analysis of developed microbial community in the anode chamber, Bioresour. Technol., 2009, 100, 5132–5139 CrossRef CAS
- D. R. Bond, D. E. Holmes, L. M. Tender and D. R. Lovley, Electrode-reducing microorganisms that harvest energy from marine sediments, Science, 2002, 295, 483–485 CrossRef CAS
- M. G. Aylmore and D. M. Muir, Thiosulfate leaching of gold—a review, Miner. Eng., 2001, 14, 135–174 CrossRef CAS
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Microbial fuel cells: methodology and technology, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS
-
G. Milazzo and S. Caroli, Tables of Standard Electrode Potentials. John Wiley and Sons, New York, 1978 Search PubMed
-
APHA, Standard Methods for Examination of Water and Wastewater, American Public Health Association, 20th edn, 1998 Search PubMed
- W. Wei, W. Chen, L. Ding, S. Cui and L. Mi, Construction of hierarchical three-dimensional interspersed flower-like nickel hydroxide for asymmetric supercapacitors, Nano Res., 2017, 10, 3726–3742 CrossRef CAS
- W. Liu, T. Yang, C. Li, P. Che and Y. Han, Regulating silver morphology via electrochemical reaction, CrystEngComm, 2015, 17, 6014–6022 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06369b |
| This journal is © The Royal Society of Chemistry 2019 |