
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of methane to C2 and C3 hydrocarbons over TiO2/ZSM-5 core–shell particles in an electric field†
Qiao Han a,
Atsuhiro Tanakaa,
Masayuki Matsumotoa,
Akira Endob,
Yoshihiro Kubotaa and
Satoshi Inagaki*ac
a,
Atsuhiro Tanakaa,
Masayuki Matsumotoa,
Akira Endob,
Yoshihiro Kubotaa and
Satoshi Inagaki*ac
aDivision of Materials Science and Chemical Engineering, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: inagaki-satoshi-zr@ynu.ac.jp; Tel: +81-45-339-3691
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, 305-8565, Japan
cPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 29th October 2019
Abstract
Catalytic conversion of methane (CH4) to light olefins is motivated by increasing recoverable reserves of methane resources, abundantly available in natural gas, shale gas, and gas hydrates. The development of effective processes for conversion of CH4 to light olefins is still a great challenge. The interface of ZSM-5 zeolite and TiO2 nanoparticles is successfully constructed in their core–shell particles via mechanochemical treatment with high shear stress. The oxidative coupling of methane at a low temperature under application of an electric field may be induced by the O2 activation via electrons running through the surface of TiO2 located at the interface of TiO2 and zeolite particles. Moreover, C3H6 was also produced by the ethylene to propylene (ETP) reaction catalyzed by Brønsted acid sites in the ZSM-5 zeolite within core–shell particles.
1. Introduction
Proven reserves of natural gas have significantly increased in the last decade mainly due to the exploitation of unconventional natural gas sources such as shale gas, coal-bed methane and gas hydrates.1 Methane (CH4), the principal component of natural gas, has been proposed as an alternative feedstock to petroleum for the production of high value-added chemicals as well as an energy resource. Methane can be conventionally converted through syngas (a mixture of CO and H2) as an intermediate2–5 to high value-added chemicals such as methanol, light olefins, and aromatics; however, this indirect route consumes a large quantity of external energy to produce syngas.5 The development of energy-saving processes for the direct conversion of methane to high value-added chemicals thus remains a significant challenge.Oxidative coupling of methane (OCM) is an efficient technology that directly converts CH4 to C2H6 and C2H4. Since Keller and Bhasin proposed this process in the 1980s,6 Hinsen and Baerns,7 Ito and Lunsford,8 and many other groups have reported various OCM catalysts.9–13 A database of high-performance OCM catalysts was reviewed by Zavyalova and co-workers.14 For most of the catalysts listed in this review, a high reaction temperature over 700 °C is necessary to realize OCM; however, this can promote the over-oxidization of reaction intermediates to CO and CO2, so that the yield of desirable products, C2H6 and C2H4, is limited.14
The formation of methyl radicals from CH4 has a high activation energy due to the high H–CH3 bond dissociation energy (435 kJ mol−1).15 On the other hand, a low reaction temperature is favorable for the selective production of desirable products (C2H6 and C2H4), due to the suppression of deeper catalytic oxidation and non-catalytic gas-phase oxidation of reaction intermediates. To overcome these issues, non-equilibrium plasma has been proposed as a promising technology to realize high electron temperatures under low gas-phase temperatures.16 Non-oxidative coupling of CH4 in non-equilibrium plasma reaction systems such as corona discharge16,17 and spark discharge18,19 have been widely investigated. Liu et al.16,17 utilized corona discharge with zeolite catalysts to produce higher hydrocarbons; C2 hydrocarbons (mostly C2H2), and trace C3+ hydrocarbons were obtained with an input power of 9 W (input voltage, 7 kV) in this reaction system. Kado et al.18 reported the non-catalytic direct conversion of CH4 to C2H2 using corona discharge. This process can also produce a large amount of C2H2 (49% yield) with a supplied power of approximately 50 W. However, a great deal of carbon deposition occurred on the electrodes during reaction. Spark discharge was also applied for producing C2H2 from CH4.19 High selectivity toward C2H2 (>85%) and lesser amounts of deposited carbon were achieved. Furthermore, the energy efficiency with spark discharge was much higher (12.1 kW h (kg-C2H2)−1) than that in dielectric barrier discharge (DBD) or corona discharge.19 Liu et al.20–22 also reported the direct conversion of CH4 and CO2 to higher hydrocarbons via DBD with zeolite catalysts. The products in this reaction system included syngas (CO + H2), light hydrocarbons (C2 and C3), liquid hydrocarbons (C4+), plasma-polymerized film, and carbon oxides (CO and CO2).21 The selectivity toward CO and C4+ was greater than 80%; however, C2H4 and C3H6 yields were extremely low (1.2% and 2.1%, respectively) with an applied power of 500 W.22
As an alternative, OCM can be achieved by application of a direct current (DC) to the semiconductive Sr-doped La2O3 catalyst without excessive electric discharge.23 Sekine and co-workers recently reported a Ce–W–O catalyst derived from CeO2 modified with Ce2(WO4)3 polyoxometalate, which exhibited high OCM activity due to a synergetic effect between the Ce2(WO4)3 structure and the electric field to produce the reactive oxygen species for the selective oxidation of CH4.24–26 The CePO4 nanorods with uniform surface Ce sites could work as a durable catalyst and showed the highest C2 yield of 18% in an electric field without the need for external heating.27 More recently, McEwen and co-workers have reviewed some of the theoretical methods that have been used to elucidate the influence of external electric fields on catalytic reactions, as well as the application of such methods to selective methane activation.28
It is well-known that oxygen vacancies existing in the surface and bulk of TiO2 (ref. 29–31) affect the catalytic activity in both the photoreactions and non-photoreactions.32,33 The 18O isotopic exchange reaction on metal oxide catalysts is a convincing method for revealing the existence of the activated oxygen species on the surface of catalyst to progress the OCM reaction. Mirodatos and co-workers34 have investigated the elementary steps dealing with the oxygen and methane activation in the OCM over lanthanum oxide catalyst using isotope transient experiments using 18O2. They suggest that the fast interaction between gaseous oxygen and lattice oxygen atoms, involving the formation of active oxygen species possible to dissociate C–H bonds, could be considered for most of the performing systems at higher reaction temperature. More recently, Ogo and co-workers have conducted the 18O2 isotope experiments and suggest that mobile oxygen over La1−xCaxAlO3−δ perovskite catalysts in an electric field promotes low-temperature OCM reaction.35
Zeolites are widely applied as a component of catalysts for the direct conversion of CH4 to CH3OH and for the methane dehydroaromatization (MDA) reaction due to their ion exchange ability, unique pore structure, and acid-catalytic properties. Cu-based zeolites36–38 and iron-containing zeolites38–40 have recently been studied for their potential in the selective oxidation of CH4 to CH3OH under oxidative conditions at low temperature. Under non-oxidative conditions, Zn,41 Mo,41–44 Mn,45 and Fe45 dispersed on zeolites have exhibited excellent catalytic performance for the MDA reaction.
In the present study, we tried to promote that the O2 activation via electrons running through the surface of TiO2 located at the interface of TiO2 and zeolite particles induces the OCM reaction at a lower temperature under application of an electric field. Mechanochemical treatment using a powder composer with high shear stress46 was employed to prepare this characteristic catalyst with a core–shell structure, where microsized zeolite cores and nanosized TiO2 shells form particles. ZSM-5 zeolite (MFI topology) with intersectional 10–10-ring channel system was selected to form the core of the particles because it has been most widely industrialized as a solid-acid catalyst for the selective production of light olefins.47–51 Moreover, the ZSM-5 catalyst has exhibited high yield in the ethylene to propylene (ETP) reaction.52,53 Therefore, the production of C2 hydrocarbons as well as the selective production of C3H6 is expected using this type of catalyst with a core–shell structure because of C2H4 produced via OCM in an electric field can be catalytically converted to C3H6 by the ETP reaction on the Brønsted acid sites of ZSM-5 within the core–shell particles.
2. Experimental
2.1. Catalyst preparation
TiO2 nanoparticles used in this study were commercially available from Aldrich. ZSM-5 zeolite (MFI topology, Si/Al = 16, JGC Catalysts and Chemicals Ltd.) was used in this study. Silicalite-1, which is a pure-silica version of MFI-type zeolite, was hydrothermally synthesized using tetrapropylammonium bromide as a structure-directing agent (see ESI† for the detailed synthetic procedure).The zeolite–TiO2 core–shell particles were mechanochemically prepared by the application of high shear stress using a high-performance powder processing machine (Nobilta NOB-mini, Hosokawa Micron Corporation). We have already confirmed that the crystallinity and microporosity of the single ZSM-5 zeolite were retained during this kind of mechanochemical treatment.54 The mechanochemical treatment was typically performed as follows: after a portion of TiO2 nanoparticles (1.2 g) was added to ZSM-5 (6.0 g), mechanochemical treatment was performed at 1000 rpm for 5 min, and then at 9000 rpm for 10 min. Another portion of TiO2 nanoparticles (1.2 g) was then added to the treated mixture, which was mechanochemically treated using the same procedure. These procedures were repeated three more times until the weight of added TiO2 was equal to that of ZSM-5. The time-courses of rotational speed and the resulting output power are shown in Fig. S1.† The obtained sample is designated as TiO2(mc)/ZSM-5. For comparison, the core–shell particles of silicalite-1 (MFI topology) and TiO2 were also prepared according to the same procedure, and the prepared samples is designated as TiO2(mc)/silicalite-1. A physical mixture of TiO2 and ZSM-5 with the same weights was prepared and is designated as TiO2(pm)/ZSM-5. The prepared core–shell particles, the physical mixture, and the simple TiO2 or ZSM-5 were calcined at 800 °C for 12 h in a muffle furnace (denoted 800 in the end of sample name; i.e., ZSM-5_800).
2.2. Characterization of catalysts
The prepared samples were measured using power X-ray diffraction (XRD, Ultima-IV, Rigaku) with Cu Kα radiation at 40 kV and 20 mA to confirm the crystallinity and phase purity. The shape and particle size of the samples were observed using field emission scanning electron microscope (FE-SEM, JSM-7001F, JEOL). Cross-sectional observation of the core–shell particles and their EDS line-scanning and mapping were performed using another FE-SEM (SU9000, Hitachi High-Technologies) equipped with energy dispersive X-ray spectroscopy (EDS; Genesis, EDAX), which was measured at an accelerating voltage of 5.0 kV. Nitrogen adsorption and desorption isotherms were collected at −196 °C with an Autosorb-iQ analyzer (Quantachrome Instruments). Prior to measurement, each sample was heated in vacuo (less than 10 Pa) at 400 °C for 12 h. The specific surface area (SBET) and micropore volume (Vmicro) were estimated using the Brunauer–Emmett–Teller (BET) and t-plot methods, respectively. The chemical composition (Si/Al molar ratio) of the zeolites was determined using inductively coupled plasma-atomic emission spectroscopy (ICP-AES; ICPE-9000, Shimadzu). The number of acid sites was measured by ammonia temperature-programmed desorption (NH3-TPD) measurement on a BELCAT-B (MicrotracBEL Corp.) with a thermal conductivity detector (TCD). The catalyst was preheated at 600 °C prior to the measurement under He flow. The TPD data were collected at a ramping rate of 10 °C min−1. The number of acid sites was determined from the area of h-peak55–57 in their profiles. Coke deposition on the used catalyst was evaluated using thermogravimetric analysis (TGA, TG-8120, Rigaku) at a ramping rate of 10 °C min−1 from room temperature to 800 °C under air flow (30 cm3 (SATP) min−1).2.3. Activity test on the ethylene-to-propylene (ETP) reaction
The catalyst was pelletized to achieve a size of 355–500 μm. The catalyst pellets were placed in a quartz tube (9.0 mm i.d.) as a fixed-bed reactor. Prior to the reaction, the catalyst was treated at 550 °C for 1 h under air flow (30 cm3 (SATP) min−1). After the pretreatment, the gas flow was changed from air to He (30 cm3 (SATP) min−1) followed by cooling to 400 °C or the other specified reaction temperature. The ETP reaction was performed at three different temperatures: 400, 450, and 500 °C. Ethylene (Wzeolite/Fethylene = 16.4 g-cat. h mol−1) was introduced into the top of the catalyst-bed with He as a carrier gas (30 cm3 (SATP) min−1). The C2H4 and products were analyzed using an online gas-chromatograph with flame ionization detector (GC-FID; GC-2014, Shimadzu) equipped with a DB-5 capillary column (60 m length; 0.53 mm i.d.; 5.0 μm thick) and an offline GC-FID (GC-2014, Shimadzu) with a KCl/Al2O3 capillary column (50 m length; 0.53 mm i.d.; 10 μm thick). The conversion of C2H4, the yield and distribution of products, and the material balance were calculated on the carbon-basis of the input amount of C2H4.2.4. Activity test on OCM reaction in an electric field
The catalytic performance of each prepared catalyst for the OCM reaction was tested with a fixed-bed continuous-flow reactor, of which a schematic image is presented in Fig. S2.† The catalyst was pelletized without any binder, roughly crushed and then sieved to obtain catalyst pellets with 355–500 μm in size. The catalyst pellets (100 mg) were charged into a quartz tube (4.0 mm i.d.) as a continuous-flow reactor. When the reaction was conducted with application of a voltage in the catalyst bed, two stainless-steel electrodes (2.0 mm i.d.) were separately set in the reactor without contact with each other; one electrode contacted with the upper side of the catalyst bed, and the other contacted the lower side. The voltage in the catalyst-bed was applied under constant-current conditions using a DC power supply (HAR-30N40, Matsusada Precision Inc.). The supplied current was controlled from 2.0 to 9.0 mA to make the electric field stable. Time courses of the input current and the resulting voltage were recorded using an oscilloscope (TDS 3052B, Tektronix). Prior to the reaction, the catalyst bed was heated at 300 °C using a furnace for 30 min under Ar flow (60 cm3 (SATP) min−1). After cooling the furnace to 150 °C, the reactants (CH4 and O2) and carrier gas (Ar) were fed into the reactor (CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:Ar = 25:15:60 cm3 (SATP) min−1, W/Fmethane = 1.6 g-cat. h mol−1). The temperature at the lower side of the catalyst-bed was monitored using a K-type sheathed thermocouple. The products after passing a cold trap were analyzed using a gas chromatograph with a thermal conductivity detector (GC-TCD; GC-8A, Shimadzu) and two GC-FIDs (GC-2014, Shimadzu). The GC-TCD was equipped with a molecular sieve 5A packed column (2.0 m length; 3.0 mm i.d.). One GC-FID was equipped with a Porapak N packed column (4.0 m length; 3.0 mm i.d.) and a methanizer (Ru/Al2O3 catalyst), while the other had a KCl/Al2O3 capillary column (50 m length; 0.53 mm i.d.; 10 μm thick).
:O2:Ar = 25:15:60 cm3 (SATP) min−1, W/Fmethane = 1.6 g-cat. h mol−1). The temperature at the lower side of the catalyst-bed was monitored using a K-type sheathed thermocouple. The products after passing a cold trap were analyzed using a gas chromatograph with a thermal conductivity detector (GC-TCD; GC-8A, Shimadzu) and two GC-FIDs (GC-2014, Shimadzu). The GC-TCD was equipped with a molecular sieve 5A packed column (2.0 m length; 3.0 mm i.d.). One GC-FID was equipped with a Porapak N packed column (4.0 m length; 3.0 mm i.d.) and a methanizer (Ru/Al2O3 catalyst), while the other had a KCl/Al2O3 capillary column (50 m length; 0.53 mm i.d.; 10 μm thick).
The conversion of CH4 and O2, product yield, and product selectivity were determined by the following eqn (1)–(4):
| CH4 conversion (%) = {(sum of C-atom moles of each product)/(C-atom moles of input CH4)} × 100 | (1) |
| O2 conversion (%) = {(moles of O2 consumed)}/(moles of input O2)} × 100 | (2) |
| Product yield (C%) = {(C-atom moles of each product)/(C-atom moles of input CH4)} | (3) |
| Product selectivity (C%) = {(product yield)/(CH4 conversion)} × 100 | (4) |
Since reliable material balance is always achieved in the range from low to high CH4 conversion which is based on the appearance of carbon-containing products (hydrocarbons and CO/CO2), the CH4 conversion is estimated by the eqn (1).
When the reaction was conducted without application of a voltage in the catalyst bed, the catalyst was heated at prescribed temperatures (from 150 to 900 °C) using a furnace and the same reaction conditions described above.
The 16O2/18O2 isotopic oxygen exchange experiment at 150 °C in an electric field was conducted. The details of experimental procedures were described in ESI.†
3. Result and discussion
3.1. Catalyst characterization
Three kinds of composed catalysts, TiO2(mc)/ZSM-5, TiO2(mc)/silicalite-1, and TiO2(pm)/ZSM-5, were prepared in this study. Fig. 1 and S3† show XRD patterns of the samples before and after thermal treatment at 800 °C. The XRD patterns of as-received and calcined ZSM-5 showed only the MFI phase without any impurities. The as-received TiO2 consisted of two crystalline polymorphs, anatase (P42/mmm) and rutile (I41/amd), whereas calcined TiO2 was only the rutile phase. This is a good consistency with a well-known phase transformation from anatase to rutile between 600 and 700 °C under atmospheric pressure.58–60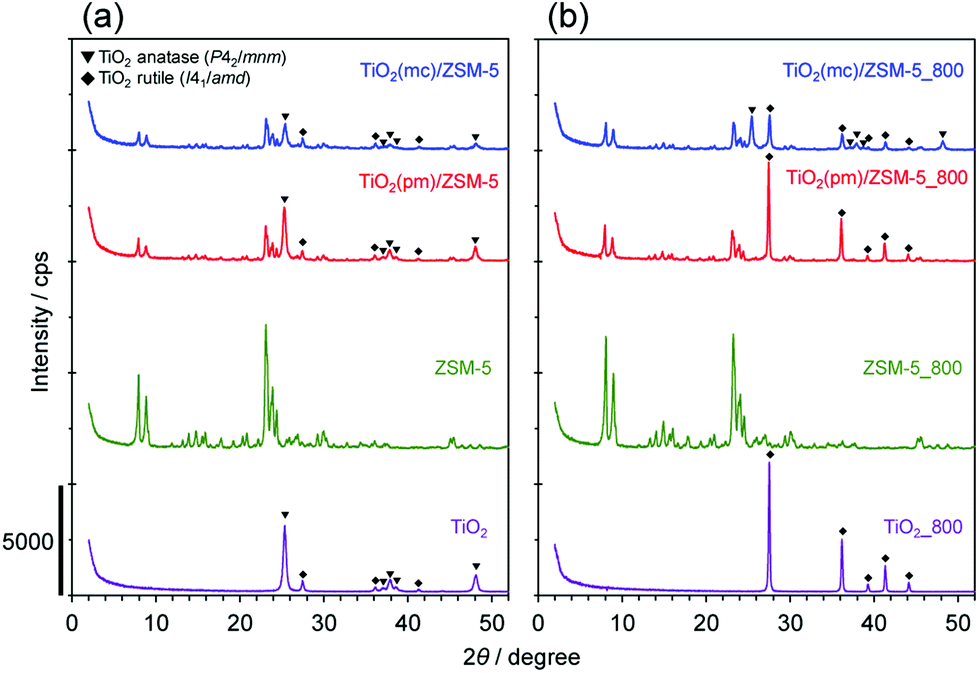 | ||
| Fig. 1 Powder XRD patterns of (a) pristine and (b) thermally-treated samples at 800 °C. | ||
In the physical mixture of TiO2 and ZSM-5, the peak intensities corresponding to an MFI structure were reasonably decreased due to the weight ratio of TiO2 to ZSM-5. In contrast, mechanochemical treatment led to a decrease in the peak intensities of both TiO2 polymorphs, although those of MFI were almost unchanged. This indicates that high shear stress during mechanochemical treatment may cause partial amorphization of TiO2 nanoparticles.54 With regard to the phase transformation of TiO2 during thermal treatment at 800 °C, TiO2 in TiO2(mc)/ZSM-5_800 was still composed of both anatase and rutile, while that in TiO2(pm)/ZSM-5_800 was only rutile. There are numerous reports describing that SiO2 or several metal oxides act as an inhibitor to the phase transformation into rutile;61–64 in particular, SiO2 species on the surface of TiO2 anatase induce a delay in the nucleation of TiO2 rutile.60,61 It is thus supposed that strong adhesion of TiO2 nanoparticles on the surface of ZSM-5 particles occurs during mechanochemical treatment.
Fig. 2 shows FE-SEM images of TiO2(pm)/ZSM-5 and TiO2(mc)/ZSM-5 before and after thermal treatment at 800 °C. Typical FE-SEM images of TiO2 nanoparticles and the parent ZSM-5 zeolite particles are shown in Fig. S4.† The particle size of parent ZSM-5 was 1.5–2.0 μm (Fig. S4b†). There were no significant differences in the morphologies of parent ZSM-5 and ZSM-5_800 (Fig. S4b and d†). On the other hand, the particle size of TiO2 was significantly increased from 25 to 100 nm by thermal treatment at 800 °C (Fig. S4a and c†). This is consistent with the sharpening of the XRD peaks that correspond to the TiO2 rutile polymorph.
 | ||
| Fig. 2 FE-SEM images of (a) TiO2(pm)/ZSM-5 and (d) TiO2(mc)/ZSM-5. (b) Low and (c) high magnification views of TiO2(pm)/ZSM-5_800. (e) Low and (f) high magnification views of TiO2(mc)/ZSM-5_800. | ||
It was evident that TiO2(pm)/ZSM-5_800 consists of ZSM-5 zeolite particles and aggregates of TiO2 nanoparticles without good contact with each other (Fig. 2b). The surface of the ZSM-5 particles in TiO2(pm)/ZSM-5_800 was still smooth (Fig. 2c), and similar to those of ZSM-5_800 (Fig. S4d†). In contrast, the surface of TiO2(mc)/ZSM-5_800 particles (Fig. 2e) was quite different from those of the parent ZSM-5 (Fig. S4b†); the former became rough (Fig. 2f) and the corners and edges of each composed particle became rounded (Fig. 2e), which indicates that the parent ZSM-5 particles were fully covered with a thin layer of TiO2 nanoparticles to give a core–shell structure. The same morphology was also observed in TiO2(mc)/silicalite-1_800 (Fig. S5c and d†).
To further verify the distribution of TiO2 within the composed particles, EDS line-scanning and EDS-mapping of core–shell particle (TiO2(mc)/ZSM-5) cross-section were performed. For line-scanning (Fig. 3b), the outer layer of the particle, which is the brightest region in the secondary electron image (Fig. 3a), is mainly composed of Ti atoms within TiO2, while the core, the central part of the slender particle, consists of Si and Al atoms, which corresponds to ZSM-5 aluminosilicates. This indicates that the prepared particles have a core–shell structure of ZSM-5 covered with a thin TiO2 layer. The EDS-mapping (Fig. 3c and d) revealed the core–shell structure more clearly.
 | ||
| Fig. 3 SEM, EDS line-scanning and EDS-mapping images of the cross-section of TiO2(mc)/ZSM-5_800. (a) Secondary electron image, (b) EDS line-scanning of Si, Al and Ti, and mapping images of (c) Si and (d) Ti. | ||
Table 1 lists the BET surface areas and micropore volumes of samples estimated from N2 adsorption–desorption isotherms (see also Fig. S6†). After the thermal treatment, the BET surface area and micropore volume slightly decreased, probably due to dealumination from the MFI framework. This consideration is supported by the lack of damage in the microporous structure of silicalite-1, because there were no changes in BET surface area and micropore volume during thermal treatment. All the composite samples of 50 wt% TiO2 and 50 wt% ZSM-5 exhibited reasonably smaller BET surface areas and micropore volumes; i.e., the BET surface area (197 m2 (g-bulk)−1) of TiO2(mc)/ZSM-5 was almost half of that (399 m2 (g-bulk)−1) of the single ZSM-5. During both mechanochemical treatment and subsequent thermal treatment, is revealed from retaining the micropore volumes corresponding to ZSM-5 zeolites within the core–shell particles (see Table 1) were unchanged.
| Sample | Specific surface areaa [m2 (g-bulk)−1] | Micropore volumeb,c [cm3 (g-zeolite)−1] |
|---|---|---|
| a Specific surface area of catalysts were calculated using the Brunauer–Emmett–Teller (BET) equation based on N2 adsorption isotherms. Data in the relative pressure range of 3–9 × 10−3 were employed for the surface area evaluation.b Micropore volumes of the catalysts were calculated by the t-plot method based on N2 adsorption isotherms. Data in the relative pressure range of 0.75–0.95 were employed for the t-plot analysis.c The ideal content of zeolite within the mixtures of TiO2 and zeolite was 50 wt%. | ||
| ZSM-5 | 399 | 0.17 |
| ZSM-5_800 | 346 | 0.14 |
| Silicalite-1 | 419 | 0.18 |
| Silicalite-1_800 | 425 | 0.18 |
| TiO2(pm)/ZSM-5 | 207 | 0.16 |
| TiO2(pm)/ZSM-5_800 | 189 | 0.14 |
| TiO2(mc)/ZSM-5 | 197 | 0.15 |
| TiO2(mc)/ZSM-5_800 | 182 | 0.13 |
| TiO2(mc)/silicalite-1 | 197 | 0.16 |
| TiO2(mc)/silicalite-1_800 | 198 | 0.16 |
It was concluded that the core–shell particles of TiO2(mc)/ZSM-5 were successfully prepared by mechanochemical treatment with high shear stress, without damage to the crystallinity.54 In these core–shell particles, small molecules (i.e., N2) can diffuse through the voids within the thin TiO2 layer into the micropores of ZSM-5.
The acidic nature of TiO2(mc)/ZSM-5_800 was evaluated by NH3-TPD measurements (see Fig. S7†). Based on the h-peak area in the NH3-TPD profile, the amount of acid sites of TiO2(mc)/ZSM-5_800 (0.24 mmol (g-zeolite)−1) was almost the same as that of ZSM-5_800 (0.25 mmol (g-zeolite)−1). Moreover, the peak-top temperature of each h-peak was almost the same. These results reveal that the acidic strength and amount of ZSM-5 zeolite within the core–shell particles was unchanged during mechanochemical treatment.
3.2. Catalytic performance on ETP reaction
The parent ZSM-5, TiO2(mc)/ZSM-5_800, and TiO2(mc)/silicalite-1_800 were evaluated as catalysts for the ETP reaction at various reaction temperatures. The product yields and distributions in the ETP reaction are shown in Fig. S8† and listed in Table S1.† As shown in Fig. S8a,† the parent ZSM-5 showed higher C2H4 conversion (85%) at 400 °C, while C2H4 conversion decreased with an increase in the reaction temperature due to the formation of a large amount of coke during the reaction. TiO2(mc)/ZSM-5_800 showed half the conversion (Fig. S8†) of the parent ZSM-5, although the contact time (Wzeolite/Fethylene, where W is the zeolite weight in the catalysts and F is the flow rate of ethylene) was set to be the same. Possible reasons for the lower conversion are lowering of C2H4 diffusion through the thin TiO2 layer to ZSM-5 and deactivation of the external acid sites on ZSM-5 due to partial amorphization of the surface of ZSM-5 (ref. 54) or covering by TiO2 fragments.TiO2(mc)/silicalite-1_800 showed no C2H4 conversion (Fig. S8c†), because there are no Brønsted acid sites, (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Al–O(H)–Si), in silicalite-1.
Al–O(H)–Si), in silicalite-1.
3.3. OCM reaction over TiO2(mc)/ZSM-5 catalyst in an electric field
The TiO2/ZSM-5 composite and the related samples were exposed to the CH4/O2 gas flow in an electric field to evaluate the catalytic properties for the OCM reaction. During the reaction, a certain amount of H2O was detected. This strongly supports that the OCM reaction successfully occurred in this catalytic system. In addition, the production of H2 was also observed. It may be caused by the partial oxidation of CH4 to give H2 and CO; however, the selectivity to H2 from CH4 was still low, which was less than 30%. The OCM reaction is thus pointed out as the main reaction in this catalytic system.Table 2 summarizes the OCM activities in an electric field over four catalysts: TiO2_800, TiO2(pm)/ZSM-5_800, TiO2(mc)/ZSM-5_800, and TiO2(mc)/silicalite-1_800. The furnace temperature was set at 150 °C; however, during OCM and the over-oxidation reaction to give CO or CO2, which are the exothermic reactions, the temperature at the bottom of the catalyst bed was autogenously increased. Although TiO2_800 catalyst made electric field stable during the input current changed from 8.0 to 4.0 mA (Fig. S9a†), the maximum of CH4 conversion was as low as 1.7%. The selectivity to CO and CO2 reached ca. 94.8% and the yields of C2H6 and C2H4 were very low (<0.1%). TiO2(pm)/ZSM-5_800 caused excessive electric discharge in the catalyst bed, even at an input current of 9.0 mA (Fig. S9b†), because ZSM-5 aluminosilicates are typical insulators with extremely lower electric conductivity; therefore, the catalytic results were not evaluated in this study. TiO2(mc)/ZSM-5_800 and TiO2(mc)/silicalite-1_800 maintained a stable voltage while a current of 9.0 mA was applied (Fig. S9c and d†), which indicates that the thin TiO2 layer within the core–shell particles enables electrons to conduct between the two electrodes set in the catalyst bed. The TiO2(mc)/ZSM-5_800 catalyst exhibited high CH4 conversion (18.4%) when the input current was 8.0 mA, and a relatively high selectivity toward C2H6 and C2H4 (8.5 and 15.7%, respectively) with lower selectivity toward CO and CO2 (64.2 and 3.4%, respectively) was achieved. The CH4 conversion over TiO2(mc)/ZSM-5 (1.5–2.0 μm) was higher than that over TiO2(mc)/silicalite-1, which exhibits a low external surface area corresponding to large particle size (ca. 15 μm). It is supposed that the active sites for the O2 activation and the subsequent CH4 activation are located on the surface of TiO2 nanoparticles, facing to zeolite particles. Therefore, the CH4 conversion is strongly affected by the external surface area of zeolite particles. Deactivation of the catalyst was not observed during the OCM reaction in electric field. In the TGA of the used catalyst, there was no significant weight loss in the range from 300 to 700 °C due to the deposited coke (Fig. S10†). This indicates that no coke formation occurs during the OCM reaction even in an electric field. In addition, the crystallinities and morphology of the used catalyst were almost remained unchanged (data not shown).
| Run | Catalyst | Input current (mA) | Temp.ca (°C) | Conversion of CH4 (%) | Selectivity (C%) | Yield (C%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | CO2 | C2H6 | C2H4 | C2H2 | C3H6 | C2b | C2H4 | C3H6 | |||||
| a Catalyst bed temperature measured using a thermocouple.b C2 yield means the sum of C2H6, C2H4, and C2H2 yields.c The intense discharge in the catalyst bed occurred intermittently. The conversion of CH4, the selectivity toward products, and the yields of C2 and C3H6 over (A) TiO2_800, (B) TiO2(pm)/ZSM-5_800, (C) TiO2(mc)/ZSM-5_800, and (D) TiO2(mc)/silicalite-1_800 in the electric field. Reaction conditions: catalyst, 100 mg; preset furnace temperature, 150 °C; feed gas, CH4:O2:Ar = 25:15:60 cm3 (SATP) min−1. Pretreatment conditions: furnace temperature, 300 °C; period, 30 min; Ar flow rate, 60 cm3 (SATP) min−1. |
|||||||||||||
| A-1 | TiO2_800 | 8.0 | 307 | 1.7 | 86.0 | 8.8 | 4.3 | 0.9 | 0.0 | 0.0 | 0.1 | 0.02 | 0.00 |
| A-2 | 6.0 | 293 | 1.5 | 88.0 | 6.9 | 4.4 | 0.7 | 0.0 | 0.0 | 0.1 | 0.01 | 0.00 | |
| A-3 | 4.0 | 274 | 0.6 | 88.4 | 6.6 | 5.0 | 0.9 | 0.0 | 0.0 | 0.0 | 0.00 | 0.00 | |
|
|||||||||||||
| B-1 | TiO2(pm)/ZSM-5_800 | Operation impossiblec | — | — | — | — | — | — | — | — | — | — | |
|
|||||||||||||
| C-1 | TiO2(mc)/ZSM-5_800 | 8.0 | 432 | 18.4 | 64.2 | 3.4 | 8.5 | 15.7 | 6.5 | 1.7 | 5.6 | 2.89 | 0.32 |
| C-2 | 6.0 | 416 | 11.9 | 61.5 | 2.8 | 12.3 | 17.4 | 3.5 | 2.6 | 3.9 | 2.06 | 0.31 | |
| C-3 | 4.0 | 350 | 2.9 | 64.5 | 3.2 | 14.5 | 8.3 | 9.5 | 0.0 | 0.9 | 0.24 | 0.00 | |
|
|||||||||||||
| D-1 | TiO2(mc)/silicalite-1_800 | 8.0 | 435 | 8.3 | 56.2 | 5.7 | 10.9 | 16.4 | 10.1 | 0.7 | 3.1 | 1.36 | 0.06 |
| D-2 | 6.0 | 418 | 7.9 | 55.2 | 5.8 | 15.1 | 17.1 | 6.0 | 0.7 | 3.0 | 1.35 | 0.05 | |
| D-3 | 4.0 | 387 | 1.1 | 65.6 | 4.4 | 15.9 | 7.3 | 3.8 | 3.1 | 0.3 | 0.08 | 0.03 | |
We tried to conduct the 18O2 isotope exchange experiment on TiO2_800 and TiO2(mc)/ZSM-5_800 catalyst in an electric field (see Fig. S11†). A certain amount of 18O16O molecules was observed over TiO2(mc)/ZSM-5_800, while TiO2 yielded only traces of 18O16O. We suppose that O2 close to an oxygen vacancy on the TiO2 surface can be activated by electrons running through the TiO2 surface, the active oxygen species subsequently dissociates a H–CH3 bond to form a methyl radical, yielding C2 hydrocarbons. This observation is just primitive; therefore, the study focusing on the active oxygen species on the TiO2 surface in an electric field will be reported elsewhere.
3.4. Electric field effect on the catalytic activity in the OCM
Table 3 and Fig. S12† show the catalytic performance of TiO2(mc)/ZSM-5_800 without application of a voltage but with heating using a furnace at 450, 600, 750, and 900 °C. At 450 °C, no catalytic reactions occurred, whereas at a high temperature of 750 °C, 2.4% of CH4 conversion was achieved, and a higher reaction temperature gave 16.9%, while high selectivity to CO and CO2 was unchanged at more than 95%, regardless of the furnace temperature. The maximum C2 yield (0.7%) with only heating was much lower than that (4.1%) under application of a voltage at a constant current of 6 mA in the catalyst bed. These results indicate that the O2 activation via electrons running through the surface of TiO2 located at the interface of TiO2 and zeolite particles under a high voltage, even at lower temperature of 150 °C, induces the OCM reaction without excessive formation of CO and CO2, compared to the temperatures higher than 750 °C.| Reaction conditions | Preset furnace temperature (°C) | Input current (mA) | Temp.ca (°C) | Conversion of CH4 (%) | Selectivity (C%) | Yield (C%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | CO2 | C2H6 | C2H4 | C2H2 | C3H6 | C2b | C3H6 | |||||
| a Catalyst bed temperature measured using a thermocouple.b C2 yield means the sum of C2H6, C2H4, and C2H2 yields. Reaction conditions: catalyst, 100 mg; feed gas, CH4 : O2 : Ar = 25 : 15 : 60 cm3 (SATP) min−1. Pretreatment conditions: furnace temperature, 300°C; period, 30min; Ar flow rate, 60 cm3 (SATP) min−1. | ||||||||||||
| Without EF | 450 | — | 463 | 0.1 | 90.3 | 9.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.00 |
| 600 | — | 612 | 0.2 | 93.0 | 7.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.00 | |
| 750 | — | 758 | 2.4 | 91.0 | 7.7 | 1.1 | 0.2 | 0.0 | 0.0 | 0.0 | 0.00 | |
| 900 | — | 901 | 16.9 | 84.9 | 11.0 | 1.5 | 2.5 | 0.0 | 0.0 | 0.7 | 0.01 | |
|
||||||||||||
| With EF | 150 | 6.0 | 417 | 11.3 | 58.4 | 2.7 | 13.6 | 15.1 | 7.8 | 2.5 | 4.1 | 0.28 |
We carefully studied the influence of the input current at the same reaction temperature (ca. 450 °C) on the OCM activity over TiO2(mc)/ZSM-5_800 catalyst in an electric field (see Table 4). When the preset temperature was fixed at 150 °C (runs 1–3 in Table 4), the catalyst-bed temperature increased as the input current increased. It is due to both exothermic reaction of OCM and joule heating within solid catalyst (detail described in Section 3.6). Hence, by tuning the preset temperature, the catalyst-bed temperature was fixed at ca. 450 °C with the different input current (runs 4–6 in Table 4). When the catalyst bed temperature was fixed, the CH4 conversion linearly increases as the input current increases. This strongly supports that the electrons through the surface of TiO2 within the composite particles induce the O2 activation to promote the OCM reaction, regardless of the reaction temperature, at least ca. 450 °C. In these reaction conditions, the selectivity to CO and CO2 was stable at ca. 70%, as described above. When the OCM reaction proceeded, C2H6 selectivity decreased while C2H4 selectivity increased; therefore, the possible reaction pathway in this catalytic system will be discussed as described below.
| Run | Input current | Preset furnace temperature | Temp.ca | Conversion of CH4 (%) | Selectivity (C%) | C2 yieldb (C%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | CO2 | C2H6 | C2H4 | C2H2 | CO + CO2 | ||||||
| a Catalyst bed temperature measured using a thermocouple.b C2 yield means the sum of C2H6, C2H4, and C2H2 yields. The selectivity of products, conversion of methane and the yield of C2 in OCM reaction over TiO2(mc)/ZSM-5_800 with an electric field. Reaction conditions: catalyst, 100 mg; feed gas, CH4:O2:Ar = 25:15:60 cm3 (SATP) min−1. Pretreatment conditions: furnace temperature, 300 °C; period, 30 min; Ar flow rate, 60 cm3 (SATP) min−1. |
|||||||||||
| 1 | 3.0 | 150 | 370 | 1.3 | 54.9 | 4.4 | 20.8 | 9.4 | 10.6 | 59 | 0.5 |
| 2 | 5.0 | 397 | 6.8 | 66.1 | 3.6 | 13.3 | 14.5 | 2.5 | 70 | 2.1 | |
| 3 | 7.0 | 415 | 10.0 | 63.3 | 2.9 | 13.2 | 16.3 | 2.5 | 66 | 3.2 | |
|
|||||||||||
| 4 | 3.0 | 374 | 454 | 0.9 | 65.3 | 5.3 | 21.2 | 8.2 | 0.0 | 71 | 0.3 |
| 5 | 5.0 | 339 | 449 | 5.1 | 71.6 | 4.2 | 7.5 | 8.4 | 8.3 | 76 | 1.2 |
| 6 | 7.0 | 290 | 448 | 10.7 | 65.2 | 3.6 | 11.9 | 15.6 | 3.7 | 69 | 3.4 |
3.5. Possible reaction pathway in electric field
The influence of the contact time (W/Fmethane) on the OCM activity over the TiO2(mc)/ZSM-5_800 catalyst under application of a constant current (7.0 mA) and lower reaction temperature (150 °C) was investigated to discuss the possible reaction pathway in this catalytic system. The dependence of OCM activity on the contact time is shown in Fig. 4 and S13,† and the overall results are listed in Table S2.† The OCM reaction was confirmed to occur as a catalytic reaction over TiO2(mc)/ZSM-5_800 in this reaction system because there is a proportional relation between W/Fmethane and each conversion of CH4 or O2, as shown in Fig. S13.† A possible reaction pathway from C1 radicals into C2 hydrocarbons (C2H6, C2H4 and C2H2) in this reaction system is much more difficult to speculate. Fig. 4 shows each yield of C2 hydrocarbons in this catalytic system. C2H2 was a minor C2 hydrocarbon product with a proportional increase dependent on the CH4 conversion. In the range of low CH4 conversion (from 4 to 8%), the yield of C2H6 was relatively higher than that of C2H4. In contrast, the yield of C2H4 jumped up beyond that of C2H6 at higher CH4 conversion (from 10 to 20%). Although it seems that the dehydrogenation of C2H6 into C2H4 occurs along with the coupling of C1 radicals into C2H6 or C2H4, the variation of each C2 yield is even more complicated to determine the reaction pathway from the results obtained here. To evaluate the mechanism of C2 hydrocarbon formations during OCM in an electric field, oxidative dehydrogenation of ethane (ODH) over TiO2(mc)/ZSM-5 catalyst was conducted in electric field at 150 °C. Reaction results are shown in Table S3.† TiO2(mc)/ZSM-5 showed high C2H6 conversion and selectivity of C2H4. The conversion of C2H6 increased with increasing the input current, but the C2H2 selectivity was still low even at the high C2H6 conversion. These results indicate the oxidative dehydrogenation of C2H6 to C2H4 easily proceeded over TiO2(mc)/ZSM-5 catalyst in an electric field. The formation of C2H2 may occur in other reaction pathways, such as micro-discharge occurred at the interface between the ZSM-5 core and the TiO2 shell of the core–shell particles, beside the dehydrogenation of C2H4. At least, we have confirmed that parent ZSM-5 zeolite does not catalyze the reaction from H2 and CO into hydrocarbons (see Table S4†) that is well known as Fischer–Tropsch reaction,65,66 regardless of reaction temperature and H2 partial pressure.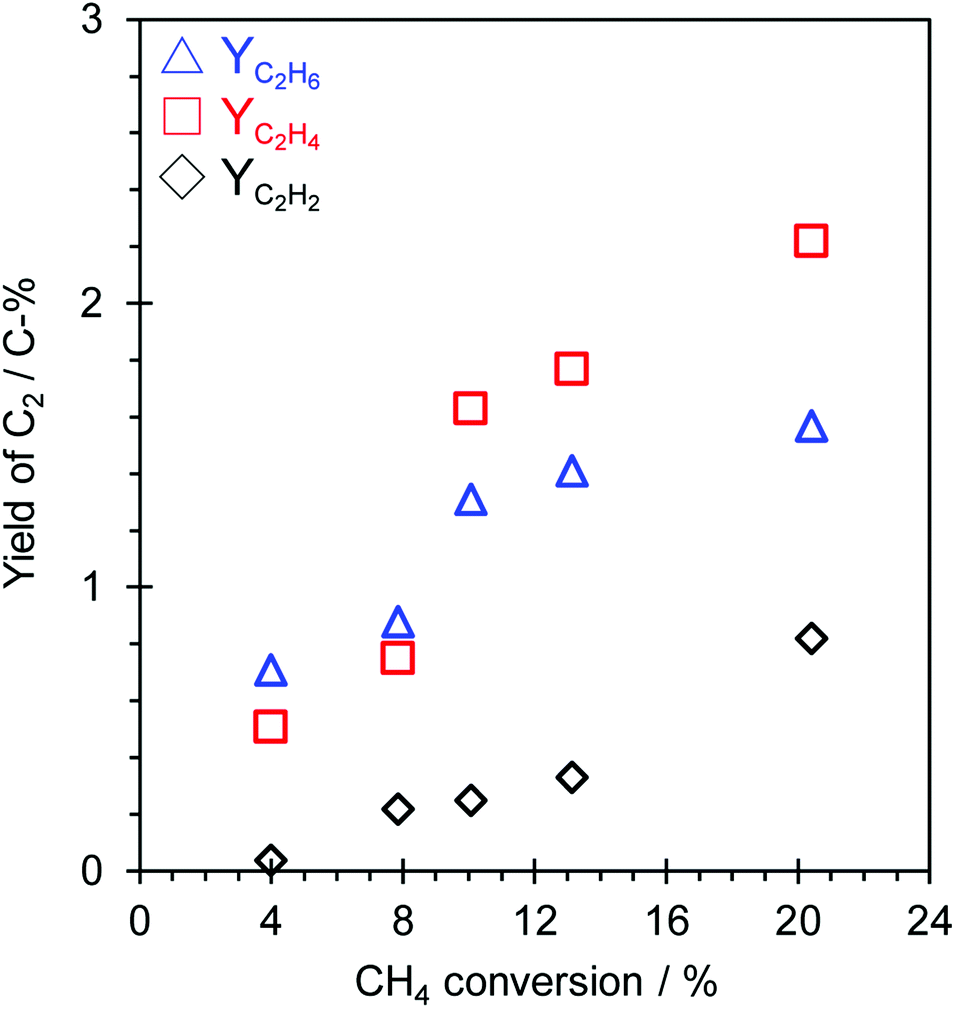 | ||
| Fig. 4 Relation between CH4 conversion and C2H6, C2H4, and C2H2 yield over TiO2(mc)/ZSM-5_800 under application of an electric field. Reaction conditions: catalyst, 100 mg; input current, 7.0 mA; feed gas, CH4:O2:Ar = 5x:3x:12x (total flow rate: 50, 75, 100, 150, 200 cm3 (SATP) min−1); preset furnace temperature, 150 °C. | ||
3.6. Influence of O2 partial pressure on the catalytic activity in an electric field
To confirm that active O2 species is necessary for dissociation of C–H bond in CH4, the influence of the O2 partial pressure on the OCM activity over the TiO2(mc)/ZSM-5_800 catalyst under application of a constant current (8.0 mA) and lower reaction temperature (150 °C) was also investigated. The results of the catalytic activity of TiO2(mc)/ZSM-5_800 are listed in Table 5. No catalytic reaction occurred in the absence of O2. In this case, the temperature at the bottom of the catalyst bed increased from 150 to 238 °C by Joule heating. On the other hand, the co-existence of CH4 and O2 in an electric field realizes the OCM reaction over this catalyst. In the low range of O2 partial pressure (3 and 5 vol%), lower CH4 conversion (<1%) was obtained, whereas the higher O2 partial pressure (10 and 15 vol%) gave higher CH4 conversion. The temperature at the bottom of the catalyst bed further increased with the increase of O2 partial pressure, due to OCM and the over-oxidation reactions to give CO or CO2, which are the exothermic reactions. For instance, when the O2 partial pressure reached 15%, the temperature at the bottom of the catalyst bed increased to 432 °C. The temperature rise due to exothermic reaction was from 238 to 432 °C, which is in good agreement with thermodynamic estimation from the conversion and selectivity.| Reactant gas (cm3 (SATP) min−1) | Temp.ca (°C) | Conversion of CH4 (%) | Selectivity (C%) | C2 yieldb (C%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | O2 | Ar | ||||||||
| CO | CO2 | C2H6 | C2H4 | C2H2 | ||||||
| a Catalyst bed temperature measured using a thermocouple.b C2 yield means the sum of C2H6, C2H4, and C2H2 yields. Reaction conditions: catalyst, 100 mg; total flow rate, 100 cm3 (SATP) min−1; input current, 8.0 mA; preset furnace temperature, 150 °C. Pretreatment conditions: furnace temperature, 300 °C; period, 30 min; Ar flow rate, 60 cm3 (SATP) min−1. | ||||||||||
| 25 | 0 | 75 | 238 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 3 | 72 | 340 | 0.6 | 84.2 | 3.8 | 10.0 | 3.8 | 0.0 | 0.1 | |
| 5 | 70 | 350 | 1.0 | 81.4 | 3.1 | 10.9 | 4.6 | 0.0 | 0.2 | |
| 10 | 65 | 405 | 8.1 | 60.7 | 2.5 | 9.9 | 12.7 | 11.8 | 2.8 | |
| 15 | 60 | 432 | 18.4 | 64.2 | 3.4 | 8.5 | 15.7 | 6.5 | 5.6 | |
3.7. Formation of C3H6 over TiO2/ZSM-5 catalyst
It should be noted that TiO2(mc)/ZSM-5_800 catalyst produced a meaningful yield of C3H6 when the input current was in the range of 4.0 to 8.0 mA, probably due to the ETP reaction catalyzed by the Brønsted acid sites in ZSM-5 within the core–shell particles. In Table 2, the yield of C3H6 increased from 0 to 0.32% with an increase in the yield of C2H4 from 0.24% to 2.89%. Such the limited C3H6 yield may be due to the subsequent oxidation by O2 into CO or CO2. When the ETP reaction with O2 over TiO2(mc)/ZSM-5 catalyst in an electric field was also performed (Fig. S14†), a relatively large amount of C3H6 as well as butenes and pentenes were detected in the lower input O2 concentration.The temperature at the bottom of the catalyst bed during the reaction reached 432 °C (when the input current was 8.0 mA), because the OCM reaction is exothermic. At such temperature, the ZSM-5 zeolite can catalyze the ETP reaction, as mentioned above (see also Fig. S8†). The TiO2(mc)/silicalite-1_800 catalyst only yielded a trace amount of C3H6, in spite of almost the same CH4 conversion over TiO2(mc)/ZSM-5_800 at 387–435 °C. These results also support that the production of C3H6 in this reaction system is caused by the catalytic ETP reaction on ZSM-5 at the desirable temperature.
Fig. 5 shows a possible reaction pathway. O2 could be activated via electrons running through the surface of TiO2 located at the interface of TiO2 and zeolite particles. Such oxygen species could convert CH4 into CH3 radical to generate C2H6 and C2H4. Moreover, C3H6 was also produced by the ETP reaction catalyzed by Brønsted acid sites in the ZSM-5 zeolite.
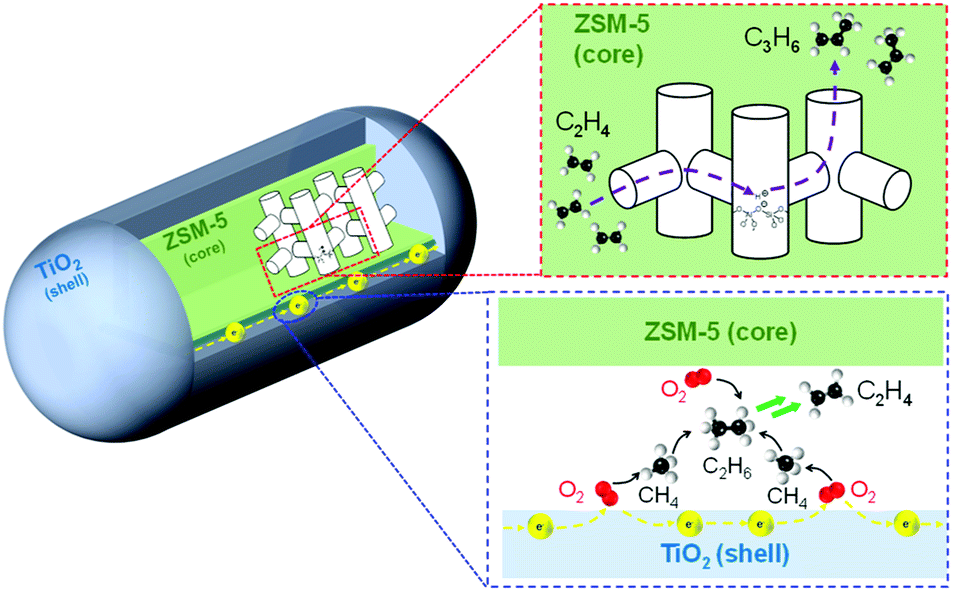 | ||
| Fig. 5 Illustration of the possible reaction scheme of OCM and ETP reactions over TiO2(mc)/ZSM-5 composite catalyst in an electric field. | ||
3.8. Temperature dependence of catalytic activity in an electric field
The catalytic performance of TiO2(mc)/ZSM-5_800 with application of a constant current of 7.0 mA with furnace heating from 150 to 700 °C was evaluated, and the results are shown in Fig. 6 and Table S5.† External heating of the catalyst bed affects the activation of CH4; the conversion of CH4 increased from 13.1 to 22.5% with an increase in the furnace temperature from 150 to 400 °C. In contrast, the CH4 conversion decreased significantly to 16.7% when the temperature was increased up to 500 °C and further decreased slightly from 500 to 700 °C. The O2 activation and the subsequent CH4 conversion may be related to the temperature dependence of the TiO2 semiconductivity,67 in that enhancement of electron conductivity in both TiO2 bulk and interface between TiO2 nanoparticles may be suppressed by that on the TiO2 surface facing to ZSM-5 particles at high temperatures (to be discussed in detail elsewhere). The product selectivity toward C2H6 and C2H4 with this catalytic system continuously decreased with an increase of the furnace temperature from 150 to 700 °C due to over-oxidation to CO and CO2. Therefore, at higher reaction temperatures, regardless of whether a current is applied in the catalyst bed, only those TiO2 nanoparticles that contact each other within the core–shell particles of TiO2(mc)/ZSM-5 may catalyze the complete oxidation of CH4 into CO and CO2, which is supported by the high selectivity toward CO and CO2 (96.6%) at 900 °C with 23.6% of CH4 conversion over the single TiO2 catalyst.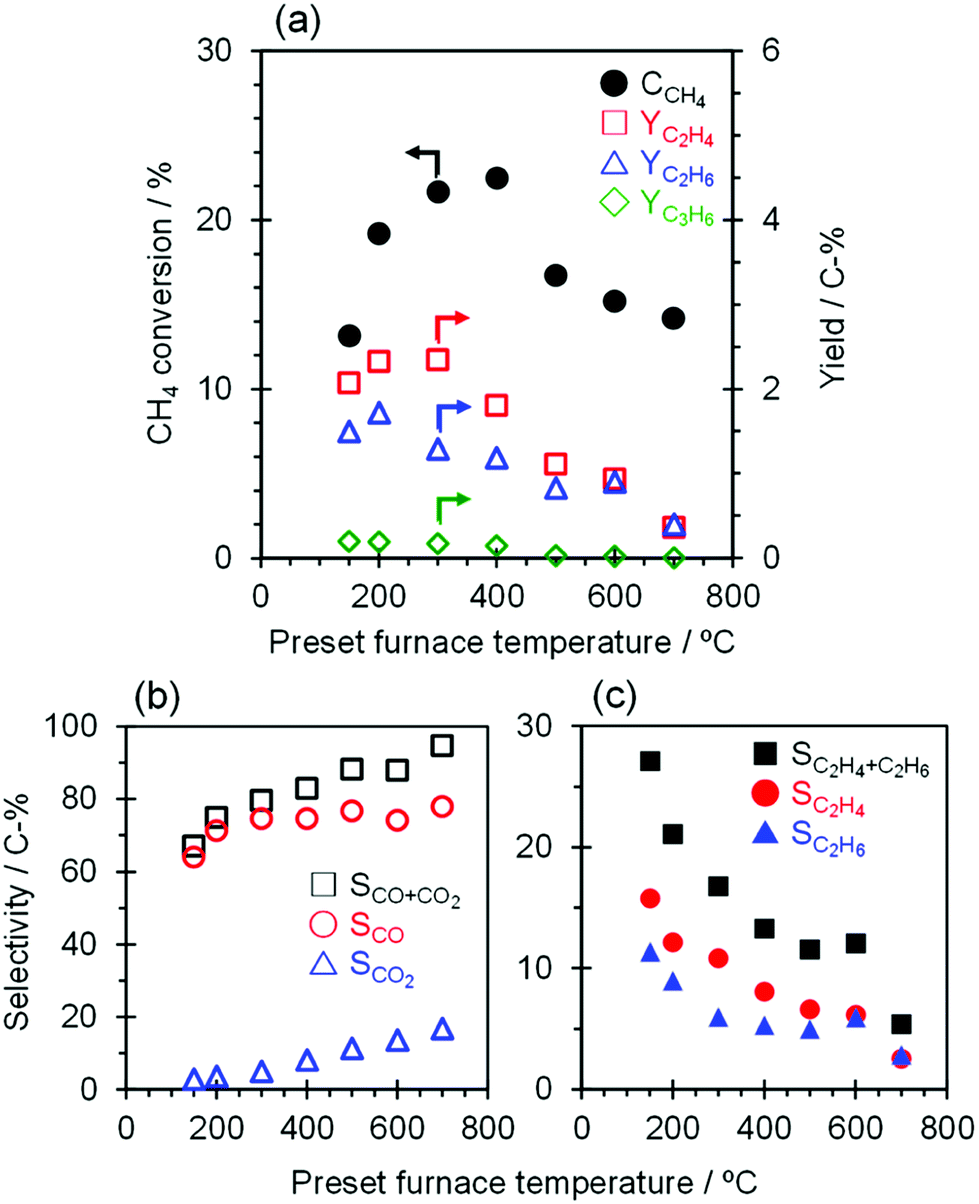 | ||
| Fig. 6 Temperature dependence over TiO2(mc)/ZSM-5_800 under application of an electric field. Relation between input furnace temperature and (a) CH4 conversion and C2 yield, (b) selectivity of CO and CO2, (c) selectivity of C2H4 and C2H6 over TiO2(mc)/ZSM-5_800. Trace amounts of propylene selectivity are not shown in graph (c). Reaction conditions: catalyst, 100 mg; input current, 7.0 mA; feed gas, CH4:O2:Ar = 25:15:60 cm3 (SATP) min−1; preset furnace temperature, 150–700 °C. | ||
4. Conclusions
Core–shell ZSM-5 zeolite particles covered with a thin shell layer of TiO2 nanoparticles were successfully prepared via mechanochemical treatment under high shear stress using a high-performance powder processing machine for application as a unique catalytic system for OCM and subsequent C3H6 production. A constant current (4.0–8.0 mA) was applied to the catalyst bed of TiO2(mc)/ZSM-5_800 at a low reaction temperature of 150 °C, which resulted in high methane conversion (18.4%) with selectivity toward C2H6 (8.5%) and C2H4 (15.7%), whereas the selectivity toward CO and CO2 was relatively low at 64.2% and 3.4%, respectively. In this catalytic system, oxidative coupling of methane at a low temperature under an electric field may be induced by O2 activation with electrons moving through the surface of TiO2 at the interface between TiO2 and zeolite particles under a high voltage. Furthermore, a good amount of C3H6 was produced during this reaction due to the ETP reaction catalyzed by Brønsted acid sites in ZSM-5 within the core–shell particles. Further improvement of the procedure for preparing a catalyst with a core–shell structure of semiconductive materials and zeolites, and a detailed investigation of the reaction pathways, will be attempted in future work, to progress this energy-sustainable catalytic system with assistance from electron semiconduction at lower reaction temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JST PRESTO program (JPMJPR16S2). We thank Prof. Yasushi Sekine, Associate Prof. Shuhei Ogo, Ms Ayaka Sato, and Ms Yuna Takeno (Waseda University) for fruitful discussion and technical assistance for 16O2/18O2 isotope oxygen exchange experiment.References
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS.
- M. L. Vanessa, A. D. Robert, K. Libor, A. L. Jair, L. K. David and R. P. Daniel, Catal. Sci. Technol., 2012, 2, 2116–2127 RSC.
- P. Tang, Q. J. Zhu, Z. X. Wu and D. Ma, Methane activation: the past and future, Energy Environ. Sci., 2014, 7(8), 2580–2591 RSC.
- T. Peng, Y. X. Wei, M. Ye and Z. M. Liu, ACS Catal., 2015, 5(3), 1922–1938 CrossRef.
- P. Schwach, X. L. Pan and X. H. Bao, Chem. Rev., 2017, 117(13), 8497–8520 CrossRef CAS.
- G. E. Keller and M. M. Bhasin, J. Catal., 1982, 73(1), 9–19 CrossRef CAS.
- W. Hinsen and M. Baerns, Chem.-Ztg., 1983, 107(7–8), 223–226 CAS.
- T. Ito and J. H. Lunsford, Nature, 1985, 314, 721–722 CrossRef CAS.
- K. Otsuka, K. Jinno and A. Morikawa, J. Catal., 1986, 100(2), 353–359 CrossRef CAS.
- K. Aika, T. Moriyama, N. Takasaki and E. Iwamatsu, J. Chem. Soc., Chem. Commun., 1986, 1210–1211 RSC.
- K. Murata, T. Hayakawa and K. Fujita, Chem. Commun., 1997, 221–222 RSC.
- A. Palermo, J. P. Hologado-Vasquez, A. F. Lee, M. S. Tikhov and R. M. Lambert, J. Catal., 1998, 177(2), 259–266 CrossRef CAS.
- K. Takanabe and E. Iglesia, Angew. Chem., Int. Ed., 2008, 47(40), 7689–7693 CrossRef CAS.
- U. Zavyalova, M. Holena, R. Schlogl and M. Baerns, ChemCatChem, 2011, 3(12), 1935–1947 CrossRef CAS.
- J. A. Kerr, Chem. Rev., 1966, 66(5), 465–500 CrossRef CAS.
- C. J. Liu, R. Mallinson and L. Lobban, J. Catal., 1998, 179(1), 326–334 CrossRef CAS.
- C. J. Liu, R. Mallinson and L. Lobban, Appl. Catal., A, 1999, 178(1), 17–27 CrossRef CAS.
- S. Kado, Y. Sekine and K. Fujimoto, Chem. Commun., 1999, 2485–2486 RSC.
- S. Kado, Y. Sekine, T. Nozaki and K. Okazaki, Catal. Today, 2004, 89(1–2), 47–55 CrossRef CAS.
- B. Eliasson, C. J. Liu and U. Kogelshatz, Ind. Eng. Chem. Res., 2000, 39(5), 1221–1227 CrossRef CAS.
- C. J. Liu, B. Z. Xue, B. Eliasson, F. He, Y. Li and G. H. Xu, Plasma Chem. Plasma Process., 2001, 21(3), 301–310 CrossRef CAS.
- T. Jiang, Y. Li, C. J. Liu, G. H. Xu, B. Eliasson and B. Z. Xue, Catal. Today, 2002, 72(3–4), 229–235 CrossRef CAS.
- K. Tanaka, Y. Sekine, K. Oshima and Y. Tanaka, Chem. Lett., 2012, 41(4), 351–353 CrossRef CAS.
- K. Sugiura, S. Ogo, K. Iwasaki, T. Yabe and Y. Sekine, Sci. Rep., 2016, 6, 25154 CrossRef CAS.
- S. Ogo, K. Iwasaki, K. Sugiura, A. Sato, T. Yabe and Y. Sekine, Catal. Today, 2018, 299, 80–85 CrossRef CAS.
- S. Ogo and Y. Sekine, Chem. Rec., 2017, 17, 1–14 CrossRef.
- A. Sato, S. Ogo, K. Kamata, Y. Takeno, T. Yabe, T. Yamamoto, S. Matsumura, M. Hara and Y. Sekine, Chem. Commun., 2019, 55, 4019–4022 RSC.
- F. Che, J. T. Gray, S. Ha, N. Kruse, S. L. Scott and J.-S. McEwen, ACS Catal., 2018, 8(6), 5153–5174 CrossRef CAS.
- G. Pacchioni, ChemPhysChem, 2003, 4, 1041–1047 CrossRef CAS.
- R. Schaub, E. Wahlström, A. Rønnau, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Science, 2003, 299, 377–379 CrossRef CAS PubMed.
- X. Y. Pan, M. Q. Yang, X. Z. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- M. K. Nowotny, L. R. Sheppard, T. Bak and J. Nowotny, J. Phys. Chem. C, 2008, 112, 5275–5300 CrossRef CAS.
- Y. Y. Yu and X. Q. Gong, ACS Catal., 2015, 5, 2042–2050 CrossRef CAS.
- S. Lacombe, H. Zanthoff and C. Mirodatos, J. Catal., 1995, 155, 106–116 CrossRef CAS.
- A. Sato, S. Ogo, Y. Takeno, J. G. Seo and Y. Sekine, ACS Omega, 2019, 4, 10438–10443 CrossRef CAS.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2010, 132(42), 14736–14738 CrossRef CAS.
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Chem. Rev., 2013, 257(2), 483–494 CAS.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. Yu and S. I. Chan, Chem. Rev., 2017, 117(13), 8574–8621 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, L. V. Pirutko, S. I. Abornev and G. I. Panov, J. Phys. Chem. C, 2011, 115(5), 2155–2161 CrossRef CAS.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–322 CrossRef CAS.
- L. S. Wang, L. X. Tao, M. S. Xie, G. F. Xu, J. S. Huang and Y. D. Xu, Catal. Lett., 1993, 21(1–2), 35–41 CrossRef CAS.
- Y. Song, Y. B. Xu, Y. Suzuki, H. Nakagome, X. X. Ma and Z. G. Zhang, J. Catal., 2015, 330, 261–272 CrossRef CAS.
- Y. Song, Q. Zhang, Y. B. Xu, Y. Zhang, K. Matsuoka and Z. G. Zhang, Appl. Catal., A, 2017, 530, 12–20 CrossRef CAS.
- J. Gao, Y. T. Zheng, J. M. Jehng, Y. D. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175(2), 347–351 CrossRef CAS.
- M. Hosokawa, K. Nogi, M. Naito and T. Yokoyama, Nanoparticle Technology Handbook, Elsevier, Oxford, U.K., 2007 Search PubMed.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 6th edn, 2007, see also: http://www.iza-structure.org/databases/ Search PubMed.
- R. J. Argauer and G. R. Landolt, US Pat., US3702886A, November 14, 1972.
- T. F. Narbeshuber, G. Vinek and J. A. Lercher, J. Catal., 1995, 157(2), 388–395 CrossRef CAS.
- H. Mochizuki, T. Yokoi, H. Imai, R. Watanabe, S. Namba, J. N. Kondo and T. Tatsumi, Microporous Mesoporous Mater., 2011, 145(1–3), 165–171 CrossRef CAS.
- S. Inagaki, S. Shinoda, Y. Kaneko, K. Takechi, R. Komatsu, Y. Tsuboi, H. Yamazaki, J. N. Kondo and Y. Kubota, ACS Catal., 2013, 3(1), 74–78 CrossRef CAS.
- B. M. Lin, Q. H. Zhang and Y. Wang, Ind. Eng. Chem. Res., 2009, 48(24), 10788–10795 CrossRef CAS.
- S. Follmann and S. Ernst, New J. Chem., 2016, 40, 4414–4419 RSC.
- S. Inagaki, K. Sato, S. Hayashi, J. Tatami, Y. Kubota and T. Wakihara, ACS Appl. Mater. Interfaces, 2015, 7(8), 4488–4493 CrossRef CAS.
- M. Niwa and N. Katada, Catal. Surv. Jpn., 1997, 1, 215–226 CrossRef CAS.
- M. Niwa and N. Katada, Chem. Rec., 2013, 13, 432–455 CrossRef CAS.
- N. Katada, Mol. Catal., 2018, 458, 116–126 CrossRef CAS.
- A. W. Czanderna, C. N. R. Rao and J. M. Honig, Trans. Faraday Soc., 1958, 54, 1069–1073 RSC.
- S. R. Yoganarasimhan and C. N. R. Rao, Trans. Faraday Soc., 1962, 58, 1579–1589 RSC.
- K. N. P. Lumar, K. Keizer, A. J. Burggraaf, T. Okubo, H. Nagamoto and S. Morooka, Nature, 1992, 358, 48–51 CrossRef.
- C. Byrne, R. Fagan, S. Hinder, D. E. McCormack and S. C. Pillai, RSC Adv., 2016, 6, 95232–95238 RSC.
- C. Anderson and A. J. Bard, J. Phys. Chem. B, 1997, 101(14), 2611–2616 CrossRef CAS.
- K. Okada, N. Yamamoto, Y. Kameshima and A. Yasumoti, J. Am. Ceram. Soc., 2001, 84(7), 1591–1596 CrossRef CAS.
- S. C. Pillai, P. Periyat, R. George, D. E. McCormack, M. K. Seery, H. Hayden, J. Colreavy, D. Corr and S. J. Hinder, J. Phys. Chem. C, 2007, 111(4), 1605–1611 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107(5), 1692–1744 CrossRef CAS PubMed.
- D. Fraenkel and B. C. Gates, J. Am. Chem. Soc., 1980, 102(7), 2478–2480 CrossRef CAS.
- D. C. Cronemeyer, Phys. Rev., 1952, 87(5), 876–886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06927e |
| This journal is © The Royal Society of Chemistry 2019 |