
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blue light photoredox-catalysed acetalation of alkynyl bromides†
Xue-Li Lyua,
Shi-Sheng Huanga,
Hong-Jian Song*a,
Yu-Xiu Liu *a and
Qing-Min Wang*ab
*a and
Qing-Min Wang*ab
aState Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wangqm@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, People's Republic of China
First published on 6th November 2019
Abstract
Herein, we report an organo-photoredox-based protocol using 2,2-diethoxyacetic acid as the acetal source to achieve acetalation of alkynyl bromides to afford various alkynyl acetal products. In addition to arylethynyl bromides, substrates bearing heteroaryl rings (thiophene, pyridine, and indole) smoothly gave the corresponding acetalation products. This mild protocol has potential utility for the synthesis of aldehydes by further protonization.
Acetals play a key role in several biological interactions,1,2 and they are widespread in natural products obtained from various sources, including insects, marine organisms, fungi, plants, and microbes.2 In addition, reports of acetal-containing drugs have recently appeared in the literature,3 and acetals are frequently used to protect carbonyl groups during complex synthesis4 (Fig. 1). Therefore, the development of new methods for the introduction of acetal moieties is highly desirable, and environmentally friendly methods are of particular interest.
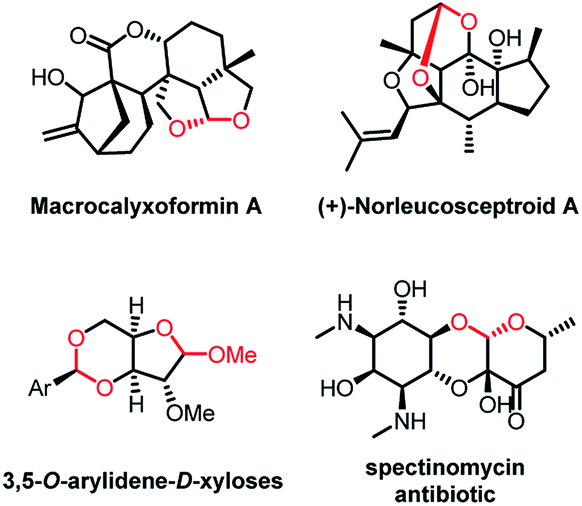 | ||
| Fig. 1 Representative compounds containing acetal moieties. | ||
In light of the growing demand for environmentally benign synthetic methods, visible-light-induced catalysis has recently garnered substantial attention in organic synthesis owing to its mild conditions and low energy requirements.5 For example, photoredox-catalyzed decarboxylation of alkyl carboxylic acids has become an important method for generating alkyl radicals.6 During decarboxylation of carboxylic acids and their derivatives under photoredox conditions, a C-centered radical can be generated via two complementary pathways: (a) a reductive quenching process in which the carboxylic acid transfers an electron to the excited-state photocatalyst to generate a C-centered radical after CO2 extrusion7 and (b) an oxidative quenching process in which carboxylic acid derivatives such as N-(acyloxy)phthalimides accept an electron from the excited-state photocatalyst to form a C-centered radical after extrusion of CO2 and a phthalimide anion (Scheme 1).8 This photoredox-catalyzed radical decarboxylative functionalization offers a novel and efficient method for constructing C–C and C–X bonds.9 Recently, Xu and co-workers10 developed a protocol for iridium-photoredox-catalyzed decarboxylative conjugated addition reactions between glyoxylic acid acetals and Michael acceptors; theirs was the first report of the introduction of acetal moieties via oxidative decarboxylation.
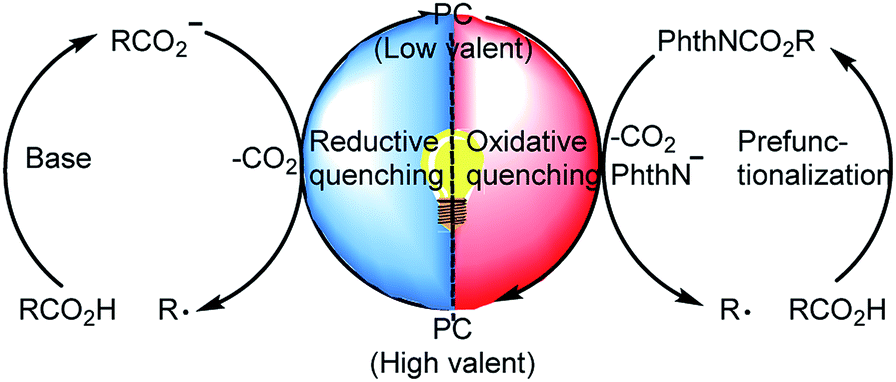 | ||
| Scheme 1 Decarboxylation of alkyl carboxylic acids and their derivatives via photoredox catalysis. | ||
Herein, we report an organo-photoredox-based protocol for acetalation of alkynyl bromides, using 2,2-diethoxyacetic acid as the acetal source and 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) as the photocatalyst. Aldehydes are reactive and therefore need to be protected against oxidation and reduction, and our operationally simple protocol provides direct access to protected aldehydes without producing large amounts of chemical waste. Considering its mild, metal-free conditions, the protocol has a considerable potential utility for the synthesis of alkynyl acetals.
We carried out reactions using alkynyl bromide 1g and 2,2-diethoxyacetic acid 2 as model reactants to optimize the conditions. When using 4CzIPN as a photocatalyst, irradiation of the substrates with 5 W blue LEDs at room temperature in the presence of Cs2CO3 under argon for 24 h, we delighted to obtain the desired product 3g in 57% yield (see the ESI† for details). Next, we varied the reaction parameters with the goal of optimizing the yield. Through control experiment, we found 4CzIPN, a blue LEDs, Cs2CO3, argon and Br as the leaving group were all indispensable. Even though benziodoxole-substituted alkynes are known to act as electrophilic alkyne acceptors,11 we found that an alkyne in which the Br atom was replaced with a benziodoxole moiety did not undergo addition of the acetal radical. By screening other photocatalysts, we found that 4CzIPN, which is a carbazole-based sensitizer reported by Zhang12a and Adachi,12b was optimal. The high reduction potential of photoexcited 4CzIPN (Ered [PC*/PC˙−] = +1.35 V vs. SCE)12a makes it suitable for promoting photo-oxidative decarboxylation reactions. Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 (Ered [IrIII*/IrII] = +1.21 V vs. SCE) also catalyzed the reaction, although the yield was substantially lower than that with 4CzIPN. Two other metallaphotoredox catalysts, Ru(bpy)3Cl2·6H2O and Ru(bpy)3(PF6)2, were screened and found to be ineffective, perhaps because the low oxidation potentials (Ered [RuII*/RuI] = +0.77 V vs. SCE) of their excited states prevented them from promoting the decarboxylation reaction. Reaction in MeCN alone or DMF alone gave a lower yield than reaction in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) MeCN/DMF. Finally, Cs2CO3 was a better base than Na2CO3 or K2CO3.
:1 (v/v) MeCN/DMF. Finally, Cs2CO3 was a better base than Na2CO3 or K2CO3.
Using the optimal conditions, we explored the substrate scope of the reaction with respect to the alkynyl bromide and found that a broad range of functional groups were tolerated (Scheme 2). Specifically, substrates with electron-donating groups on the aromatic ring of the alkynyl bromide gave moderate yields (34–64%) of the expected products (3a–3k) upon reaction with 2. For example, reactions of phenylacetylene bromides with Me, Et, nPr, nBu, tBu, and Ph groups at the para position of the benzene ring afforded 3b–3g, respectively. Some heteroatom-containing electron-donating groups were also explored. In addition to a methoxy group (3h), a 3,4-methylenedioxy group, a 3,4-ethylenedioxy group were acceptable, and acetals 3i and 3j, respectively, were obtained in 43% yield. A phenylacetylene bromide with an amide-substituted benzene ring also reacted smoothly, giving a 49% yield of 3k. Next we examined compounds with halogen-substituted benzene rings. Phenylacetylenes with a para F or Br atom, could give 3m and 3q in 44% and 49% yields, respectively. 2,2-Diethoxyacetic acid also reacted with ortho-, meta-, and para-Cl-substituted phenylacetylene bromides to afford desired products 3n–3p in 48–55% yields. Substrates with strongly electron-withdrawing CF3 (3r, 42%) and CN (3s, 43% and 3t, 55%) substituents on the benzene ring were satisfactory as well, and the location of the CN group slightly affected the yield: the ortho-substituted compound gave a higher yield (55%) than the para-substituted compound (43%). Carbonyl substituents were also tolerated: ketone- and ester-bearing phenylacetylene bromides yielded the corresponding products (3u and 3v) in 30–45% yields. We also found that the presence of multiple Cl atoms on the benzene ring did not affect the yield; 3w was obtained in 40% yield. We also explored the reactivity of substrates in which the phenyl ring had been replaced by a heterocycle and found that electron-rich thiophene (3x, 34%) and indole (3z, 40%) rings and an electron-deficient aromatic pyridine ring (3y, 56%) were tolerated.
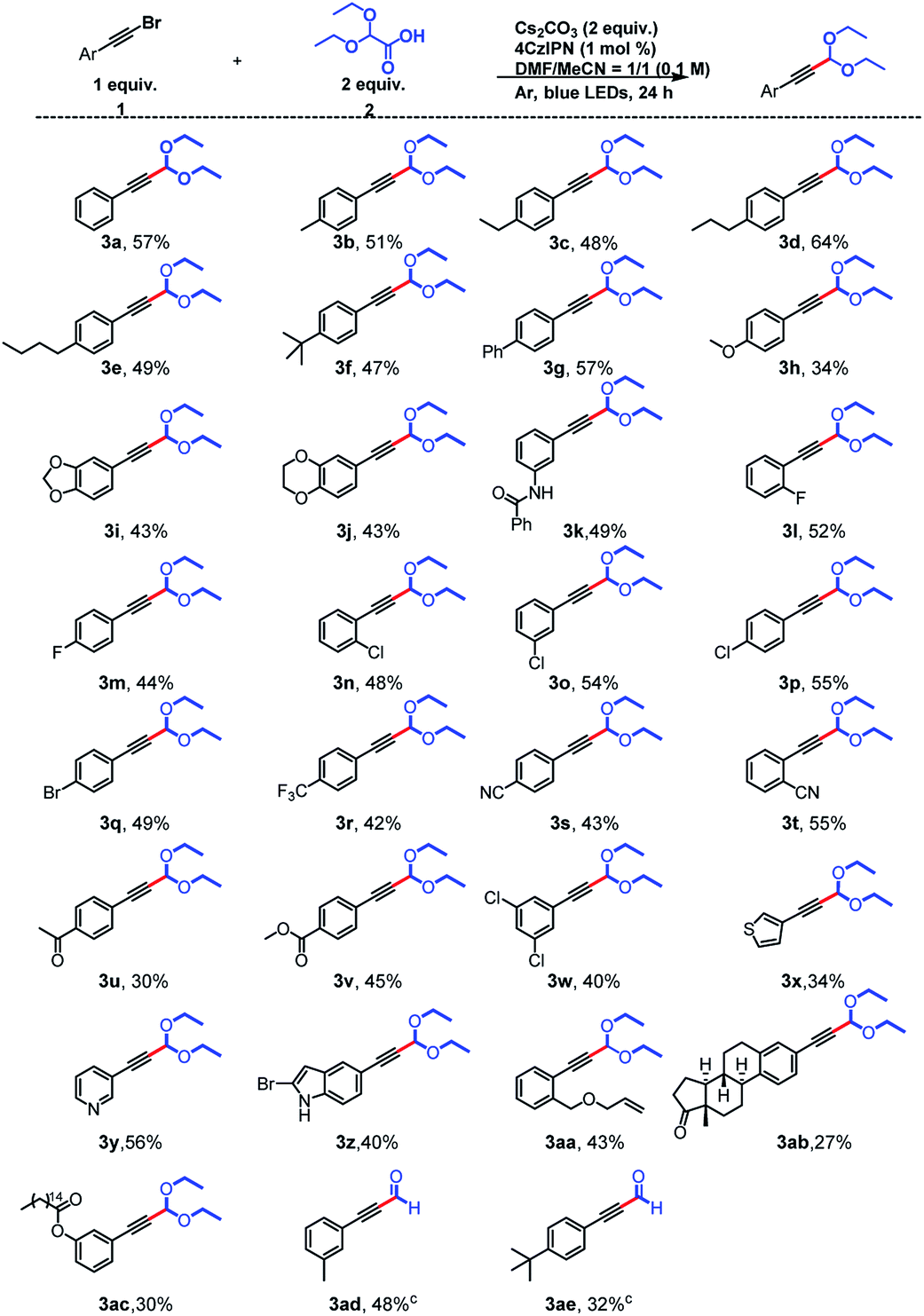 | ||
| Scheme 2 Scope of the reaction with respect to the alkynyl bromide. aStandard conditions: a mixture of 1g (0.3 mmol, 1.0 equiv.), 2 (0.6 mmol, 2.0 equiv.), 4CzIPN (0.003 mmol, 1 mol%), and Cs2CO3 (0.6 mmol, 2.0 equiv.) in 1:1 (v/v) DMF/MeCN (3 mL total) was irradiated with 5 W blue LEDs at room temperature under argon for 24 h. bIsolated yields are provided. cAcetals were not separated, and the subsequent removal of the acetal group was directly carried out, and the yield is the total yield of the two-step reaction. | ||
Alkenyl groups can also act as acceptors for acetal radicals, as reported by Xu10 and Wang.13 However, when we carried out the reaction between 2 and an olefin-containing substrate, we isolated (3aa, 43%), which indicates that the olefin did not participate in the reaction. In addition, polycyclic product (3ab, 27%) was prepared from a Br-substituted derivative of estrone, which is a steroid hormone and an endogenous estrogen. We were also able to obtain 3ac (30%) from a substrate derived from palmitic acid, which is an important component of blood lipids. The derivatization of these biologically relevant compounds indicates the potential utility of this protocol. Finally, when the products of the acetalation reactions of 1-(bromoethynyl)-3-methylbenzene and 1-(bromoethynyl)-4-(tert-butyl)benzene were directly protonated,14 the corresponding hydroformylation products (3ad and 3ae) were obtained with yield of 48% and 32%.
To gain insight into the reaction mechanism, we conducted some radical-trapping and radical-inhibition experiments (Scheme 3). When the reaction of alkynyl bromide 1g was carried out in the presence of the radical inhibitor TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl), no reaction occurred and none of the desired product was observed, a result that supports the involvement of radical intermediates (eq. (1)). In addition, the acetal radical could be trapped by ethene-1,1-diyldibenzene (eq. (2)), as confirmed by high-resolution mass spectrometry. Surprisingly, when 2 is omitted, 1g of debromination occurs in the presence of Cs2CO3 and a photocatalyst, which consumes a portion of the alkynyl bromide, wherein the terminal alkyne may be derived from less water in the solvent as its proton hydrogen source15 and it may be the main cause of low yield. Because terminal alkynes are known to act as radical acceptors,16 we wondered whether a terminal alkyne could undergo the acetalation reaction. To explore this possibility, we carried out two experiments with 1-ethynyl-4-phenylbenzene. When this substrate was subjected to the standard conditions, the acetalation reaction did not proceed (eq. (4)). However, when N-bromosuccinimide and AgNO3 were present in the reaction mixture (eq. (5)), 3g was obtained in 52% yield, which confirms that an alkynyl bromide was the true reactive species.
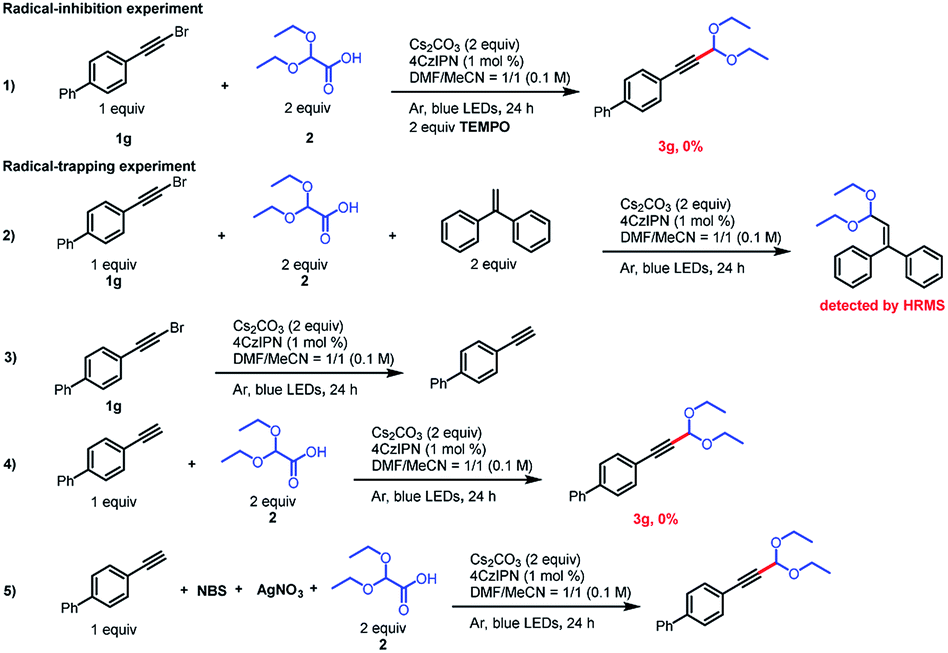 | ||
| Scheme 3 Mechanistic experiments. | ||
On the basis of our mechanistic studies, we propose the mechanism outlined in Scheme 4. First, the metal-free photocatalyst 4CzIPN is photoexcited to 4CzIPN* by irradiation with the 5 W blue LEDs. The cesium salt of 2,2-diethoxyacetic acid (Eox = +0.95 V vs. SCE)17 readily undergoes a single-electron transfer reaction with 4CzIPN* to form carboxyl radical intermediate I. Intermediate I then loses a molecule of CO2 to form acetal radical II, which adds to arylethynyl bromide 1g to give stable bromoalkenyl radical III.18 Collapse of this radical gives product 3g and a bromine radical.19 The bromine radical (E1/2red [Br/Br−] = +0.80 V vs. SCE in DME)20 is reduced to a bromine anion by the reduced-state photocatalyst (E [PC/PC˙−] = −1.21 V vs. SCE),12 which completes the catalytic cycle. However, we cannot exclude another mechanism involving alkynyl radicals, whose generation has been reported under similar conditions.21
 | ||
| Scheme 4 Possible reaction mechanism. | ||
Conclusions
In summary, we have developed a protocol for visible-light-promoted acetalation of alkynyl bromides with readily available 2,2-diethoxyacetic acid as the acetal source. The alkynyl bromide substrates can be synthesized easily, and the acetalation reaction proceeds without the need for an additional oxidant and is therefore compatible with various functional groups. In addition to arylethynyl bromides, alkynyl bromides bearing heteroaryl rings (thiophene, pyridine, and indole) were also effective substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Key Research and Development Program of China (2018YFD0200100) and the National Natural Science Foundation of China (21732002, 21672117, 21772102) for generous financial support for our programs. The authors thank Yuanqiong Huang (Nankai University) for helpful discussion.References
- E. Lenci, G. Menchi, F. I. Saldívar-Gonzalez, J. L. Medina-Franco and A. Trabocchi, Org. Biomol. Chem., 2019, 17, 1037–1052 RSC.
- For compounds containing spiroacetal moieties, see: (a) A. F. Kluge, Heterocycles, 1986, 24, 1699–1740 CrossRef CAS; (b) F. Perron and K. F. Albizati, Chem. Rev., 1989, 89, 1617–1661 CrossRef CAS; (c) W. Francke and W. Kitching, Curr. Org. Chem., 2001, 5, 233–251 CrossRef CAS; (d) M. A. Brimble and D. P. Furkert, Curr. Org. Chem., 2003, 7, 1461–1484 CrossRef CAS; (e) J. E. Aho, P. M. Pihko and T. K. Rissa, Chem. Rev., 2005, 105, 4406–4440 CrossRef CAS; (f) S. Favre, P. Vogel and S. Gerber-Lemaire, Molecules, 2008, 13, 2570–2600 CrossRef CAS; (g) Y. K. Booth, W. Kitching and J. J. De Voss, Nat. Prod. Rep., 2009, 26, 490–525 RSC; (h) J. Sperry, Z. E. Wilson, D. C. K. Rathwell and M. A. Brimble, Nat. Prod. Rep., 2010, 27, 1117–1137 RSC; (i) R. Quach, D. F. Chorley and M. A. Brimble, Org. Biomol. Chem., 2014, 12, 7423–7432 RSC; (j) F.-M. Zhang, S.-Y. Zhang and Y.-Q. Tu, Nat. Prod. Rep., 2018, 35, 75–104 RSC For compounds containing bridged bicyclic acetals moieties, see: D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1–49 CAS.
- (a) E. M. Seward, E. Carlson, T. Harrison, K. E. Haworth, R. Herbert, F. J. Kelleher, M. M. Kurtz, J. Moseley, S. N. Owen, A. P. Owens, S. J. Sadowski, C. J. Swain and B. J. Williams, Bioorg. Med. Chem. Lett., 2002, 12, 2515–2518 CrossRef CAS; (b) Y. Ohtake, T. Sato, T. Kobayashi, M. Nishimoto, N. Taka, K. Takano, K. Yamamoto, M. Ohmori, M. Yamaguchi, K. Takami, S.-Y. Yeu, K.-H. Ahn, H. Matsuoka, K. Morikawa, M. Suzuki, H. Hagita, K. Ozawa, K. Yamaguchi, M. Kato and S. Ikeda, J. Med. Chem., 2012, 55, 7828–7840 CrossRef CAS; (c) L.-G. Milroy, G. Zinzalla, F. Loiseau, Z. Qian, G. Prencipe, C. Pepper, C. Fegan and S. V. Ley, ChemMedChem, 2008, 3, 1922–1935 CrossRef CAS.
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 3rd edn, 1999 CrossRef; (b) P. J. Kocienski, Protecting Groups, George Thieme, Stuttgart, 1st edn, 1994 Search PubMed.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xie, H.-M. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC; (c) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (e) P. Xu, W.-P. Li, J. Xie and C.-J. Zhu, Acc. Chem. Res., 2018, 51, 484–495 CrossRef CAS.
- For selected papers, see: (a) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS; (c) M. Rahman, A. Mukherjee, I. S. Kovalev, D. S. Kopchuk, G. V. Zyryanov, M. V. Tsurkan, A. Majee, B. C. Ranu, V. N. Charushin, O. N. Chupakhin and S. Santra, Adv. Synth. Catal., 2019, 361, 2161–2214 CrossRef CAS; (d) Y.-F. Liang, X.-H. Zhang and D. W. C. MacMillan, Nature, 2019, 559, 83–88 CrossRef; (e) J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522–6526 CrossRef CAS; (f) N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 CrossRef CAS; (g) C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 CrossRef CAS; (h) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS; (i) Z. Huang, R. Shang, Z.-R. Zhang, X.-D. Tan, X. Xiao and Y. Fu, J. Org. Chem., 2013, 78, 4551–4557 CrossRef CAS; (j) B. Zhao, R. Shang, W.-M. Cheng and Y. Fu, Org. Chem. Front., 2019, 5, 1782–1786 RSC.
- For selected papers, see: (a) Z.-W. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed; (b) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS; (c) L. Candish, L. Pitzer, A. Goméz-Suárez and F. Glorius, Chem.–Eur. J., 2016, 22, 4753–4756 CrossRef CAS; (d) L. Candish, E. A. Standley, A. Goméz-Suárez, S. Mukherjee and F. Glorius, Chem.–Eur. J., 2016, 22, 9971–9974 CrossRef CAS; (e) A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464–13468 CrossRef CAS PubMed.
- For selected papers, see: (a) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 CrossRef CAS; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (c) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS; (d) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (e) C. Jin, Z.-Y. Yan, B. Sun and J. Yang, Org. Lett., 2019, 21, 2064–2068 CrossRef CAS PubMed.
- W.-G. Kong, C.-J. Yu, H.-J. Anand and Q.-L. Song, Org. Lett., 2018, 20, 349–352 CrossRef CAS.
- S. Zhang, Z.-M. Tan, H.-N. Zhang, J.-L. Liu, W.-T. Xu and K. Xu, Chem. Commun., 2017, 53, 11642–11645 RSC.
- (a) H.-C. Huang, G.-J. Zhang and Y.-Y. Chen, Angew. Chem., Int. Ed., 2015, 127, 7983–7987 CrossRef; (b) H.-C. Huang, G.-J. Zhang, L. Gong, S.-Y. Zhang and Y.-Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS; (c) X.-S. Liu, Z.-T. Wang, X.-M. Cheng and C.-Z. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 CrossRef CAS.
- (a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (b) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS; (c) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- H. Huang, C.-G. Yu, Y.-T. Zhang, Y.-Q. Zhang, P. S. Mariano and W. Wang, J. Am. Chem. Soc., 2017, 139, 9799–9802 CrossRef CAS.
- The accomplishment of protonization see ESI.†.
- M. Zhao, C.-X. Kuang, Q. Yang and X.-Z. Cheng, Tetrahedron Lett., 2011, 52, 992–994 CrossRef CAS.
- (a) H. Jiang, Y.-Z. Cheng, Y. Zhang and S.-Y. Yu, Org. Lett., 2013, 15, 4884–4887 CrossRef CAS; (b) S. Sumino, M. Uno, T. Fukuyama, I. Ryu, M. Matsuura, A. Yamamoto and Y. Kishikawa, J. Org. Chem., 2017, 82, 5469–5474 CrossRef CAS.
- H. Huang, X.-M. Li, C.-G. Yu, Y.-T. Zhang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 CrossRef CAS.
- (a) Z.-Y. Song, C.-L. Zhang and S. Ye, Org. Biomol. Chem., 2019, 17, 181–185 RSC; (b) J.-J. Zhang, P.-H. Li and L. Wang, Org. Biomol. Chem., 2014, 12, 2969–2978 RSC.
- L. Eberson, M. P. Hartshorn, J. J. McCullough, O. Persson and F. Radner, Acta Chem. Scand., 1998, 52, 1024–1028 CrossRef CAS.
- V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS.
- (a) Y.-C. Zhao, J.-W. Jin and P. W. H. Chan, Adv. Synth. Catal., 2019, 361, 1313–1321 CrossRef CAS; (b) K. Ni, L.-G. Meng, K. Wang and L. Wang, Org. Lett., 2018, 20, 2245–2248 CrossRef CAS . Another proposed mechanism see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06596b |
| This journal is © The Royal Society of Chemistry 2019 |