
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of a novel WO3/Ag2MoO4 particles-on-plate staggered type II heterojunction with improved visible-light photocatalytic activity in removing environmental pollutants†
Huanli Wang *ab,
Yafei Wanga,
Ailing Xua,
Qipeng Yanga,
Fujun Taoc,
Mingliang Mad,
Zhiwen Song*a and
Xiaobo Chen*b
*ab,
Yafei Wanga,
Ailing Xua,
Qipeng Yanga,
Fujun Taoc,
Mingliang Mad,
Zhiwen Song*a and
Xiaobo Chen*b
aSchool of Environmental and Municipal Engineering, Qingdao University of Technology, China. E-mail: whl_r@126.com; songzhiwen@qut.edu.cn
bUniversity of Missouri, USA. E-mail: chenxiaobo@umkc.edu
cShanghai Maritime University, China
dSchool of Civil Engnieering, Qingdao University of Technology, China
First published on 29th October 2019
Abstract
A novel WO3/Ag2MoO4 heterojunction has been synthesized through a facile precipitation method with Ag2MoO4 particles firmly deposited on the surface of WO3 nanoplates, forming “particles-on-plate” type II heterojunction structures. This heterojunction exhibited improved photocatalytic activities for the degradation of rhodamine B (RhB), 4 chlorophenol (4-CP) and tetracycline hydrochloride (TC) under visible-light irradiation compared to pure Ag2MoO4 and WO3. In addition, the heterojunction with 10 wt% Ag2MoO4 displays the best photocatalytic performance, which was about 2 times better than that of pure WO3 or Ag2MoO4. The TC photodegradation rate reaches up to 91% within 90 min visible light irradiation. Furthermore, the photocatalytic efficiency of the Ag2MoO4/WO3 heterojunction is 1.3 times higher than that of the mixture of the two individual photocatalysts. This remarkable enhanced photocatalytic performance results from the staggered bandgap between Ag2MoO4 and WO3, which can suppress the recombination of electron–hole pairs efficiently. Moreover, based on the radical trapping experiment, the superoxide radical anions (·OH) and photogenerated holes (h+) are the crucial active oxidizing species.
1. Introduction
Photocatalysis has aroused significant attention recently due to its efficiency, environmental friendliness and convenience for solving the energy and environment crisis. The key to photocatalysis is to construct an efficient and stable photocatalyst.1,2 To date, various kinds of photocatalysts have been reported such as TiO2,3,4 Ag3PO4,5–7 Bi2WO6,8–11 MoS2,12,13 C3N4,14,15 BiVO4 (ref. 16) and WO3.17–19 Among them, WO3 is regarded as a promising photocatalyst due to its strong adsorption in visible light, photocorrosion resistance, and stable physicochemical properties. Nevertheless, its practical application is largely restricted due to its comparatively narrow band gap (2.4 to 2.8 eV) and lower light energy conversion efficiency.20 The commonly used strategies to improve the photocatalytic performance include ion doping,21 heterojunctions,22–25 and morphological modulation.10,26 Among those strategies, the heterojunction is proven to be most widely used because of its effectiveness and convenience for improving the photocatalytic activity, as highlighted in our recent review.22 Furthermore, we have also constructed some nanojunction systems such as Bi2O3–Bi2WO6 (ref. 11) and Ag3PO4–GQDs composites;7 these heterojunction photocatalysts exhibited higher photocatalytic activity than their bare components. Therefore, constructing WO3 based heterojunction photocatalyst should be an effective way to improve the photocatalytic performance of WO3. Up to date, various WO3-based heterojunctions have been reported, such as WO3/BiVO4,18 WO3–C3N4,27,28 WO3/CdS,29 WO3/Ag,30 WO3/RGO31 and CNTs–WO3.32 Compared with bare WO3, the construction of these heterojunctions enhanced the photocatalytic performance in different degrees. However, it is still highly desirable to design novel WO3-based heterojunctions to further improve the photocatalytic performance of pure WO3. Over the last decade, silver molybdate (Ag2MoO4) has attracted extensive attentions in photoluminescence,33 biological sterilization34 and electrocatalytic oxygen reduction reaction,35 and it has been shown as an excellent semiconductor to modify other semiconductors to improve their photocatalytic performance,36–38 such as Ag/AgCl/Ag2MoO4,39 Ag@Ag2MoO4–AgBr composite,40,41 Ag2MoO4–Ag3PO4.42 As the band edges of Ag2MoO4 match well with those of WO3, it can be expected that the WO3/Ag2MoO4 heterojunction would be a photocatalyst with a staggered bandgap and therefore exhibit a high performance. Also, it is desirable to construct such heterojunction, as it has never been done so far.In this study, Ag2MoO4–WO3 heterojunctions with various Ag2MoO4 mass percentages have been developed with a facile precipitation method. Their photocatalytic activities have been investigated with the photodegradation of rhodamine B (RhB), 4 chlorophenol (4-CP) and tetracycline hydrochloride (TC) under visible-light irradiation. And the mechanism of the improved photocatalytic performance of the heterojunction photocatalysts has also been proposed based on the characterization and analysis results.
2. Experimental
2.1. Chemicals
Degussa TiO2 with particle size 25 nm (P25), TC and 4-CP were purchased from Shanghai Macklin Biochemical Co., Ltd. Na2MoO4·2H2O, Na2WO4·2H2O, AgNO3, HCl, ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd.2.2. Preparation of tungsten oxide (WO3) nanoplates
WO3 nanoplates were synthesized according to ref. 31. In a typical synthesis, Na2WO4·2H2O (200 mg) were dissolved in deionized water (10 mL). Subsequently, 5 mL HCl (35%) was slowly added to the above solution with continuous stirring. The suspension was then transferred to a 45 mL Teflon-lined stainless-steel autoclave and heated at 140 °C for 8 h. The sample was collected by centrifugation with ethanol and deionized water for several times and dried by vacuum freeze drier. Finally, the samples obtained were annealed at 300 °C for 30 min.2.3. Preparation of silver molybdate (Ag2MoO4) and Ag2MoO4–WO3
The surface decoration of WO3 with Ag2MoO4 nanoparticles (NPs) was accomplished by a simple in situ deposition method. Briefly, a certain amount of WO3 (0.375, 0.75, 1.125 and 1.5 g) was dispersed into 40 ml deionized water and sonicated for 30 min to give a WO3 aqueous suspension. Then, AgNO3 (5 mM, 40 mL) was dissolved in the above suspension under constant stirring. After that, Na2MoO4·2H2O (2.5 mM, 40 mL) was added drop by drop into the above solution. After continually stirring for 4 h in the dark, the obtained yellow precipitate of Ag2MoO4–WO3 was separated by centrifugation with deionized water and ethanol for several times and dried at 30 °C for 6 h in vacuum drier. For comparison, pure Ag2MoO4 was also prepared with this method without WO3.2.4. Characterization
Scanning Electron Microscopy (SEM) and Transmittance Electron Microscopy (TEM) were performed on field emission scanning electron microscope (Hitachi S-4800) and field emission electron microscope (FEI TECNAI G2 F20 200 kV) to reveal the morphology of the catalysts. Powder X-ray diffractometer (XRD) patterns were recorded on Bruker D8 ADVANCE to analysis the phases of the catalysts. UV-vis diffuse reflectance spectra (DRS) were recorded on a UV-vis spectrophotometer (Shimadzu Corporation, UV-3600), and the specific surface area (BET) measurements were performed on a N2 adsorption specific surface & pore size analysis instrument (3H-2000PS1, Beishide Instrument S&T). Photoluminescence (PL) spectra were measured on the F-7000, Hitachi spectrophotometer at room temperature, and the wavelength of excitation light was 325 nm.2.5. Photocatalytic test
The photocatalytic performances were evaluated with the photodegradation of RhB, 4-CP and TC under visible-light irradiation (300 W Xe lamp with a cut-off filter, λ > 420 nm). The photo of the reaction setting up was shown in Fig. S1.† In each typical experiment, 100 mg catalyst was added into 100 mL RhB, 4-CP or TC solution (10 mg L−1). To establish the adsorption–desorption equilibrium between the catalysts and the pollutants, the suspension was magnetically stirred in dark for 60 min before the light irradiation. During irradiation, 2.0 mL suspension was collected at the regular intervals and centrifuged to remove the catalysts in RhB solution or filtered through a Navigator syringe filter of 0.22 μm in TC and 4-CP solution. The concentrations of RhB were determined by UV-6000PC spectrophotometer at the characteristic absorption peak of RhB (553 nm), and the concentrations of 4-CP and TC were recorded with the high-performance liquid chromatography (HPLC, Agilent 1100 series) equipped with a UV detector at 280 nm. The flow rate of mobile phase (TC: 8% methanol, 72% oxalic acid and 20% acetonitrile; 4-CP: 80% methanol and 20% water) was set at 0.5 mL min−1.To determine the free radical in the progress of photodegradation, 6 mM AgNO3, 1 mM disodium ethylene diamine tetraacetate (Na2-EDTA), 1 mM p-benzoquinone (BQ) or 1 mM isopropanol (IPA) were employed as sacrifice agents in the RhB (100 mL, 10 mg L−1) photocatalytic process.
2.6. Photoelectrochemical measurements
The photoelectrochemical measurements were conducted on a CHI660E electrochemical workstation using a standard three-electrode system in which a saturated calomel electrode (SCE), a platinum sheet (2 cm2) and a FTO glass (2 cm2) coated with Ag2MoO4, WO3 and 10% Ag–W film were used as reference, counter and working electrodes, respectively. While 0.5 M Na2SO4 solution was used as the electrolyte. In addition, the potential of the EIS measurements was the open-circuit potential. The photocurrent responses of the samples were determined under inconsecutive irradiation by 300 W Xe lamp. During the experiment, the test potential was 0.5 V vs. SCE.3. Results and discussion
3.1. Preparation of Ag2MoO4/WO3 heterojunctions
A series of Ag2MoO4/WO3 heterojunctions were prepared by a facile precipitation–deposition method, as shown in Fig. 1. Firstly, WO3 plates were synthesized by the reaction of Na2WO4·2H2O and HCl during the hydrothermal treatment. Subsequently, Ag2MoO4 NPs were in situ anchored onto the plate-like WO3 substrate by the reaction of Na2MoO4 and AgNO3 at room temperature. The resulting catalysts with different Ag2MoO4 weight percentage of 5, 10, 15 and 20% are abbreviated as 5, 10, 15, and 20% Ag–W respectively. | ||
| Fig. 1 Schematic illustration for the preparation of Ag2MoO4/WO3 heterojunction. | ||
3.2. Characterization of photocatalysts
The morphology of WO3 and Ag–W heterojunctions have also been characterized by SEM. As shown in Fig. 2A and B, the WO3 displays a uniform nanoplate morphology, with an average length of 150–200 nm, which can also be further demonstrated by the TEM image (Fig. 2C) and the HRTEM (Fig. 2D) which showed the facet (200) is the dominating facet. After the surface decoration with Ag2MoO4, the obtained Ag–W heterojunction retain the nanoplate morphology (Fig. 3A). Notably, TEM images of Ag–W heterojunctions shows that there are plenty of small Ag2MoO4 nanoparticles distributed on WO3 nanoplates (Fig. 3B and C). In addition, the high-resolution TEM (HRTEM) image (Fig. 3D) clearly displays two sets of lattice fringes with inter-plane spacings of 0.364 nm and 0.233 nm, which can be well assigned to the values for the (200) crystal plane of monoclinic WO3 (JCPDS card no. 43-1035) and the (400) crystal plane of cubic Ag2MoO4 (JCPDS card no. 08-0473), respectively. Furthermore, the EDS pattern (Fig. 4A) reveals that Ag–W heterojunctions were composed of W, Ag, O and Mo elements. The molar ratio of W to Mo is 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.1, suggesting that the weight ratio of Ag2MoO4 to WO3 species is equal to 0.09:1. The above results confirm the formation of Ag–W heterojunctions.
:2.1, suggesting that the weight ratio of Ag2MoO4 to WO3 species is equal to 0.09:1. The above results confirm the formation of Ag–W heterojunctions.
 | ||
| Fig. 2 (A) SEM, (B) enlarged SEM of WO3 nanoplates, (C) TEM image and (D) HRTEM images of WO3 nanoplates with dominating 200 facets. | ||
 | ||
| Fig. 3 (A) SEM, (B and C) TEM, and (D) HRTEM images of 10% Ag–W heterojunction: Ag2MoO4 nanoparticles were selectively deposited on 200 facets. | ||
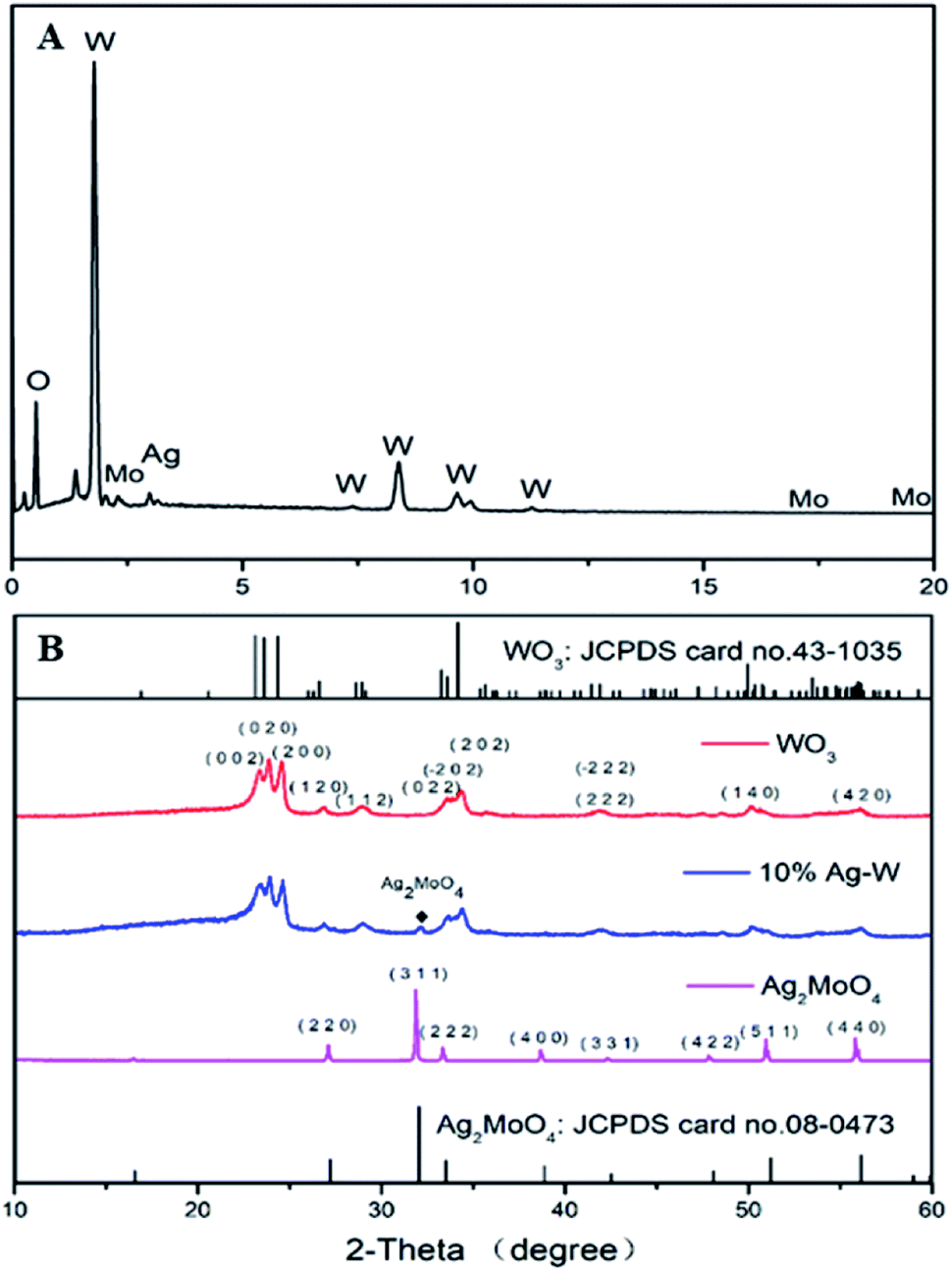 | ||
| Fig. 4 (A) EDS of 10% Ag–W heterojunction; (B) XRD patterns of as-prepared 10% Ag–W heterojunction compared with JCPDS of Ag2MoO4 and WO3. | ||
The crystal phases of all as-prepared photocatalysts were tested by the XRD to demonstrate the phase and crystallinity (Fig. 4B). The standard XRD pattern of Ag2MoO4 and WO3 have also been supplied. All the diffraction peaks of pure WO3 can be well indexed to monoclinic WO3 (JCPDS card no. 43-1035), while all diffraction peaks of pure Ag2MoO4 can be well assigned to cubic Ag2MoO4 (JCPDS card no. 08-0473). For Ag–W heterojunctions, they showed the co-existence of Ag2MoO4 and WO3. Major diffraction peaks with 2θ values of 23.1°, 23.6°, 24.4°, 26.6°, 28.9°, 34.2° and 49.9° could be perfectly indexed to the (002), (020), (200), (120), (112), (202) and (140) crystal planes of monoclinic WO3 (JCPDS card no. 43-1035), respectively. Besides, additional diffraction peaks with 2θ values of 27.1°, 31.8°, 33.3°, 38.6°, 50.9° and 55.8° could be attributed to the (220), (311), (222), (400), (511) and (440) crystal planes of cubic Ag2MoO4 (JCPDS card no. 08-0473), respectively.
The UV-vis diffuse reflectance spectra of pure Ag2MoO4, WO3 and 10% Ag–W heterojunction have been recorded in order to investigate the capacity of visible light absorption, as shown in Fig. 5A. Clearly, pure Ag2MoO4 shows a typical absorption at wavelength shorter than 400 nm, which accords previous reports.35 Pure WO3 exhibits an obviously absorption edge at ca. 500 nm, consistent with the previous reports.31 Interestingly, 10% Ag–W heterojunction displays a broad-spectrum photo absorption with an edge at ca. 530 nm, reflecting a red shift of photo absorption. This red shift of photoabsorption maybe resulting from the partial interfacial charge transfer between Ag2MoO4 and WO3 which is caused by the staggered bandgap.11 Therefore, VLD photocatalytic activity of 10% Ag–W heterojunction can be expected to be superior to that of Ag2MoO4 and WO3.
 | ||
| Fig. 5 UV-vis diffuse reflectance spectra (A) and linear transformation (hν)–(Ahν)2 curves (B) of Ag2MoO4, WO3 and 10% Ag–W heterojunction. | ||
The Eg (energy band gaps) of Ag2MoO4 and WO3 can be estimated from a linear transformation (hν)–(Ahν)2 curves, as Fig. 5B presents. Accordingly, Eg of Ag2MoO4 and WO3 are calculated to be 3.38 and 2.54 eV, severally. In addition, the positions of the valence band (VB) and conduction band (CB) of photocatalysts have also been figured out by Mulliken electronegativity:35
| ECB = X − EC − 1/2Eg | (1) |
| EVB = ECB + Eg | (2) |
The N2 isothermal adsorption desorption curves of WO3, Ag2MoO4 and 10% Ag–W heterostructures have also been recorded. As shown in Fig. 6, WO3 and 10% Ag–W heterostructure exhibit a similar BET surface area of about 12 m2 g−1. In addition, according to the desorption branches in the inset of Fig. 6, the pore size distribution presents nanopores (∼26.5 nm) in 10% Ag–W heterostructure, which is usually benefit for the improvement of the photocatalytic activity.
 | ||
| Fig. 6 N2 adsorption–desorption isotherms and the corresponding pore size distribution (inset) of WO3, Ag2MoO4 and 10% Ag–W heterojunction. | ||
3.3. Photocatalytic activity
The photocatalytic activities of Ag–W heterostructures with different weight ration of Ag2MoO4 and WO3 were evaluated by the photodegradation of dye RhB, colorless 4-CP and antibiotic TC under visible light irradiation at room temperature. For comparison, pure WO3, Ag2MoO4 and P25 have also been used as photocatalysts.Fig. 7A presents the photodegradation efficiency of RhB over different photocatalysts. Clearly, all the Ag–W heterostructures (5% Ag–W, 10% Ag–W, 15% Ag–W and 20% Ag–W) exhibited remarkable enhanced photocatalytic performance in comparison with bare WO3 and Ag2MoO4, and the RhB photodegradation efficiencies reached up to 95%, 97%, 89% and 73%, respectively within 160 min of reaction. Among these photocatalysts, 10% Ag–W heterostructures showed the most excellent photocatalytic performance, it's photocatalytic activity was more than 2 and 1.9 times than that of Ag2MoO4 and WO3 respectively. In addition, the photodegradation efficiency of TC and 4-CP over Ag–W heterostructures with different Ag2MoO4 content have also been investigated respectively, 10% Ag–W heterostructures also showed the most excellent photocatalytic performance (Fig. S2 and S3†). Moreover, the load content of Ag2MoO4 had a prominent effect on the photocatalytic performance, too low or high content of Ag2MoO4 loading could not improve the separation of charges effectively, leading to an unsatisfactory photocatalytic performance. Similar results have also been observed by previous reports.31,43,44 Furthermore, the photodegradation efficiency of 10% Ag–W (97%) was almost 3.88 times higher than that of P25.
 | ||
| Fig. 7 Degradation curves (A) and (B) rate constants (k) under the visible light irradiation (λ > 420 nm) over different photocatalysts. | ||
Moreover, the degradation efficiency were further analyzed by using the pseudofirst-order model,45 −ln(C/C0) = kt. In this formula, C0 represents the initial concentration of RhB, C represents the concentration of RhB at time t, and k represents the reaction rate constant. As shown in Fig. 7B, clearly, 10% Ag–W exhibited the highest k value (0.01991 min−1), which was extremely higher than those of Ag2MoO4 (0.00482 min−1) and WO3 (0.00489 min−1), and is more than 10 times that of TiO2 (0.00158 min−1). In addition, to further confirm the effect of the heterojunctions in Ag–W heterojunction, the degradation of RhB by using the mixture of two single components of Ag–W heterostructures with the same weights (90 mg WO3 + 10 mg Ag2MoO4) as photocatalysts have also been conducted under visible light irradiation. The photodegradation efficiency over 10% Ag–W was almost 1.43 times than that of the mechanical mixture of WO3 and Ag2MoO4 (68%), and the k over 10% Ag–W was more than 3 times than that of the mixture (0.00628 min−1). All of these results demonstrated that there was a synergic effect between the two components (WO3 and Ag2MoO4) in Ag–W heterostructures.
To preclude the dye sensitization effect during the photocatalytic degradation, colorless antibiotic TC and 4-CP have also been chosen as model pollutants. As shown in the illustrated HPLC chromatograms of TC and 4-CP solution at the regular intervals times (Fig. 8A). When TC was chosen as model pollutants, the characteristic peak of TC solution at retention time (5.8 min) decreased quickly (Fig. 8A). After 210 minutes irradiation, the degradation efficiency of TC reached up to 99%. Similar with the result of the HPLC chromatograms of 4-CP solution, the degradation efficiency of 4-CP reached up to 70% within 320 minutes (Fig. 8B), demonstrating the remarkable photodegradation of 10% Ag–W heterojunction.
 | ||
| Fig. 8 HPLC chromatograms of (A) TC (100 mL 5 mg L−1) and (B) 4-CP (100 mL 5 mg L−1) solutions over the 10% Ag–W heterojunction for different photocatalytic reaction times, inset: the photodegradation efficient column of (A) TC and (B) 4-CP under visible light irradiation (λ > 420 nm). | ||
The stability of photocatalysts is one of most significant factors in practical applications. Hence, the photocatalytic stability of the 10% Ag–W heterojunction was researched by repeatability experiments for photodegradation of RhB under visible-light irradiation (Fig. 9A). After four consecutive runs, the photodegradation efficiency of 10% Ag–W heterojunction maintains 93%, and the k decrease slightly from 0.01991 min−1 to 0.01679 min−1 (Fig. S4†). The slight decrease of its activity should result from the loss of samples in the recycle experiments. All these results demonstrate the excellent recyclability of Ag–W heterojunction.
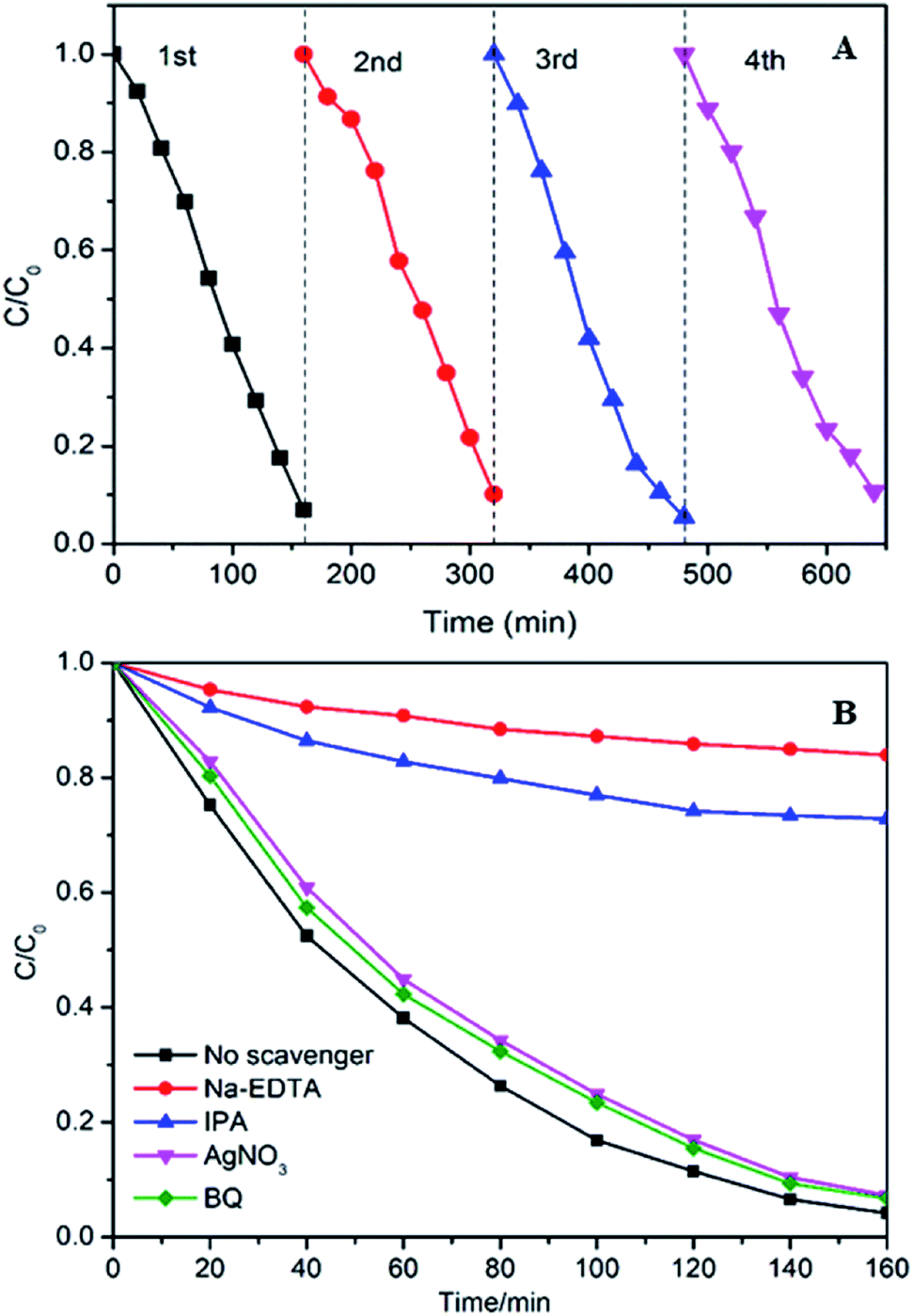 | ||
| Fig. 9 (A) Cycling runs in photocatalytic photodegradation of RhB (10 mg L−1, 100 mL) over 10% Ag–W heterojunction (100 mg) under visible light (λ > 420 nm); (B) effect of different scavengers on the degradation of RhB (10 mg L−1, 100 mL) efficiencies over 10% Ag–W heterojunction. | ||
3.4. Photocatalytic photodegradation mechanism
During the photocatalysis process, the photogenerated electrons (e−) and holes (h+) can transfer to the surface of catalyst and will generate sorts of reactive species, such as hydroxyl radicals (·OH), superoxide radicals (·O2−) and so on, which play an important role in photodegrading organic contaminant.46–48 Herein, we evaluated the dominating active species during the process of photodegradation of RhB. In the photodegradation experiments, AgNO3, disodium ethylene diamine tetraacetate (Na2-EDTA), benzoquinone (BQ) and isopropanol (IPA) which acted as the scavengers of e−, h+, ·O2−, ·OH38,49 were added into RhB suspensions before visible light irradiation respectively. Just as shown in Fig. 9B, when Na2-EDTA and IPA were added into the RhB solution, the photocatalytic efficiency decreases sharply, the k value decrease to less than 1/10 of the original one (Fig. S5†), and the RhB degradation efficiency decreases from 94.9% to 50% or 19.3% respectively after 160 min of reaction. While the photocatalytic activities are influenced slightly with the addition of AgNO3 and BQ. These results illustrate that h+ and ·OH are the primary active species which conduce to the photodegradation of RhB.To investigate the electronic interaction between Ag2MoO4 and WO3, the photocurrents of 10% Ag–W heterojunction, Ag2MoO4 and WO3 electrodes have also been measured. To the best of our knowledge, the intensity of photocurrent reflects the transference speed of photogenerated electrons and holes. Clearly, all the electrodes showed fast and uniform photocurrent responses in Fig. 10A. Under visible light irradiation, the intensity photocurrent of 10% Ag–W heterojunction was almost 6 and 2 times than that of Ag2MoO4 and WO3, respectively (Fig. 10A). The photocurrent enhancement of 10% Ag–W heterojunction manifested that Ag–W heterojunction can efficiently accelerate the separation of photogenerated electrons and holes due to the synergetic effect of Ag2MoO4 and WO3 semiconductor. In addition, EIS analysis has also been measured to further study the electron transport property of the 10% Ag–W heterojunction, Ag2MoO4 and WO3 electrodes. Usually, a smaller circular radius in EIS Nyquist plot represents a lower charge transfer resistance in the electrode.50 Obviously, the EIS Nyquist plot of 10% Ag–W heterojunction showed the smallest circular radius among the three electrodes (Fig. 10B), suggesting the lowest charge transfer resistance. To further confirm the charge transfer property, the PL emission spectrum which can be used to evaluate the separation efficiency of photogenerated electron–hole pairs was employed (Fig. 10C). Generally, a weaker intensity of the PL emission spectra represents a lower charge recombination rate.51 As shown in Fig. 10C, 10% Ag–W heterojunction display the lowest intensity under the excitation of light at λ = 325 nm, further indicating the vital role in the suppression of the recombination of photogenerated charge carriers.
 | ||
| Fig. 10 Photocurrent density (A), EIS plots (B) and PL plots (C) of WO3, Ag2MoO4 and 10% Ag–W heterojunction. | ||
Based on the above results, the possible photodegradation mechanism for the enhanced photocatalytic activity of Ag–W heterojunction are analyzed. Two main reasons contributing to the improved photocatalytic performance of Ag–W heterojunction. On one hand, the improvement of the photoabsorption of visible-light enhances the utilization of solar energy and hence product more photogenerated holes and electrons, as shown in Fig. 11. On the other hand, the staggered bandgap and intimate contact between Ag2MoO4 and WO3 greatly promote the charge separation and transfer in the Ag–W heterojunction.52–59 Under visible light irradiation, both Ag2MoO4 and WO3 can be excited to photogenerated electron–hole pairs. Since the potentials for CB and VB of Ag2MoO4 are more negative than that of Ag2MoO4, photogenerated electrons in the CB of Ag2MoO4 can be transferred to that of WO3 while holes in the VB of WO3 can be transferred to that of Ag2MoO4. Therefore, such charge transfer process can suppress the recombination of electron–hole pairs effectively, which has also been confirmed by the photocurrent measurements, EIS and PL emission spectrum. Consequently, numerous h+ accumulated on the VB of Ag2MoO4 can directly oxidize organic contaminant adsorbed on the surface of the photocatalyst or mineralized them indirectly through hydroxyl radicals (·OH) generated by the reaction of holes and H2O or OH−.
 | ||
| Fig. 11 Schematic diagrams of electron–hole pair separation and the possible photodegradation mechanism over Ag–W heterojunction under visible-light irradiation. | ||
4. Conclusions
In summary, we designed and prepared a novel the Ag–W heterojunction with different mass percentage though a facile precipitation method. These heterogeneous photocatalysts showed extremely superior photocatalytic performance than bare Ag2MoO4 and WO3 for RhB/4-CP/TC photodegradation, and the 10% Ag–W exhibits the most excellent photocatalytic activity. Moreover, the mechanism of the enhanced photocatalytic activity of Ag–W heterojunctions has been analyzed based on the energy band positions and the free radical capture experiments. The enhancement of the photocatalytic performance is mainly attributed to the stagger band gap structure, improved the separation of photogenerated electron–hole pairs. Furthermore, the Ag–W heterojunction exhibits good stability with inappreciable loss after four cycling runs. Therefore, this work may provide new insight for developing VLD photocatalyst on application of environmental renovation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Qingdao Science and Technology Program (16-5-1-32-jch, 17-3-3-77-nsh), the China Postdoctoral Science Foundation (2016M602110), the National Natural Science Foundation of China (31570541, 31170509, 21506109), the Key R&D project of Shandong province (GG201809130031) and the Primary Research and Development Plan of Shandong Province (2017GSF220001).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS.
- X. Chen and S. S. Mao, J. Nanosci. Nanotechnol., 2006, 6, 906–925 CrossRef CAS.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS.
- R. Qu, W. Zhang, N. Liu, Q. Zhang, Y. Liu, X. Li, Y. Wei and L. Feng, ACS Sustainable Chem. Eng., 2018, 6, 8019–8028 CrossRef CAS.
- Y. Wang, H. Wang, A. Xu and Z. Song, J. Mater. Sci.: Mater. Electron., 2018, 29, 16691–16701 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS.
- C. Zhang and Y. Zhu, Chem. Mater., 2005, 17, 3537–3545 CrossRef CAS.
- L. Zhang, H. Wang, Z. Chen, P. K. Wong and J. Liu, Appl. Catal., B, 2011, 106, 1–13 CAS.
- H. Wang, S. Li, L. Zhang, Z. Chen, J. Hu, R. Zou, K. Xu, G. Song, H. Zhao and J. Yang, CrystEngComm, 2013, 15, 9011–9019 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- D. Chen and J. Ye, Adv. Funct. Mater., 2008, 18, 1922–1928 CrossRef CAS.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928–1933 CrossRef CAS.
- P. M. Rao, L. Cai, C. Liu, I. S. Cho, C. H. Lee, J. M. Weisse, P. Yang and X. Zheng, Nano Lett., 2014, 14, 1099–1105 CrossRef CAS.
- S. Chen, Y. Hu, S. Meng and X. Fu, Appl. Catal., B, 2014, 150, 564–573 CrossRef.
- B. Roose, K. C. Gödel, S. Pathak, A. Sadhanala, J. P. C. Baena, B. D. Wilts, H. J. Snaith, U. Wiesner, M. Grätzel and U. Steiner, Adv. Energy Mater., 2016, 6, 1501868 CrossRef.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- S. Xu, H. Lu, L. Chen and X. J. R. A. Wang, RSC Adv., 2014, 4, 45266–45274 RSC.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef.
- T. Zhang, M. Liu, Y. Meng, B. Huang, X. Pu and X. Shao, Sep. Purif. Technol., 2018, 206, 149–157 CrossRef CAS.
- G. Wu, J. Ma, S. Li, J. Guan, B. Jiang, L. Wang, J. Li, X. Wang and L. Chen, J. Colloid. Interf. Sci., 2018, 528, 360–371 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- J. Zhang and Z. J. R. A. Ma, RSC Adv., 2017, 7, 2163–2171 RSC.
- J. Jin, J. Yu, D. Guo, C. Cui and W. Ho, Small, 2015, 11, 5262–5271 CrossRef CAS.
- W. Zhu, J. Liu, S. Yu, Y. Zhou and X. Yan, J. Hazard. Mater., 2016, 318, 407–416 CrossRef CAS.
- W. Zhu, F. Sun, R. Goei and Y. Zhou, Appl. Catal., B, 2017, 207, 93–102 CrossRef CAS.
- L. Tian, L. Ye, J. Liu and L. Zan, Catal. Commun., 2012, 17, 99–103 CrossRef CAS.
- Y.-Y. Bai, Y. Lu and J.-K. Liu, J. Hazard. Mater., 2016, 307, 26–35 CrossRef CAS.
- Z. Wang, J. Zhang, J. Lv, K. Dai and C. Liang, Appl. Surf. Sci., 2017, 396, 791–798 CrossRef CAS.
- A. Esfandiar, A. Irajizad, O. Akhavan, S. Ghasemi and M. R. Gholami, Int. J. Hydrogen Energy, 2014, 39, 8169–8179 CrossRef CAS.
- M. Pandiri, R. Velchuri, R. Gundeboina and V. Muga, J. Photochem. Photobiol., A, 2018, 360, 231–241 CrossRef CAS.
- Z. Jiao, J. Zhang, Z. Liu and Z. Ma, J. Photochem. Photobiol., A, 2019, 371, 67–75 CrossRef CAS.
- W. Cao, Y. An, L. Chen and Z. Qi, J. Alloys Compd., 2017, 701, 350–357 CrossRef CAS.
- Y. Bai, Y. Lu and J. Liu, J. Hazard. Mater., 2016, 307, 26–35 CrossRef CAS.
- Y.-Y. Bai, Y. Lu and J.-K. Liu, J. Hazard. Mater., 2016, 307, 26–35 CrossRef CAS.
- Z. Wang, J. Lv, J. Zhang, K. Dai and C. Liang, Appl. Surf. Sci., 2018, 430, 595–602 CrossRef CAS.
- W. Cao, Y. An, L. Chen and Z. Qi, J. Alloys Compd., 2017, 701, 350–357 CrossRef CAS.
- R. Dhanabal, S. Velmathi and A. C. Bose, Catal. Sci. Technol., 2016, 6, 8449–8463 RSC.
- W. Liu, J. Shen, X. Yang, Q. Liu and H. Tang, Appl. Surf. Sci., 2018, 456, 369–378 CrossRef CAS.
- X.-j. Wang, Q. Wang, F.-t. Li, W.-y. Yang, Y. Zhao, Y.-j. Hao and S.-j. Liu, Chem. Eng. J., 2013, 234, 361–371 CrossRef CAS.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef.
- L. Cai, H. Jiang and L. Wang, Appl. Surf. Sci., 2017, 420, 43–52 CrossRef CAS.
- J. Lin, Y. Liu, Y. Liu, C. Huang, W. Liu, X. Mi, D. Fan, F. Fan, H. Lu and X. Chen, ChemSusChem, 2019, 12, 961–967 CrossRef CAS.
- N. Shao, J. Wang, D. Wang and P. Corvini, Appl. Catal., B, 2017, 203, 964–978 CrossRef CAS.
- J. Yang, W. Li, J. Li, D. Sun and Q. Chen, J. Mater. Chem., 2012, 22, 17744–17752 RSC.
- Y. Hao, X. Dong, S. Zhai, X. Wang, H. Ma and X. Zhang, Chem. Commun., 2016, 52, 6525–6528 RSC.
- M. Green, Z. Liu, P. Xiang, Y. Liu, M. Zhou, X. Tan, F. Huang, L. Liu and X. Chen, Light: Sci. Appl., 2018, 7, 87 CrossRef.
- L. Guan and X. Chen, ACS Appl. Energy Mater., 2018, 1, 4313–4320 CrossRef CAS.
- R. Shen, J. Xie, P. Guo, L. Chen, X. Chen and X. Li, ACS Appl. Energy Mater., 2018, 1, 2232–2241 CrossRef CAS.
- X. Chen, F. Liu, X. Yan, Y. Yang, Q. Chen, J. Wan, L. Tian, Q. Xia and X. Chen, Chem.–Eur. J., 2015, 21, 18711–18716 CrossRef CAS.
- F. Liu, X. Chen, Q. Xia, L. Tian and X. Chen, RSC Adv., 2015, 5, 77423–77428 RSC.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Chem. Eng. J. DOI:10.1016/j.cej.2019.123034.
- Y. Zhang, L. Luo, Z. Shi, X. Shen, C. Peng, J. Liu, Z. Chen, Q. Chen and L. J. C. Zhang, ChemCatChem, 2019, 11, 2855–2863 CrossRef CAS.
- Y. Liu, X. Mi, J. Wang, M. Li, D. Fan, H. Lu and X. Chen, Inorg. Chem. Front., 2019, 6, 948–954 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07175j |
| This journal is © The Royal Society of Chemistry 2019 |