
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitched reaction specificity in polyesterases towards amide bond hydrolysis by enzyme engineering†
Antonino Biundo‡
a,
Raditya Subagiab,
Michael Maurerc,
Doris Ribitsch *ab,
Per-Olof Syrén*d and
Georg M. Guebitzab
*ab,
Per-Olof Syrén*d and
Georg M. Guebitzab
aAustrian Centre of Industrial Biotechnology (ACIB), Konrad Lorenz Strasse 20, 3430 Tulln an der Donau, Austria. E-mail: doris.ribitsch@boku.ac.at
bInstitute of Environmental Biotechnology, University of Natural Resources and Life Sciences (BOKU), Konrad Lorenz Strasse 20, 3430 Tulln an der Donau, Austria
cDepartment of Bioengineering, University of Applied Sciences, Mauerbachstrasse 43, 1140 Vienna, Austria
dScience for Life Laboratory, Department of Fibre and Polymer Technology and Department of Protein Science, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Tomtebodavägen 23, Box 1031, 17165 Solna, Sweden. E-mail: per-olof.syren@biotech.kth.se
First published on 7th November 2019
Abstract
The recalcitrance of plastics like nylon and other polyamides contributes to environmental problems (e.g. microplastics in oceans) and restricts possibilities for recycling. The fact that hitherto discovered amidases (EC 3.5.1. and 3.5.2.) only show no, or low, activity on polyamides currently obstructs biotechnological-assisted depolymerization of man-made materials. In this work, we capitalized on enzyme engineering to enhance the promiscuous amidase activity of polyesterases. Through enzyme design we created a reallocated water network adapted for hydrogen bond formation to synthetic amide backbones for enhanced transition state stabilization in the polyester-hydrolyzing biocatalysts Humicola insolens cutinase and Thermobifida cellulosilytica cutinase 1. This novel concept enabled increased catalytic efficiency towards amide-containing soluble substrates. The afforded enhanced hydrolysis of the amide bond-containing insoluble substrate 3PA 6,6 by designed variants was aligned with improved transition state stabilization identified by molecular dynamics (MD) simulations. Furthermore, the presence of a favorable water-molecule network that interacted with synthetic amides in the variants resulted in a reduced activity on polyethylene terephthalate (PET). Our data demonstrate the potential of using enzyme engineering to improve the amidase activity for polyesterases to act on synthetic amide-containing polymers.
Introduction
Since their introduction in the twentieth century, polymers have found applications extensively in packaging, in the manufacturing of vehicles, airplanes and advanced medicinal and electronic devices and are indispensable for our everyday life. Although the proportion of bio-based monomers in the annual 300 Mton production of synthetic polymers is increasing1 (currently representing 1%), many man-made polymers are non-biodegradable.2 Discarded polymers get fragmented through abiotic mechanisms into smaller and inert pieces (≤5 mm) referred to as microplastics.3 As only 1–2% of all plastics are recycled in Europe and in the US and up to 43% of the plastic used worldwide is disposed of in landfills,4,5 the impact of post-consumer polymer waste on our environment and health can be tremendous.6,7 Innovative approaches to valorize this source of secondary raw material and minimize our impact on the environment are urgently needed.6,7One approach consists of capitalizing on mild enzymatic hydrolysis to depolymerize macromolecular architectures.8 Due to the high specificity of enzymes, this would allow for step-wise recovery of pure hydrolysis products even from polymer mixtures or composite materials. Enzymatic hydrolysis of the surface of polymers by extracellular hydrolases was reported to be the first step of the biotic degradation of polyesters by microorganisms.8 Thus, several studies have focused on the enzymatic hydrolysis of polyesters using members of the α/β-hydrolase superfamily, including cutinases and lipases, to increase wettability and dye staining, and to decrease pilling and hydrophobicity.9–11 Recently, it was reported that the bacterium Ideonella sakaiensis is able to degrade and assimilate the polyester polyethylene terephthalate (PET).12
Due to resonance stabilization, chemical hydrolysis of an amide bond proceeds three orders of magnitude slower than that of an ester bond.13 Moreover, even if amide and peptide bonds share the same features, amide bonds in polyamides (PA) are separated by extended carbon chains (Fig. S1†), which permit the formation of a more rigid and crystalline structure. Nylon 6 (polycaprolactam) (Fig. S1B, left†) forms intermolecular H-bonds which are longer and therefore weaker, while nylon 6,6(poly(hexamethylene adipamide)) (Fig. S1B, right†) contains short H-bonds which yield a strong and dense molecular architecture with a high melting temperature (255–265 °C in comparison with 210–220 °C for nylon 6).14 Thus, mild enzymatic depolymerization of inert polyamide backbones is challenging. Some enzymes have been reported to hydrolyze nylon oligomers, which are by-products of nylon production.15–17 Efficient enzymatic hydrolysis of polyamides seems to be considerably more difficult.18 Since nylon is used in fashion and as light-weight metal-replacements it has been found to significantly contribute to the ∼10–20 Mtons of plastics that end up in the oceans each year.3,5 Therefore, nylon-degrading biocatalysts would constitute one important class of designer enzymes allowing recycling of wastes to valuable building blocks. To meet this formidable challenge, enzyme engineering and design tools are greatly needed as evolution did not yet have had sufficient time to develop enzymes efficient towards hydrolysis of synthetic polyamides. In this work, enzyme engineering afforded polyesterase variants with up to 15-fold higher activity in hydrolysis of the insoluble amide model substrate N1,N6-dihexyladipamide (3PA 6,6).
Cutinases are extracellular serine hydrolases produced by bacteria and fungi to attack the plant cuticle and especially to degrade the natural polyester cutin.19–22 Cutinases and lipases have been extensively analyzed in order to increase their activity on bulky substrates.23–25 The ability of cutinases to adsorb onto hydrophobic surfaces has been widely used to hydrolyze biodegradable and non-biodegradable polyesters,26,27 such as poly(lactic acid) (PLA) and PET, respectively, which results in surface functionalization.28,29 Hydrolysis of bulky substrates can be achieved after adsorption of the enzyme on the surface of the material.25 In cutinases, as previously reported for cutinase 1 from Thermobifida cellulosilytica (TcC, formerly known as Thc_Cut1),28 hydrophobic patches, which are present on the surface of these enzymes, can improve the adsorption onto polymers. The latter has been demonstrated to enhance the activity of the enzyme, as shown for the introduction of heterologous domains30 or the truncation of enzyme domains to improve the hydrophobicity of the protein surface.24 Moreover, cutinases possess an exposed active site, as compared to lipases and this feature would allow the polymer chain to be attacked by the enzyme.
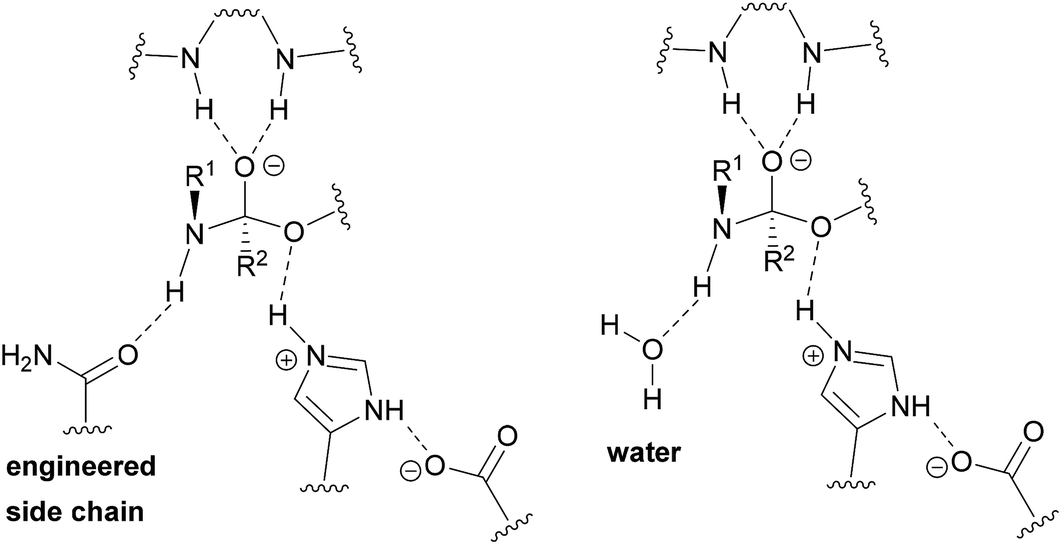
Enlarging the cavity of the active site of biocatalysts that hydrolyze natural polyesters like cutin, for a better fitting of the synthetic polymer chain, is a common design method to improve enzymatic activity on bulky, insoluble hydrophobic substrates.31,32 Structural analysis is used to identify specific residues, which when replaced by enzyme engineering, can lead to improved affinity and accelerated biocatalysis.33,34 However, introducing the missing key hydrogen bond acceptor in (poly)esterases, required for facilitated nitrogen inversion,35 and thus efficient (poly)amidase activity, by traditional enzyme design constitutes a bottleneck.36 In this study, we developed a novel approach for which a water network in the enzyme is adapted to the synthetic substrate to enable specific and catalytically favorable interactions.37 With their solvent exposed active site, we reasoned that cutinases would constitute suitable starting templates to achieve optimized transition state (TS) stabilization by water-restructuring mutations,38 to unlock the chemistry and spatial constraints dictated by protein side chains and, thus, to afford accelerated amide-bond hydrolysis.
We aimed at introducing amidase activity in two model cutinases that were previously reported to hydrolyze PET (Fig. 1E), namely Humicola insolens cutinase (HiC) and TcC. To study the activity on soluble substrates, p-nitrobutyranilide (pNBA) and p-nitrophenyl butyrate (pNPB) (Fig. 1A and B, respectively), which contained either an amide or ester bond, respectively, were used to evaluate enzyme kinetics. In order to identify “hot-spot” positions that could be important in dictating interactions between water and the transition state (TS), molecular dynamics (MD) simulations were performed using the model substrate 3PA 6,6 (Fig. 1C) (i.e. representing an oligomer of nylon 6,6, Fig. 1D). Experimentally determined activity enhancements of variants were in agreement with the extent of transition state stabilization obtained from molecular modeling.
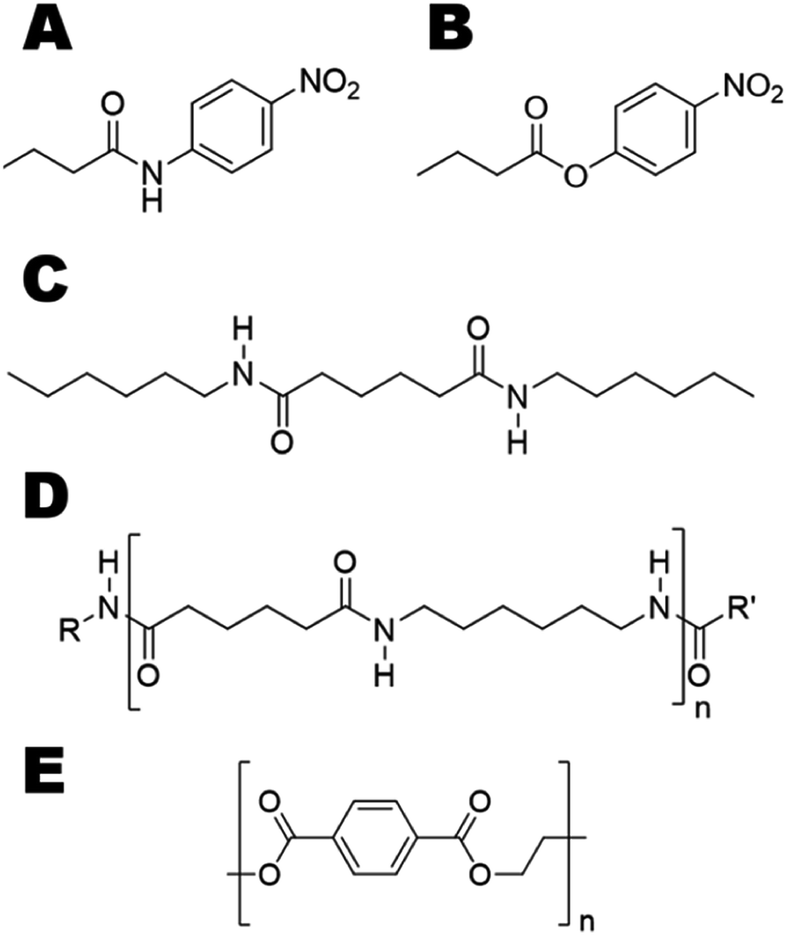 | ||
| Fig. 1 Chemical structures of the investigated model substrates used to study switched reaction specificity of polyesterases. Substrates ranged from a small aliphatic amide (A, p-nitrobutyranilide), ester (B, p-nitrophenyl butyrate) to representative amide oligomer (C, N1,N6-dihexyladipamide (3PA 6,6)). The corresponding polyamide (D, polyhexamethylene adipamide (nylon 6,6)) is shown for reference. The polyester (polyethylene terephthalate (PET)) is shown in (E). | ||
Materials and methods
Chemicals, films and reagents
All chemicals and reagents used in this work were of analytical grade. The model substrate N1,N6-dihexylhexanediamide (3PA 6,6) was produced according to Heumann et al.39 Films of nylon 6,6 and polyethylene terephthalate (PET) were purchased from Goodfellow (UK) (thickness 50 μm). Buffer components, bovine serum albumin (BSA), para-nitrophenol (pNP), para-nitrophenyl butyrate (pNPB), para-nitroaniline (pNA), and methanol were purchased from Sigma-Aldrich (USA). The substrate for amidase activity assay para-nitrobutyranilide (pNBA) was purchased from AKoS GmbH (Germany).Sequence analysis and sequencing
DNA and protein sequences were analyzed by CLC Main Workbench, version 7.0.3 (Qiagen, Netherlands). DNA sequencing was performed as a customer at LGC genomics (Germany) using Sanger sequencing.40Preparation of variants
All DNA manipulations described in this work were performed by standard methods.41 Megaprimers for the TcC variants, possessing the specific mutation (Table S1†) were purchased from Microsynth AG (Switzerland) and used in two-stage PCR reactions to introduce mutations using Pfu DNA polymerase (Promega GmbH, Germany) and pET26b(+)_TcC as template. Restriction enzymes were purchased from New England Biolabs and used following the manufacturer's protocol. Plasmid DNA was prepared using PureYield™ Plasmid Miniprep and Plasmid Midiprep system (Promega GmbH, Germany). The amplified products were chemically transformed in Escherichia coli XL-10.For HiC the codon-optimized genes (i.e. E. coli codon usage) of wild-type, I167Q and L64H/I167Q variants were purchased from GeneArt® (Life Technologies, USA). The genes were cloned into the vector pET26b(+) and chemically transformed by heat shock into E. coli XL-10.
Bacterial strains, plasmid and media
Bacterial cells of E. coli XL-10 and E. coli BL21-Gold(DE3) (Agilent Technologies, USA) strains were used for subcloning and expression, respectively. The vector pET26b(+) (Novagen, Germany) was used for expression of HiC and TcC wild-type and variants. Luria-Bertani (LB) medium, supplemented by 40 μg mL−1 kanamycin as a selective agent, was used to culture the plasmid-carrying cells. In order to determine the cell density at OD600 Hach DR3900 Spectrophotometer (USA) was used.Enzyme expression and purification
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da MWCO (Sartorius AG, Germany).
000 Da MWCO (Sartorius AG, Germany).Protein analysis
Bradford assay44 using Bio-Rad Protein Assay kit was carried out to measure the protein concentration using bovine serum albumin (BSA) (Sigma-Aldrich, USA) as standard. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to Laemmli using precast gels purchased from Bio-Rad (USA), run at 200 V for 30 min. Prestained protein marker IV (Peqlab, Germany) was used as a molecular mass marker. Coomassie method was used to stain the protein bands.Enzyme activity assay
Hydrolysis of polyethylene terephthalate (PET)
In order to determine the activity towards bulky polyesters, 5 μM HiC and TcC wild-type and variants were incubated with 0.5 × 1 cm amorphous PET films (50 μm thickness) in 0.1 M potassium phosphate buffer pH 7 for 72 h at 50 °C and 100 rpm. In order to remove impurities, PET films were previously washed with three different washing steps at 50 °C and 100 rpm, each step for 30 min, firstly with 5 g L−1 Triton X-100, then with 0.1 M sodium carbonate and finally with MQ water. After incubation the supernatant was treated to precipitate the enzyme by pouring ice-cold methanol in a ratio of 1:1. The sample was then acidified with 6 M HCl to a pH 4 and then centrifuged at 14000×g at 0 °C for 15 min. Finally the supernatant was filtered through a 0.22 μm filter and analyzed by high performance liquid chromatography (HPLC) in a Hewlett Packard series 1050 system, coupled with an Agilent Poroshell 120 Ec-C18, 30 × 50 mm, 2.7 μm column with a precolumn Poroshell 120 Ec-C18, 3.0 × 5.0 mm, 2.7 μm, UHPLC guard column (Agilent Technologies, USA) heated at 40 °C with a fluorescence detector. The samples were eluted using a non-linear gradient with a constant flow rate of 0.75 mL min−1. Concentration of formic acid (A) was always kept at 0.01%. The gradient started with 10% methanol (B) and 80% water (C) at 1 min, 50% B and 40% C at 8 min, 90% B and 0% C at 11 min. The injection volume of the sample was 10 μL. The release products were detected via UV spectroscopy at 241 nm.47
Hydrolysis of model substrate 3 PA 6,6
To investigate the activity of the enzymes and their variants towards amide bonds, 10 mg 3PA 6,6 model substrate was incubated in the presence of 5 μM enzyme in 0.1 M potassium phosphate buffer pH 7 at 50 °C and 100 rpm. After hydrolysis the samples were frozen and lyophilized for 48 h, until complete removal of water. The powder was resuspended in 100% methanol. After centrifugation at 14000 rpm the solution was filtered through a 0.2 μm PSTF filter and placed in a glass vial. Calibration standards of 3PA 6,6 in methanol for gas chromatography-flame-ionization detection (GC-FID) were prepared in a range of 1 to 250 mg L−1. The GC-FID analyses were performed using a Hewlett-Packard (Palo Alto, CA). A J&W Scientific (Folson, CA) DB-17MS capillary column (30 m × 250 μm × 0.25 μm) was used with nitrogen as the carrier gas. A splitless injection mode (1 μL) was used with the following GC temperatures: injection port, 300 °C; initial column temperature, 40 °C; initial hold time, 0.5 min; first temperature ramp, 20 °C min−1; second column temperature, 70 °C; second hold time, 1 min; second temperature ramp, 30 °C min−1; final column temperature, 340 °C; final hold time, 2 min. The total GC-FID run time was 14 min.
Gas chromatography-mass spectrometry (GC-MS)
In order to determine possible release of building blocks from the bulky substrate nylon 6,6, after incubation of the enzyme with the polymer, the supernatant of the reactions was lyophilized using a Christ Freeze dryer Beta 1-16, 220 V, 50 Hz, 1.2 kW. The lyophilized samples were resuspended in ethyl acetate (Sigma-Aldrich, USA) and filtered through a 0.2 μm PSTF filter and placed in a glass vial. The GC-MS analyses of the supernatant were performed using an Agilent 7890A (Santa Clara, CA) equipped with a mass selective detector 5975C VL MSD with triple axis. The system was equipped with a PAL-xt autosampler (CTC Analytics AG, Switzerland). A J&W Scientific (Folson, CA) DB-17MS capillary column (30 m × 250 μm × 0.25 μm) was used with nitrogen as the carrier gas. A deactivated glass wool tapered bleed temperature optimized (BTO) was used as an inlet liner and a splitless injection mode (1 μL) was used with the following GC temperatures: injection port, 300 °C; initial column temperature, 50 °C; initial hold time, 2 min; first temperature ramp, 7 °C min−1; second column temperature 100 °C; second hold time, 1 min; second temperature ramp, 6 °C min−1; third column temperature 220 °C; third hold time, 2 min; third temperature ramp, 20 °C min−1; final column temperature, 340 °C; final hold time, 5 min; total run time, 43 min. The MSD source was kept at 230 °C, the quadrupole at 190 °C, the transfer line at 300 °C with a scan range between 25 and 500 AMU with a gain factor of 5. The solvent was delayed for 2.8 min.Molecular dynamic (MD) simulations
Molecular dynamics simulations on Humicola insolens cutinase was based on the PDB file 4OYY48 using YASARA49 version 14.5.21. For Thermobifida cellulosilytica cutinase 1, the PDB file 5LUI was used.50 For both enzyme structures, missing hydrogens were added and the corresponding hydrogen network was optimized by using Amber03 force field and keeping all heavy atoms fixed. Crystallographic waters were kept. For the van der Waals interactions, a cut-off of 7.86 Å was used and PME accounted for long-range electrostatics.51 The structures were minimized through repeated steps of short molecular dynamics and energy minimizations, initially by releasing fixed waters and then on all atoms. The obtained structures were finally subjected to simulated annealing. The second tetrahedral intermediate formed during acylation of the catalytic serine (S131 for TcC and S103 for HiC, respectively) represented the TS for nitrogen inversion.35 The tetrahedral intermediate was constructed by covalent attachment of the 3PA 6,6 substrate. For HiC, the construction of the tetrahedral intermediate was guided by a previously generated homology model.36 Force field parameterization for the tetrahedral intermediate was obtained by the AUTOSMILES methodology as implemented in YASARA.52 All simulations were performed in a water box that contained approximately 4000 (for HiC) and 5000 (for TcC) explicit water molecules. The pH was set to 7.4 and corresponding protonation states of enzymes side chains was predicted by the built-in empirical method53 in YASARA. The introduced His in the HiC L64H/I167Q variant was kept unprotonated throughout the MD simulation (predicted pKa 7.2). The simulation cell was neutralized through the addition of 0.9% NaCl. All simulations were performed under standard conditions under the canonical ensemble at 298 K using a Berendsen thermostat (and the Amber03 force field). MD-simulations were performed for 120 ns (with 20 ns equilibration phase and 100 ns production phase) and in duplicate. For the duplicates, different initial random seeds were obtained by a slight change in the simulation temperature (by 0.0012 K). All possible water networks in proximity of the reacting amide group of the substrate were analyzed. A weak hydrogen bond was defined as an interaction between donor and acceptor with a hydrogen bond distance of ≤3 Å, and angle ≥120°.Results and discussion
Enzyme design and molecular dynamics analysis of variants on insoluble substrate
We hypothesized that de-shielding the protein backbone to afford enhanced polarity and reduced steric hindrance at identified sites would be beneficial in providing enhanced access of “spectator” water molecules to the TS, as previously discussed in the context of protein unfolding.18 From a basal energy minimization of the second tetrahedral intermediate representing the TS for nitrogen inversion,35 potential hot-spot residues were identified. For TcC (Fig. 2A), the side chain of I179 (shown in balls) obstructs access of water molecules to the modelled transition state. For HiC (Fig. 2B), the side chains of L64 and I167 (shown in balls) prevents the interaction between water and the TS representing hydrolysis of 3PA 6,6. We reasoned that mutating these identified hydrophobic sites to small, polar residues would enable water to access the TS and to engage in hydrogen bonding to the reacting NH-group of the amide substrate. In order to test the different hot-spots, in the case of HiC, two different variants were constructed, HiC_I167Q and HiC_L64H_I167Q. In the case of TcC, three single variants were constructed, TcC_I179A, TcC_I179N, and TcC_I179Q. 100 ns MD-simulations run in duplicate suggested that variants of TcC (Fig. 2C, left) were capable of weak hydrogen bond formation accepted by water in the modeled TS, which was associated with higher experimentally determined activity of 3PA 6,6 hydrolysis. In contrast, HiC variants (Fig. 2C, right) did not display significantly higher abundances of productive water clusters.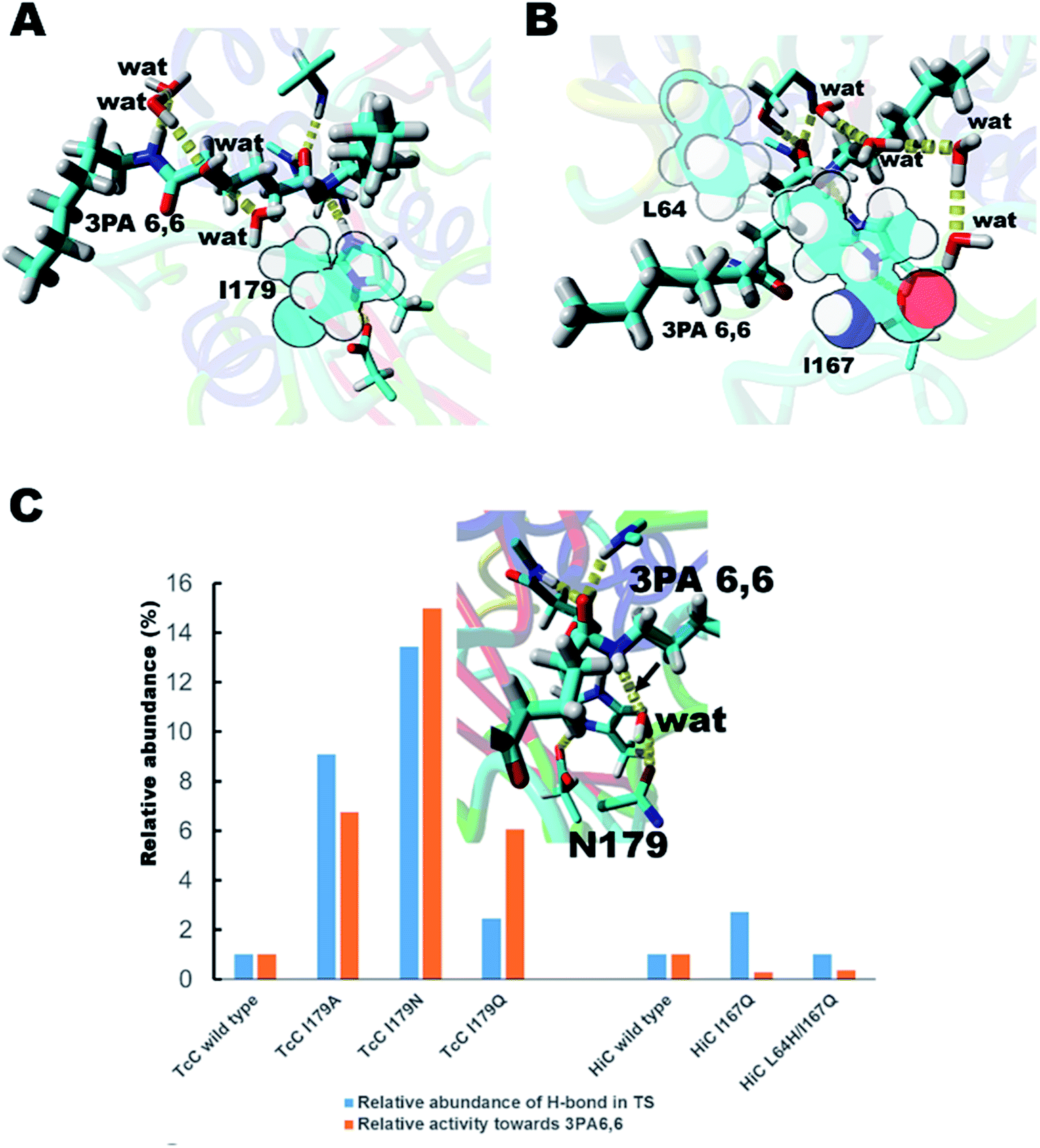 | ||
| Fig. 2 Enzyme design for identification of hot-spot residues for TcC (A) and HiC (B). The catalytic triad (S131/H209/D177) of TcC and parts of the oxyanion hole (Y61, M132) are shown. The catalytic triad (S103/H171/D158) of HiC and parts of the oxyanion hole (S26, Q104) are labeled. (C) In silico and experimental evaluation of engineered polyesterases. For reference, the relative abundance of hydrogen bond formation and experimentally determined hydrolysis activities (Table 1) are normalized to wild type in each case. The inset shows a snapshot from an MD-simulation of the TcC I179N variant with the key hydrogen bond indicated by the arrow. The substrate 3PA 6,6 is shown in enlarged sticks and water molecules are labeled “wat”. | ||
Expression and biochemical characterization of variants
The expression of the corresponding codon-optimized genes in Escherichia coli BL21-Gold(DE3) was first assayed in shake flasks revealing that TcC wild type and variants were expressed in high amounts (Fig. 3A). In contrast, HiC wild type and designed variants did not show overexpression notwithstanding codon optimization and thus expression by fed-batch bioreactors was required, to reach sufficient amounts of enzyme to be used for downstream processing (Fig. 3A). Initial experimental analysis of purified variants on the selected small substrates (pNPB and pNBA) revealed a general trend: variants targeted for redesigned water networks (Fig. 2), displayed enhanced specificity for amides and decreased specificity for esters (Fig. 3B, Fig. S2†). The most prominent effect was observed for HiC L64H/I167Q that showed increased catalytic efficiency towards pNBA (5-fold increase in kcat/KM) and a significant decrease in esterase activity (13-fold) as compared to that of wild-type HiC, which together resulted in a 66-fold change in relative reaction specificity in favor of amide bond hydrolysis. For TcC, only the variant I179Q displayed a higher catalytic efficiency for amide bond hydrolysis (10-fold) as compared to that of wild-type (Fig. 3B), whereas all three variants showed a 5 to 10 fold in loss of their esterase activity. Among the three variants, TcC I179Q showed the highest change in relative reaction specificity (Fig. 3B).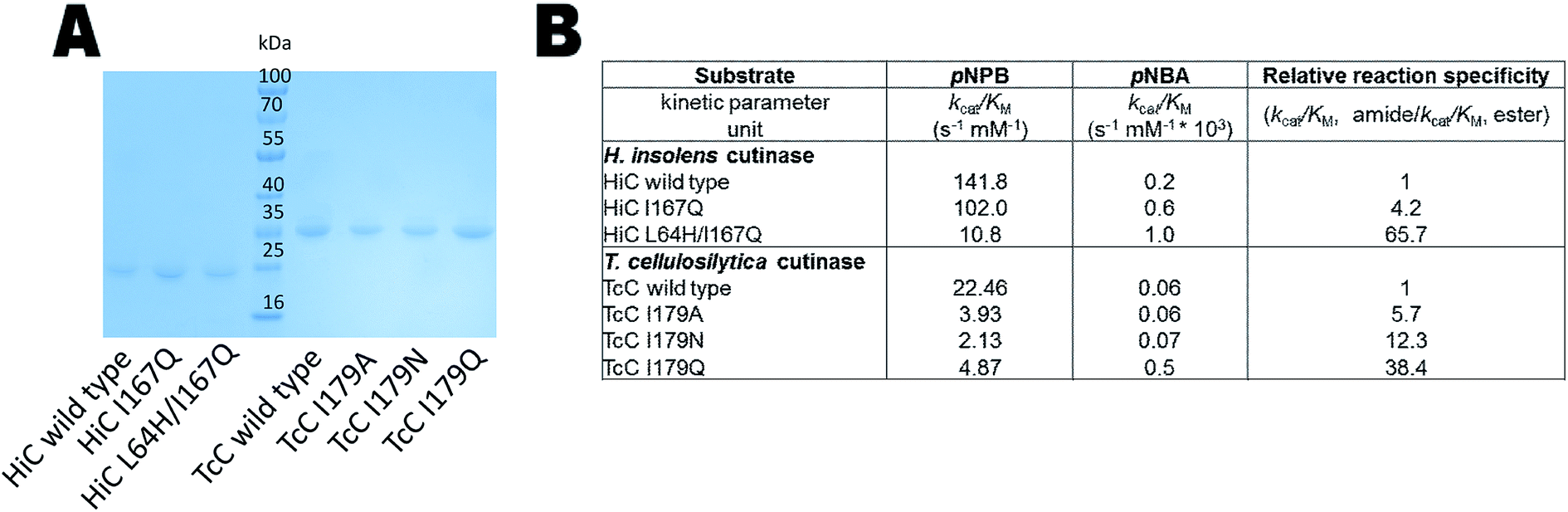 | ||
| Fig. 3 Biochemical analysis of HiC and TcC wild type and variants. (A) SDS-PAGE analysis of HiC and TcC enzymes (purification level > 90%, 23 and 30 kDa, respectively) after immobilized metal ion affinity chromatography. (B) Kinetic analysis of wild-type enzymes and variants in the hydrolysis of a small ester- and amide-containing substrates (Fig. S2†). Values of relative reaction specificities were compared and normalized against each wild-type enzyme. | ||
Enhanced amidase activity towards oligomers
In order to test the ability of the HiC and TcC variants to act on amide-containing insoluble substrates, the hydrolysis of the small substrate 3PA 6,6 (Fig. 1C and S3†) was analyzed both experimentally and computationally (Fig. 2C and Table 1). Remarkably, all designed TcC variants showed higher activity with 3PA 6,6 hydrolysis rates being accelerated 6- to 15- fold (Table 1). Indeed, it was found that the relative abundance of weak hydrogen bonds to the reacting amide group in the modeled TS correlated with the experimentally determined hydrolytic activity, especially in the case of TcC variants (Fig. 2C and Table 1), which points towards an important role for chemistry in contributing to the rate-limiting step. For HiC, engineered variants did not show significantly altered propensities of hydrogen bond formation compared to wild type (up to 3-fold change), in contrast to TcC for which the probability increased one order of magnitude (up to 14-fold). This observation is aligned with the lower activities displayed by HiC variants. It is possible that our strategy to capitalize on a homology model previously published by us for HiC impacted the design.36 MD analysis confirmed that the preferred hydrogen bond acceptor in the modeled TS of the variants was a water molecule network (Fig. S4 and Tables S2–S4†), in line with our hypothesis that water-restructuring mutations (Fig. 2) dictate interactions between water and the TS for oligomer hydrolysis. In fact, for TcC a water network was the only possibility for the I179A variant (Table 1, Fig. S4†). Moreover, the introduced mutations were found to impact the density of water around the TS for 3PA 6,6 hydrolysis (Table S4†). Analysis of root mean square deviation (RMSD) throughout the 100 ns simulation did not reveal conformational changes in the variants (Fig. S5†). The RMSF values were 0.5 Å for TcC wild type and variants, whereas the corresponding values were 0.7 Å for HiC wild type and I167Q variant and 1.0 Å for the double mutant respectively. This could point towards a more plastic structure for HiC and we cannot rule out dynamical effects contributing to catalysis.
|
||||
|---|---|---|---|---|
| Variant | Relative abundance of a hydrogen bond donated by the reacting NH-group and accepted by the engineered side chain in TSa (%) | Relative abundance of a hydrogen bond donated by the reacting NH-group and accepted by water in TSa (%) | Total relative abundance of a hydrogen bond in TSb (%) | Experimental activity mol mol−1 c |
| a Refers to weak hydrogen bonds. Average value from analysis of duplicate 100 ns MD-simulations. For each trajectory of wild type and variants, 38000 snapshots were analyzed for TcC and 40000 snapshots for HiC.b Total relative probability of formation of a weak key hydrogen bond to the scissile NH-group.c The experiments were run in potassium phosphate buffer pH 7 at 50 °C and 100 rpm. |
||||
| TcC wild type | — | 0.3 | 0.3 | 80 ± 37 |
| TcCI1 79A | — | 2.5 | 2.5 | 540 ± 32 |
| TcCI1 79N | 2.4 | 1.4 | 3.7 | 1196 ± 43 |
| TcCI1 79Q | 0.2 | 0.5 | 0.7 | 485 ± 27 |
| HiC wild type | — | 0.5 | 0.5 | 1227 ± 327 |
| HiCI167Q | — | 1.4 | 1.4 | 543 ± 122 |
| HiCL64H/I167Q | — | 0.6 | 0.6 | 634 ± 203 |
Experimental tests of the activity of the designer enzymes on nylon 6,6 showed that this polymer was not able to be hydrolyzed in a high yield and only small amounts of release products were detected by GC/MS (Fig. S6–S7†). This was probably due to the strong hydrogen bonding present in the structure of nylon 6,6.
Loss of polyesterase activity
Although, cutinases have been widely reported to be able to hydrolyze the polyester PET (Fig. S8†), the two cutinases used in this work, HiC and TcC, showed different hydrolysis activity on PET. On the one hand, HiC wild type showed a 25 fold higher activity on PET than that of TcC, which corresponds to the release of almost 200 moles of products per mol of biocatalyst (Fig. 4A and B), similarly to what was shown towards the soluble substrate pNPB (Fig. 3B). Remarkably, the variant HiC L64H/I167Q showed a complete loss of activity on PET, while HiC I167Q showed a striking decrease in activity of almost 94%. Instead, TcC variants did not show a complete loss of polyesterase activity. The establishment of a water network for stabilization of the nitrogen inversion needed for the hydrolysis of the amide bond was able to reduce their ability to hydrolyze ester bond. A possible explanation is electrostatic repulsion between water and the oxygen atoms of the ester-containing substrate as previously discussed.54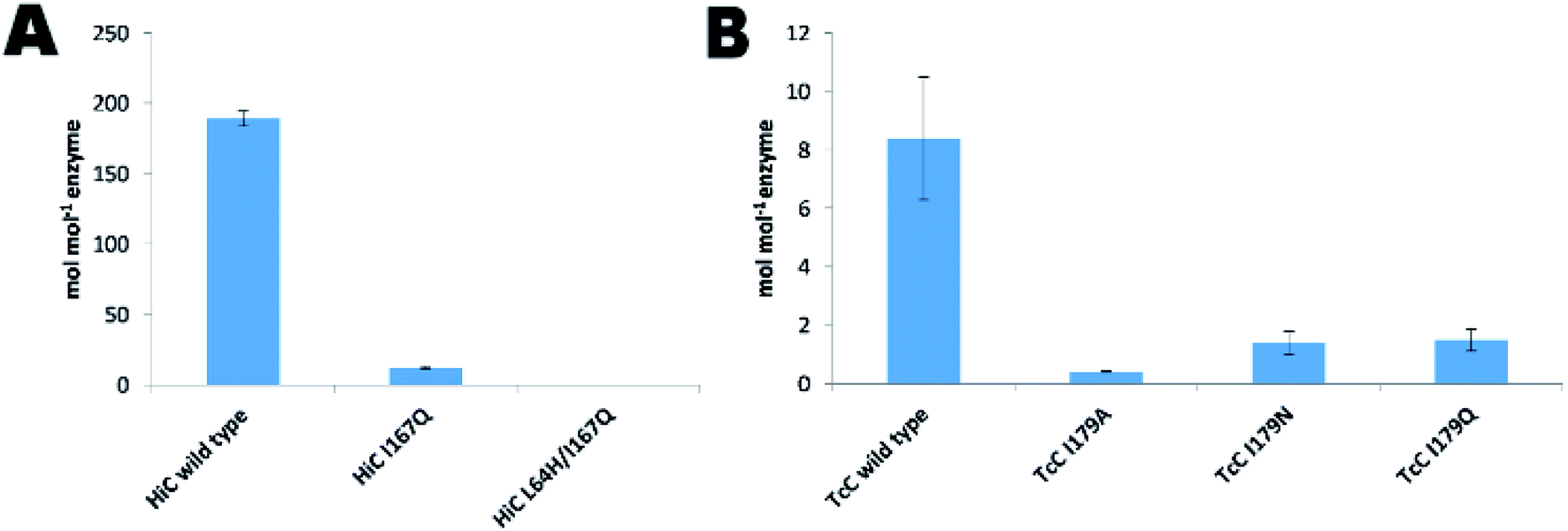 | ||
| Fig. 4 PET hydrolysis of wild-type and designer enzymes. (A) HiC and (B) TcC wild type and variants. The released product terephthalic acid was measured. The data are the mean value of three different measurements and the bars represent the standard deviation. | ||
Conclusions
In this study, we have successfully demonstrated that the constitution of a water network to facilitate the nitrogen inversion mechanism by H-bonding, which is present in amidases and proteases, can increase promiscuity of polyesterases to act on amide-containing substrates. Cutinases, and especially HiC and TcC, have been widely used for the hydrolysis of polyesters due to their ability to hydrolyze the natural hydrophobic polymer cutin, thanks to their active site present on the surface of the enzyme. The substitution of selected hydrophobic amino acids to polar residues enabled water to access the transition state and stabilize it by hydrogen bond formation. This approach was explored by analysis of different substrates which contain either amide- or ester-bonds and the ability of the variants to accept both soluble and insoluble substrates was confirmed. We identified several variants of TcC that showed significantly accelerated rates of hydrolysis of insoluble amides represented by 3PA 6,6. Both HiC variants showed improved amidase activity on the small substrate pNBA, which reached, in the case of L64H/I167Q, almost 66-fold increased relative reaction specificity compared to the HiC wild-type. Interestingly, the latter variant, L64H/I167Q, showed a complete loss of its polyesterase activity on the bulky substrate PET. However, all the other tested variants retained a partial activity on the recalcitrant polymer, probably due to the lacking combination of the two substitutions. To the best of our knowledge, no microorganism has been identified so far, which is able to degrade the industrial bulky polymer nylon 6,6. Herein, we have shown that a reconfigured water network adapted to the synthetic polymer backbone and obtained by enzyme design can turn polyester-degrading enzymes into amidases by providing efficient transition state stabilization. Further improvements of the variants which carry the amidase activity may increase their activity and could pave the way towards the formation of a “nylonidase” and the recovery of valuable polyamide building blocks from complex plastic mixtures, composites and blends55 through selective depolymerization of the polyamide constituents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly acknowledge funding from FORMAS young research leader fellowship (Grant no. 2017-01116). The PDC Center for High Performance Computing at the Royal Institute of Technology (KTH) is greatly acknowledged. This work has received further support from the Federal Ministry of Science, Research and Economy (BMWFW), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, the Government of Lower Austria and ZIT – Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG. The authors also acknowledge the Gunnar Sundblad Research Foundation, the INTEGRATE project from the Swedish Research council (VR) under the project number 2016-06160 and the Swedish Energy Agency/VINNOVA under the project Re:Mix 2017-002010.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- L. Shen, E. Worrell and M. Patel, Biofuels, Bioprod. Biorefin., 2010, 4, 25–40 CrossRef CAS.
- M. Claessens, S. De Meester, L. Van Landuyt, K. De Clerck and C. R. Janssen, Mar. Pollut. Bull., 2011, 62, 2199–2204 CrossRef CAS.
- E. Huerta Lwanga, H. Gertsen, H. Gooren, P. Peters, T. Salánki, M. van der Ploeg, E. Besseling, A. A. Koelmans and V. Geissen, Environ. Sci. Technol., 2016, 50, 2685–2691 CrossRef CAS.
- J. Raynaud, J. Richens, A. Russell, United Nations Environment Programme and Plastic Disclosure Project. and Trucost (Firm), Valuing plastic: the business case for measuring, managing and disclosing plastic use in the consumer goods industry, UNEP, 2014 Search PubMed.
- R. Sussarellu, M. Suquet, Y. Thomas, C. Lambert, C. Fabioux, M. E. J. Pernet, N. Le Goïc, V. Quillien, C. Mingant, Y. Epelboin, C. Corporeau, J. Guyomarch, J. Robbens, I. Paul-Pont, P. Soudant and A. Huvet, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2430–2435 CrossRef CAS.
- M. Bergmann, M. B. Tekman and L. Gutow, Nature, 2017, 544, 297 CrossRef CAS.
- R.-J. Müller, Process Biochem., 2006, 41, 2124–2128 CrossRef.
- G. Fischer-Colbrie, S. Heumann, S. Liebminger, E. Almansa, A. Cavaco-Paulo and G. M. Guebitz, Biocatal. Biotransform., 2004, 22, 341–346 CrossRef CAS.
- A. Eberl, S. Heumann, T. Brückner, R. Araujo, A. Cavaco-Paulo, F. Kaufmann, W. Kroutil and G. M. Guebitz, J. Biotechnol., 2009, 143, 207–212 CrossRef CAS.
- M. A. M. E. Vertommen, V. A. Nierstrasz, M. van der Veer and M. M. C. G. Warmoeskerken, J. Biotechnol., 2005, 120, 376–386 CrossRef CAS.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS.
- B. Zerner, R. P. M. Bond and M. L. Bender, J. Am. Chem. Soc., 1964, 86, 3674–3679 CrossRef CAS.
- A. J. Oshinski, H. Keskkula and D. R. Paul, Rubber toughening of polyamides with functionalized block copolymers: 1. Nylon-6, 1992 Search PubMed.
- S. Kakudo, S. Negoro, I. Urabe and H. Okada, Appl. Environ. Microbiol., 1993, 59, 3978–3980 CAS.
- I. D. Prijambada, S. Negoro, T. Yomo and I. Urabe, Appl. Environ. Microbiol., 1995, 61, 2020–2022 CAS.
- T. Deguchi, Y. Kitaoka, M. Kakezawa and T. Nishida, Appl. Environ. Microbiol., 1998, 64, 1366–1371 CAS.
- Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS.
- C. M. L. Carvalho, M. R. Aires-Barros and J. M. S. Cabral, Electron. J. Biotechnol., 1998, 1, 160–173 CrossRef.
- S. Chen, L. Su, J. Chen and J. Wu, Biotechnol. Adv., 2013, 31, 1754–1767 CrossRef CAS.
- G. M. Guebitz and A. Cavaco-Paulo, Trends Biotechnol., 2008, 26, 32–38 CrossRef CAS.
- R. Wei, T. Oeser and W. Zimmermann, Adv. Appl. Microbiol., 2014, 89, 267–305 Search PubMed.
- D. Ribitsch, E. Herrero Acero, A. Przylucka, S. Zitzenbacher, A. Marold, C. Gamerith, R. Tscheließnig, A. Jungbauer, H. Rennhofer, H. Lichtenegger, H. Amenitsch, K. Bonazza, C. P. Kubicek, I. S. Druzhinina and G. M. Guebitz, Appl. Environ. Microbiol., 2015, 81, 3586–3592 CrossRef CAS.
- A. Biundo, D. Ribitsch, G. Steinkellner, K. Gruber and G. M. Guebitz, Biotechnol. J., 2017, 12(8), 1600450 Search PubMed.
- A. Biundo, D. Ribitsch and G. M. Guebitz, Appl. Microbiol. Biotechnol., 2018, 102, 3551–3559 CrossRef CAS PubMed.
- E. Herrero Acero, D. Ribitsch, G. Steinkellner, K. Gruber, K. Greimel, I. Eiteljoerg, E. Trotscha, R. Wei, W. Zimmermann, M. Zinn, A. Cavaco-Paulo, G. Freddi, H. Schwab and G. Guebitz, Macromolecules, 2011, 44, 4632–4640 CrossRef CAS.
- D. Ribitsch, E. H. Acero, K. Greimel, I. Eiteljoerg, E. Trotscha, G. Freddi, H. Schwab and G. M. Guebitz, Biocatal. Biotransform., 2012, 30, 2–9 CrossRef CAS.
- A. Pellis, L. Silvestrini, D. Scaini, J. M. Coburn, L. Gardossi, D. L. Kaplan, E. Herrero Acero and G. M. Guebitz, Process Biochem., 2017, 59, 77–83 CrossRef CAS.
- A. Ortner, A. Pellis, C. Gamerith, A. Orcal Yebra, D. Scaini, I. Kaluzna, D. Mink, S. de Wildeman, E. Herrero Acero and G. M. Guebitz, Green Chem., 2017, 19, 816–822 RSC.
- D. Ribitsch, A. O. Yebra, S. Zitzenbacher, J. Wu, S. Nowitsch, G. Steinkellner, K. Greimel, A. Doliska, G. Oberdorfer, C. C. Gruber, K. Gruber, H. Schwab, K. Stana-Kleinschek, E. H. Acero and G. M. Guebitz, Biomacromolecules, 2013, 14, 1769–1776 CrossRef CAS.
- R. Araújo, C. Silva, A. O'Neill, N. Micaelo, G. Guebitz, C. M. Soares, M. Casal and A. Cavaco-Paulo, J. Biotechnol., 2007, 128, 849–857 CrossRef.
- E. Herrero Acero, D. Ribitsch, A. Dellacher, S. Zitzenbacher, A. Marold, G. Steinkellner, K. Gruber, H. Schwab and G. M. Guebitz, Biotechnol. Bioeng., 2013, 110, 2581–2590 CrossRef CAS.
- C. Silva, R. Araújo, M. Casal and G. M. Gübitz, Enzyme Microb. Technol., 2007, 40, 1678–1685 CrossRef CAS.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS.
- P.-O. Syrén, FEBS J., 2013, 280, 3069–3083 CrossRef.
- P.-O. Syrén, P. Hendil-Forssell, L. Aumailley, W. Besenmatter, F. Gounine, A. Svendsen, M. Martinelle and K. Hult, ChemBioChem, 2012, 13, 645–648 CrossRef.
- M. J. Fink and P.-O. Syrén, Curr. Opin. Chem. Biol., 2017, 37, 107–114 CrossRef CAS.
- J. M. Fox, K. Kang, M. Sastry, W. Sherman, B. Sankaran, P. H. Zwart and G. M. Whitesides, Angew. Chem., Int. Ed., 2017, 56, 3833–3837 CrossRef CAS.
- S. Heumann, A. Eberl, H. Pobeheim, S. Liebminger, G. Fischer-Colbrie, E. Almansa, A. Cavaco-Paulo and G. M. Gübitz, J. Biochem. Biophys. Methods, 2006, 69, 89–99 CrossRef CAS.
- F. Sanger, S. Nicklen and A. R. Coulson, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5463–5467 CrossRef CAS.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, 1989 Search PubMed.
- K. Marisch, K. Bayer, M. Cserjan-Puschmann, M. Luchner and G. Striedner, Microb. Cell Fact., 2013, 12, 58 CrossRef CAS PubMed.
- V. Perz, M. T. Zumstein, M. Sander, S. Zitzenbacher, D. Ribitsch and G. M. Guebitz, Biomacromolecules, 2015, 16, 3889–3896 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- D. Ribitsch, S. Heumann, E. Trotscha, E. Herrero Acero, K. Greimel, R. Leber, R. Birner-Gruenberger, S. Deller, I. Eiteljoerg, P. Remler, T. Weber, P. Siegert, K.-H. Maurer, I. Donelli, G. Freddi, H. Schwab and G. M. Guebitz, Biotechnol. Prog., 2011, 27, 951–960 CrossRef CAS.
- A. Biundo, A. Hromic, T. Pavkov-Keller, K. Gruber, F. Quartinello, K. Haernvall, V. Perz, M. S. Arrell, M. Zinn, D. Ribitsch and G. M. Guebitz, Appl. Microbiol. Biotechnol., 2016, 100, 1753–1764 CrossRef CAS.
- A. Biundo, J. Reich, D. Ribitsch and G. M. Guebitz, Sci. Rep., 2018, 8, 1–7 CrossRef CAS PubMed.
- D. Kold, Z. Dauter, A. K. Laustsen, A. M. Brzozowski, J. P. Turkenburg, A. D. Nielsen, H. Koldsø, E. Petersen, B. Schiøtt, L. De Maria, K. S. Wilson, A. Svendsen and R. Wimmer, Protein Sci., 2014, 23, 1023–1035 CrossRef CAS.
- E. Krieger, T. Darden, S. B. Nabuurs, A. Finkelstein and G. Vriend, Proteins: Struct., Funct., Bioinf., 2004, 57, 678–683 CrossRef CAS.
- D. Ribitsch, A. Hromic, S. Zitzenbacher, B. Zartl, C. Gamerith, A. Pellis, A. Jungbauer, A. Łyskowski, G. Steinkellner, K. Gruber, R. Tscheliessnig, E. Herrero Acero and G. M. Guebitz, Biotechnol. Bioeng., 2017, 114, 2481–2488 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS.
- E. Krieger, J. E. Nielsen, C. A. E. M. Spronk and G. Vriend, J. Mol. Graphics Modell., 2006, 25, 481–486 CrossRef CAS.
- P.-O. Syrén, F. Le Joubioux, Y. Ben Henda, T. Maugard, K. Hult and M. Graber, ChemCatChem, 2013, 5, 1842–1853 CrossRef.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. LaPointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07519d |
| ‡ Current affiliation: School of Engineering Sciences in Chemistry, Biotechnology and Health, Science for Life Laboratory, Department of Fibre and Polymer Technology, and Department of Protein Science, KTH Royal Institute of Technology, 17165 Solna, Sweden |
| This journal is © The Royal Society of Chemistry 2019 |