
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh effectiveness of pure polydopamine in extraction of uranium and plutonium from groundwater and seawater†
Valery N.
Bliznyuk
 *ae,
Kamila
Kołacińska
b,
Alexander A.
Pud
c,
Nikolay A.
Ogurtsov
c,
Yuriy V.
Noskov
c,
Brian A.
Powell
ade and
Timothy A.
DeVol
*ae
*ae,
Kamila
Kołacińska
b,
Alexander A.
Pud
c,
Nikolay A.
Ogurtsov
c,
Yuriy V.
Noskov
c,
Brian A.
Powell
ade and
Timothy A.
DeVol
*ae
aDepartment of Environmental Engineering and Earth Sciences, Clemson University, Clemson, SC 29634-0919, USA. E-mail: vblizny@clemson.edu; devol@clemson.edu
bInstitute of Nuclear Chemistry and Technology in Warsaw, Poland
cV. P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry, NAS of Ukraine, 50 Kharkivske Shose, Kyiv 02160, Ukraine
dDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA
eNuclear Environmental Engineering Sciences and Radioactive Waste Management (NEESRWM) Center, Clemson University, Clemson, SC 29634-0942, USA
First published on 24th September 2019
Abstract
Sorption properties of polydopamine (PDA) for uranium and plutonium from an aqueous environment are reported at three different pH values (2, 4 and 6.5–7). In addition to deionized (DI) water, artificial groundwater (GW) and seawater (SW) were used with U uptake close to 100% in each case. PDA polymer has been identified as a material with extremely high sorption capacity Qmax ∼500 mg g−1 of the polymer at pH 6.5 and high selectivity for uranium. Similar high sorption properties are revealed for plutonium uptake. PDA-uranyl and PDA-plutonium interactions responsible for the observed adsorption processes have been addressed with a set of experimental techniques including FTIR spectroscopy, electron microscopy and cyclic voltammetry.
1. Introduction
Since the first few references on polydopamine (PDA) in 1970–2006 (ref. 1–4) and an encouraging paper on mussel-inspired multifunctional PDA coatings in 2007,5 interest in this polymer has grown significantly due to a rapidly expanding range of potential applications of PDA and its composites in energy storage, catalysis, filtration, biomedical systems (sensors, drug delivery, theranostics), environmental cleaning and heavy metal accumulation, etc.6–8 PDA potentialities stem from its fascinating combination of catecholamine with other functionalities and, paradoxically, from a diverse, not quite understood yet supramolecular organization and composition. These two major factors hopefully promise the bright future and applications of this polymer. In fact, the uncertainty of the PDA qualitative and quantitative composition is predetermined by both the diversity of insoluble or poorly soluble products of dopamine oxidation/polymerization (the covalently bound polymer, oligomers and noncovalent stacked (dopamine)2/5,6-dihydroxyindole (DHI) trimers) and their ability to self-organize due to intermolecular interactions (H-bonding, van der Waals, π–π and charge transfer interactions, etc.) (Fig. 1).9,10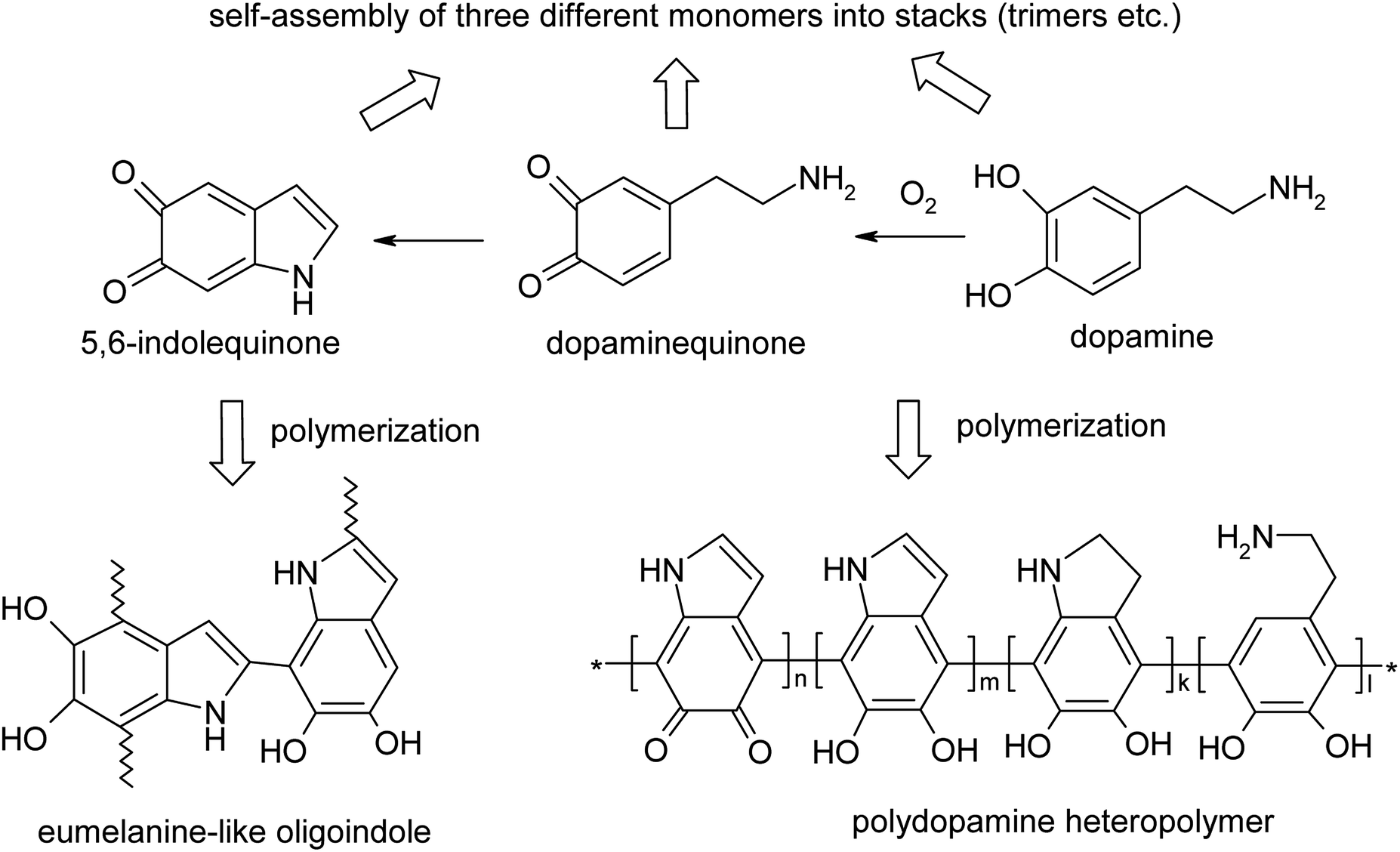 | ||
| Fig. 1 Alternative structures of PDA with covalent versus hydrogen bond self-assembly structures.5–9 | ||
In turn, this diversity fits well with some applications that need a wide set of functional groups facilitating PDA ability for chemical transformation/tailoring8,11,12 as well as in removal of organic compounds or removal/accumulation of heavy metals/radionuclides from aqueous media.7,13–16 Among currently known PDA applications, the environmental field can be considered as extremely important because of an inevitable release of pollutants from industry, nuclear stations and waste disposal facilities, as well as of a possible recovery of radioactive elements from natural sources in GW and SW. Initially, such PDA applicability was suggested on the basis of the above-mentioned mussel-like functionalities, which are prone to form adhesion bonds with different surfaces through noncovalent metal coordination or chelating, hydrogen bonding, π–π stacking, cation–π interactions and quinhydrone charge–transfer complexes.6,7,17 Naturally, due to the similar interactions and the rich surface chemistry, the insoluble PDA can quite effectively adsorb/absorb many pollutants. While only few papers demonstrate the applicability of pure PDA for the heavy metal removal,16,18 in most cases it is used in a form of its (nano)composites to get high surface contacts with solutions of the pollutants and probably synergistic enhancement of the absorption – as compared with the pure PDA.6,7,13–15,19,20 Quite impressive results in the nanocomposites case have been demonstrated, especially when it comes for removal of some radionuclides (e.g. uranium and plutonium) from water media. In particular, for the nanocomposites of mSiO2/PDA and waste paper derived carbon coated with PDA (WPC@PDA) the calculated maximum capacities for uranium U(VI) were 332.3 mg g−1 at T = 318.5 K, pH = 5.5 (287.5 mg g−1 at T = 298.15, pH = 5.5)21 and 384.6 mg g−1 at T = 298 K, pH = 7.0,22 respectively. The only (to the best of our knowledge) comparison of a PDA nanocomposite and pure PDA was made by Zhu et al. at lower pH = 4.5 and T = 298 K.15 They calculated that the adsorption capacities of the core–shell polydopamine@MgAl-layered double hydroxide (PDA@MgAl-LDH) and pure PDA were 142.86 mg g−1 and 54.05 mg g−1, respectively.15 This paper confirmed the importance of high surface area and possible synergy of the different components of the nanocomposite for the effective pollutants' removal.15 Nevertheless, keeping in mind the significance of these factors, we have to remember that a substantial part of the PDA functional groups (amine or catechol) in the composite shells are involved in formation of adhesion bonds with the surface of the other component and, therefore, cannot interact with the pollutant ions/molecules. In the case of core–shell nanocomposites this adhesion effect can be decreased due to increasing the PDA shell thicknesses that supplies more binding sites for the actinide adsorption.23 Therefore, to find the best effective absorbing material we have to determine an optimal ratio of the components in a PDA based (nano)composite. To realize this, we need to know more about a pure PDA binding ability.15 In this communication we report on properties of PDA as a sorbent for actinides (U and Pu) under laboratory and real-life environmental conditions of GW and SW.
2. Experimental section
2.1. Synthesis of polydopamine
PDA was synthesized through the self-polymerization of dopamine in tris(hydroxymethyl)aminomethane (Tris)–HCl buffer (20 mM, pH ∼8.5) as described elsewhere.5 In short, dopamine hydrochloride (0.3 g, Acros) was dissolved in 60 mL of distilled water. Then previously prepared 3 M Tris–HCl water solution (0.4 mL) was added to this dopamine solution. The reaction mixture was stirred for 4 days. The final dark-brown dispersion was then purified by dialysis through a tubing cellulose membrane (D9777, MWCO = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) for 72 h against distilled water. Then PDA was separated from the dispersion by centrifugation at 3000 rpm for 15 min followed by drying at ambient conditions for 48 h and then in dynamic vacuum for 3 h. The resulting yield of PDA was around 20%.
000) for 72 h against distilled water. Then PDA was separated from the dispersion by centrifugation at 3000 rpm for 15 min followed by drying at ambient conditions for 48 h and then in dynamic vacuum for 3 h. The resulting yield of PDA was around 20%.
2.2. Preparation of the actinide solutions
Sorption properties of PDA for uranium and plutonium were studied at three different pH 2, 4 and 6.5–7.For GW extraction experiments, an artificial GW solution was prepared in accordance to the composition described elsewhere.23 To achieve required concentrations of ions, 1 L of GW was prepared by 7.5 times dilution of mineral water “Muszynianka” (Poland) followed by addition of salts: NaF (20 mg), Na2SO4 (6 mg), KCl (9 mg), (NH4)Cl (11 mg), (NH4)NO3 (8 mg). The pH was adjusted with NH3 up to 7.02. Ion concentrations were controlled by ion-chromatography and confirmed the desired composition of the GW solution. In the last step, GW was spiked with 10 mg of natural uranium (taken from a standard solution of U (2 g L−1) in nitric acid). The addition of uranium increased to some extent the concentration of nitrates above the typical value. The pH value was re-adjusted with NH3 up to 7.05. The GW was left for 48 h for stabilization.
Uranium-spiked SW (described in literature)24 was prepared by dissolving 35.1 g of Instant Ocean Salt25 in 1 L of DDI water. Uranium solution of 1 Bq mL−1 concentration was prepared by adding 0.43 mL of uranium-233 of 500 Bq mL−1 concentration in 215 mL of sea water (50 μg L−1). The pH of the solution was neutral.
To measure uptake of plutonium at different pH values, its solutions were diluted from a 6.0 × 105 Bq mL−1 stock solution in 8 M HNO3 from the Clemson University Environmental Radiochemistry Laboratory inventory. The plutonium was originally purchased as PuO2(s) from ORNL and a 10 mg aliquot dissolved in HNO3/HF with heating. The resulting solution was evaporated to dryness and reconstituted in 8 M HNO3 for purification on a TEVA resin column. The Pu solution was loaded from 8 M HNO3 and eluted in a mixture of 1 M HCl and 0.25 M HF. The elution was evaporated to incipient dryness. Small aliquots (0.5 mL) of 8 M HNO3 were added and evaporated to remove trace chloride and fluoride then the final solution was reconstituted in approximately 40 mL of 8 M HNO3. Determination of the oxidation state of the stock solution using solvent extraction indicated 92% Pu(IV), 5% Pu(V), and 3% Pu(VI). To prepare experimental working solutions, the stock solution was diluted to 25 Bq mL−1 (7.02 × 10−7 M) and adjusted to the desired pH with small volumes of HCl and NH4OH solutions.
2.3. Adsorption experiments
In a typical sorption experiment 50 mL polypropylene centrifuge tube was loaded with 20 mg of dry PDA powder and 10 mL of the aqueous actinide solution and was then shaken on a rotor for at least 48 h. After centrifuging at 6000 rpm for 15 min, 5 mL top fraction (in some experiments 1 mL) was taken out and replaced with 5 mL of the fresh UO2(NO3)2 solution of the original concentration. The extracted 5 ml (or 1 mL) solution was used for analysis and the remaining main portion with the PDA sample was put for the next 48 h extraction (with continuous mixing). A 1 mL top portion of the 5 mL eluent was filtered through 0.45 μm Teflon syringe filter (to remove dispersed particles of PDA) and added to 20 mL borosilicate glass scintillation vials with 10 mL of Ultima Gold™ AB scintillation cocktail and counted on a PerkinElmer 1220 Quantulus ultra low-level liquid scintillation spectrometer for 15 or 90 minutes (depending on the activity of the initial solution). Comparison of the count rate for eluent after adsorption with that of the original stock solution (reference sample) provided the amount of the radionuclide adsorbed by PDA. The whole procedure was repeated until saturation on the dependence of accumulated radionuclide versus mixing step has been observed.In the case of plutonium, we could not work with high concentration solutions and measured retention of 242Pu from 25 Bq mL−1 activity solutions (170 ppb) in a single step experiment.
The GW extraction experiments were run by a little different protocol and only in a single step experiment. Specifically, approximately 20 mg of PDA was mixed with 40 mL of GW solution and shaken for 48 h on the rotor. After this time, the sample was centrifuged (20 min, 6000 rpm) and filtered with standard syringe filter (0.45 μm). A blank sample taken to the experiment was only GW solution without any sorbent. Filtrates were directly measured by ion chromatography. For ICP-MS detection of 238U and Si, filtrates were diluted 66 times (0.15 mL to 10 mL). The pH values were measured before and after sorption experiment.
2.4. Characterization
Electron microscopy measurements in scanning transmission electron microscopy (STEM) mode and the elemental analysis of the PDA sample with the absorbed radionuclides energy dispersion spectroscopy-(EDS) were performed with the help of Hitachi HD-2000 microscope, 20 kV accelerating voltage. To realize these measurements PDA sample with pre-adsorbed uranium (280 mg 238U per 1 g of polymer) from sorption capacity test and specially prepared PDA with pre-adsorbed plutonium (65 μg 239Pu per 1 mg of polymer) were used for sample preparation. After sorption from 0.0125 mM (239Pu; 6890 Bq mL−1) concentration solution for one week some amount of PDA precipitate was transferred in a separate vial, dispersed in water and then deposited on copper grids covered with carbon films.Fourier transform infra-red spectra in attenuated total reflection mode (ATR-FTIR) of PDA samples without and with the absorbed radionuclides were obtained with the help of Thermo Scientific 6700 spectrometer equipped with a mercury cadmium telluride narrow band detector and Smart iTR single bounce diamond ATR crystal.
Cyclic voltammetry (CV) studies were performed via a Wave-Driver 20 Bipotentiostat/Galvanostat using a BASi VC-2 voltammetry cell (20 mL), working glassy carbon (bare and coated with the samples with and without radionuclides), Pt counter and Ag/AgCl reference electrodes, correspondingly. In short, CV measurements were performed as follows: a pre-cleaned glassy carbon (BASi MF-2012) electrode (d = 3.0 mm) was put in a vertical position, small amount of PDA (∼20 μg) with pre-adsorbed 238U (6 μg), or 239Pu (∼0.8 μg) was deposited on its clean horizontal surface from water dispersion and the electrode was left for drying for one day. Such preparation procedure insured good adhesion and electrical contact between the PDA film and glassy carbon electrode surface. Typically, 6–7 mL of electrolyte (water with [Na][ClO4] or acetonitrile with [Bu4N] [PF6] supporting electrolytes in 0.1 M concentration) were used in 20 mL glass vial under argon. The cyclic voltammograms were scanned in the potential windows from −0.6 to +1.2 V (water-based electrolyte) and from −1.8 V to +1.8 V (acetonitrile-based electrolyte) at sweeping rate 50 mV s−1 with 5 scans for each scan rate after conditioning for 2 h with 100 mV s−1 potential scanning. The acetonitrile-based electrolyte was used to visualize at high potentials redox processes of Pu adsorbed by PDA.
3. Results and discussion
3.1. The actinide moieties uptake from the model aqueous solutions
The applied sorption method of the uptake measurements involves simultaneously changes in two parameters (radionuclides concentration and time) of the system and gives the direct measurement of the maximum sorption capacity (Qmax). Specifically, in this method Qmax is not a result of a typical mathematical extrapolation of values measured from a low concentration to a high concentration but represents a real experimental fact of saturation of the uptake process after several steps of the radionuclide sorption with the same portion of the sorbent (PDA). In particular, we have found that pure PDA has really strong ability to absorb significant amount of actinides, which however strongly depends on pH of the solution. Thus, as one can see from Fig. 2 and Table 1, Qmax values for uranium and retention for plutonium sharply increase from pH 2 to pH 4 and then at pH 6–6.5 quite strongly grow in the former and very weakly grow for the latter. This tendency is well illustrated in Fig. 2 showing strong dependence of the development of the uranium uptake on pH, which in turn is predetermined by HNO3 concentration.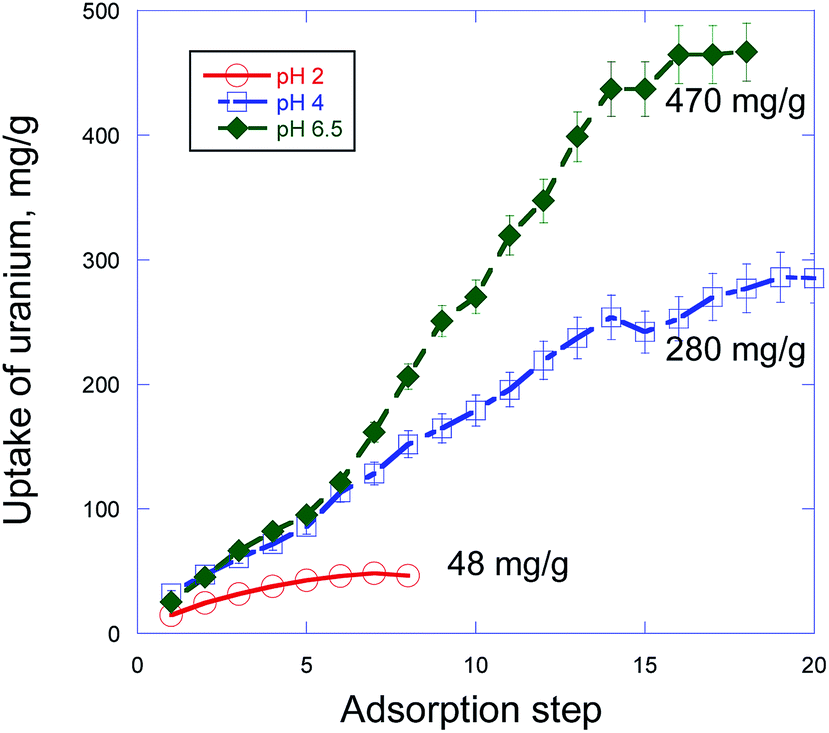 | ||
| Fig. 2 Uranium sorption capacity test by PDA from aqueous solutions via step-by-step accumulation at three different pH values. | ||
| Isotope(s) | pH | Concentration | Activity | Matrix media | Detection method | Retention in one adsorption step% |
|---|---|---|---|---|---|---|
| a Liquid scintillation counting (1220 Quantulus). b Inductively coupled plasma mass spectrometry (ICP-MS, Thermo X Series II). | ||||||
| 238U/233U | 2 | 100 ppm | 10 Bq mL−1 | DI water with HNO3 | LSCa | 70 |
| 4 | 100 ppm or 200 ppm | 10 Bq mL−1 | DI water with HNO3 | 90 | ||
| 6.5 | 100 ppm or 200 ppm | 1.24 Bq mL−1 | Mineral water | ICP-MSb | 100 | |
| 2.5 Bq mL−1 | ||||||
| 242Pu or 239Pu | 2 | 170 ppb | 25 Bq mL−1 | DI water with HNO3 | LSC and ICP-MS | 60 |
| 4 | 170 ppb | 25 Bq mL−1 | DI water with HNO3 | 99 | ||
| 6 | 170 ppb | 25 Bq mL−1 | DI water with HNO3 | 100 | ||
| 238U | 7 | 10.4 ppm | 0.124 Bq mL−1 | Spiked GW | ICP-MS | 100 |
| 233U | 7 | 40 ppb | 1.3 Bq mL−1 | Spiked SW | LSC | 100 |
| 233U | 7 | 130 ppb | 45.9 Bq mL−1 | Spiked SW | LSC | 100 |
Dependence of the radionuclides sorption on pH both for PDA containing core–shell composites15,21,22,26 and other materials27–29 is well known and is typically assigned to change of charge state of both the radionuclide bearing species and the sorbent surface. In particular, the main U(VI) species in aqueous solution were predominated by UO22+ cations at pH < 4. At higher pH around 5–8 the mixture of complex cations UO2(OH)+, (UO2)3(OH)5+ and (UO2)4(OH)7+ and neutral UO2(OH)2 species prevail.15,21 Importantly that in our case due to high carbonate content (200 ppm to 2 mM Na2CO3, see Experimental methods) at pH 6.5 in the solution there present UO2CO3(aq) and [UO2(CO3)2]2− species, which obviously also effect U(VI) adsorption on PDA.30
Inevitably, in the case of core–shell particulate sorbents with a PDA shell and of pure PDA, the adsorption of the radionuclide species will be affected by the sorbent surface charge. Indeed, it was demonstrated that at pH higher than ∼3.7 the ζ-potential of the surface of Fe3O4@PDA nanoparticles was negative that facilitated adsorption of the complex uranyl hydrated cations.26 At lower pH the surface charge was positive that suppressed adsorption of UO22+ cations26 and, obviously, of PuO22+ cations.
At first view these physico-chemical differences in all participants of the sorption processes in the PDA based system can explain dependence of the radionuclides maximum sorption capacity (Qmax) or retention on pH (Table 1). However, despite the differences, the sorption processes at pH 4 and 6.5 proceed similarly with a very close U(VI) uptake rates up to 6th step (336 h) (Fig. 2) under investigation. This result strongly differs from the known fact that adsorption of U(VI) at PDA hybrid core–shell nanocomposites is much higher at pH > 5 than that of at pH 4.15,21,22,26 In particular, in accord with Fig. 2 in ref. 26 U(VI) adsorption efficiency at core–shell Fe3O4@PDA nanocomposite for 200 h sharply increased (about triplicated) when transition from pH 4 to pH 6. The found proximity of the U(VI) adsorption behavior at pH 4 and 6.5 up to 6th step (Fig. 2) suggests independence of a quantity and origin of binding sites in PDA (at these pH) and a similar kinetics of their interaction with existing in the solutions different U(VI) species. However, after these steps the U(VI) sorption at pH 6.5 accelerates about twice (can be estimated from slopes of the curves) compared with that of at pH 4 with the next linear uptake growth up to 14th step followed by tendency to saturation (Fig. 2). Reasons of this significant change in the interaction rate at pH 6.5 are not clear yet completely and need to be studied separately. Nevertheless, taking into account the fact that under investigation at pH 6.5 there coexist different U(VI) neutral or charged species bearing one or few uranium atoms [UO2(OH)+, (UO2)3(OH)5+, (UO2)4(OH)7+, UO2(OH)2, UO2CO3(aq), [UO2(CO3)2]2−],15,21,30 as one of possible explanations we suggest different kinetics of adsorption of these species at negatively charged surface of PDA. Obviously, smaller species with one uranium atom are more movable and can diffuse/adsorb to/at the PDA surface faster than larger ones with more quantity of uranium atoms that gives slower U(VI) uptake rate in the former case. After subsequent decrease of the concentration of smaller species in the solution (even despite addition of small portion of the fresh U(VI) solution under investigation) the heavier species begin to prevail in the U(VI) uptake. This leads to a formal increase of the uptake rate (but just for account of the heavier species) which is revealed in the slope of the sorption curve at pH 6.5 after 6th step in Fig. 2. This explanation matches well with the practically similar U(VI) uptake at pH 4 up to 6th step for account obviously of adsorption of prevailing in this medium UO22+ species bearing only one uranium atom and with invariability of the uptake rate with actually linear uptake U(VI) growth up to 14th step followed by tendency to saturation.
While in the case of plutonium such sorption measurements were not performed, we found that its retention from the low concentrated solutions also was higher at pH ≥ 4 (up to 100%) and drops at lower pH (60% at pH 2) (Table 1, column 7).
From our view the low uptake of U and Pu bearing species at pH 2 can be caused not only by positive charge of PDA surface at this pH26 but probably also with oxidizing action of 0.01 M HNO3 on PDA. This suggestion is supported by FTIR spectra of PDA powders after treatment in HNO3 solutions at pH 2, 4 and 6.5 for 16 days (Fig. S1†). In particular, the PDA bands encompassing catechol O–H stretching vibrations at about 3214 cm−1 and 1286 cm−1 display a weak but quite clear irregular trend to decrease with increasing HNO3 concentration as compared with pristine PDA. This change matches well with a small simultaneous increase of the pronounced shoulder at 1720 cm−1, which encompasses carbonyl bands of both carboxylic groups and quinone units (see FTIR discussion below). All changes suggest an increase of these groups content in PDA due to oxidation of catechol hydroxyls under treatment in the HNO3 solutions.
3.2. The U(VI) moieties uptake from the GW and SW
The high pure PDA efficiency in the actinides uptake from the model water solutions was confirmed on the example of U(VI) uptake from simulated GW and SW. These media are highly important in terms of the ecological and possible commercial aspects even despite the fact that e.g. natural concentration of U in the ocean is very low (∼3.3 ppb).31,32Specifically, we have found that PDA completely removes uranium from the simulated GW with no significant retention of the background ions (Table 2). The similar high selectivity to uranium in natural SW was shown earlier for Fe3O4@PDA magnetic nanoparticles.26
It should be emphasized here that during the last two decades a number of organic and inorganic ligands have been developed for extraction of uranium from GW and SW.33–37 The reported values of sorption capacity vary depending on the composition of the ligand, pH conditions, concentration of uranium, and presence of other metallic ions. Very often comparison between the results obtained in different laboratories is hardly possible due to large number of parameters, which might influence the result. Moreover, the uptake property depends strongly on carrying the experiments under conditions of real uranium contaminated water (field experiments) versus performing such tests under model conditions in the lab. The former case gives at least 10 times lower Qmax in comparison to the latter due to extremely small concentration of the analyte (ppb range in seawater) and associated with this fact additional kinetical restrictions.37 Generally, the developed polymeric sorbents have higher sorption capacity for uranium extraction (e.g., 395 mg g−1 phosphonic acid derivatives, up to 750 mg g−1 polyamidoxime33) in comparison to inorganic analogs (e.g., 5.6 mg g−1 hematite and 97.5 mg g−1 graphene oxide34). Specially developed metal organic frameworks (MOF) modified with functional groups show very impressive values of Qmax (up to 1684 mg g−1) at pH4.35 However, the same MOF do not work so well at neutral conditions of natural water (188–278 mg g−1 at pH 8 (ref. 35)). Considered as a “golden standard” for uranium extraction from SW, polyamidoxime polymer and its derivatives demonstrate high Qmax values in laboratory tests (350, 440, and even 750 mg g−1 in synthetic uranium contaminated water33), while much less impressive values of ∼2 mg g−1, or 25.9 mg g−1 are reported for field experiments.33,36 PDA was tested mainly as a shell in polymer-inorganic composites before with the reported Qmax values of 384.6 mg g−1 (pH 7) or below.22 Based on our experiments we can conclude that previously reported Qmax for PDA were underestimated and that it is promising sorbent for uranium under natural water conditions with Qmax values approaching the best reported values for polyamidoxime and other recently developed sorbents.
Fig. 3 shows linear uptake of U from the spiked SW with the adsorption step number and fast kinetics most of the retention happens during the first 2 hours followed by slow increase due to exhaust of the U(VI) concentration. Sorption capacity was not measured in these experiments but can be assumed to be like the value in the model water solution obtained for pH 6.5 (see Fig. 2).
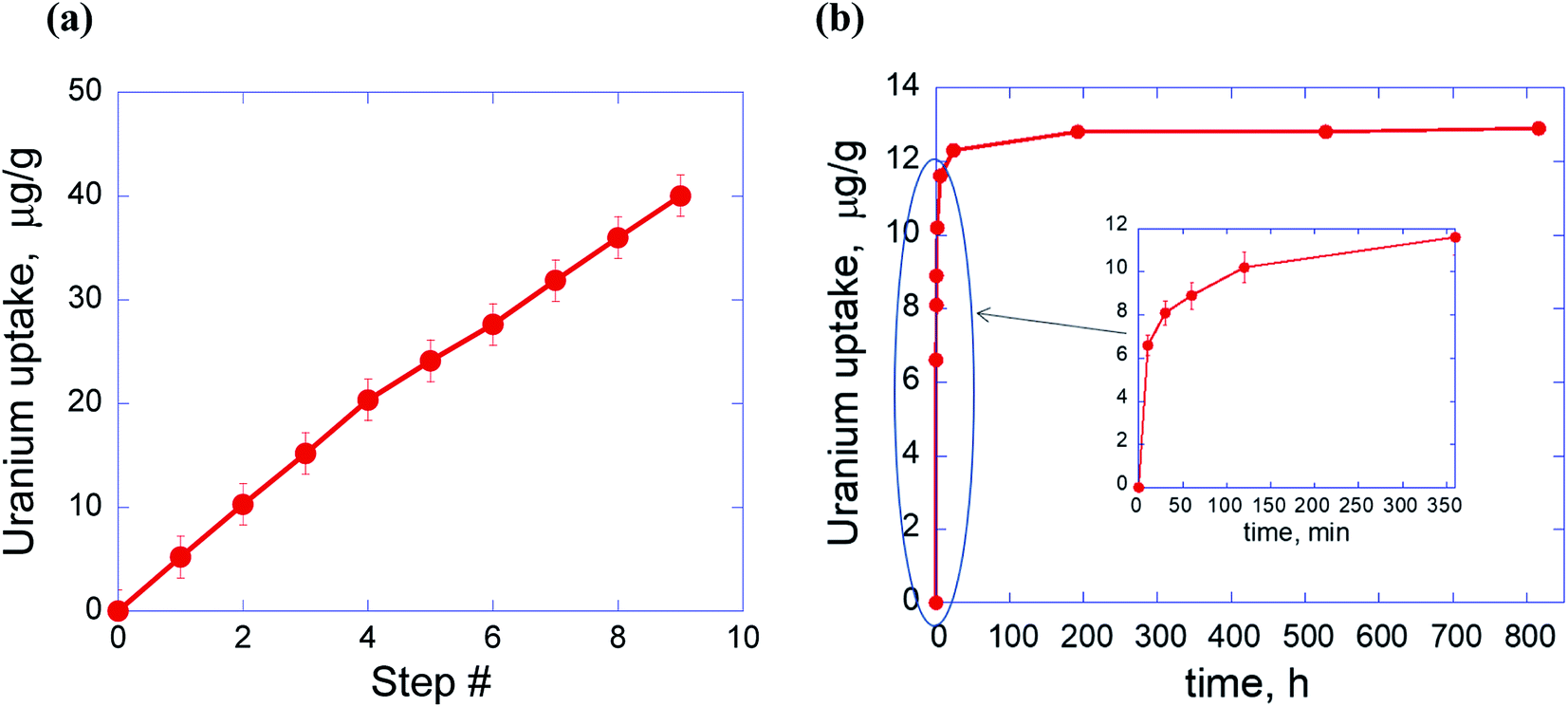 | ||
| Fig. 3 Uptake of uranium by PDA from the spiked SW as a function of the adsorption step (each step lasts for 48 h) (a) and kinetics of the adsorption during one-step procedure (b). Inset shows the first six hours of the adsorption experiment. | ||
At the low concentration limit, isotherm shown in Fig. 3a can be best described by linear or Freundlich fit. The corresponding constants are: Kd = 45565 (linear fit) and Kf = 1418; n = 0.52 (Freundlich fit) (Fig. S2†).
The kinetics of uranium sorption (Fig. 3b) is essentially nonlinear with the fast first adsorption process and significant reversed (i.e., desorption) process. Therefore, the kinetics is best described with the first order reversible reaction with the first rapid uptake step approximated as being instantaneous (Fig. S3†). This sorption behavior can be obviously explained from positions of the coexistence and adsorption at neutral pH (see above discussion of Fig. 2) of uranyl bearing species in monomer, dimer or multimer forms,15,21,27 which differ, therefore, by molecular weight and size. In line with this the rapid uptake stage begins from very fast adsorption of uranium carbonate or uranium hydroxide monomers (as more mobile species) followed by adsorption of corresponding dimers and multimers.
3.3. Further characterization of the radionuclides uptake on the PDA sorbent
 | ||
| Fig. 4 (A and C) STEM image of agglomerates formed by PDA particles with 239Pu containing nano-clusters (brighter color due to Z-contrast) formed during extraction of Pu based moieties from the aqueous solution at pH 7 (A) and 238U radionuclides uniformly distributed on the PDA surface (C); (B and D) EDS maps showing distribution of Pu (B) and U (D) radioisotopes the same areas as shown in (A) and (C) correspondingly. | ||
Fig. 5 shows the FTIR spectra of PDA and the PDA samples are completely saturated with the adsorbed U or Pu bearing moieties separated at the end of the sorption processes from the model aqueous solutions at pH 4 and pH 7, respectively.
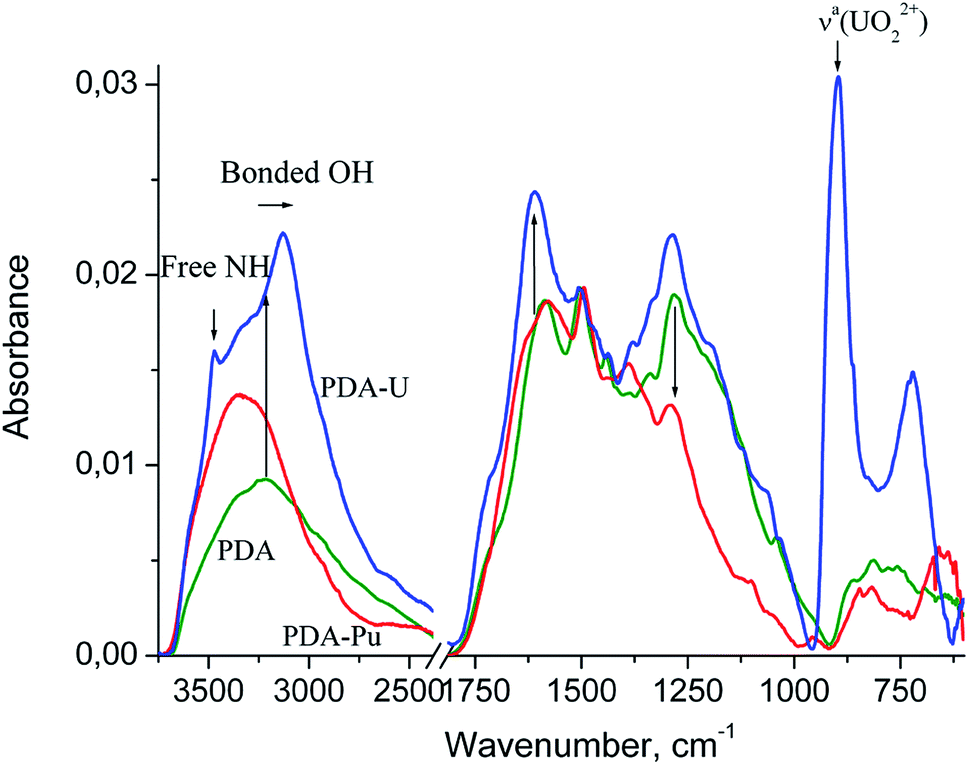 | ||
| Fig. 5 ATR-FTIR spectra of PDA samples with U or Pu extracted from aqueous solution at pH 4 and pH 7 respectively. The spectra are normalized at the maximum of the strongest band of PDA at around 1500 cm−1. The arrows indicate changes in the intensity and position of PDA bands after accumulation of radionuclides. | ||
The structure of the PDA spectrum is very similar to that of parent dopamine hydrochloride (see ref. 38, SDBS no: 1654), and differs from that mainly in the broadening and merging of narrow bands. One can see a broad medium band at about 3214 cm−1 (at 3143–3371 cm−1 in DA), which can be assigned to hydrogen bonded O–H stretching vibrations,39–42 1588 cm−1 (1579, 1600 and 1612 cm−1 assigned to aromatic νaC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and δNH2), 1503 cm−1 (1471, 1499 and 1519 cm−1, aromatic νsCC), NH vib. in DHIN (see ref. 38, SDBS no: 1358 and 717; ref. 39–43), 1443 cm−1 (1471 cm−1, δCH2 vib. in DA and DHIN), 1338 cm−1 (1341 and 1320 cm−1, δaC–O–H) and 1282 cm−1 (1286, 1260, 1190, 1176, 1161 and 1081 cm−1, δsC–O–H, νC–O and νC–C, νaC–C–H. and νsC–C–H).
C and δNH2), 1503 cm−1 (1471, 1499 and 1519 cm−1, aromatic νsCC), NH vib. in DHIN (see ref. 38, SDBS no: 1358 and 717; ref. 39–43), 1443 cm−1 (1471 cm−1, δCH2 vib. in DA and DHIN), 1338 cm−1 (1341 and 1320 cm−1, δaC–O–H) and 1282 cm−1 (1286, 1260, 1190, 1176, 1161 and 1081 cm−1, δsC–O–H, νC–O and νC–C, νaC–C–H. and νsC–C–H).
Strong band at about 1588 cm−1 in PDA is spreading to higher wavenumbers up to 1800 cm−1 and exhibits a shoulder at 1720 cm−1, which is characteristic of CO stretching vibration for carboxylic acid.39,40 Their appearance confirms the presence of pyrrolecarboxylic acid units. The above-mentioned broadening of the band is consistent with possible contribution from quinone units (o-quinones exhibit carbonyl band at about 1660 cm−1).39
The most intensive band in the PDA-U spectrum (not observed in case of pure PDA) can be seen at 896 cm−1. This band can be ascribed to uranyl complex. The characteristic absorption frequency of the asymmetric vibration (ν3) of free UO22+ is shifted to lower wavenumbers from the free ion value of 965 ± 1 cm−1.44 This shift indicates coordination of the uranyl with strongly donating ligand group.45 Moreover, the band at 896 cm−1 shows a twofold enhancement of the full width at half maximum compared to the band of free UO22+.44 This can indicate variation of the existing ligand structures and stoichiometries of the complexes.
The low-frequency band at 721 cm−1 can be attributed to aquo (H2O) complexes of the uranyl cation. Except for the three fundamental modes of the free water molecules (νaOH and νsOH vib. at 3550–3200 cm−1 and δHOH at 1630–1600 cm−1), coordinated water can exhibit some libration modes in low-frequency region 1000–200 cm−1.46,47 The frequencies of the libration modes vary largely and depend on the bond length and orientation of the metal to water oxygen bond relative to the water molecule, the symmetry and nature of hydrogen bonds and some other geometrical factors.47 For example, the bands of medium intensity at 677, 520, 463 and 421 cm−1 were ascribed to water libration and water twist in IR spectrum of the natural uranium carbonate andersonite.48 The appearance of shoulders around 1122, 1063 and 1035 cm−1 in the spectrum of PDA-U and 1101 cm−1 in the spectrum of PDA-Pu may indicate the formation of hydroxyl complexes, which exhibit the metal–OH bending mode in the region 1200–700 cm−1.39,46,49
In comparison to pristine PDA, PDA-U sample demonstrates a twofold enhancement of the band at 3214 cm−1 and its red shift to 3130 cm−1. The band at 1588 cm−1 shifts to 1610 cm−1 and increases distinctly in the region of CO stretching vibrations of the carboxylic acid groups and the quinone units. The appearance the band at 1380 cm−1 can be assigned to νsCO vibrations of the ionized carboxyl group, which is absent in the spectrum of the unionized carboxylic acids.39,40 In addition, the intensity of the broad band at 1282 cm−1 is enhanced mainly in the region of antisymmetric and symmetric O–H deformation vibrations at about 1321 and 1261 cm−1. Such changes in O–H, CO bands can be induced by a change in the charge distribution on the oxygen atom when the coordinative bond formed between an oxygen donor and the uranium atom.
The PDA-U spectrum also shows sharp band at 3472 cm−1, which can be assigned to free N–H stretching vibrations of DHI blocks as the indole and indoline NH groups show accordingly these vibrations at 3493 and 3376 cm−1 (see ref. 38, SDBS no: 717 and 1358). It is very unlikely that this band corresponds to νNH2 and νOH vibrations of DA and various catecholamine moieties. Indeed, primary amines have two bands of the N–H asymmetric and symmetric vibrations at lower wavenumbers39,40 while OH groups involved in the hydrogen and coordinative bonding are revealed at lower absorption frequencies. Therefore, the NH groups of indole have less significant role in uranium uptake and release from initial hydrogen bonding in PDA. This reveals some different mechanism of chelation between PDA moieties and uranyl in comparison to polyamidoxime case, where simultaneous interaction of UO22+ with amino groups and hydroxyls of the polymer was found to be responsible for coordination of the latter.37,50 Nevertheless, we believe that some influence of the amine groups in coordination of uranyl cannot be completely excluded.
For the sake of convenience, the repeating unit of PDA can be simplified to just 5,6-dihydroxyindole (DHI) unit. In this case, compositions of PDA-U and PDA-Pu samples can be described with following ratios:
| U:DHI = 1:5.66 (mol) and Pu:DHI = 1:24.3 (mol) |
Importantly, PDA-U sample prepared at pH = 6.5 contains 1.7 times more of uranium in comparison to the one that obtained at pH = 4 (see Table 1). Therefore, formation of polynuclear uranyl complexes, which follows from the above-discussed adsorption measurements, is quite likely. Considering linear geometry of uranyl O–U–O, we can propose the following possible structures of the uranyl complexes with equatorial coordination of ligands:
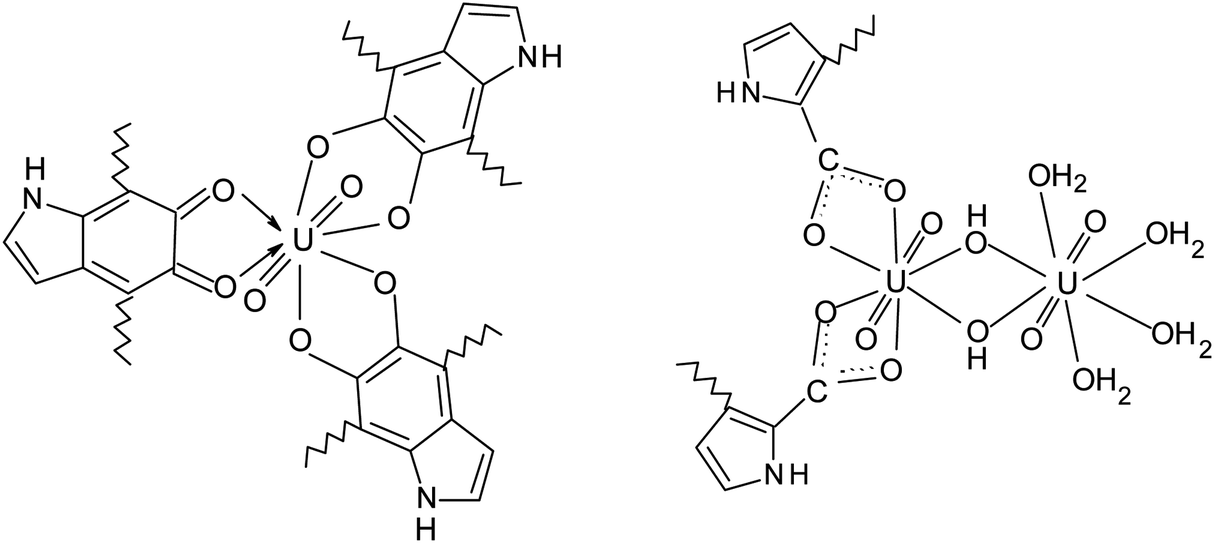
The uranyl ions can form some complexes with carbonate anions in the solutions. However, the complexity of the IR spectra (Fig. 5) does not allow to identify the peaks associated with these complexes.
In the PDA-Pu spectrum, the weak band at 957 cm−1 can be ascribed to ν3 of PuO22+ because the asymmetric vibration band of free ion is located at 962 cm−1.44 Corresponding experimental PuO2+ frequency is still unknown, although some calculations were performed.51,52 However, considering the functional dependence of the MO force constant on the atomic number of a transuranium element, we can assume with a high probability that ν3 of PuO2+ should be observed at lower wavenumber (close to ν3 of NpO2+ at 824 cm−1).44 The low intensity of the band at 817 cm−1 (PDA-Pu spectrum) and close proximity to the band 812 cm−1 (PDA spectrum) indicate absence or low content of Pu(V). As seen in Fig. 5, the intensity ratio of the bands I(ν3 of UO22+)/I(ν3 of PuO22+) is approximately 49, whereas the ratio [U]/[Pu] is equal to 4.3. Taking into account the comparability of the band intensities of uranyl and plutonyl for solutions with similar ion concentrations,44 it can be concluded that plutonium accumulated basically in PDA in more reduced form Pu(IV) or Pu(III).
PDA-Pu sample is also characterized by lower intensity of the band at ∼1282 cm−1, which is, to a large extent, related with δOH and νC–O vibrations. This change is obviously due to the partial oxidation of the catechol OH groups. This is evidenced by an appreciable increase of shoulder at about 1630 cm−1, where the quinones moieties absorb. One of the possible reactions is shown below, where dihydroxyindole represents catecholamine moieties of the PDA for simplicity:

The disappearance of the CH2 scissor vibrations (1443 cm−1) in PDA-Pu IR spectrum also provides clear evidences for the oxidation of the DA and DHIN blocks to give in the latter case DHI block. The concert of such oxidation reactions is consistent with the drop of the absorption intensity in the region of 3070–2450 cm−1, where stretching vibrations of the strongly bounded OH groups and C–H bonds typically observed.
 | ||
| Fig. 6 Representative cyclic voltammograms recorded for PDA with pre-adsorbed U or Pu bearing species and pure PDA placed on the GC electrode. Background electrolytes: 0.1 M NaClO4 in water electrolyte (in case of PDA-U) and 0.1 M Bu4NPF6 in acetonitrile electrolyte (in case of PDA-Pu and PDA). CV curves for PDA and PDA-U are shifted vertically for clarity. | ||
The observed positions and quantity of the redox couples differ from the previously reported ones for U or Pu in literature for bare GC electrode and various conditions.53–55 In particular, uranyl(VI) nitrate in conditions close to those of this study showed only one redox couple of U(VI)/U(V) in cathodic region at −0.01(ox) V/−0.25(red) V (vs. Ag/AgCl).53 A quite similar behavior with one redox couple of U(VI)/U(V) at −0.075/−0.2 V (vs. Ag/AgCl) was found for uranyl(VI) perchlorate on Pt electrode in aqueous solution of 0.1 M LiClO4.56 However, in the more acidic medium (0.1 M HClO4) there were observed on GC electrode both a quasi-reversible redox couple U(VI)/U(V) at −0.09/−0.2 V (vs. Ag/AgCl) and the irreversible anodic peak at 0.27 V assigned to oxidation of U(IV) to U(VI).57
In general, both uranium and plutonium electrochemistry on bare electrodes depends on pH and composition of the background solution.58 Specifically, in strong acidic media Pu multiple redox transitions Pu(VI)/Pu(V), Pu(VI)/Pu(IV), Pu(V)/Pu(IV), Pu(IV)/Pu(III) overlapping in a quite narrow potential range (with formal potentials 0.9–1.2 V vs. saturated hydrogen electrode or about 0.68–0.98 V vs. different kinds of Ag/AgCl electrodes).58–61 This overlapping can lead to the fact that on cyclic voltammograms the Pu species give wide or poorly shaped redox responses. In neutral media these processes shift to lower potentials.58,60
The situation is quite different when the uranium or plutonium electrochemical processes run on the electrode surface covered by an adsorbent layer (e.g. graphene oxide (GO) or conducting polymer PEDOT-PSS) with or without pre-adsorbed radionuclide species.53–55 In particular, in the case of GO layer with the pre-adsorbed uranyl deposited on GC electrode the position of U(VI)/U(V) redox couple shifts to anodic region by around 205–250 mV in comparison to bare GC.53 Moreover, the shape of the peaks became more complicated and additional weakly pronounced redox couple at 0.21 V/0.13 V appeared that suggested both change of valence state of U and altering the electronic configuration of uranyl(VI) cations due to their coordination with functional groups on the GO surface.53 Unlike the bare GC electrode case, in 1 M H2SO4, U(VI) (UO22+) shows on the PEDOT-PSS/GC electrode a well-defined cathodic peak at −0.158 V revealing its reduction to U(IV) through the ec (electron transfer-chemical step) stage including formation of U(V) (UO2+) and its very fast disproportionation into UO22+ and U(IV).55
While to our knowledge there is no published information on electrochemistry of highest valence state of Pu on adsorbent coated electrodes, it is found that the GO or PEDOT:PSS layers on GC showed enhanced electrocatalytic activity towards Pu(IV)/Pu(III) redox couple and significantly decreased the cathodic and anodic peak potentials in acidic media (1 M H2SO4).52,54,55 In particular, due to the GO influence this redox couple shifted from 0.666 V/0.092 V to 0.605 V/0.398 V (ref. 54) while due to the PEDOT:PSS influence the couple shifted from 0.674 V/0.132 V on the bare GC electrode to 0.540 V/0.496 V.62
If compare the known electrochemical transitions of U and Pu bearing species in different valence states53–62 on bare and coated electrodes with positions of the redox couples of the PDA-U and PDA-Pu samples (Fig. 6) one can see that in the case of PDA the shifts of these transitions along the potential axis and, therefore, specific interactions of PDA with the actinides species are significantly stronger than in the cases of GO or PEDOT:PSS. This electrochemical specificity of pure PDA in interactions with the radionuclides matches well with the discussed above high adsorption ability of PDA and formation of uranium and plutonium complexes. Interaction with PDA and resulting oxidation of PDA moieties by actinides in highest oxidation states (VI) causing appearance of reduced forms of the actinides in PDA. Such redox processes lead obviously to the existence in the PDA-U and PDA-Pu samples of U and Pu in different valence states (VI, V, IV, III), which are revealed in the poorly resolved three redox couples encompassing in fact the redox transitions between these states in their cyclic voltammograms. An additional input in shape and quantity of these couples can be probably made by involvement in the electrochemical reactions of the actinide's complexes with some PDA moieties (e.g. hydroxyl or amino groups). However, to more precisely identify these redox couples a detailed characterization in a separate electrochemical work is needed.
4. Conclusions
As a result of our study the pure PDA polymer has been identified as a material with extremely high sorption capacity that is capable of concentrating soluble uranium and plutonium bearing species from GW and SW at weakly acidic and near-neutral pH. Its binding ability for uranium is realized probably due to formation of uranyl complexes with catechol- and amino- and other functional groups. This effect is synergistically enhanced especially at neutral pH by involvement in such complexation process of uranyl bearing species in monomer, dimer or multimer forms.In particular, the uptake of uranium bearing species starts as a rapid uptake step on the order of minutes followed by a slow equilibration that can be approximated as a first-order reversible reaction. The strong interaction between U(VI) and PDA was demonstrated by the high sorption capacity 480 mgU/gPDA, even in complex media such as e.g. SW. The high uptake of plutonium bearing species can be explained not only by the same reasons but also through the partial oxidation of the catechol OH groups by Pu(V/VI) accompanied with appearance of Pu(IV) bearing species.
The results of this study strongly confirm that the pure PDA polymer has a significant potential to be used for removal of the radionuclides from different contaminated waters (e.g. GW, seawater), for the mining of uranium from SW and moreover for development of new radioanalytical methods. Nevertheless, the known variability of the PDA composition suggest that further characterization is still necessary to assess its full potential.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Ms. Amruta Pujari for her help with uranium uptake experiments. This work was partly funded under a NATO Science for Peace and Security Program, G5094 grant entitled: Reliable nuclear materials identification technology from spectrometry data. Research in this manuscript dealing with plutonium was funded by the Office of Basic Energy Sciences of the U.S. Department of Energy as part of the Heavy Element Chemistry Program (DE-SC0010355) and by the Department of Energy National Nuclear Security Administration Stewardship Science Academic Alliances (#DE-NA-0002920). Dr Kamila Kołacińska would like to acknowledge her support from the Fulbright Scholars Program.References
- M. Deng, H. Zhao, S. Zhang, C. Tian, D. Zhang, P. Du, C. Liu, H. Cao and H. Li, J. Mol. Catal. B: Enzym., 1970, 112, 15–24 CrossRef.
- M. F. Mineyeva, D. Tummler, E. A. Kuznetsova, A. E. Vasiliev and K. S. Rayevsky, Ann. Ist. Super. Sanita, 1982, 18, 45–48 CAS.
- E. Herlinger, R. F. Jameson and W. Linert, J. Chem. Soc., Perkin Trans. 2, 1995, 259–263 RSC.
- K. Liu, W. Z. Wei, J.-X. Zeng, X.-Y. Liu and Y.-P. Gao, Anal. Bioanal. Chem., 2006, 385, 724–729 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 3180, 426–430 CrossRef.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS.
- M. d'Ischia, A. Napolitano, V. Ball, C.-T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS.
- J. Park, T. F. Brust, H. J. Lee, S. C. Lee, V. J. Watts and Y. Yeo, ACS Nano, 2014, 8, 3347–3356 CrossRef CAS PubMed.
- R. Luo, L. Tang, J. Wang, Y. Zhao, Q. Tu, Y. Weng, R. Shen and N. Huang, Colloids Surf., B, 2013, 106, 66–73 CrossRef CAS.
- M. Lee, J. Rho, D. E. Lee, S. Hong, S. J. Choi, P. B. Messersmith and H. Lee, ChemPlusChem, 2012, 77, 987–990 CrossRef CAS.
- Z. Iqbal, S. Alsudir, M. Miah and E. P. C. Lai, Electrophoresis, 2011, 32, 2181–2187 CrossRef CAS.
- K. Zhu, S. Lu, Y. Gao, R. Zhang, X. Tan and C. Changlun, Appl. Surf. Sci., 2017, 396, 1726–1735 CrossRef CAS.
- N. Farnad, K. Farhadi and N. H. Voelcker, Water, Air, Soil Pollut., 2012, 223, 3535–3544 CrossRef CAS.
- C. Lim, J. Huang, S. Kim, H. Lee, H. Zeng and D. S. Hwang, Angew. Chem., Int. Ed., 2016, 55, 3342–3346 CrossRef CAS.
- X. Zhang, X. Jia, G. Zhang, J. Hu, W. Sheng, Z. Ma, J. Lu and Z. Liu, Appl. Surf. Sci., 2014, 314, 166–173 CrossRef CAS.
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CrossRef CAS.
- C. M. Hui, J. Pietrasik, M. Schmitt, C. Mahoney, J. Choi, M. R. Bockstaller and K. Matyjaszewski, Chem. Mater., 2014, 26, 745–762 CrossRef CAS.
- L. Bai, S. Duan, W. Jiang, M. Liu, S. Wang, M. Sang, X. Gong, J. Li and S. Xuan, Appl. Surf. Sci., 2017, 426, 1121–1132 CrossRef CAS.
- J. Zhu, Q. Liu, J. Liu, R. Chen, H. Zhang, M. Zhang, P. Liu, R. Li and J. Wang, J. Taiwan Inst. Chem. Eng., 2018, 91, 266–273 CrossRef CAS.
- D. Langmuir, Aqueous Environmental, Geochemistry Prentice Hall, NJ, 1997 Search PubMed.
- J. Y. Zeng, H. Zhang, Y. Sui, N. Hu, D. Ding, F. Wang, J. H. Xue and Y. D. Wang, Ind. Eng. Chem. Res., 2017, 56, 5021–5032 CrossRef CAS.
- J. Bidwell and S. Spotte, Artificial Seawaters: Formulas and Methods, Jones and Barlett, Boston, 1985 Search PubMed.
- F. Wu, N. Pu, G. Ye, T. Sun, Z. Wang, Y. Song, W. Wang, X. Huo, Y. Lu and J. Chen, Environ. Sci. Technol., 2017, 51, 4606–4614 CrossRef CAS.
- W. Dong and J. Wan, Environ. Sci. Technol., 2014, 48, 6569–6577 CrossRef CAS.
- B. A. Powell, A. B. Kersting, M. Zavarin and P. Zhao, Development of a Composite Non-Electrostatic Surface Complexation Model Describing Plutonium Sorption to Aluminosilicates, U.S. Department of Energy by Lawrence Livermore National Laboratory, report under contract DE-AC52-07NA27344, 2011, DOI:10.2172/1019063.
- A. Zhang, T. Asakura and G. Uchiyama, React. Funct. Polym., 2003, 57, 67–76 CrossRef CAS.
- T. Wen, X. Wang, J. Wang, Z. Chen, J. Li, J. Hu, T. Hayat, A. Alsaedi, B. Grambow and X. Wang, Inorg. Chem. Front., 2016, 3, 1227–1235 RSC.
- H. J. Schenk, L. Astheimer, E. G. Witte and K. Schwochau, Sep. Sci. Technol., 1982, 17, 1293–1308 CrossRef CAS.
- F. Wang, H. Li, Q. Liu, Z. Li, R. Li, H. Zhang, L. Liu, G. A. Emelchenko and J. Wang, Sci. Rep., 2016, 6, 19367 CrossRef CAS.
- M. M. Aly and M. F. Hamza, J. Dispersion Sci. Technol., 2013, 34, 182–213 CrossRef CAS.
- Y. Zou, P. Wang, W. Yao, X. Wang, Y. Liu, D. Yang, L. Wang, J. Hou, A. Alsaedi, T. Hayat and X. Wang, Chem. Eng. J., 2017, 330, 573–584 CrossRef CAS.
- W. Yang, Q. Pan, S. Song and H. Zhang, Inorg. Chem. Front., 2019, 6, 1924–1937 RSC.
- L. Chen, Z. Bai, L. Zhu, L. Zhang, Y. Cai, Y. Li, W. Liu, Y. Wang, L. Chen, J. Diwu, J. Wang, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32446–32451 CrossRef CAS.
- B. F. Parker, Z. Zhang, L. Rao and J. Arnold, Dalton Trans., 2018, 47, 639–644 RSC.
- Spectral Database for Organic Compounds SDBS, National Institute of Advanced Industrial Science and Technology (AIST), Japan [Online], https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi Search PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, Chichester, 2001 Search PubMed.
- L. J. Bellamy, The infra-red spectra of complex molecules, Chapman and Hall, London, 1975 Search PubMed.
- S. Gunasekaran, R. T. Kumar and S. Ponnusamy, Indian J. Pure Appl. Phys., 2007, 45, 884–892 CAS.
- F. Yu, S. Chen, Y. Chen, H. Li, L. Yang, Y. Chen and Y. Yin, J. Mol. Struct., 2010, 982, 152–161 CrossRef CAS.
- D. G. O'Sullivan, J. Chem. Soc., 1960, 3278–3284 RSC.
- L. H. Jones and R. A. Penneman, J. Chem. Phys., 1953, 21, 542–544 CrossRef CAS.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, Chichester, Brisbane, Toronto, Singapore, 1986 Search PubMed.
- V. P. Tayal, B. K. Srivastava, D. P. Khandelwal and H. D. Bist, Appl. Spectrosc. Rev., 1980, 16, 43–134 CrossRef CAS.
- N. Kalashnyk, D. L. Perry, F. Massuyeau and E. Faulques, J. Phys. Chem. C, 2018, 122, 7410–7420 CrossRef CAS.
- H. P. Emerson and B. A. Powell, Radiochim. Acta, 2015, 103, 553–563 CAS.
- D. S. Bolotin, N. A. Bokach and V. Yu. Kukushkin, Coord. Chem. Rev., 2016, 313, 62–93 CrossRef CAS.
- J. Hay, R. L. Martin and G. Schreckenbach, J. Phys. Chem. A, 2000, 104, 6259–6270 CrossRef.
- G. A. Shamov and G. Schreckenbach, J. Phys. Chem. A, 2005, 109, 10961–10974 CrossRef CAS.
- V. N. Bliznyuk, N. A. Conroy, Y. Xie, R. Podila, A. M. Rao and B. A. Powell, Phys. Chem. Chem. Phys., 2018, 20, 1752–1760 RSC.
- S. K. Guin, A. S. Ambolikar and J. V. Kamat, RSC Adv., 2015, 5, 59437–59446 RSC.
- R. Agarwal, M. K. Sharma, K. Jayachandran, J. S. Gamare, D. M. Noronha and K. V. Lohithakshan, Anal. Chem., 2018, 90, 10187–10195 CrossRef CAS.
- K. Mizuguchi, Y.-Y. Park and H. Tomiyasu, J. Nucl. Sci. Technol., 1993, 30, 542–548 CrossRef CAS.
- Y. Suzuki, T. Nankawa, T. Ozaki, T. Ohnuki, A. J. Francis, Y. Enokida and I. Yamamoto, J. Nucl. Sci. Technol., 2007, 44, 1227–1232 CrossRef CAS.
- D. L. Clark, S. S. Hecker, G. D. Jarvinen and M. P. Neu, in The Chemistry of the Actinides and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, 2006, pp. 813–1264 Search PubMed.
- S. Kihara, Z. Yoshida, H. Aoyagi, K. Maeda, O. Shirai, Y. Kitatsuji and Y. Yoshida, Pure Appl. Chem., 1999, 71, 1771–1807 CAS.
- B. Allard, H. Kipatsi, J. O. Liljenzin and J. Inorg, Nucl. Chem., 1980, 42, 1015–1027 CrossRef CAS.
- A. Fallet, N. Larabi-Gruet, S. Jakab-Costenoble and P. Moisy, J. Radioanal. Nucl. Chem., 2016, 308, 587–598 CrossRef CAS.
- R. Agarwal and M. K. Sharma, Electrochim. Acta, 2017, 224, 496–502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI file contains additional figures showing variation of the PDA structure under different pH (FTIR spectra), analysis of uranium sorption from seawater (kinetics and adsorption isotherm fits with different models), morphology of pure PDA from STEM, and EDX spectra confirming accumulation of U and Pu elements in the PDA matrix from aqueous environments. See DOI: 10.1039/c9ra06392g |
| This journal is © The Royal Society of Chemistry 2019 |